

Abstract
Tuberous sclerosis complex is a disease caused by mutations in the TSC1 or TSC2 genes, which encode a protein complex that inhibits mTOR kinase signaling by inactivating the Rheb GTPase. Activation of mTOR promotes the formation of benign tumors in various organs while the mechanisms underlying the neurological symptoms of the disease remain largely unknown. Here, we report that in mice Tsc2 haploinsufficiency causes aberrant retinogeniculate projections that suggest defects in EphA receptor-dependent axon guidance. We also show that EphA receptor activation by ephrin-A ligands in neurons leads to inhibition of ERK1/2 kinase activity and decreased inhibition of Tsc2 by ERK1/2. Thus, ephrin stimulation inactivates the mTOR pathway by enhancing Tsc2 activity. Furthermore, Tsc2 deficiency and hyperactive Rheb constitutively activate mTOR and inhibit ephrin-induced growth cone collapse. Our results demonstrate that TSC2-Rheb-mTOR signaling cooperates with the ephrin-Eph receptor system to control axon guidance in the visual system.
Tuberous sclerosis complex (TSC) is an autosomal dominant disease characterized by the presence of benign tumors called hamartomas, which can affect virtually every organ system of the body including the brain (where hamartomas are known as cortical tubers)1. Most TSC patients also develop epilepsy, and 25–50% are diagnosed with autism spectrum disorders. Although it has been proposed that the cortical tubers cause seizures and cognitive deficits, increasing evidence suggests a poor correlation between cortical tubers and the incidence of epilepsy or autism in TSC patients2. Furthermore, animal models of TSC have increased susceptibility to seizures in the absence of cortical tubers, supporting the notion that tubers are not responsible for epilepsy. Hence, other mechanisms – such as miswiring of neuronal connections – may contribute to the pathogenesis of epilepsy, autism and intellectual disabilities in TSC patients.
TSC is caused by mutations in either of two genes: TSC1 or TSC2, whose protein products form a complex that plays a major role in the phosphatidylinositol 3-kinase (PI3K)-Akt-mTOR pathway. Binding of a growth factor such as insulin to its cell surface receptor leads to activation of PI3K, which in turn activates the Akt kinase. Phosphorylation of TSC2 by Akt releases the inhibitory effect of the TSC1/TSC2 complex on the Ras family GTPase, Rheb3, 4. Rheb and its downstream effector mTOR are master regulators of cell growth. When Rheb is activated, the protein synthesis machinery is turned on, most likely via mTOR, and cell growth programs are initiated. Therefore, the TSC1/TSC2 complex keeps cell size in check by inhibiting mTOR-mediated mRNA translation. Cells with insufficient TSC1 or TSC2 function grow beyond their normal size and form hamartomas, but the pathophysiology of the neurological symptoms in TSC patients remains poorly understood.
The establishment of neural circuits in vivo requires a precise interplay between extending axons and guidance cues in their environment. One of the best-characterized axon pathways in the central nervous system is the projection of retinal ganglion cells (RGCs) from the eye to their targets in the brain. Many proteins – such as neurotrophins, semaphorins, slits and ephrins – regulate retinal axon pathfinding and topographic mapping in target regions such as the dorsal lateral geniculate nucleus (dLGN)5. Interactions between EphA receptors and ephrin-A ligands expressed in gradients in retinal neurons and across the dLGN play prominent roles in initial topographic map formation in the dLGN6. Spontaneous retinal activity then contributes to map refinement during postnatal stages7–9.
Binding of ephrin ligands triggers Eph receptor clustering, autophosphorylation and downstream signaling cascades that cause cytoskeletal rearrangements and changes in cell adhesion10. Through these mechanisms, Eph receptors control axon turning, retraction and branching. Local regulation of protein synthesis and degradation in the axon also contributes to the rapid changes in growth cone dynamics that occur during axonal navigation11–15. Both repulsive and attractive cues can alter local protein translation in an mTOR-dependent manner, suggesting that guidance cues might affect axon growth and navigation at least in part by modulating mTOR activity14, 16.
We have identified a new role for Tsc1/Tsc2 in axon guidance by using mouse models of TSC. We found that components of the Tsc-mTOR pathway are highly expressed in developing RGC axons and that Tsc2+/− mice, which have elevated mTOR activity in RGCs, develop aberrant retinogeniculate projections. Consistent with this phenotype, in vitro Tsc2+/− RGCs are less sensitive to ephrin-A repulsive effects. Furthermore, EphA receptor signaling inhibits the mTOR pathway and reduces local protein synthesis in neurons. Our findings reveal a new mode of regulation of the Tsc-mTOR pathway by cell surface receptor tyrosine kinases through the ERK1/2 kinases and shed light on the mechanism by which EphA receptors control mTOR activity and growth cone dynamics.
RESULTS
Increased retinal mTOR activity in Tsc2+/− mice
We recently demonstrated that components of the Tsc-mTOR pathway are preferentially localized in the axons of embryonic hippocampal neurons and that homozygous Tsc1 inactivation causes the formation of multiple axons17. Given that homozygous Tsc2 knockout results in embryonic lethality18, and that TSC is an autosomal dominant disease where most cells in the brain of patients are likely heterozygous for TSC1 or TSC2 mutations, we investigated whether Tsc2 haploinsufficiency might also lead to a multi-axon phenotype. Double-labeling for the axonal marker Tau1 and the somato-dendritic marker MAP2 revealed similar numbers of neurons with multiple axons in cultures of E16 Tsc2+/− and wild-type cortical neurons (Supplementary Fig. S1), suggesting that Tsc2 haploinsufficiency may cause more subtle axonal abnormalities. Immunofluorescence staining of purified RGCs from the early postnatal retina confirmed that, similar to hippocampal neurons17, wild-type RGCs have higher axonal than dendritic levels of Tsc2 phosphorylated at the inhibitory Thr1462 site, total Rheb, and active S6K1 (phosphorylated at Thr389) (Supplementary Fig. S2). This indicates that the Tsc2-Rheb-mTOR pathway is preferentially activated in the axonal compartment of different types of neurons, including RGCs. We therefore examined axon guidance and connectivity in Tsc2+/− mice, focusing on the developing retinogeniculate projection as a model system.
We first analyzed mTOR activity in the Tsc2-deficient retina. To facilitate analysis of RGCs, retinal sections from P23 mice were stained for βIII-tubulin or the RGC markers Brn3a and Brn3b, in combination with phospho-S6 (Ser235/236), a reliable marker for mTOR activity (Fig. 1 and Supplementary Fig.S3). The percentage of Brn3a- or Brn3b-positive RGCs that were also positive for phospho-S6 was significantly higher in Tsc2+/− mice than wild-type littermates. Quantification of phospho-S6 fluorescence intensity also revealed significantly higher phospho-S6 levels in Tsc2+/− than wild-type RGCs (Fig. 1d). Therefore, RGCs with heterozygous Tsc2 inactivation have elevated mTOR signaling in vivo.
Figure 1. Increased mTOR activity in Tsc2+/− retina in vivo.

(a) Flat mount (first and third row) or horizontal sections (second and fourth row) of retinas of Tsc2+/− and wild-type littermates on postnatal day 23 (P23) were co-stained with antibodies against phospho-S6 (Ser235/236, green) and Brn3a (red). Blue represents nuclear DAPI staining. Arrows denote double-labeled RGCs and asterisks indicate phospho-S6 positive, but Brn3a-negative cells. Scale bars, 25 ⎧m in flat mounts and 50 ⎧m in cross-sections. (b) Flat mount retinas of Tsc2+/− and wild-type littermates (P23) were stained with antibodies against phospho-S6 (Ser235/236, green) and Brn3b (red). Arrows denote phospho-S6 and Brn3b double-labeled RGCs. Scale bar, 20 ⎧m. (c) Quantification of the phospho-S6 positive RGCs as shown in (a) and (b). In both Brn3a- and Brn3b-positive populations, numbers of phospho-S6+ RGCs are significantly increased in Tsc2+/− compared to wild-type littermates (**P < 0.01 and *P < 0.05 by Mann-Whitney tests). (d) Quantification of phospho-S6 (Ser235/236) fluorescence intensity as shown in (a) and (b) from Tsc2+/− versus wild-type (WT) mouse retinas. There was a significant increase in phospho-S6 fluorescence intensity in Tsc2+/− RGCs versus wild-type cells. Data are expressed as mean ± s.e.m. (WT: 34.80 ± 1.909, n = 44 RGCs; Tsc2+/−: 50.76 ± 1.343, n = 114 RGCs; *** P < 0.0001, unpaired t test). A.U. = arbitrary units.
Abnormal retinogeniculate projections in Tsc2+/− mice
To determine whether Tsc2+/− mice have axon guidance defects, we labeled RGCs and their axonal projections with the anterograde tracer cholera-toxin B (CTB). We injected one eye with CTB-594 (red) and the other with CTB-488 (green) at P14, and visualized the binocular retinogeniculate projections in dLGN sections at P16 (Fig. 2a,b and Supplementary Fig. S4a,b). We noted that Tsc2+/− dLGNs occasionally displayed more than one ipsilateral patch (Fig. 2c), a defect similar to that described in ephrin-A2 and ephrin-A5 double knockout mice7. Analysis of ipsilateral and contralateral projections19, 20 revealed a small but significant increase in the percentage of the dLGN occupied by projections from the ipsilateral eye in Tsc2+/− mice compared to wild-type littermates (Fig. 2d). In contrast, similar portions of the Tsc2+/− and wild-type dLGN were occupied by contralateral projections or received overlapping inputs (Fig. 2e,f). By using a line scan technique to calculate the mean pixel intensity along the dorsal-medial to ventral-lateral axes of the dLGN21, we found that the ipsilateral projections were shifted towards the ventral-lateral region in Tsc2+/− mice (Fig. 2g), similar to what was reported for ephrin-A knockout mice21.
Figure 2. Tsc2+/− mice have defects in ipsilateral retinogeniculate projections.

(a–b) A series of coronal sections from the anterior (top) to the posterior (bottom) showing retinogeniculate projections into dLGN in P16 Tsc2+/+ (a) and Tsc2+/− (b) mice. Alexa594 (red) and Alexa488 (green) conjugated CTB are injected to left and right eyes, respectively. D, dorsal; L, lateral. Dashed lines represent the borders of dLGN in sections displaying the ipsilateral projections. Scale bar, 100 µm. (c) In some Tsc2+/− mice, ipsilateral projection formed multiple patches rather than a single central patch. Axons from the ipsilateral eye are in green on top and red at the bottom. Scale bar, 100 µm. (d) Multi-threshold analysis of the percentage of ipsilateral projections in dLGN in wild-type versus Tsc2+/− littermates at P16. Tsc2+/− mice have larger ipsilateral territories than wild-type littermates, regardless of threshold. Data are expressed as mean ± s.e.m. (n = 6 mice for each genotype; * P < 0.05 by t-test). (e) Quantification of the percentage of contralateral projections in dLGN. No significant difference exists at any threshold between wild-type and Tsc2+/− littermates. (f) Quantification of dLGN receiving contra-ipsi overlapping projections. No significant difference exists at any threshold between wild-type and Tsc2+/− littermates. (g) The center of the ipsilateral projection in the dLGN was shifted ventrally and laterally in Tsc2+/− mice. Line scan technique was used to calculate the mean pixel intensity along the dorsomedial (DM, 0%) to ventrolateral (VL, 100%) axes in 10 wild-type and 11 Tsc2+/− littermates. Error bars represent s.e.m. and ** P = 0.0014 by Mann Whitney test.
The topography of RGC projections to the dLGN is regulated by the opposing gradients of EphA receptors expressed on RGC axons and ephrin-A ligands expressed on target cells6, 22. Thus, the retinogeniculate topographic mapping defects in the Tsc2+/− mice suggest that RGCs with defective Tsc2 signaling are less sensitive to ephrin-A-dependent growth cone repulsion or axon pruning. To distinguish between these two possibilities, we injected eyes with cholera-toxin B at P4, before the onset of pruning and map refinement7. We found that the dLGN area occupied by ipsilateral projections was larger in Tsc2+/− than wild-type mice (P = 0.0017 by ANOVA, n =12 wild-type and 12 Tsc2+/− mice). While we cannot completely rule out a contribution of pruning or crossing defects, these results indicate that the RGC projections to the dLGN are already aberrantly exuberant before pruning begins.
Since the ventral-lateral shift and the exuberance of the ipsilateral retinogeniculate projections in Tsc2+/− mice resemble the defects observed in ephrin-A-deficient mice21, we examined in more detail the topography of the RGC termination zones in the dLGN. We selectively labeled subsets of RGC axons with focal DiI-crystal placement in the ventral-temporal quarter of the retina. Single termination zones restricted to the dorsal-medial end of the dLGN were observed in wild-type mice, as expected (Fig. 3a). In contrast, the termination zones in Tsc2+/− littermates showed a ventral-lateral shift and sometimes more than one distinct termination zone (Fig. 3b). Quantification of the RGC termination zones along the dorsal-medial to ventral-lateral axis of the dLGN demonstrated a significantly larger extension of the Tsc2+/− RGC projections along the DM-VL axis compared to controls (Fig. 3c). Overall, the mapping defects observed in the Tsc2+/− ventral-temporal projections mimic those described in ephrin-A knockout mice, suggesting attenuation of EphA receptor signaling in Tsc2-deficient RGCs.
Figure 3. Tsc2+/− mice have defects in topographic mapping of contralateral projections in dLGN.

(a) Representative series of fluorescent micrographs showing termination territories in contralateral dLGN from DiI-labeled ventro-temporal RGCs of Tsc2+/+ versus Tsc2+/− littermates at P19. Anterior is to the left and posterior to the right in each series of sections. Dashed lines represent the borders of dLGN. Arrows indicate DiI labeled RGC termination zones. Insets represent tracings of the flat-mounted retina and injection site for each mouse. OT: optic tract; D: dorsal; V: ventral; T: temporal; N: nasal; and L: lateral. (b) Schematic drawings and colored dots show the position and area of DiI labeled termination in each corresponding dLGN section from wild-type (in red) and Tsc2+/− (in green) littermates. In Tsc2+/−, the lower border of the termination extended erroneously into the area that is innervated by the ventral-nasal RGCs in wild-type mice8. Scale bar represents 100 µm. (c) Quantification of the percentages of DM to VL extension to length of dLGN along the same axis. Data are represented as mean ± SEM (19.1 ± 2.1% in wild-type, n = 4 mice; and 50.3% ± 9.7 % in Tsc2+/− littermates, n = 6 mice. **: P < 0.01 by t-test).
Loss of Tsc2 inhibits ephrin-induced growth cone collapse
After confirming that Tsc2 protein expression is reduced in Tsc2+/− whole retina lysates and isolated cortical neurons (Supplementary Fig. S4c,e), we used Tsc2+/− RGCs to examine the sensitivity of their growth cones to ephrin-induced collapse23. Ephrin-A1 stimulation caused the collapse of 70–80% wild-type RGC growth cones but only 35- 40% Tsc2+/− growth cones (Fig. 4a,b). Wild-type and Tsc2+/− growth cones collapsed similarly in response to LPA, however, indicating that Tsc2 deficiency specifically impairs EphA receptor-mediated collapse (Fig. 4b). The decreased sensitivity of Tsc2+/− RGCs to ephrin-As is consistent with the aberrant pattern of retinogeniculate projections observed in vivo.
Figure 4. Tsc2 deficiency and hyperactive Rheb inhibit ephrin-A-induced growth cone collapse.
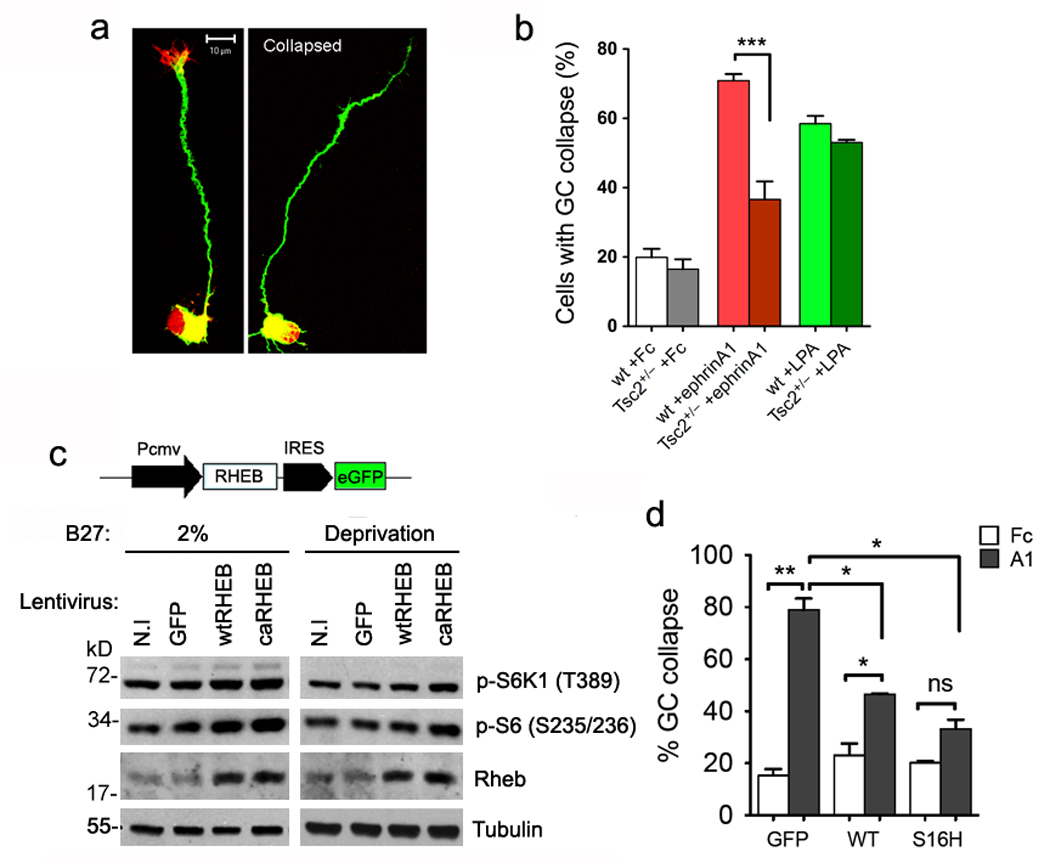
Purified RGCs from Tsc2+/− and wild-type littermates at P8 were stimulated with LPA (1µM) or pre-clustered ephrin-A1-Fc or Fc (control) proteins (5 µg/ml). Growth cone collapse was determined by phalloidin staining. (a) Representative micrographs showing staining with rhodamine-phalloidin (red) and anti-Tuj1 (green) of RGCs with intact versus collapsed growth cone. Scale bar, 10 µm. (b) Quantification of the percentage of RGCs with collapsed growth cone. Data represent mean ± s.e.m.. Ephrin-A1-Fc stimulated collapse was dramatically reduced in Tsc2+/− compared to wild-type RGCs (***: P < 0.001 by t-test). There is no significant difference in LPA-treated growth cones from the two genotypes. (c) The lentiviral construct was used to over-express either wild-type Rheb (wtRHEB) or a constitutively active mutant (caRHEB, S16H), or GFP-alone (GFP) as the control in Tsc2+/+ cells. Western analyses verified higher levels of Rheb, phospho-S6K1 (Thr389) and phospho-S6 (Ser235/6) in wtRHEB and caRHEB infected hippocampal neurons than those in GFP or non-infected (N.I) neurons, in either 2% B27 medium or B27-lacking medium for 16 hours. Full-length gels are presented in Supplementary Figure S13. (d) Quantification of the percentage of growth cone collapse in purified RGC cultures infected with wtRHEB, caRHEB or GFP lentivirus. Results represent mean ± s.e.m. from 3 independent experiments. RGCs with over-expression of wtRHEB or caRHEB have significantly less collapse than GFP expressing RGCs (by t-test **P < 0.01 comparing GFP infected cells in Fc versus ephrin-A1 stimulation; *P < 0.05 comparing wtRHEB infected cells in Fc versus ephrin-A1 stimulation for; *P < 0.02 comparing ephrin-A1 stimulations between GFP and wtRHEB, or GFP and caRHEB infected neurons; and ns = not significant).
To verify that the decreased sensitivity of Tsc2-deficient growth cones to ephrin-A1 was not due to lower EphA expression or signaling, we used a lentiviral shRNA17, 24 to knock down Tsc2 in cultured neurons. Tsc2 downregulation not detectably affect EphA4, ephrin-A2 and ephrin-A5 protein levels (Supplementary Fig. S5a). Furthermore, cell surface biotinylation did not reveal a significant effect of Tsc2 knockdown on EphA4 cell surface levels (Supplementary Fig. S5b,c). Because ephrins induce Eph receptor clustering, which regulates signaling, we also verified that the density of EphA clusters was not altered in Tsc2 knockdown compared to control lentivirus-infected neurons (Supplementary Fig. S6). Thus, the effects of Tsc2 deficiency on the retinogeniculate projection in vivo and ephrin-A responsiveness of RGCs in vitro are not due to aberrant ephrin-A/EphA expression, localization or clustering.
Because Tsc2 is a GTPase-activating protein (GAP) that inhibits mTOR by inactivating the small GTPase Rheb, we also investigated the role of Rheb in growth cone collapse. We used lentiviral vectors to express the constitutively active RhebS16H mutant, which is insensitive to Tsc2, and wild-type Rheb, which is also substantially activated when overexpressed (Fig. 4c). Increased mTOR signaling was verified by measuring phosphorylation of endogenous S6K1 and S6 in HEK293 cells infected with the Rheb lentiviruses (Supplementary Fig. S7). Ephrin-A1 induced significant collapse in growth cones expressing wild-type but not constitutively active RhebS16H. However, collapse was reduced in neurons expressing wild-type Rheb or RhebS16H as compared to control neurons expressing only GFP (Fig. 4d). Therefore, similar to Tsc2-deficiency, Rheb hyperactivation inhibits ephrin-A-induced growth cone collapse.
Ephrin-As depress ERK and mTOR activities in neurons
We next examined whether there is crosstalk between EphA signaling and the mTOR pathway in neurons. Stimulation with ephrin-A1 led to EphA phosphorylation, which was detectable within 2 minutes and peaked at 30 minutes (Fig. 5a). We also observed an inverse correlation between EphA activation and ERK1/2 phosphorylation/activation, in agreement with previous findings in non-neuronal cells25. Ephrin-A1-treated neurons also displayed rapid dephosphorylation of S6K1 within 2–5 minutes and a delayed decrease in phospho-S6 after ~15 minutes of stimulation (Fig. 5a,b). 4E-BP1, another effector of mTOR, was also dephosphorylated upon ephrin-A1 stimulation. Furthermore, ephrin-A1 inhibited phosphorylation of ERK1/2, S6K1 and S6 in a dose-dependent manner (Fig. 5c). Stimulation of cortical neurons with ephrin-A5, the predominant ephrin-A in the dLGN, also decreased ERK1/2, S6K1 and S6 phosphorylation, but with a slower time course (Fig. 5d,e). Interestingly, the levels of activated Akt (phosphorylated at Ser473) and TSC2 phosphorylated at Thr1462, a site phosphorylated by Akt, remained constant throughout the period of ephrin stimulation. Thus, activation EphA receptor signaling inhibits both ERK1/2 and the mTOR pathway in neurons, the latter independently of the upstream mTOR regulator Akt.
Figure 5. Inactivation of ERK1/2 and mTOR pathway in ephrin stimulated neurons.
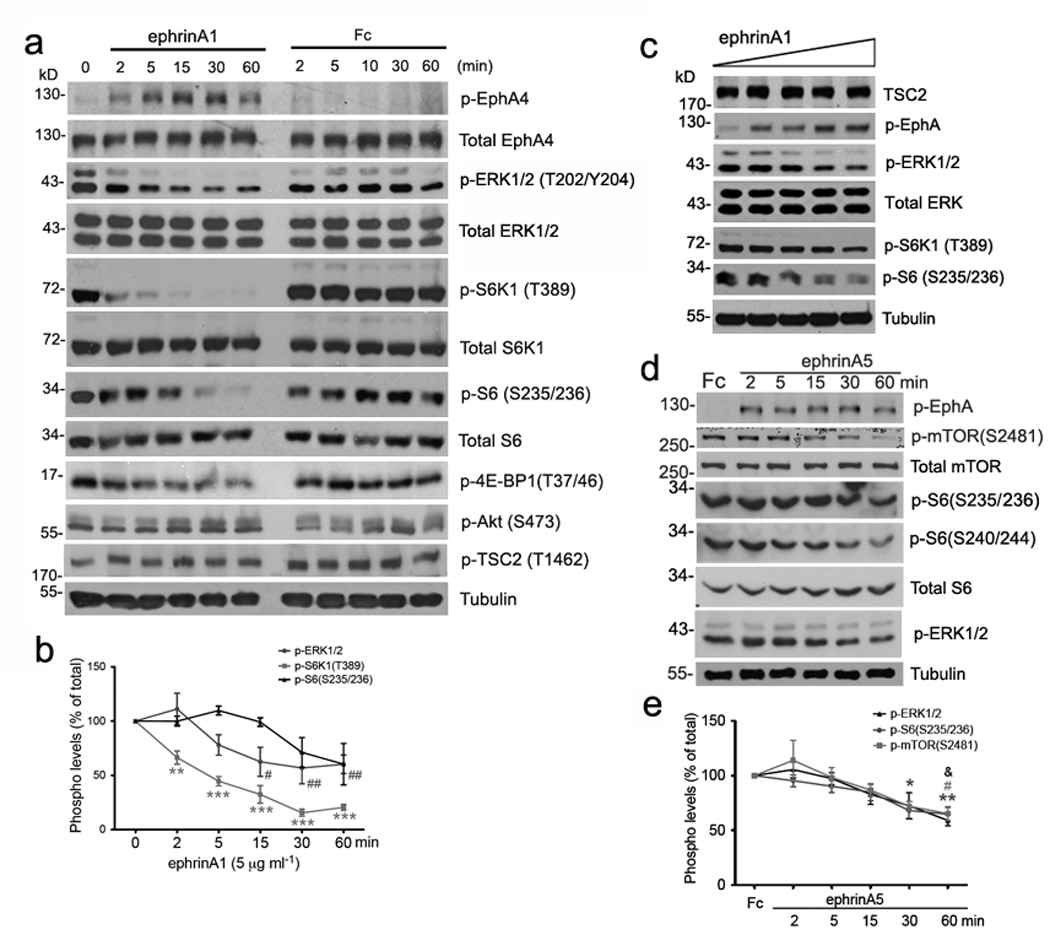
(a) Tsc2+/+ cortical neurons maintained in culture for 7 days were stimulated with either pre-clustered ephrin-A1-Fc or Fc as the control. Western analyses of neurons stimulated with 5 µg/ml proteins for the indicated minutes. Total EphA4, ERK1/2, S6K1, S6 and tubulin are used as loading controls. (b) Quantitative analysis of phosphor-protein levels over the stimulation course as in (a). Significant reduction in phosphorylation levels was found by t-test at each time point versus 0 min (** P < 0.01 comparing phospho-S6K1 (Thr389) at 2 min and *** P < 0.001 at 5, 15, 30 or 60 min; # P < 0.05 comparing phospho-ERK1/2 at 15 min and ## P < 0.01 at 30 or 60 min). (c) Western analyses of cortical neurons stimulated for 30 minutes with ephrin-A1 at ascending concentrations (0.1, 1, 5, 10 and 20 µg/ml). Total ERK1/2 and tubulin are used as loading controls. (d) Western analyses of Tsc2+/+ cortical neurons grown in culture for 12 days and stimulated with either pre-clustered Fc (as the control) for 60 minutes or ephrin-A5-Fc for the indicated minutes. Total mTOR, S6 and tubulin are used as loading controls. (e) Quantification of the phosphorylation levels of mTOR, S6 and ERK1/2 in Fc versus ephrinA5-Fc (5 ⎧g /ml) stimulations. Significant reduction in phosphorylation levels was found by t-test comparing ephrin-A5 at each time point versus Fc stimulation (* P < 0.05 comparing phospho-S6 at 30 min and ** P < 0.01 at 60 min; # P < 0.05 comparing phospho-mTOR at 60min; & P < 0.05 phospho-ERK1/2 at 60min versus Fc). Full-length gels are presented in Supplementary Figure S13.
Ephrin-As inhibit protein synthesis in isolated neurites
Inhibition of mTOR activity by ephrin-As would be expected to repress protein synthesis in neurites and growth cones. We used the modified Boyden chamber system to harvest neurite-enriched lysates from neuronal cultures and measure metabolic incorporation of 35S-labeled amino acids into newly synthesized proteins (Fig. 6a). Ephrin-A1 and the mTOR inhibitor rapamycin both caused a similar 25–35% reduction in protein synthesis while translation inhibitors such as UO126 and anisomycin almost completely blocked 35S incorporation (Fig. 6b,c). Thus, mTOR inhibition by rapamycin or ephrin-A1 appears to significantly reduce protein synthesis in neurites.
Figure 6. Ephrin inhibits axonal protein synthesis and the local translation of a beta-actin reporter in growth cones.

(a) Schematic representation of the experimental design using [35S]-labeling and collection of axon-enriched lysates using Boyden transwells (see Material and Methods). (b) Cell bodies of rat hippocampal neurons grown in Boyden transwells were removed, and the neurites in the lower chamber were stimulated immediately with preclustered Fc, ephrin-A1-Fc (5 ⎧g/ml), UO126 (20 µM), rapamycin (20 nM) or anisomycin (40 ⎧M) for 30 minutes. Quantification of the [35S] incorporation (CPM/⎧g) in TCA-precipitated lysate shows that ephrin-A1 inhibits de novo protein synthesis as compared to Fc, but to a lower extent than UO126, rapamycin, or anisomycin. Results represent mean ± SEM from 3 independent experiments (**P < 0.01, ***P < 0.001, and ****P < 0.0001 by t-tests). (c) A representative [35S] autoradiograph showing reduction in the amount of newly synthesized proteins from axons treated with ephrin-A1, UO126, rapamycin or anisomycin compared with control Fc-treated axons. (d) Hippocampal neurons were transfected with dGFPmyr-bACT-3’UTR and frames with dashed lines represent region of bleaching. Representative images show the fluorescence recovery after photobleaching in terminal axons transfected neurons over a period of 20min, in the presence of ephrin-A1-Fc or Fc as control. Arrows indicate axonal growth cones. Scale bar, 20 ⎧m. (e) Quantification of GFP fluorescence intensity pre- and post-bleaching in ephrin-A1-Fc versus Fc treatments. Data at each time point represent average % of pre-bleach levels ± s.e.m. (n = 3 for Fc and n = 6 for ephrin-A1-Fc). Significant difference in signal recovery was assessed by two-way ANOVA when comparing ephrinA1 versus Fc treatments at each time point post-bleaching (*P< 0.05 at 6 min; **P< 0.01 at 4 min; and ***P< 0.001 at 7~19 min). (f) Quantification of GFP signal recovery in anisomycin versus DMSO treatments. *** P < 0.001 by two-way ANOVA.
Local translation and degradation of β-actin and proteins that regulate the actin cytoskeleton play critical roles in controlling cytoskeletal dynamics in axons and growth cones13–16. To monitor local translation in ephrin-A1-stimulated neurons, we used the construct dGFPmyr-bACT 3’UTR, encoding a destabilized and myristoylated GFP from a transcript fused to the 3’UTR of β-actin26 (Supplementary Fig. S8a). This 3’UTR contains a sequence for localization to neuronal processes. In fluorescence recovery after photo-bleaching experiments, we photo-bleached axonal terminal portions including the growth cones of transfected neurons, and monitored signal recovery over a period of ~20 min. In general, this time scale is not long enough for transport or diffusion of myristoylated dGFP protein from the soma to the growth cone27, 28. The dGFPmyr-bACT 3’UTR fluorescence signal recovered rapidly after bleaching in control neurons, while fluorescence recovery was minimal for the co-transfected control d2-mCherry (data not shown). dGFP fluorescence recovery in axons pre-treated with the translation inhibitor anisomycin (100 ⎧M) was dramatically delayed and repressed, as expected (Fig. 6d–f, Supplementary Fig. S8b,c). Importantly, fluorescence recovery in the presence of ephrin-A1-Fc was significantly reduced compared to Fc control. These findings suggest that ephrin-A1 stimulation inhibits fast and localized translation of β-actin and other mRNAs in the growth cone. Nevertheless, we did not detect significant effects of anisomycin or rapamycin on ephrin-A1-induced growth cone collapse (data not shown), as previously reported29. This suggests that the inhibition of mTOR activity and local translation induced by ephrin stimulation is already sufficient for maximal growth cone collapse.
Loss of Tsc2 blocks mTOR inactivation by EphA receptors
Consistent with its inhibitory effects on ephrin-dependent growth cone collapse, Tsc2 knockdown abolished ephrin-A1-dependent downregulation of S6K1 and S6 phosphorylation in neurons (Fig. 7a,b). The levels of phospho-S6K1 and phospho-S6 remained unchanged over a 60-minute time course of ephrin stimulation, excluding the possibility of a delayed dephosphorylation (Fig. 7c). Cre-mediated inactivation the Tsc1/Tsc2 complex in cortical neurons isolated from Tsc1flox/flox or Tsc2floxlflox mice also prevented the ephrin-A-dependent S6 dephosphorylation (Supplementary Fig. S9). Furthermore, ephrin-A1 stimulation decreased S6K1 and S6 phosphorylation in wild-type but not Tsc2−/− mouse embryonic fibroblasts (Supplementary Fig. S10a), indicating that EphA signaling can also regulate the mTOR pathway in non-neuronal cells. These results suggest that Tsc2 is both necessary and sufficient for the negative regulation of mTOR activity by ephrin-As, likely through a mechanism that does not involve Tsc2 phosphorylation by Akt at Thr1462 (see above). Interestingly, ERK1/2 inactivation by ephrin-A1 still occurred in Tsc2 knockdown neurons, even though baseline ERK1/2 phosphorylation was lower than in control neurons (Fig. 7a,b). Thus, ERK1/2 might be responsible for EphA-dependent Tsc2 activation.
Figure 7. Tsc2 is required for the ephrin-A1-stimulated mTOR inactivation.

Tsc2+/+ cortical neurons were infected with lentivirus expressing shRNA against Tsc2 (T2sh1) or a firefly luciferase (GL3, control). Non-infected (N.I.) as well as the infected neurons were then stimulated with pre-clustered ephrin-A1-Fc versus Fc for 30 minutes. (a) Representative western blots for indicated proteins. Downregulation of Tsc2 decreased Tsc1 levels and increased SAD-A/B levels, consistent with our previous studies17, 24. There were no significant differences in EphA4 and EphA5 expression, ephrin-induced EphA tyrosine phosphorylation or in ephexin phosphorylation between Tsc2 knockdown and control neurons. Tsc2 knockdown abolished the ephrin-A1-dependent downregulation of S6K1 and S6 phosphorylation observed in control neurons. Inactivation of ERK1/2 by ephrin-A1 was still observed in the Tsc2 knockdown neurons, even though the baseline level of phospho-ERK1/2 was lower in these neurons than in the controls. Total ERK1/2, S6 are used as loading controls. Full-length gels are presented in Supplementary Figure S14. (b) Quantification of the western blot analyses. Relative intensities for each protein were normalized according to the tubulin loading control and expressed as mean ratios of GL3 or T2sh1 to non-infected (NI) neurons in each independent experiment. Data represent mean ± SEM from 4 independent experiments (*P < 0.05; ** P < 0.01; *** P < 0.001 and ns=not significant). (c) T2sh1-infected neurons were stimulated with pre-clustered ephrin-A1-Fc or control Fc proteins for the time periods as indicated. Phospho-S6K1 and phospho-S6 remained unchanged over the time course of stimulation. Tubulin, total ERK, S6K and S6 were used as loading controls.
Guidance cues differentially modulate Tsc2 activity
Tsc2 activity can be inhibited not only by Akt-dependent phosphorylation at Thr1462 and Ser939, but also by ERK-dependent phosphorylation at Ser6644, 30. BDNF stimulation dramatically increased Akt and ERK1/2 phosphorylation in cultured neurons, whereas ephrin-A1-Fc significantly suppressed ERK1/2 but not Akt phosphorylation (Fig. 8a). We therefore used phospho-specific antibodies to assess the modulation of Tsc2 by Akt and ERK1/2 in neurons stimulated with BDNF or ephrin-A1. As expected, S6K1 phosphorylation was up-regulated by BDNF and down-regulated by ephrin-A1 stimulation (Fig. 8a). BDNF also increased Tsc2 phosphorylation on both Thr1462 and Ser664 ~2 fold compared to control-treated neurons (Fig. 8a,b). In contrast, Tsc2 Ser664 phosphorylation was approximately 35% lower in ephrin-A1-stimulated cells than in control-treated cells. These results suggest that in cultured neurons, BDNF increases Tsc2 phosphorylation through activation of both Akt and ERK, which synergistically inhibit Tsc2 GAP function towards Rheb, whereas ephrin-A1 selectively decreases phosphorylation of Tsc2 at the ERK site. Hence, the Tsc-mTOR pathway can be positively or negatively regulated by distinct axon guidance cues.
Figure 8. Regulation of TSC2 (Ser664) phosphorylation by ephrin-A1 is involved in growth cone dynamics.
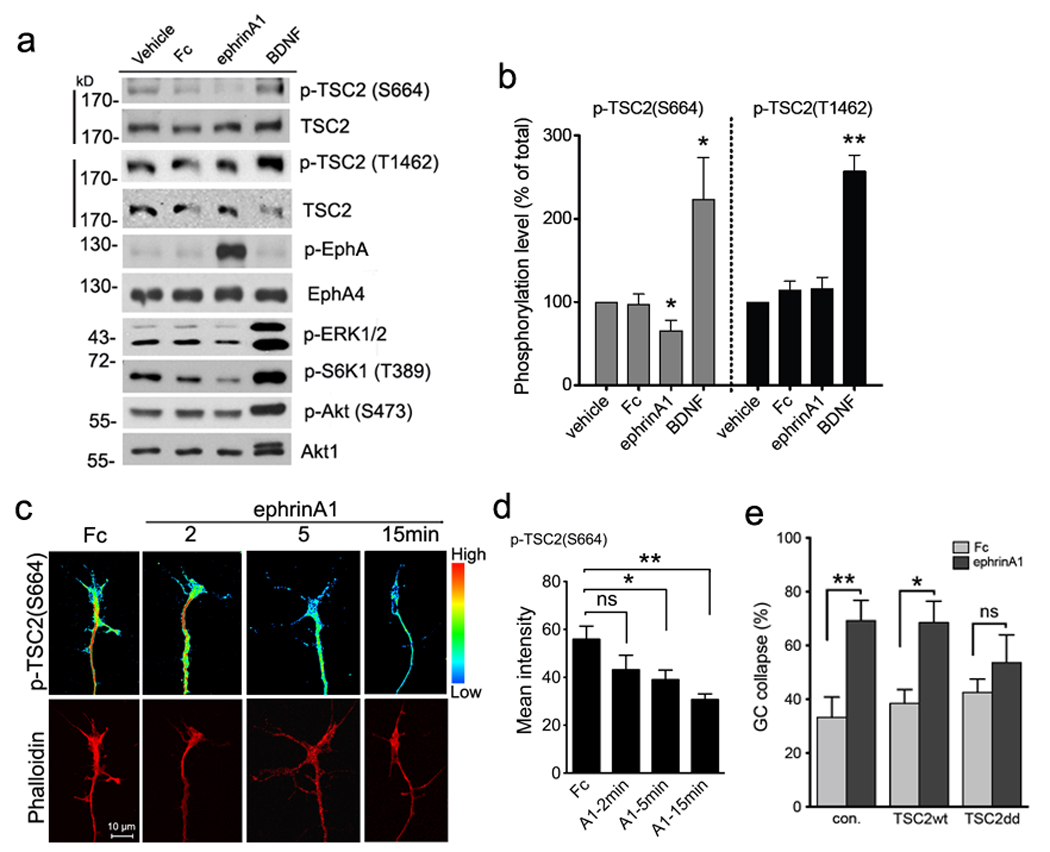
(a) Tsc2+/+ cortical neurons maintained in culture for 12 days were stimulated as indicated for 30 minutes. Phospho-TSC2 (Ser664) or phospho-TSC2 (Thr1462) along with total TSC2 was probed on the same blots and total Akt1 used as the loading control. Full-length gels are presented in Supplementary Figure S14. (b) Intensities of phospho-TSC2 (Ser664) and phospho-TSC2 (Thr1462) were normalized to total TSC2. Data are expressed as % of vehicle-treated intensity ± s.e.m. (* P < 0.05 comparing p-TSC2 (Ser664) in ephrin-A1 versus vehicle, or BDNF versus vehicle; ** P < 0.01 comparing phospho-TSC2 (Thr1462) in BDNF versus vehicle treatment, t-test). (c) Purified RGC cultures from P7 rats were stimulated as indicated and then stained with anti-phospho-TSC2 (Ser664) and rhodamine-phalloidin. Phospho-TSC2 (Ser664) staining was converted to pseudo-colored spectrum display to distinguish intensity change. Scale bar 10 µm. (d) Quantification of pixel intensity of phospho-TSC2 (Ser664) labeling in RGC growth cones. Data represent mean ± s.e.m. (n = 4 – 10 growth cones; * P < 0.05 and ** P < 0.01 and ns =not significant by t-test). (e) Transfection of neurons with phospho-mimic mutant of TSC2 partially blocked growth cone collapse. Rat hippocampal neurons transfected with wild-type TSC2, mutant TSC2 (S664D/S540D) or empty control plasmids were stimulated with Fc or ephrin-A1-Fc for 30 min. Growth cone collapse was determined by phalloidin staining. Data represent mean % collapse ± SEM from 3 independent experiments with duplicates in each experiment (* P < 0.05, ** P < 0.01 and ns =not significant, by t-test).
We then examined whether ephrin-A stimulation can regulate Tsc-mTOR signaling locally in the axon. We detected high phospho-Tsc2 (S664) immunofluorescence in growth cones and filopodia (Supplementary Fig. S11a–c). Upon ephrin-A1 stimulation, the signal diminished rapidly from the growth cone and axon tips. At 5 min, when the majority of growth cones (83%) are not yet collapsed, ephrin-A1-treated growth cones displayed a significant reduction in phospho-Tsc2 (S664) labeling (Fig. 8c,d). Phospho-S6K1 immunofluorescence showed a similar reduction upon ephrin-A1 treatment (Supplementary Fig. S11d,e). Ephrin-A1 stimulation for 15 minutes led to complete growth cone collapse, coincident with only a faint residual phospho-Tsc2 (S664) and phospho-S6K (T389) signal in the axon tips. Thus, ephrin-A1 induces fast dephosphorylation of Tsc2 on Ser664 and S6K1 on Thr389 within the growth cone, consistent with the timing of inhibition of local translation in growth cones measured with the β-actin reporter.
To confirm that the rapid change in Tsc2 phosphorylation in the growth cone plays a role in ephrin-dependent growth cone collapse, we transfected wild-type Tsc2 or the phospho-mimic S540D/S664D mutant in neurons30. We observed partial blockade of ephrin-A1-induced growth cone collapse by the phospho-mimic Tsc2 mutant but not wild-type Tsc2 (Fig. 8e). This implicates regulation of Tsc2 activity by ERK in the cellular machinery that controls growth cone dynamics in response to ephrin stimulation.
DISCUSSION
Here we demonstrate that the Tsc1/Tsc2 complex is required for ephrin-A-induced growth cone collapse and elucidate a mechanism by which ephrins regulate the Tsc-mTOR pathway in axons. We also show that Tsc2-deficient RGCs project aberrantly to the dLGN, indicating that the Tsc-mTOR pathway plays a critical role in Eph-mediated axon guidance in vivo. Therefore, Tsc2-mediated regulation of mTOR is crucial for growth cone dynamics and proper axon pathfinding (Supplementary Fig. S12).
Eph receptors in neural development, plasticity and repair
The role of Eph receptors and ephrins in the central nervous system has been extensively studied and a striking feature is the remarkable diversity of their functions10. The Eph receptors represent the largest family of receptor tyrosine kinases in the mammalian genome and regulate various signaling pathways through a number of downstream effectors, including guanine nucleotide exchange factors (Ephexins, Kalirin, Intersectin, and Tiam1), GTPase-activating proteins (p120RasGAP, α2-chimaerin, SPAR), tyrosine kinases (Src, Abl, Fak), phosphatases (LMW-PTP, Ship2), and adaptor proteins (Nck, Grb2, Grb4 and Crk)10. Here, we uncover a novel pathway whereby Eph receptors regulate mTOR via ERK inhibition, which leads to Tsc2 activation. Likely, Eph forward signaling simultaneously modulates this and other signaling pathways to modify cytoskeletal dynamics in the growth cone. We demonstrated that ephrin-As inhibit mTOR activity in neurons, and it will be interesting to examine whether ephrins of the B class can also regulate mTOR.
Eph/ephrin signaling is important in the formation of not only retinal connections but also other axonal projections, such as corticospinal and thalamocortical connections and the corpus callosum. Our findings in the retinogeniculate connections may therefore have general relevance for the mapping of axonal connections in the developing central nervous system. Recently, the Eph system was also shown to play a crucial role in radial column formation in the neocortex, implicating these molecules in neuropsychiatric disorders associated with abnormal columnar organization31. Whether TSC mouse models have abnormal columnar organization has not yet been investigated. Eph receptors and ephrins also persist in the adult brain, particularly in regions where neuronal circuits continue to be remodeled, such as the hippocampus32. The hippocampal LTP and learning abnormalities reported in Tsc-deficient mouse models could be explained, at least in part, by the role of the Tsc-mTOR pathway in Eph signaling33. Finally, multiple Eph receptors and ephrins are upregulated at sites of CNS injury, where they seem to hinder axon regeneration through their repulsive signaling34. In particular, EphA4 is emerging as an inhibitor of nerve regeneration, possibly by interacting with both ephrin-B2 in reactive astrocytes and ephrin-B3 in myelin. Recently, mTOR activation by Pten or Tsc1 knockout was shown to promote the regenerative capacity of injured axons35. Our data suggest that one of the possible mechanisms by which mTOR activation could promote axon regeneration is by opposing Eph repulsive signaling. Further experiments involving genetic and chemical manipulations of Eph and mTOR signaling are needed to better delineate their interplay during axon regeneration.
The role of mTOR in axon guidance
We have shown that upon Eph activation, ERK activity is inhibited. This leads to activation of TSC2, which inhibits Rheb. In agreement with these findings, loss of Tsc2 and overexpression of constitutively active Rheb decrease the responsiveness of growth cones to repulsive ephrin signals. To date, mTOR is the only known downstream target of Rheb. There are two distinct mTOR complexes: mTORC1, which mainly regulates the translational machinery, and mTORC2, which mainly regulates the actin cytoskeleton. mTORC1 is acutely sensitive to rapamycin while mTORC2 is not, although mTORC2 can be inhibited by long-term rapamycin treatment. Rheb can activate mTORC1 by disrupting the binding of the inhibitory protein FKBP38, but reportedly inhibits mTORC236. Thus, the similar reduction in growth cone collapse by suppression of Tsc2 or over-expression of Rheb strongly suggests that this phenotype is mediated by mTORC1. Interestingly, previous studies demonstrated that growth cone collapse induced by semaphorins or Slit requires rapamycin-sensitive mTOR activity16, 37, suggesting that either too much or too little mTORC1 activity may interfere with growth cone dynamics and result in aberrant axon guidance.
The mTOR pathway is a critical regulator of protein synthesis, and we indeed found that ephrin stimulation reduces protein synthesis in neurons. However, mTOR might mediate additional functions downstream of Eph receptors, including transcription, ubiquitin-dependent proteolysis, autophagy, membrane trafficking, and microtubule and actin cytoskeleton dynamics. Additional experiments are needed to determine whether the role of mTOR in ephrin-dependent growth cone guidance involves regulation of protein synthesis or additional mechanisms, such as regulation of cytoskeletal dynamics.
Accumulating evidence suggests that the machineries for protein synthesis and degradation are both present and locally active in the growth cone and can be regulated by guidance cues11, 38. Axon guidance molecules like netrin-1 and semaphorins can trigger protein synthesis in retinal growth cones isolated from their cell bodies16 and inhibition of mTOR has been shown to interfere with proper RGC axon pathfinding. Two recent studies have shown that β-actin mRNA is subject to local translation in the growth cone. In Xenopus, the growth cone turning activity of BDNF requires local protein synthesis and is abolished by inhibitors of mRNA translation15. Local BDNF exposure induces co-localization of β-actin mRNA with the asymmetrically localized ZBP1 protein, resulting in asymmetric distribution of newly translated β-actin and growth cone turning. Similarly, netrin induces transport of Vg1RBP (Xenopus homolog of ZBP1) into filopodia and increases β-actin translation13. This requires the 3’UTR of the β-actin mRNA and mTOR activity. Our results uncovered further molecular links between axon guidance cues and local mRNA translation in the axon, including the Tsc1/Tsc2 complex and Rheb. Furthermore, we show that ephrin-dependent growth cone collapse involves ERK inactivation and consequent Tsc2 activation, resulting in mTOR suppression. Therefore, the mTOR kinase can be utilized by diverse extracellular cues to modulate growth cone dynamics.
There are at least two mechanisms by which mTORC1 regulates the translational machinery: phosphorylation of S6K, which increases the translation of mRNA transcripts containing a tract of pyrimidine (TOP) motif, and phosphorylation of the eIF4E-binding proteins, which relieves the inhibitory effect of 4EBP1 on cap-dependent translation initiation. The roles of S6K and 4EBP1 in axons are poorly characterized, although we have recently demonstrated that activated S6K1 is preferentially localized in the growing axon when neurites are first developing17. Identification of the repertoire of mRNAs whose translation is regulated by the Tsc-mTORC1 pathway in axons will provide important insights into the role of translational control in growth cone dynamics.
Pathogenesis of TSC disease
While significant progress has been made in identifying the molecular and cellular mechanisms underlying tuber formation in TSC patients, a key unresolved issue is what causes seizures and autistic features in TSC patients2, 39–41. Advances over the past few decades have suggested that dysregulation of axon growth and guidance plays a significant role in the pathogenesis of epilepsy, autism and intellectual disabilities. Deficient axonal growth has been associated with cerebral dysgenesis and intellectual disability42 while excessive growth of neuronal processes has been associated with epilepsy43. The role of neuronal connectivity has also become apparent in autism. MRI studies have also demonstrated increased white matter volume44 and aberrant white matter adjacent to brain regions implicated in social cognition in autistic individuals45, suggesting that abnormal connections between brain regions involved in social functioning may contribute to impaired social cognition. It will be interesting to see if TSC patients with autism can be distinguished from TSC patients without autism on the basis of abnormal connectivity in regions involved in social cognition.
Individuals affected by TSC carry heterozygous mutations in either the TSC1 or the TSC2 genes. Loss of heterozygosity (LOH) has been demonstrated in lesions such as cardiac rhabdomyomas, renal angiomyolipomas and subependymal giant cell astrocytomas but not in cortical tubers46. In animal models, acute inactivation of the Tsc1 gene in hippocampal slice cultures results in increased soma size, decreased spine density, and increased spine length in pyramidal neurons47. Moreover, Tsc1+/− mice have deficits in hippocampal-dependent learning. They also spend less time with unfamiliar mice and are worse at nest building compared to wild-type littermates, two lines of evidence indicating abnormal social behavior. However, analysis of adult Tsc1+/− brains failed to detect any changes in cell soma size, spine density or dendritic arborization48. Therefore, the neuropathology associated with the cognitive and behavioral deficits remains unclear. Our data demonstrate that Tsc2 haploinsufficiency causes defects in axonal connectivity. It will be interesting to investigate whether the abnormalities that we have detected in the projections of retinal neurons may also be present in other neuronal populations and may contribute to the neurobehavioral phenotype of TSC mouse models and TSC patients.
Material and Methods
Antibodies, Constructs and Animals
Anti-phospho-TSC2 (S664)30, anti-EphA4, anti-phospho-EphA 49, anti-phospho-ephexin23 antibodies were described earlier. Antibodies to phospho-ERK1/2, total ERK1/2, phospho-TSC2 (T1462), Rheb, phospho-S6K1 (T389), total S6K1, phospho-S6 (S235/236), total S6, phospho-Akt (S473), phospho-4E-BP1 (T37/46), phospho-mTOR (S2481) (Cell Signaling), TSC2, EphA4, ephrin-A2, ephrin-A5, EphA5, Brn3b (H-18), (Santa Cruz), phospho-TSC2 (S664) (BioLegend), TSC1 (Zymed), TSC2 (Biosource), Tau1 (Chemicon), MAP2 (sigma), chimaerin1 (Abnova), Brn3a, SAD-A/B (Millipore) as well as recombinant ephrin-A1-Fc, ephrin-A5-Fc (R&D systems), BDNF (Biosource), and chemicals PMA, U0126, rapamycin (Sigma), anisomycin (Calbiochem) were obtained commercially.
The dGFPmyr-bACT 3’UTR and d2-mCherry constructs were generous gifts from Dr. J Twiss (University of Delaware, USA) and Dr. G Bassell (Emory University, USA), respectively. The lentiviral Tsc2 shRNA construct was custom made by Cellogenetics 17. Tsc2+/− and Tsc2f/f mice were previously described18, 50. All procedures were carried out in accordance with the Guide for the Humane Use and Care of Laboratory Animals, and the study was approved by the Animal Care and Use Committee of Children’s Hospital Boston.
Site-directed mutagenesis
Total RNA was isolated from cultured rat cortical neurons with Qiagen RNeasy Kit according to manufacturer’s instruction. Complementary DNA was then synthesized with Superscript II reverse transcriptase. From cDNA pool, rat Rheb was PCR-amplified with the primer set: 5’-AAGGAAAAAAGCGGCCGC-ACCATGCCTCAGTCCAAGTCCCGG-3’ (forward) and 5’-CGGGATCCTCACATCACCGAGCACGAAGACTTCCCTTG-3’ (reverse). For packaging lentivirus, Rheb PCR product was further cloned into the pHAGE-CMV-IRES lentiviral vector at Not I/Bam HI sites. Rheb S16H mutant was generated using a QuikChange II XL site-directed mutagenesis kit (Stratagene) with primers: 5'-CCATCCTGGGCTATCGGCATGTGGGAAAGTCCTCAT-3' and 5'-ATGAGGACTTTCCCACATGCCGATAGCCCAGGATGG-3'.
Lentivirus infection, neuronal culture and immunocytochemistry
Lentiviri were pseudo-typed in HEK293T cells that were co-transfected with packaging vectors (rev, tat, gag/pol, vsv-g) and each lentiviral construct. Six hours later, transfection solutions were replaced with DMEM containing 1% FBS. Viral supernatants were collected twice at 48 and 60 hr after transfection, filtered and frozen.
Retinal ganglion cells (RGCs) were prepared from P7 rat or P8 mouse pups according to the immuno-panning method as previously described 23. Cortical and hippocampal neurons were isolated from E18 rat or E16 mouse embryos. Briefly, cortex and hippocampi were dissected out and dissociated in papain for 5 min in HEPES-buffered Hank’s solution. Cells were grown on poly-D-Lysine and mouse laminin-coated glass coverslips or only poly-D-Lysine treated plates/dishes in Neurobasal medium with B27 (Invitrogen). The day after plating, neurons were infected with lentivirus in the presence of 0.6µg/ml polybrene for 6 hr. To over-express Rheb, the infection was done twice at the 2nd and 3rd day after plating.
For immunocytochemistry, neurons were fixed with 4% paraformaldehyde/4% sucrose in PBS for 10 min. Fixed cells were permeabilized in 0.1% Triton X-100 for 15 minutes and blocked in 4% normal goat serum plus 2% BSA in PBS. Typically, neurons were incubated with the primary antibody at 4°C overnight and fluorescent secondary antibody for 2 hours at room temperature. Images were captured using Zeiss LSM5 confocal microscope system.
Nucleofection
E18 rat hippocampal and cortical neurons were transfected immediately after dissociation using Amaxa Nucleofector (Amaxa Biosystems) system and plated at 50,000 per each 13-mm2 coverslip. 2 days after plating/transfection, neurons were used for growth cone collapse assay or live imaging.
Ephrin-A1 stimulation and growth cone collapse assay
Ephrin-A1 stimulation and quantification of growth cone collapse was performed as previously described 23. Briefly, to pre-aggregate proteins, recombinant ephrin-A1-Fc, ephrin-A5-Fc (R&D system) or human IgG Fc (Jackson Immunoresearch Labs) was incubated with goat anti-human Fc (Jackson Immnoresearch Labs) at ratio of a 1:4.5. The volume was adjusted with HBSS to 5×. After shaking for 45 minutes at room temperature, proteins were diluted in conditioned culture media and added to the neuronal cultures. Generally, cells were stimulated for 30 minutes before lysis or fixation unless otherwise specified. LPA-stimulated collapse was performed at 1 µM for 10 min prior to fixation. To visualize growth cone collapse, fixed neurons were stained with rhodamine-phalloidin (Cytoskeleton Inc. Denver). Uncollapsed growth cones were identified by the presence of lamellipodia and two or more filopodia. Collapsed growth cones were distinguishable from their non-collapsed counterparts by the presence of a collapsed bulb and having fewer than two filopodia at the leading edge. Counting of growth cone collapse was performed blind to genotype or treatment.
EphA receptor clustering study
Hippocampal neurons infected with GL3 against luciferase or TSC2 shRNA (T2sh1) lentivirus were treated with pre-aggregated ephrin-A1-Fc versus Fc (1–2 µg/ml) for 5 min at 37°C. Neurons were fixed, washed, permeablized (0.1% TritonX-100), and stained with cy3-anti human IgG Fc and Tau-1 antibodies. EphA clusters were imaged with Leica LSM5 confocal system. EphA cluster/punta were counted blind to treatments.
Cholera toxin (CTB) injection and DiI labeling
CTB labeling and analyses were performed as previously described 20. Briefly, mouse pups at postnatal day 14 were injected binocularly with fluorescence-labeled Cholera Toxin B subunit (Alexa-488 or Alexa-594; Molecular Probes) and allowed to recover for 48 hr. Dissected brains were fixed in 4% (v/v) paraformaldehyde for 48 hr at 4°C. Coronal sections (100 µm) were imaged with a 10× objective. All of the image acquisition and analyses were performed blind to genotype. The same gains and image exposure times were used for each label in all specimens to limit variability caused by imaging procedures. Raw images of the dLGN were imported to Photoshop (Adobe). The size of the ipsilateral, the contralateral and the degree of overlap were quantified with the multi-threshold protocol, which is designed to compare overlap across a range of signal–noise values in control versus mutant mice 19, 20.
To compare the distribution of the ipsilateral projection pattern, we employed a line scan technique to calculate the mean pixel intensity of projection terminals in wild-type and Tsc2+/− mice along the long (dorsomedial (DM) to ventrolateral (VL)) axis. The location of the center of the ipsilateral projection was calculated with DM border of the LGN set as 0% and VL border as 100%.
To focally label RGCs in the ventro-temporal quarter of retina, ~ 1 mm3 DiI crystal (Molecular Probe) was embedded below the cornea-sclera border along the VT -> DN orientation into the right eyeball. Mice were sacrificed 5 days later and intercardially perfused with 4% paraformaldehyde in PBS. The eyeballs were extracted and post-fixed in 4% paraformaldehyde for 1 hour while the brains were post-fixed overnight. Retinal whole-mounts were dissected out for checking DiI labeling position and area. Brains corresponding to retinas that had comparable DiI labeling were further sectioned with a Leica cryostat and the contralaterally-labeled dLGN serial sections were imaged with a Nikon fluorescence microscope with a 10× objective and mounted Hamamatsu ORCA-ER camera. Further image processing was performed using Adobe Photoshop 7.0 and the DM-VL extension of dLGN labeling was measured using ImageJ (NIH).
[35S] Metabolic labeling of nascent proteins
De novo protein synthesis was quantified by measuring the incorporation of 35S-Met/Cys (Perkin Elmer) in trichloroacetic acid (TCA) precipitated proteins. Cortical neurons (12 DIV) in Boyden transwells were washed with and switched to Met/Cys/serum-free DMEM (Invitrogen) for 30 min at 37°C to deplete intracellular methionine and cysteine. Prior to stimulation and/or 35S labeling, cell bodies were removed in the top chamber and further washed with equilibrated Met/Cys/serum-free DMEM. The bottom chamber was incubated with pre-clustered Fc or ephrin-A1-Fc (5 µg/ml) in the presence of 35S-Met/Cys (40uCi/ml) for 30 min, or with UO126 (20 µM), rapamycin (20 nM) or anisomycin (40 µM) for 10 min before adding 35S-Met/Cys for another 20 min. Axon-enriched lysates were harvested in 100 µl of RIPA buffer (with protease inhibitors and phosphotase inhibitors). 30 µl of each lysate was precipitated with 1ml of 10% TCA and protein pellets were further dissolved in 150 µl NaOH (1N; 37°C) and pH-adjusted by adding 50 µl of 0.33N HCl. 3× 50 µl aliquots were each added to 10 ml Ultima Gold XR (Perkin Elemer) scintillation fluid and read by a liquid scintillation counter. CPM (counts per minute)/µg protein was obtained by dividing average CPM of triplicates by protein amount (µg) measured with Bradford kit (Bio-rad). Lysates were also added to 3× loading buffer and boiled for 5 min. The gel samples were then resolved on 10% SDS-PAGE gel, fixed in 3% glycerol/10% glacial acetic acid/20% methanol for 1 hour and stained with EZblue solution (Sigma) to confirm the equal loading. Gels were then dried completely (80°C for 2.5 hours) and exposed to a storage phosphor screen for 3 days. [35S] autoradiographs were scanned to ImageQuant 5.0 (GE Healthcare) with a PhosphorImager (Molecular Dynamics).
Live cell imaging and fluorescence recovery after photobleaching (FRAP)
E18 hippocampal neurons were transfected with a destabilized GFP containing a myristoylation sequence and further fused to 3’-untranslated region of beta-actin (referred to as dGFPmyr-bACT 3’UTR thereafter) and d2-mCherry constructs (at ratio 4.5:3) using Amaxa neucleofection. Beta-actin 3’UTR drives axonal expression of dGFP26 and the myristoylation sequence restrict side diffusion of the GFP upon translation. For live imaging, transfected neurons on 35-mm bottom glass dishes (MatTek) were washed and switched to pre-equilibrated Hibernate E Low Fluorescence medium (Brain Bits LLC). FRAP was performed using a Zeiss LSM5 upright confocal microscope fitted with a heating stage and 63× water-immersion objective (numeric aperture = 0.9). Pinhole was set to ~ 7.05 for 488 line and ~ 6.33 for 568 line (corresponding to ~5 µm optic slice) to ensure signal collection from entire thickness of axons. A rainbow spectrum display was employed to adjust the gain and off-set to ensure that the highest pixel intensity is just saturating. Chosen axons were first pre-bleached by scanning (Zoom = 0.9) at 31 sec/frame with 29 sec interval for 10 frames (~ 10min) using 15% laser power (488 line). ROI (region of interest) containing axon terminal/growth cone was bleached using 100% laser power for 120 iterations (< 2min), followed by regular imaging using 15% laser power at speed of 7.86 sec/scan with a 52.3 sec interval over a 20min course. For guidance cue stimulation, pre-aggregated Fc or ephrin-A1-Fc was added at 5 µg/ml final concentration immediately before imaging and FRAP. For the study of translation inhibition, neurons were pretreated with 100 µM of anisomycin or DMSO for 20 ~ 30min prior to FRAP study. Fluorescence intensity was measured using ImageJ and statistic analyses were performed in Prism 5 using two-way ANOVA to compare significant signal recovery over the course of treatment and among different treatments.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Beth Stevens and Dori Shafer for assistance with multi-threshold LGN analysis, Jeffrey Twiss and Tanuja Merianda for assistance with FRAP, Mark Bear, Emily Osterweil and Dilja Krueger for assistance with metabolic labeling experiments. The Tsc2flox/flox mice were gift of Michael Gambello, and Tsc1flox/flox mice were gift of David Kwiatkowski. We are also grateful to Beth Stevens, Chinfei Chen, Zhigang He and members of the Sahin lab for critical reading of the manuscript. This work is supported in part by grants from the NIH (NS58956 to M.S. and HD025938 to E.P.), the John Merck Scholars Fund, Tuberous Sclerosis Alliance, the Manton Foundation and Children’s Hospital Boston Translational Research Program to M.S., and the Children’s Hospital Boston Mental Retardation and Developmental Disabilities Research Center (P01 HD18655). D.Y.N. is supported by a Mentor Based Postdoctoral Fellowship from Autism Speaks. A.D.N. is supported by a grant from the Hearst Foundation. H.B. is supported by an HHMI Research Training Fellowship for Medical Students. We thank Laura Mariani, Abbey Sadowski and Samuel Goldman for technical assistance.
Footnotes
AUTHOR CONTRIBUTIONS
D.Y.N. performed most of the experiments; A.D.N. performed preliminary biochemistry experiments on the relationship between Eph and Tsc pathways; D.Y.N., I.K., T.H. and M.S. conducted and analyzed the retinal projection experiments. J.M.H. and H.B. performed the quantification on the growth cone collapse assays and immunocytochemical staining. S.D. provided guidance with the initial experiments on the Tsc2+/− mice. P.P.P. provided reagents and trouble-shooting for experiments on Tsc2 Ser664 phosphorylation. S.C. and E.B.P. contributed unpublished preliminary data on the Eph-Tsc interaction. M.S. supervised the project. D.Y.N., E.B.P. and M.S. wrote and edited the manuscript.
Reference List
- 1.Crino PB, Nathanson KL, Henske EP. The tuberous sclerosis complex. N Engl J Med. 2006;355:1345–1356. doi: 10.1056/NEJMra055323. [DOI] [PubMed] [Google Scholar]
- 2.Wong M. Mechanisms of epileptogenesis in tuberous sclerosis complex and related malformations of cortical development with abnormal glioneuronal proliferation. Epilepsia. 2008;49:8–21. doi: 10.1111/j.1528-1167.2007.01270.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Manning BD, Tee AR, Logsdon MN, Blenis J, Cantley LC. Identification of the tuberous sclerosis complex-2 tumor suppressor gene product tuberin as a target of the phosphoinositide 3-kinase/akt pathway. Mol Cell. 2002;10:151–162. doi: 10.1016/s1097-2765(02)00568-3. [DOI] [PubMed] [Google Scholar]
- 4.Inoki K, Li Y, Zhu T, Wu J, Guan KL. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat Cell Biol. 2002;4:648–657. doi: 10.1038/ncb839. [DOI] [PubMed] [Google Scholar]
- 5.Flanagan JG. Neural map specification by gradients. Curr Opin Neurobiol. 2006;16:59–66. doi: 10.1016/j.conb.2006.01.010. [DOI] [PubMed] [Google Scholar]
- 6.Feldheim DA, et al. Topographic guidance labels in a sensory projection to the forebrain. Neuron. 1998;21:1303–1313. doi: 10.1016/s0896-6273(00)80650-9. [DOI] [PubMed] [Google Scholar]
- 7.Pfeiffenberger C, et al. Ephrin-As and neural activity are required for eye-specific patterning during retinogeniculate mapping. Nat Neurosci. 2005;8:1022–1027. doi: 10.1038/nn1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pfeiffenberger C, Yamada J, Feldheim DA. Ephrin-As and patterned retinal activity act together in the development of topographic maps in the primary visual system. J Neurosci. 2006;26:12873–12884. doi: 10.1523/JNEUROSCI.3595-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Torborg CL, Feller MB. Spontaneous patterned retinal activity and the refinement of retinal projections. Prog Neurobiol. 2005;76:213–225. doi: 10.1016/j.pneurobio.2005.09.002. [DOI] [PubMed] [Google Scholar]
- 10.Pasquale EB. Eph-ephrin bidirectional signaling in physiology and disease. Cell. 2008;133:38–52. doi: 10.1016/j.cell.2008.03.011. [DOI] [PubMed] [Google Scholar]
- 11.Lin AC, Holt CE. Function and regulation of local axonal translation. Curr Opin Neurobiol. 2008;18:60–68. doi: 10.1016/j.conb.2008.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brittis PA, Lu Q, Flanagan JG. Axonal protein synthesis provides a mechanism for localized regulation at an intermediate target. Cell. 2002;110:223–235. doi: 10.1016/s0092-8674(02)00813-9. [DOI] [PubMed] [Google Scholar]
- 13.Leung KM, et al. Asymmetrical beta-actin mRNA translation in growth cones mediates attractive turning to netrin-1. Nat Neurosci. 2006;9:1247–1256. doi: 10.1038/nn1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wu KY, et al. Local translation of RhoA regulates growth cone collapse. Nature. 2005;436:1020–1024. doi: 10.1038/nature03885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yao J, Sasaki Y, Wen Z, Bassell GJ, Zheng JQ. An essential role for beta-actin mRNA localization and translation in Ca2+-dependent growth cone guidance. Nat Neurosci. 2006;9:1265–1273. doi: 10.1038/nn1773. [DOI] [PubMed] [Google Scholar]
- 16.Campbell DS, Holt CE. Chemotropic responses of retinal growth cones mediated by rapid local protein synthesis and degradation. Neuron. 2001;32:1013–1026. doi: 10.1016/s0896-6273(01)00551-7. [DOI] [PubMed] [Google Scholar]
- 17.Choi YJ, et al. Tuberous sclerosis complex proteins control axon formation. Genes Dev. 2008;22:2485–2495. doi: 10.1101/gad.1685008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Onda H, Lueck A, Marks PW, Warren HB, Kwiatkowski DJ. Tsc2(+/−) mice develop tumors in multiple sites that express gelsolin and are influenced by genetic background. J Clin Invest. 1999;104:687–695. doi: 10.1172/JCI7319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Torborg CL, Feller MB. Unbiased analysis of bulk axonal segregation patterns. J Neurosci Methods. 2004;135:17–26. doi: 10.1016/j.jneumeth.2003.11.019. [DOI] [PubMed] [Google Scholar]
- 20.Stevens B, et al. The classical complement cascade mediates CNS synapse elimination. Cell. 2007;131:1164–1178. doi: 10.1016/j.cell.2007.10.036. [DOI] [PubMed] [Google Scholar]
- 21.Ellsworth CA, Lyckman AW, Feldheim DA, Flanagan JG, Sur M. Ephrin-A2 and -A5 influence patterning of normal and novel retinal projections to the thalamus: conserved mapping mechanisms in visual and auditory thalamic targets. J Comp Neurol. 2005;488:140–151. doi: 10.1002/cne.20602. [DOI] [PubMed] [Google Scholar]
- 22.Huberman AD, Murray KD, Warland DK, Feldheim DA, Chapman B. Ephrin-As mediate targeting of eye-specific projections to the lateral geniculate nucleus. Nat Neurosci. 2005;8:1013–1021. doi: 10.1038/nn1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sahin M, et al. Eph-dependent tyrosine phosphorylation of ephexin1 modulates growth cone collapse. Neuron. 2005;46:191–204. doi: 10.1016/j.neuron.2005.01.030. [DOI] [PubMed] [Google Scholar]
- 24.Di Nardo A, et al. Tuberous sclerosis complex activity is required to control neuronal stress responses in an mTOR-dependent manner. J Neurosci. 2009;29:5926–5937. doi: 10.1523/JNEUROSCI.0778-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Miao H, et al. Activation of EphA receptor tyrosine kinase inhibits the Ras/MAPK pathway. Nat Cell Biol. 2001;3:527–530. doi: 10.1038/35074604. [DOI] [PubMed] [Google Scholar]
- 26.Willis DE, et al. Extracellular stimuli specifically regulate localized levels of individual neuronal mRNAs. J Cell Biol. 2007;178:965–980. doi: 10.1083/jcb.200703209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Aakalu G, Smith WB, Nguyen N, Jiang C, Schuman EM. Dynamic visualization of local protein synthesis in hippocampal neurons. Neuron. 2001;30:489–502. doi: 10.1016/s0896-6273(01)00295-1. [DOI] [PubMed] [Google Scholar]
- 28.Yudin D, et al. Localized regulation of axonal RanGTPase controls retrograde injury signaling in peripheral nerve. Neuron. 2008;59:241–252. doi: 10.1016/j.neuron.2008.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Roche FK, Marsick BM, Letourneau PC. Protein synthesis in distal axons is not required for growth cone responses to guidance cues. J Neurosci. 2009;29:638–652. doi: 10.1523/JNEUROSCI.3845-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ma L, Chen Z, Erdjument-Bromage H, Tempst P, Pandolfi PP. Phosphorylation and functional inactivation of TSC2 by Erk implications for tuberous sclerosis and cancer pathogenesis. Cell. 2005;121:179–193. doi: 10.1016/j.cell.2005.02.031. [DOI] [PubMed] [Google Scholar]
- 31.Torii M, Hashimoto-Torii K, Levitt P, Rakic P. Integration of neuronal clones in the radial cortical columns by EphA and ephrin-A signalling. Nature. 2009;461:524–528. doi: 10.1038/nature08362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Filosa A, et al. Neuron-glia communication via EphA4/ephrin-A3 modulates LTP through glial glutamate transport. Nat Neurosci. 2009;12:1285–1292. doi: 10.1038/nn.2394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ehninger D, et al. Reversal of learning deficits in a Tsc2+/− mouse model of tuberous sclerosis. Nat Med. 2008;14:843–848. doi: 10.1038/nm1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Benson MD, et al. Ephrin-B3 is a myelin-based inhibitor of neurite outgrowth. Proc Natl Acad Sci U S A. 2005;102:10694–10699. doi: 10.1073/pnas.0504021102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Park KK, et al. Promoting axon regeneration in the adult CNS by modulation of the PTEN/mTOR pathway. Science. 2008;322:963–966. doi: 10.1126/science.1161566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yang Q, Inoki K, Kim E, Guan KL. TSC1/TSC2 and Rheb have different effects on TORC1 and TORC2 activity. Proc Natl Acad Sci U S A. 2006;103:6811–6816. doi: 10.1073/pnas.0602282103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Piper M, et al. Signaling mechanisms underlying Slit2-induced collapse of Xenopus retinal growth cones. Neuron. 2006;49:215–228. doi: 10.1016/j.neuron.2005.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Merianda TT, et al. A functional equivalent of endoplasmic reticulum and Golgi in axons for secretion of locally synthesized proteins. Mol Cell Neurosci. 2009;40:128–142. doi: 10.1016/j.mcn.2008.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bolton PF, Park RJ, Higgins JN, Griffiths PD, Pickles A. Neuro-epileptic determinants of autism spectrum disorders in tuberous sclerosis complex. Brain. 2002;125:1247–1255. doi: 10.1093/brain/awf124. [DOI] [PubMed] [Google Scholar]
- 40.Weber AM, Egelhoff JC, McKellop JM, Franz DN. Autism and the cerebellum: evidence from tuberous sclerosis. J Autism Dev Disord. 2000;30:511–517. doi: 10.1023/a:1005679108529. [DOI] [PubMed] [Google Scholar]
- 41.Wong V, Khong PL. Tuberous sclerosis complex: correlation of magnetic resonance imaging (MRI) findings with comorbidities. J Child Neurol. 2006;21:99–105. doi: 10.1177/08830738060210020901. [DOI] [PubMed] [Google Scholar]
- 42.Volpe JJ. Neuronal Proliferation, Migration, Organization, and Myelination. In: Volpe JJ, editor. Neurology of the newborn. Philadelphia: W.B. Saunders; 2001. pp. 45–99. [Google Scholar]
- 43.Sutula T, Cascino G, Cavazos J, Parada I, Ramirez L. Mossy fiber synaptic reorganization in the epileptic human temporal lobe. Ann Neurol. 1989;26:321–330. doi: 10.1002/ana.410260303. [DOI] [PubMed] [Google Scholar]
- 44.Courchesne E, et al. Unusual brain growth patterns in early life in patients with autistic disorder: an MRI study. Neurology. 2001;57:245–254. doi: 10.1212/wnl.57.2.245. [DOI] [PubMed] [Google Scholar]
- 45.Barnea-Goraly N, et al. White matter structure in autism: preliminary evidence from diffusion tensor imaging. Biol Psychiatry. 2004;55:323–326. doi: 10.1016/j.biopsych.2003.10.022. [DOI] [PubMed] [Google Scholar]
- 46.Henske EP, et al. Allelic loss is frequent in tuberous sclerosis kidney lesions but rare in brain lesions. Am J Hum Genet. 1996;59:400–406. [PMC free article] [PubMed] [Google Scholar]
- 47.Tavazoie SF, Alvarez VA, Ridenour DA, Kwiatkowski DJ, Sabatini BL. Regulation of neuronal morphology and function by the tumor suppressors Tsc1 and Tsc2. Nat Neurosci. 2005;8:1727–1734. doi: 10.1038/nn1566. [DOI] [PubMed] [Google Scholar]
- 48.Goorden SM, van Woerden GM, van der Weerd L, Cheadle JP, Elgersma Y. Cognitive deficits in Tsc1+/− mice in the absence of cerebral lesions and seizures. Ann Neurol. 2007;62:648–655. doi: 10.1002/ana.21317. [DOI] [PubMed] [Google Scholar]
- 49.Shamah SM, et al. EphA receptors regulate growth cone dynamics through the novel guanine nucleotide exchange factor ephexin. Cell. 2001;105:233–244. doi: 10.1016/s0092-8674(01)00314-2. [DOI] [PubMed] [Google Scholar]
- 50.Hernandez O, Way S, McKenna J, 3rd, Gambello MJ. Generation of a conditional disruption of the Tsc2 gene. Genesis. 2007;45:101–106. doi: 10.1002/dvg.20271. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.