

Abstract
Engineering extracellular matrices that utilize the body's natural healing capacity enable the progression of regenerative therapies. Fibrin, widely used as a surgical sealant, is one such matrix that may be augmented by the addition of protein factors to promote cell infiltration and differentiation. The thrombin-catalyzed conversion of fibrinogen to fibrin exposes N-terminal fibrin knobs that bind to C-terminal pockets to form the fibrin network. Here, we have created a platform system for the production of therapeutic proteins that capitalize on these native knob:pocket interactions for protein delivery within fibrin matrices. This system enables the retention of therapeutic proteins within fibrin without additional enzymatic or synthetic crosslinking factors. Using an integrin-binding fibronectin fragment as a model protein, we demonstrate that engineered knob-protein fusions bind consistently and specifically to fibrin(ogen). Equilibrium dissociation constants (KD) obtained using surface plasmon resonance indicate that these fusions have μM binding affinities, comparable to the native knob-containing fibrin fragments. The specificity of these interactions was verified by ELISA in the presence of molar excess of competing knob mimics. Release profiles and real-time confocal imaging demonstrate that the fusions were retained within fibrin matrices, even under the stringent continuous perfusion conditions used in the latter. In summary, this work explores the benefits and limitations of engaging native, biologically-inspired, non-covalent knob-pocket interactions within fibrin(ogen) for the retention of therapeutic proteins in fibrin matrices and provides insight into the stability of native knob:pocket interactions within fibrin networks.
Keywords: Fibrin, Fibrinogen, Molecular biology, Recombinant protein, Affinity, Controlled drug release
INTRODUCTION
Fibrin, formed in response to vascular injury, constitutes the protein polymeric scaffold known as the provisional extracellular matrix. Fibrin serves as the temporary initial scaffolding for the invasion of inflammatory and repair cells into wound environments. Fibrinogen, the inactive precursor to fibrin, is a 340 kDa plasma glycoprotein that circulates in blood plasma at concentrations of 2 to 4 mg/mL. As a part of its normal functions, fibrin(ogen) features a variety of binding sites for self-regulatory enzymes (including thrombin, factor XIII, plasminogen, tissue plasminogen activator), ECM components, growth factors, cytokines and cell receptors; components that are critical in the remodeling of the matrix as part of the wound healing response [1, 2]. Due to the ease of purification of this highly expressed plasma protein and its native role in the guidance of wound repair, the fibrinogen/fibrin system is employed in a number of surgical hemostats and tissue sealants (FDA-approved products include Tisseel®, EvicelTM) and is actively used as a biomaterial for developing therapeutic strategies in regenerative medicine (reviewed in [3-5]). Similarly, we are interested in utilizing fibrin as a platform biomaterial and investigated a novel method to incorporate non-fibrin binding proteins within fibrin matrices to enhance the efficacy of fibrin in guiding regenerative processes.
Fibrinogen is a symmetrical, rod-shaped molecule comprising two sets of three polypeptides (Aα-, Bβ-, and γ-chains) assembled lengthwise and crosslinked at their N-termini via multiple disulfide bonds, forming a single central N-terminal disulfide knot (NDSK) and two distal globular domains (D domains). During the activation of fibrinogen to fibrin monomer, the serine protease thrombin cleaves the N-termini of both the Aα- and Bβ-chains, exposing new cryptic residues (i.e. A- and B-knobs) in the NDSK domain that bind to complementary sites (i.e. a- and b-pockets) near the two distal C-termini of the γ- and Bβ-chains within the D domains (Fig. 1). These non-covalent knob:pocket interactions lead to the assembly of fibrin protofibrils that make up the fibrin network [6, 7]. These networks are further stabilized by activated transglutaminase factor XIII (factor XIIIa) that covalently crosslinks γ- and α-chains of adjacent fibrin monomers within the protofibril [8].
Fig. 1.
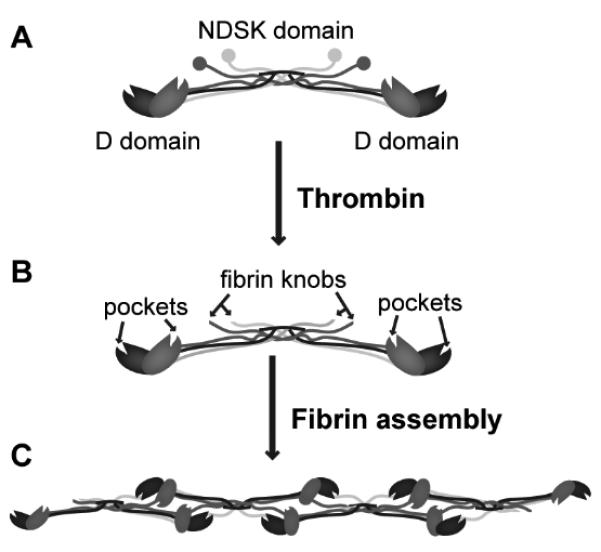
Schematic of fibrinogen and its activation. The activation of fibrinogen (A) by thrombin exposes fibrin knobs in the NDSK domain that bind to complementary pockets in the D domain of the molecule (B). Such knob:pocket interactions are key in the assembly of fibrin protofibrils (C) in fibrin network formation.
Significant efforts have been made to develop and enhance fibrin as a tissue scaffold and drug delivery vehicle through both modification of the molecule and engagement of the native polymer system biology. For instance, functionalized polyethylene glycol was explored as a means to covalently tether therapeutic proteins to fibrinogen's backbone thus enabling their delivery in fibrin polymers [9]. Alternatively, factor XIIIa, also responsible for the covalent attachment of other non-fibrin proteins (e.g. fibronectin, α2-plasmin inhibitor) to the C-terminus of the Aα chains in vivo, has been shown to be a reproducible method of crosslinking RGD-containing peptides into fibrin gels during coagulation [10]. The factor XIIIa substrate sequence has subsequently been used for the incorporation of recombinantly-produced growth factors, cell adhesion molecules and morphogens into fibrin matrices, usually with an additional protease-sensitive cleavage site to facilitate their release from fibrin [11-15]. As an intriguing spin-off, bi-domain peptides comprising the factor XIIIa substrate sequence and the heparin-binding domain have been used to increase the concentration of fibrin-bound heparin, thereby improving the retention characteristics of heparin-binding growth factors [16-19]. In particular, such biomimetic affinity-based systems are advantageous for the local delivery of factors that are internalized by cells as part of the signaling process.
In this work, we have taken an alternative approach to the delivery of proteins from fibrin matrices by capitalizing on the native knob:pocket interactions involved in fibrin assembly. Short synthetic peptides modeled after the fibrin knob sequences are known to bind to the C-terminal pockets of fibrin(ogen), as illustrated by numerous binding studies and X-ray crystallographic structures [20-23]. Specifically, various tetrapeptides have significantly different affinities for fibrin(ogen) pockets, with GPRP (Gly-Pro-Arg-Pro; A-knob tetrapeptide mimetic) having the greatest affinity followed by GPRV (Gly-Pro-Arg-Val; native A-knob tetrapeptide), then GHRP (Gly-His-Arg-Pro; native B-knob tetrapeptide). Therefore, an affinity-based scheme stemming from an inherent non-covalent interaction such as the knob:pocket interaction should retain proteins within fibrin matrices without intermediary molecules or enzymes, like factor XIIIa.
We hypothesized that the fusion of tetrapeptide fibrin knob sequences to proteins will endow fibrin-binding capacities to non-fibrin-binding proteins via knob:pocket interactions. We have developed a platform system to rapidly clone and express recombinant proteins displaying different tetrapeptide sequences on their N-terminus, thus mimicking knobs displayed on activated fibrin molecules. The integrin-binding fibronectin 9th and 10th type III repeats (FnIII9-10) are amenable to recombinant protein production, do not bind fibrin(ogen), and have previously been shown to induce osteogenic differentiation in mesenchymal stem cells [24, 25]. Using this protein domain for our model protein, we demonstrate that knob-protein fusions bind stably to fibrin(ogen) via specific knob:pocket interactions. Retention of these fusion proteins within fibrin matrices was evaluated through the use of ELISA-based protein release assays as well as real-time confocal imaging of perfused fibrin matrices.
MATERIALS AND METHODS
Modification of bacterial expression vector
The original thrombin cleavage site in the pGEX4T-1 vector (GE Healthcare, Piscataway, NJ) was modified from LVPR↓GSPE to LVPR↓GPRV, LVPR↓GPRP and LVPR↓GHRP using the QuikChange® II-E Site-Directed Mutagenesis Kit (Stratagene, La Jolla, CA). All plasmids were introduced to and maintained in the electro-competent XL-1 Blue E. coli strain provided and cultured in LB + ampicillin plates at 37°C. Plasmids were extracted from cultures using the QIAquick Spin Miniprep Kit (QIAGEN, Valencia, CA) and verified via sequencing (Johns Hopkins Synthesis & Sequencing Facility, Baltimore, MD).
Amplification and insertion of FnIII9-10 into expression vectors
Fibronectin's cell-binding 9th and 10th type III repeats (FnIII9-10) was used as our model protein. The SalI site in the multiple cloning site (MCS) was used to insert the FnIII9-10 open reading frame into the pGEX4TGSPE, pGEX4TGPRV, pGEX4TGPRP and pGEX4TGHRP vectors obtained above using standard molecular cloning techniques (enzymes purchased from NEB, Ipswich, MA). Briefly, the FnIII9-10 open reading frame was amplified in a high fidelity polymerase chain reaction with Phusion High Fidelity DNA polymerase from the previously established pGEX4T-1-FnIII9-10 plasmid [25] using primers with flanking SalI sequences (underlined) and introducing Gly-Gly-Cys (bold) on the C-terminal if necessary: ACTGGTCGACTGGGTCTTGATTCCCCAACT and ACTGGTCGACTCAGCAACCACCTGTTCGGTAATTAATGGA. Vectors and insert were digested with SalI. The vectors were additionally dephosphorylated using heat-deactivate-able Antartic phosphatase to prevent self-ligation. The respective vectors and insert were ligated using T4 DNA Ligase and transformed into electro-competent XL-1 Blue cells. Selection for successfully ligated plasmids containing the insert was made on LB + ampicillin plates. The orientation of the FnIII9-10 insert was verified by in-colony PCR-screening using forward or reverse primers on the plasmid in combination with an internal primer within the insert. Colonies with the insert in the correct orientation were grown up for plasmid extraction. All plasmids were verified via sequencing.
Protein production and purification
Plasmids containing the FnIII9-10 insert were transformed into electro-competent BL21 E. coli and protein production of the Glutathione S-transferase (GST) -tagged proteins stimulated as recommended by the manufacturer. Following appropriate culture, cells were pelleted by centrifugation at 4°C and resuspended in ice-cold PBS supplemented with protease inhibitor (Roche, Indianapolis, IN), then lysed by adding 1 mg/mL of lysozyme followed by sonication. 1% Triton X and 10 U/mL of DNase was added and the lysate further incubated for 30 min with gentle agitation. The cell lysate was cleared of cellular debris by centrifugation followed by filtration through a 0.22 μm pore filter. Purification of the recombinant proteins was performed following the manufacturer's recommendation using the ÄKTAFPLC with a GST Prep FF 16/10 column (GE Healthcare) for affinity purification of GST-tagged proteins. Washing and binding steps were done in filter-sterilized PBS and elution completed with glutathione (GSH) buffer (50 mM Tris-HCl, 10 mM reduced GSH, pH 8.0). The GSH buffer was then exchanged to PBS using an Amicon Ultra-15 centrifugal filter with MWCO 10,000 (Millipore, Billerica, MA). The GST-tagged protein was incubated overnight with bovine thrombin (MP Biomedicals, Solon, OH) dosed at 10 U per mg recombinant protein and the cleaved protein solution was reintroduced on the GST Prep FF 16/10 column to remove the GST tag, followed by the HiTrap Benzamidine FF column (GE Healthcare) to remove thrombin. Gxxx-FnIII9-10-(C) proteins were assessed for purity by SDS-PAGE and quantitated at Abs280nm using the Nanodrop 1000 (Thermo Scientific, Wilmington, DE) using extinction coefficients calculated using an online peptide property calculator (found at http://www.basic.northwestern.edu/biotools/proteincalc.html), then aliquoted and stored at −80°C until use.
Preparation of fibrinogen fragment D
Human fibrinogen (#FIB3, Enzyme Research Laboratories, South Bend, IN) at 2 mg/mL was digested with 0.1 U/mL human plasmin (Enzyme Research Laboratories) in HEPES+Ca buffer (150 mM NaCl, 5 mM CaCl2, 25 mM HEPES, pH 7.4) overnight at room temperature. Fragment D (or D-domain) was isolated as previously described, with slight modifications, using a polymeric resin with covalently linked GPRPAA peptide [23]. Briefly, the plasmin-digested fibrinogen and GPRPAA-beads were incubated for 30 min, with occasional agitation. The unbound protein fragments were removed with exhaustive washing with HEPES+Ca buffer. Fragment D was eluted using 1 M sodium bromide, 50 mM sodium acetate, pH 5.3 and exchanged back into HEPES+Ca buffer using a centrifugal filter with MWCO 10,000.
Surface Plasmon Resonance (SPR) binding assays
The Biacore 2000 was used to perform SPR experiments to determine the equilibrium dissociation constants (KD) of the various Gxxx- FnIII9-10 proteins to fibrinogen fragment D covalently immobilized to gold-coated coverslips via self-assembled monolayer (SAM) surface chemistry. Mixed SAMs were generated on gold-coated chips as described previously by incubating with a 1 mM mixture of tri(ethylene glycol)-terminated alkanethiol (HS-(CH2)11–(OCH2CH2)3–OH; EG3) and carboxylic acid-terminated alkanethiol (HS-(CH2)11–(OCH2CH2)6– OCH2COOH; EG6-COOH) thiols for 4 h [24]. After mounting the gold chip in the Biacore 2000, the EG6-COOH component of the SAM was activated with 200 mM EDC and 50 mM NHS in 0.1 M 2-(N-morpho)-ethanesulfonic acid and 0.5 M NaCl, pH 6.0 (5 μL/min for 10 min). Fragment D was then immobilized via NHS-amine linkages (5 μL/min for 10 min). Unreacted NHS groups were quenched with 20 mM ethanolamine (10 μL/min). Upon stabilization of the baseline signal, varying concentrations of the Gxxx-FnIII9-10 were flowed at 25 μL/min for 4 min immediately followed by a 10 min dissociation phase; the fragment D surface was regenerated between analyte runs. All sensorgram response curves were normalized to a reference flow cell in which fragment D was not immobilized. SPR sensorgrams were analyzed and equilibrium constants were determined with Scrubber 2 and ClampXP software (Center for Biomolecular Interactions Analysis, University of Utah) [26].
ELISA binding assays
A modified ELISA technique was used to establish the affinity of fibrinogen for the various Gxxx-FnIII9-10-C proteins covalently immobilized on maleimide-activated plates via their C-terminal cysteines. Sulfhydryl-containing proteins/peptides were added to pre-blocked maleimide-activated 96-well plates (Pierce, Thermo Scientific, Rockford, IL) at 10 μg/mL in conjugation buffer (CB) comprising 150 mM NaCl, 100 mM phosphate, 10 mM EDTA, pH 7.2, with attachment occurring via maleimide-sulfhydryl (thioether) linkages. TCEP (1 mM) was added to maintain sulfhydryl groups in the reduced form. Following, unreacted maleimide groups were quenched with 20 μg/mL cysteine in CB. Human fibrinogen (25 μg/mL) was incubated with the substrates and unbound protein removed by washing. Bound fibrinogen was detected using HRP-conjugated goat anti-fibrinogen antibody (MP Biomedicals #55239) and 1-StepTM Ultra TMB-ELISA (Pierce). The TMB reaction was quenched with 1 M H2SO4 before measuring the Abs450nm using the SpectraMax M2 (Molecular Devices, Sunnyvale, CA). All intervening wash steps were conducted using washing buffer (WB) comprising 150 mM NaCl, 100 mM phosphate, 0.05% Tween-20, pH 7.2; all binding steps were conducted using binding buffer (BB) comprising PBS + 1% BSA. For basic affinity assays, the concentration of the fusion proteins was varied. For specificity assays, varying concentrations of Gxxx tetrapeptides (Genscript, Piscataway, NJ) were added to the fibrinogen solution during incubation with the covalently immobilized proteins.
Biotinylation of Gxxx-FnIII9-10 proteins
Gxxx-FnIII9-10-C proteins were conjugated via the sulfhydryl group of the C-terminal cysteine to maleimide-functionalized biotin (maleimide-PEG2-biotin; Pierce #21902) following the manufacturer's recommended protocol. Unreacted maleimide-PEG2-biotin was removed using Slide-A-Lyzer dialysis cassettes with MWCO 3,500 (Pierce). The extent of biotinylation was determined using the Pierce Biotin Quantitation Kit (Pierce).
Fibrin clotting assays
Clotting was initiated in a 96-well plate format and the Abs350nm used as a standard measure of turbidity. Briefly, increasing doses of the Gxxx-FnIII9-10-biotin proteins were preincubated with 4 mg/mL human fibrinogen (ERL #FIB3) in Tris+Ca buffer (140 mM NaCl, 5 mM CaCl2, 20 mM Tris, pH 7.4). Following the 1 h preincubation, clotting was initiated by adding 1 U/mL of human thrombin (ERL) or batroxobin moojeni (Centerchem, Norwalk, CT), and 1 U/mL factor XIIIa (kindly donated by Baxter AG, Vienna Austria). Real time measurements of clot turbidity were taken every minute for 1 h. To determine the amount of unclotted protein, the clots were spun down and the supernatant or clot liquor was analyzed for total protein using the Quant-iT protein assay (Invitrogen, Carlsbad, CA).
Rheological assays
The Bohlin CVO 120 high resolution rheometer (Malvern Instruments, Westborough, MA) with plate-plate geometry was used to assess the viscoelastic characteristics of fibrin clots at room temperature. Briefly, fibrinogen (preincubated with or without Gxxx-FnIII9-10-biotin as indicated) and enzyme mixtures with identical compositions as those used in the turbidity and clottability assays were mixed by pipetting and immediately added to the bottom plate. The upper plate (14 mm diameter) was immediately lowered to a gap size of 1 mm and the mixture was allowed to polymerize for 30 min in a humid chamber. Following, oscillating measurements were taken over a frequency range of 0.05 to 1.0 Hz at a constant strain of 0.5%.
Protein release assays
Analogous to the clotting assays, Gxxx-FnIII9-10-biotin was preincubated with fibrinogen in Tris+Ca buffer in 2 mL conical bottom tubes. Clotting was initiated by adding thrombin or batroxobin, and factor XIIIa. After a one-hour incubation, the resulting clot was overlaid with 1 mL Tris+Ca buffer. The entire volume of buffer was removed and replaced with fresh buffer at 1, 4, 8, 12, 24, 48, 72, 96-hour timepoints for analysis. Gxxx-FnIII9-10-biotin proteins in the removed supernatant samples were quantitated using a sandwich ELISA. Briefly, 96-well ELISA plates were coated with 5 μg/mL streptavidin, then washed and blocked with 1% BSA. The sample, mouse monoclonal anti-FnIII9-10 antibody (HFN7.1; Developmental Studies Hybridoma Bank, Iowa City, IA) and HRP-conjugated goat anti-mouse antibody (Pierce #1858413) were added sequentially to the plate with intervening wash steps. TMB was added to react with the HRP and the reaction quenched with H2SO4 before measuring the Abs450nm.
Perfusion assays with confocal microscopy
To examine the retention of knob fusion proteins within fibrin polymer matrices, we coupled microfluidic techniques with confocal microscopy. Microfluidic chamber templates (Y-shaped: 4 cm length, 1 mm width, 60-75 μm height) were fabricated with standard photolithography techniques on silicon wafers. Polydimethylsiloxane (PDMS; Sylgard 184; Dow Corning, Midland, MI) was cast onto the silicon wafer template, cured, and removed from the wafer to obtain the microfluidic chambers. Individual PDMS devices were mounted onto clean coverslips immediately after receiving plasma treatment (negative polarization for 1 min at 25 mAmp). Fibrinogen was covalently immobilized to the interior surface of the PDMS channels to promote uniform fibrin matrix formation on all chamber wall surfaces, thereby preventing the separation between the chamber surface and fibrin matrix during perfusion. Briefly, the device channels were treated with 1.5% 3-methacryloxypropyltrimethoxysilane in methanol (20 min), then 0.5% gluteraldehyde (30 min), and then incubated with fibrinogen (100 μg/ml; 30 min). After subsequent rinsing with Tris buffer, a 2 mg/mL fibrinogen solution containing 10% labeled fibrinogen (Alexa Fluor-555; Invitrogen), 1 U/mL thrombin, 1 U/mL factor XIIIa, and labeled Gxxx-FnIII9-10 (Alexa Fluor-633; Invitrogen) in a 1:10 molar ratio (Gxxx-FnIII9-10:fibrinogen) was injected into the chamber and allowed to polymerize for 1 h. Following polymerization, Tris+Ca buffer was perfused through the chamber at 10 μL/min for 30 min. Confocal z-stack micrographs (2 μm thick stack with 0.5 μm intervals; Zeiss LSM 510) were taken before the Tris+Ca buffer rinse and then every 5 min after commencement of perfusion. Throughout the duration of the experiment and across sample groups, the image acquisition settings remained constant (i.e. pinhole, master gain) for both fluorophores. Colocalization and release of the Gxxx-FnIII9-10 was evaluated and quantitated with confocal software ZEN (Carl Zeiss International). Using the colocalization analysis tool in ZEN, baseline thresholds for both fibrin and Gxxx-FnIII9-10 signal intensity were held constant for each set of images. Calculated parameters included the relative area of colocalization (colocalized area relative to total area), mean signal intensity for each channel above the threshold, colocalization coefficients for each fluorescent channel (total number of colocalized pixels relative to total number of pixels above the threshold for a given channel; 0 = no co-localization and 1 = all pixels colocalized), and Pearson's correlation coefficient (R; 1 = high correlation; < 0 = no correlation).
Statistics
All experimental data are reported as mean ± SEM of at least 3 independent triplicate experiments. Results were analyzed using GraphPad PRISM 5.0. Statistical comparisons for all experimental sets were based on one-way ANOVA using the Tukey post-hoc test for pair-wise comparisons with significance defined by p < 0.05.
RESULTS
Gxxx-FnIII9-10 recombinant protein production
We developed an expression vector system allowing the simple and rapid cloning of any gene-of-interest into a series of vectors thereby enabling the creation of any desired protein endowed with an N-terminal fibrin knob sequence. The pGEX4T-1 expression vector allows the expression of proteins with an N-terminal 26 kDa GST tag. This tag can then be removed via thrombin cleavage, analogous to the process of fibrinogen activation wherein protein segments are cleaved from the N-termini of the Aα- and Bβ-chains. Using the original pGEX4T-1 vector, the N-terminal sequence exposed upon thrombin cleavage of the GST-tagged protein is GSPE (Gly-Ser-Pro-Glu), which does not bind to fibrin pockets. We modified the corresponding coding sequence on the expression vector to each of three knob tetrapeptide sequences, namely GPRP, GPRV and GHRP, using site-directed mutagenesis (Fig. 2A). Following, we inserted the coding sequence for our model protein, the 20 kDa FnIII9-10 fragment, using standard molecular cloning techniques. The model protein has a known crystal structure, can be produced using standard bacterial expression systems, and may be detected via immunological assays using a number of commercially available antibodies [24, 25, 27]. Fig. 2B shows a representative Coomassie-stained SDS-PAGE gel illustrating product purity at the different stages of protein purification using affinity purification via the ÄKTApurifier system. Notably, the GST tag serves dual purposes as affinity tag and a measure of thrombin cleavage efficacy. Using this platform cloning and expression system we successfully expressed and purified the following knob-protein fusions: GSPE-FnIII9-10 (non-binding control), GPRP-FnIII9-10, GPRV-FnIII9-10 and GHRP-FnIII9-10.
Fig. 2.

Gxxx-FnIII9-10 protein production and purification. (A) Molecular engineering of the pGEX4T-1 vector to express proteins with variable thrombin cleavage sites, LVPR↓Gxxx. Site-directed mutagenesis was used to modify the coding sequence for the thrombin cleavage site (T). Next, the open reading frame of the protein-of-interest, FnIII9-10, was inserted into the multiple cloning site (MCS). The expressed proteins comprise an N-terminal glutathione S-transferase (GST) tag, followed by the thrombin cleavage site and protein-of-interest. (B) Coomassie-stained SDS-PAGE gel demonstrating protein purity at the different stages of purification using affinity chromatography. GST-tagged Gxxx-FnIII9-10 (lane 2) was purified from contaminating proteins in the bacterial cell lysate (lane 1) via the GST affinity column. Thrombin-catalyzed cleavage releases the 26 kDa GST-tag from the 20 kDa Gxxx-FnIII9-10 protein (lane 3). Next, the thrombin and GST-tag were removed using benzamidine and GST affinity columns respectively, leaving the desired protein-of-interest (lane 4).
Evaluating Gxxx-FnIII9-10 fibrinogen-binding affinity using SPR
The affinity of Gxxx-FnIII9-10 proteins for immobilized fibrinogen fragment D was examined using surface plasmon resonance (SPR). Fibrinogen fragment D, comprising the C-terminal polymerization pockets of fibrinogen (i.e. D-domain) without the N-terminal knobs, was covalently immobilized on gold chips via self-assembled monolayers (SAMs). Following, solutions of Gxxx-FnIII9-10 (the analyte) were flowed across the fragment D surface, with protein-protein interactions registering as an increase in response units, as measured using the Biacore 2000 system (Fig. 3A). For each protein, the equilibrium response at each concentration was plotted against concentration and fit to a single-site binding model to determine the equilibrium dissociation constant, KD (Fig. 3B). GPRP-FnIII9-10 had the greatest affinity for immobilized fibrinogen fragment D, with a KD of 3.0 ± 0.1 μM. This is congruent with recently presented SPR data reporting KDs for knob-containing NDSK fragments in similar μM range [28]. In contrast, GPRV-FnIII9-10 generated a KD of 125 ± 6 μM while both GHRP-FnIII9-10 and the control GSPE-FnIII9-10 demonstrated no detectable binding to fragment D. Additionally, the specificity of the GPRP-FnIII9-10 interaction with the pocket was demonstrated by adding a 10-molar excess of soluble GPRP peptide to the flow analyte solution (Fig. 3B). We observed a significant increase in KD from 3.0 μM to 43 ± 4 μM under these competing conditions, suggesting that that the fibrin(ogen)-binding capacities of GPRP-FnIII9-10 is attributed to its N-terminal knob sequence. Based on the promising SPR data, further investigations were focused on the high affinity GPRP-FnIII9-10 protein and the non-binding control GSPE-FnIII9-10.
Fig. 3.
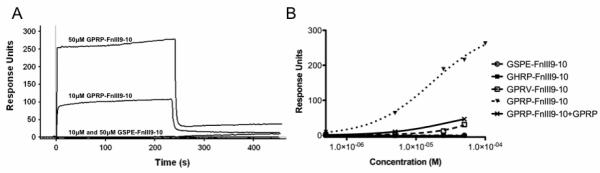
SPR studies demonstrating Gxxx-FnIII9-10 affinity for fibrinogen fragment D. (A) Representative sensorgrams showing protein-protein interaction between immobilized fragment D and GPRP-FnIII9-10 but not the non-binding control GSPE-FnIII9-10. Binding interactions were observed as an increase in response units (1 RU = 1 pg/mm2). (B) At equilibrium, the sensorgram responses for each analyte were plotted against the injection concentration and fit to a single site equilibrium model. Global fits were performed to determine KD. The highest affinity was observed with GPRP-FnIII9-10.
Evaluating GPRP-FnIII9-10 fibrinogen-binding affinity using ELISA
In addition to the Gxxx-FnIII9-10 recombinant proteins described above, we have expressed and purified equivalent versions with an additional C-terminal cysteine. The Gxxx-FnIII9-10-C proteins were immobilized on maleimide-activated 96-well plates via maleimide-sulfhydryl chemistry, allowing the unidirectional presentation of their N-terminal knobs to fibrinogen-containing solutions in the wells. Using standard ELISA techniques, the bound fibrinogen was detected using an anti-fibrinogen HRP-conjugate, with stronger signals corresponding to increased amounts of bound fibrinogen. Complementing the SPR data, the ELISA results indicate that soluble fibrinogen bound to immobilized GPRP-FnIII9-10-C but not the control, GSPE-FnIII9-10-C (Fig. 4A). This demonstrates the mechanism of action, that fibrinogen is binding the knob-protein fusion via the knob sequences and not via interaction with the fibronectin domains in our model protein.
Fig. 4.

ELISA studies analyzing GPRP-FnIII9-10 affinity for fibrinogen. (A) Affinity assays demonstrating binding between immobilized GPRP-FnIII9-10-C, but not GSPE-FnIII9-10-C, and fibrinogen. ELISA plate readouts (Abs450nm) were normalized against the maximum signal obtained using GPRPFPAC peptide (Genscript) at a coating concentration of 20 μg/mL. (B) Specificity assays demonstrating that GPRP and GPRV peptides compete with immobilized GPRP-FnIII9-10 for fibrinogen. ELISA plate readouts were normalized against the signals obtained in the presence of the non-binding GSPE peptide to correct for osmolarity.
To further demonstrate specificity, soluble tetrapeptides modeled after fibrin knob sequences were added in large molar excess (102- to 104-fold) to the fibrinogen solution that was to be incubated with immobilized GPRP-FnIII9-10-C. As expected, free GPRP tetrapeptides competed with the immobilized GPRP-FnIII9-10-C for binding to fibrinogen, resulting in a dose-dependent inhibition of fibrinogen binding following a one-hour pre-incubation (Fig. 4B). This result suggests that the GPRP sequence is the mediator of the interaction between the immobilized protein and fibrinogen. The tetrapeptides GPRV and GHRP are also known to bind fibrinogen via knob:pocket interactions, albeit with reduced affinities [21]. Our results indicate that GPRV competed with GPRP-FnIII9-10 for binding to fibrinogen, but to a lesser extent as compared to GPRP, while GHRP was not a significant competitor to GPRP-FnIII9-10 under the experimental conditions. Notably, these results agree with our SPR data (vide supra) indicating that the affinity constant of GPRV-FnIII9-10 was at least an order of magnitude smaller than that of GPRP-FnIII9-10, while GHRP-FnIII9-10 had little affinity for fibrinogen fragment D. These results support the interpretation that the interaction between the GPRP-FnIII9-10 protein and fibrinogen is primarily mediated by the respective N-terminal tetrapeptide sequences through specific interactions with fibrin(ogen) pockets.
Fibrin assembly in the presence of GPRP-FnIII9-10
Since knob:pocket interactions are involved in fibrin assembly, the addition of non-native knob-protein fusions could potentially interfere with this process and impede matrix formation, thereby limiting the utility of this affinity system for the retention of proteins within fibrin matrices. We were therefore interested in evaluating the impact of such knob-protein fusions on fibrin assembly. GPRP-FnIII9-10-C was selectively conjugated to maleimide-functionalized biotin, generating GPRP-FnIII9-10-biotin conjugates that can be quantitated via sandwich ELISA. GPRP-FnIII9-10-biotin proteins were preincubated with fibrinogen before clotting was induced by the addition of factor XIIIa and either thrombin (exposes both A- and B- knobs) or batroxobin (exposes the A-knobs). The process of fibrin assembly can be inferred from the gross absorbance of the fibrinogen-containing mixture since the lateral aggregation of protofibrils during clotting results in a rapid rise in turbidity [29]. The turbidity curves obtained suggest that fibrin assembly in the presence of thrombin (Fig. 5A) or batroxobin (Fig. 5B) was not significantly impacted by the presence of GPRP-FnIII9-10-biotin, or the control GSPE-FnIII9-10-biotin, at the conjugate:fibrinogen molar ratio of 1:10. Rheological assaying of clots formed following a 30-minute clotting time suggests that the presence of either conjugate did not significantly affect the native viscoelastic properties of the clots (Fig. 5C). Moreover, quantitation of the soluble proteins in the clot liquor (the remaining supernatant after spinning down the insoluble fibrin) also indicates that the presence of GPRP-FnIII9-10-biotin, or the control GSPE-FnIII9-10-biotin, did not significantly impact the clottability of the mixture (Fig. 5D). These results agree with past research showing that at least a 100-fold excess of the GPRP tetrapeptide was necessary to completely inhibit fibrin assembly [30] and suggest that the presence of knob-protein fusions will not significantly interfere with the formation of the fibrin matrix when present at the nanogram concentrations relevant for use in protein delivery systems for growth factors.
Fig. 5.

Fibrin assembly in the presence of GPRP-FnIII9-10 and GSPE-FnIII9-10. Clots were formed from 6 μM (= 2 mg/mL) fibrinogen solutions containing 0.6 μM GPRP-FnIII9-10-biotin or GSPE-FnIII9-10-biotin upon the addition of 1 U/mL factor XIIIa and 1 U/mL human α-thrombin or batroxobin moojeni. Control clots were formed in the absence of the conjugates. (A) Turbidity curves obtained in the presence of thrombin. (B) Turbidity curves obtained in the presence of batroxobin. (C) Complex moduli of the clots after 30 min clotting time. Measurements were taken at a frequency of 0.22 Hz and strain of 0.5%. (D) Quantitation of soluble protein in the clot liquor from clots after 1 h clotting time.
Evaluating GPRP-FnIII9-10 retention in fibrin matrices using release assays
We have established that GPRP-FnIII9-10-(biotin) binds fibrin(ogen) pockets and does not interfere with the clotting process at the loading concentrations desired for our targeted application. We then proceeded to examine the retention of GPRP-FnIII9-10-biotin conjugates within three-dimensional fibrin matrices by monitoring protein release from the clots into the surrounding buffer reservoir. In particular, we were interested in observing the release profiles from both normal thrombin-catalyzed clots and batroxobin-catalyzed clots; batroxobin selectively exposes the A-knobs but not the B-knobs, theoretically leaving an excess of binding pockets that should improve retention characteristics. As described in the previous section, the GPRP-FnIII9-10-biotin conjugates were preincubated with fibrinogen at a 1:10 molar ratio (conjugate:fibrinogen), before clotting was induced by the addition of factor XIIIa and either thrombin or batroxobin. After a one-hour incubation period, the clot was overlaid with pre-warmed buffer and incubated at 37°C with agitation. Samples taken at regular intervals were evaluated for the presence of fibrin(ogen) and conjugate via sandwich ELISAs. In particular, the HFN7.1 conformation-specific antibody was used against the conjugates to further demonstrate that the conformational integrity of the FnIII9-10 was preserved during the release assay. Additionally, GPRP-FnIII9-10-biotin is capable of binding to ELISA plates coated with fibrinogen and being detected by the same antibody (data not shown), indicating that the conformational integrity of the FnIII9-10 domain is preserved even while bound to fibrin(ogen).
The amount of soluble fibrin(ogen) in all the samples remained below the detection limit (≈10 ng/mL) of the fibrin(ogen) ELISA, indicating that the clots remained intact during the four-day incubation period (data not shown). From the FnIII9-10-biotin ELISA, we found that less GPRP-FnIII9-10-biotin was released within the first 24 hours as compared to GSPE-FnIII9-10-biotin for both the thrombin (Fig. 6A,B) and batroxobin (Fig. 6C,D) clots, suggesting that the affinity between GPRP-FnIII9-10-biotin and fibrin(ogen) was able to retard the initial ‘burst release’ of protein from the fibrin matrix, resulting in an overall greater retention of the loaded protein within the clot. Nonetheless, statistical comparisons of the absolute quantity of protein released at each time point did not reveal major differences between GPRP-FnIII9-10-biotin and GSPE-FnIII9-10-biotin conjugates with respect to the amount of protein released, particularly after the initial 24-hour burst release. Rather, statistically significant differences were found between the amounts of protein released from thrombin- versus batroxobin-catalyzed clots for both GPRP-FnIII9-10 (Fig. 7A, B) and GSPE-FnIII9-10 (Fig. 7C, D), suggesting that other factors may be involved in mediating protein retention past the 24-hour time point.
Fig. 6.

Release of GPRP-FnIII9-10 versus GSPE-FnIII9-10 from thrombin- or batroxobin-catalyzed fibrin clots. Clot compositions as described in Fig. 5. 200 μL clots were overlaid with 1 mL Tris+Ca buffer and samples were taken at the designated timepoints over 4 days. (A, B) GPRP/GSPE-FnIII9-10-biotin measured in the samples taken from thrombin-catalyzed clots and the compiled release profiles. (C, D) Corresponding data from clots formed in the presence of batroxobin.
Fig. 7.

Release of GPRP-FnIII9-10 and GSPE-FnIII9-10 from thrombin- versus batroxobin-catalyzed fibrin clots. Clot compositions as described in Fig. 5. 200 μL clots were overlaid with 1 mL Tris+Ca buffer and samples were taken at the designated timepoints over 4 days. (A, B) GPRP-FnIII9-10-biotin measured in samples taken from thrombin- or batroxobin-catalyzed clots and the compiled release profiles. (C, D) Corresponding data for GSPE-FnIII9-10-biotin release.
Evaluating GPRP-FnIII9-10 retention in fibrin matrices using perfusion assays
We designed a perfusion system to visually examine the real-time dynamics of protein retention within three-dimensional fibrin matrices using confocal microscopy. GPRP-FnIII9-10-C (and the control GSPE-FnIII9-10-C) was fluorescently labeled by conjugation to maleimide-activated Alexa Fluor-633; while fibrinogen was labeled via NHS-amine chemistry with carboxylic acid terminated Alexa Fluor-555. Similar to the release assays, GPRP-FnIII9-10-AF633 or GSPE-FnIII9-10-AF633 was pre-incubated with fibrinogen at 1:10 molar ratio (conjugate:fibrinogen) for 1 h prior to initiating clot formation with thrombin and factor XIIIa within a microfluidic channel. Confocal images were taken following polymerization (prior to perfusion) and then every five minutes following the initiation of buffer perfusion at a rate of 10 μL/min. Representative confocal micrographs illustrate that the GPRP-FnIII9-10-AF633 conjugates were incorporated into the fibrin matrix, were retained with within the clots under significant perfusion and slowly released over a span of 30 min at a buffer flow rate of 10 μL/min (Fig. 8). Meanwhile GSPE-FnIII9-10-AF633 displayed minimal to no fluorescent signal associated with fibrin fibers at any timepoint (Fig. 9). Colocalization analysis for each set of confocal micrographs further demonstrated that 94.2% of fibrin matrix was initially colocalized with GPRP-FnIII9-10 (Fig. 8M), whereas in the negative control only 0.6% fibrin colocalized with GSPE-FnIII9-10 (Fig. 9M). These results indicate that the GPRP-FnIII9-10 fusion protein, but not GSPE-FnIII9-10 was incorporated into the fibrin network. Moreover, 12.8% of the fibrin matrix still retained GPRP-FnIII9-10 after undergoing a rigorous perfusion of buffer demonstrating a robust interaction between the fusion protein and fibrin.
Fig. 8.

Incorporation and retention of GPRP-FnIII9-10. A 6 μM fibrinogen and 0.6 μM GPRP-FnIII9-10 solution was incubated for 30 min. Upon adding 1 U/mL thrombin and 1 U/mL factor XIIIa, the solution was injected into the microfluidic channel and allowed to clot for 1 h. Confocal micrographs were taken before perfusion (A-C), 10 min (D-F), 20 min (G-I), and 30 min (J-L) after perfusing Tris+Ca buffer at a rate of 10 μL/min. Colocalization of the fibrin and GPRP-FnIII9-10 (A-L) was immediately observed after polymerization. Subsequent perfusion of buffer through the matrix diminished the GPRP-FnIII9-10 signal over a 30 min period. (M) Quantitation of signal intensity and colocalization at each time point. Red = Fibrin conjugated to Alexa Fluor-555; Green = GPRP-FnIII9-10 conjugated to Alexa Fluor-633. Scale bar = 10 μm.
Fig. 9.

Limited incorporation of GSPE-FnIII9-10 in fibrin clots. Following the same protocol as describe in Fig. 8, minimal GSPE-FnIII9-10 was incorporated into the fibrin clot. Confocal micrographs were taken before perfusion (AC), 10 min (D-F), 20 min (G-I), and 30 min (J-L) after perfusing Tris+Ca buffer at a rate of 10 μL/min. (M) Quantitation of signal intensity and colocalization at each time point. Red = Fibrin conjugated to Alexa Fluor-555; Green = GSPE-FnIII9-10 conjugated to Alexa Fluor-633. Scale bar = 10 μm.
DISCUSSION
We were able to successfully produce and purify recombinant fibrin-binding proteins with the generalized structure GxRx-protein-(C), capitalizing on the N-terminal GxRx sequence for protein retention within fibrin and the C-terminal cysteine as a functional handle. The structure of such fusion proteins provides direct control over the displayed tetrapeptide sequence and capitalizes on the myriad sulfhydryl-targeting protein modification/tagging chemistries available. As proof-of-concept, we have shown that the high affinity GPRP sequence enables the direct and specific binding of such fusion proteins to fibrin(ogen) without requiring any additional proteins or enzymes, in contrast with previous technologies.
Our SPR and ELISA binding assays demonstrate conclusively that the interaction between our GxRx-FnIII9-10 fusions and fibrinogen is mediated through the GxRx sequence via knob:pocket interactions. As expected, the strength of this interaction is dependent upon the exact tetrapeptide sequence used; specifically, the affinity decreased in the order GPRP-FnIII9-10 (KD: 3.0 ± 0.1 μM), GPRV-FnIII9-10 (KD: 125 ± 6 μM), GHRP-FnIII9-10 (minimal binding). Previous binding affinity studies performed using radiolabeling techniques on the equivalent synthetic tetrapeptides reported the following KDs - 25 μM for GPRP, 100 μM for GPRV, 140 μM for GHRP [30]. Assessing the affinities in the presence of calcium ions significantly lowered the affinities to: 20 μM for GPRP, 80 μM for GPRV, 16 μM for GHRP [31]. All of our experiments were performed with a 5 mM calcium concentration, so it is somewhat surprising that we did not observe a measurable interaction with GHRP-FnIII9-10, even though B-knob is traditionally perceived as a much weaker binder compared to the A-knob in the context of fibrin fragments [28, 32, 33].
This discrepancy between the tetrapeptide and fusion protein affinities suggests that the adjacent protein domain on the fusion protein may influence the functionality of tetrapeptide sequences. In particular, the residues immediately following the N-terminal tetrapeptide sequences could affect the conformational stability and/or charge distribution of the displayed knob, thereby affecting the system of electrostatic and hydrogen bonds that mediate its affinity for fibrin(ogen) [23, 33, 34]. Based on these previous studies and our findings, it appears that the GPRP sequence provides the strongest and most consistent binding characteristics, both as a free tetrapeptide and as part of a protein, across most experimental conditions. In contrast, the affinities of the native GPRV (A-knob) and GHRP (B-knob) sequences for fibrin(ogen) appear to have been negatively affected when presented in the context of our knob-protein fusion.
Following confirmation of the fibrin-binding capacity of GPRP-FnIII9-10 in standard two-dimensional affinity systems with minimal competition from knob tetrapeptides, we progressed to more physiologically relevant three-dimensional systems. Since knob-protein fusions inherently exploit the knob:pocket interactions involved in fibrin assembly, we first verified that the presence of the GSPE-FnIII9-10-biotin and GPRP-FnIII9-10-biotin conjugates did not significantly impede the clotting process or alter global matrix characteristics at the dosages used. Fibrin matrix characteristics are intrinsically linked to its formation kinetics and can be inferred from clot turbidities. Our turbidity curves show much overlap between clots formed in the presence or absence of the conjugates, suggesting that the clotting process was not significantly affected. Rheological measurements and quantitation of unclotted (i.e. soluble) proteins of the resulting matrices further demonstrate that the conjugates do not have a discernible effect on key characteristics commonly used in the study of fibrin clots. Rather, we observed consistent differences between clots formed in the presence of thrombin versus batroxobin, which is a reflection of the mechanistic differences between the enzymes. In particular, thrombin exposes both A- and B-knobs, while batroxobin exposes only the A-knobs of fibrinogen to induce clotting, a difference that was exploited in the design of the release assays.
We then proceeded to examine the protein release profiles from pre-loaded thrombin- or batroxobin-catalyzed fibrin clots. We initially anticipated the release of GPRP-FnIII9-10-biotin conjugates from these fibrin clots to be significantly slower than that of the non-fibrin-binding GSPE-FnIII9-10-biotin control. Moreover, we expected better retention characteristics in the batroxobin-catalyzed clots due to the presence of excess free fibrin pockets in the absence of the B-knobs. Our results suggest that knob:pocket interactions were able to retard the release of GPRP-FnIII9-10-biotin to some extent during the initial 24-hour burst release but not significantly thereafter (Fig. 6). This could be a reflection of the limited sensitivity of the sandwich ELISA (detection limit ≈ 10 ng/mL) used for protein detection, particularly with the low concentrations encountered during the later timepoints. We also speculate that differences in clot structure could have contributed to some of the variability observed after the initial release phase since it is known that fibrin clots formed under the catalysis of different enzymes and at different enzyme concentrations are structurally dissimilar, potentially affecting gel porosity and protein diffusion characteristics [35, 36].
As an alternative interpretation of the release data, it is also possible that the fibrin-binding capacity of the GPRP-FnIII9-10-biotin conjugate was diminished when presented in a three-dimensional system of both fibrin pockets and competing fibrin knobs. Therefore, we proceeded to develop a microfluidic system for the real-time monitoring of protein incorporation, retention, and release within fibrin matrices to assist with our interpretation of the system's strengths and limitations and to clarify whether the release data were subject to ELISA detection limits. Fibrin clots, pre-loaded with GPRP-FnIII9-10-AF633 conjugate, were formed under similar conditions as used in the previous release assays and vigorously perfused to create a temporally-accelerated release system that can be monitored in real-time using confocal microscopy. Results from these studies support our earlier 2-D SPR and ELISA binding results indicating that GPRP-FnIII9-10 specifically and significantly bind fibrin polymerization pockets. Representative images confirmed that the GPRP-FnIII9-10-AF633, but not the control GSPE-FnIII9-10-AF633, bound specifically to fibrin within a three-dimensional fibrin network (Fig. 8, 9) under static conditions. Furthermore, GPRP-FnIII9-10, but not the control, displayed retention within the matrix, specifically associated with individual fibers. These data strongly support the interpretation that the specific affinity between our fusion proteins and fibrin(ogen) is translatable from a two-dimensional to a three-dimensional platform. Additionally, since clots in vivo are rarely subject to such high perfusion rates, this provides strong support for our driving hypothesis – that knob:pocket interactions can be used to augment protein retention within fibrin clots.
We speculate that the retention characteristics of such knob-protein fusions may be further improved by increasing the overall affinity constant through the use of stronger fibrin-binding sequences or by introducing multivalency. In particular, we note that previous research using a heparin-based affinity system for the retention of proteins in fibrin matrices via non-covalent interactions with similar affinities (KDs in the μM range) suggest that a large (~thousand-fold) excess of binding sites is necessary to successfully retain such proteins within the fibrin matrix [37]. This poses a potential limitation for the use of single knob:pocket interactions for the retention of proteins in fibrin matrices since the number of available pockets is limited by fibrinogen concentration as well as the presence of competing knobs. On the other hand, this work offers valuable insight into the significance behind the multimeric nature of the native fibrin(ogen) molecule, comprising up to four fibrin knobs (two A-knobs and two B-knobs) per fully activated molecule. Specifically, suitably tagged recombinant proteins with pre-defined N-terminal sequences can be used as tools to bridge the current informational gap between studies utilizing short synthetic peptides and those focusing on multimeric fibrin fragments, particularly since the physiological relevance of each type of knob:pocket interaction (A:a, A:b, B:a, B:b) to fibrin assembly remains controversial to this day.
CONCLUSIONS
We have capitalized on modern molecular biology techniques to create a platform system for the rapid cloning, expression, and purification of recombinant proteins with defined N-terminal tetrapeptide sequences. In this paper, we have conclusively shown that such knob-protein fusions bind consistently and specifically to fibrin(ogen) in a variety of contexts, including standard two-dimensional affinity systems (SPR, ELISA), and more physiologically relevant three-dimensional setups (release and perfusion systems). Our results demonstrate that knob:pocket interactions can be used for protein retention within fibrin clots and suggest that similar experimental setups may be designed for studying knob:pocket interactions in fibrin clots by the appropriate design and tagging of proteins-of-interest.
ACKNOWLEDGEMENTS
This work was supported by the Coulter Foundation (GTF125000120) and NIH (1R21EB008463) to T.H.B. and the NIH FIRST Fellowship (K12 GM000680) to S.E.S. The authors acknowledge technical assistance with the microfluidic device preparation from Edward Park and Dr. Hang Lu.
HFN7.1 antibody was obtained from the Developmental Studies Hybridoma Bank (NIHCD; maintained by the University of Iowa, Department of Biological Sciences, Iowa City, IA 52242).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Laurens N, Koolwijk P, de Maat MP. Fibrin structure and wound healing. J Thromb Haemost. 2006;4(5):932–939. doi: 10.1111/j.1538-7836.2006.01861.x. [DOI] [PubMed] [Google Scholar]
- 2.Mosesson MW. Fibrinogen and fibrin structure and functions. J Thromb Haemost. 2005;3(8):1894–1904. doi: 10.1111/j.1538-7836.2005.01365.x. [DOI] [PubMed] [Google Scholar]
- 3.Spotnitz WD, Burks S. Hemostats, sealants, and adhesives: components of the surgical toolbox. Transfusion. 2008;48(7):1502–1516. doi: 10.1111/j.1537-2995.2008.01703.x. [DOI] [PubMed] [Google Scholar]
- 4.Ahmed TA, Dare EV, Hincke M. Fibrin: a versatile scaffold for tissue engineering applications. Tissue Eng Part B Rev. 2008;14(2):199–215. doi: 10.1089/ten.teb.2007.0435. [DOI] [PubMed] [Google Scholar]
- 5.Breen A, O'Brien T, Pandit A. Fibrin as a Delivery System for Therapeutic Drugs and Biomolecules. Tissue Eng Part B Rev. 2009;15(2):201–214. doi: 10.1089/ten.TEB.2008.0527. [DOI] [PubMed] [Google Scholar]
- 6.Lord ST. Fibrinogen and fibrin: scaffold proteins in hemostasis. Curr Opin Hematol. 2007;14(3):236–241. doi: 10.1097/MOH.0b013e3280dce58c. [DOI] [PubMed] [Google Scholar]
- 7.Weisel JW. Which knobs fit into which holes in fibrin polymerization? J Thromb Haemost. 2007;5(12):2340–2343. doi: 10.1111/j.1538-7836.2007.02794.x. [DOI] [PubMed] [Google Scholar]
- 8.Ariens RA, Lai TS, Weisel JW, Greenberg CS, Grant PJ. Role of factor XIII in fibrin clot formation and effects of genetic polymorphisms. Blood. 2002;100(3):743–754. doi: 10.1182/blood.v100.3.743. [DOI] [PubMed] [Google Scholar]
- 9.Barker TH, Fuller GM, Klinger MM, Feldman DS, Hagood JS. Modification of fibrinogen with poly(ethylene glycol) and its effects on fibrin clot characteristics. J Biomed Mater Res. 2001;56(4):529–535. doi: 10.1002/1097-4636(20010915)56:4<529::aid-jbm1124>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 10.Schense JC, Hubbell JA. Cross-linking exogenous bifunctional peptides into fibrin gels with factor XIIIa. Bioconjug Chem. 1999;10(1):75–81. doi: 10.1021/bc9800769. [DOI] [PubMed] [Google Scholar]
- 11.Ehrbar M, Metters A, Zammaretti P, Hubbell JA, Zisch AH. Endothelial cell proliferation and progenitor maturation by fibrin-bound VEGF variants with differential susceptibilities to local cellular activity. J Control Release. 2005;101(13):93–109. doi: 10.1016/j.jconrel.2004.07.018. [DOI] [PubMed] [Google Scholar]
- 12.Schmoekel HG, Weber FE, Schense JC, Gratz KW, Schawalder P, Hubbell JA. Bone repair with a form of BMP-2 engineered for incorporation into fibrin cell ingrowth matrices. Biotechnol Bioeng. 2005;89(3):253–262. doi: 10.1002/bit.20168. [DOI] [PubMed] [Google Scholar]
- 13.Pittier R, Sauthier F, Hubbell JA, Hall H. Neurite extension and in vitro myelination within three-dimensional modified fibrin matrices. J Neurobiol. 2005;63(1):1–14. doi: 10.1002/neu.20116. [DOI] [PubMed] [Google Scholar]
- 14.Arrighi I, Mark S, Alvisi M, von Rechenberg B, Hubbell JA, Schense JC. Bone healing induced by local delivery of an engineered parathyroid hormone prodrug. Biomaterials. 2009;30(9):1763–1771. doi: 10.1016/j.biomaterials.2008.12.023. [DOI] [PubMed] [Google Scholar]
- 15.Hall H, Hubbell JA. Matrix-bound sixth Ig-like domain of cell adhesion molecule L1 acts as an angiogenic factor by ligating alphavbeta3-integrin and activating VEGF-R2. Microvasc Res. 2004;68(3):169–178. doi: 10.1016/j.mvr.2004.07.001. [DOI] [PubMed] [Google Scholar]
- 16.Willerth SM, Johnson PJ, Maxwell DJ, Parsons SR, Doukas ME, Sakiyama-Elbert SE. Rationally designed peptides for controlled release of nerve growth factor from fibrin matrices. J Biomed Mater Res A. 2007;80(1):13–23. doi: 10.1002/jbm.a.30844. [DOI] [PubMed] [Google Scholar]
- 17.Wood MD, Sakiyama-Elbert SE. Release rate controls biological activity of nerve growth factor released from fibrin matrices containing affinity-based delivery systems. J Biomed Mater Res A. 2008;84(2):300–312. doi: 10.1002/jbm.a.31269. [DOI] [PubMed] [Google Scholar]
- 18.Sakiyama-Elbert SE, Hubbell JA. Development of fibrin derivatives for controlled release of heparin-binding growth factors. J Control Release. 2000;65(3):389–402. doi: 10.1016/s0168-3659(99)00221-7. [DOI] [PubMed] [Google Scholar]
- 19.Sakiyama-Elbert SE, Hubbell JA. Controlled release of nerve growth factor from a heparin-containing fibrin-based cell ingrowth matrix. J Control Release. 2000;69(1):149–158. doi: 10.1016/s0168-3659(00)00296-0. [DOI] [PubMed] [Google Scholar]
- 20.Kawasaki K, Hirase K, Miyano M, Tsuji T, Iwamoto M. Amino acids and peptides. XVI. Synthesis of N-terminal tetrapeptide analogs of fibrin alpha-chain and their inhibitory effects on fibrinogen/thrombin clotting. Chem Pharm Bull (Tokyo) 1992;40(12):3253–3260. doi: 10.1248/cpb.40.3253. [DOI] [PubMed] [Google Scholar]
- 21.Laudano AP, Doolittle RF. Studies on synthetic peptides that bind to fibrinogen and prevent fibrin polymerization. Structural requirements, number of binding sites, and species differences. Biochemistry. 1980;19(5):1013–1019. doi: 10.1021/bi00546a028. [DOI] [PubMed] [Google Scholar]
- 22.Yang Z, Mochalkin I, Doolittle RF. A model of fibrin formation based on crystal structures of fibrinogen and fibrin fragments complexed with synthetic peptides. Proc Natl Acad Sci U S A. 2000;97(26):14156–14161. doi: 10.1073/pnas.97.26.14156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kostelansky MS, Betts L, Gorkun OV, Lord ST. 2.8 A crystal structures of recombinant fibrinogen fragment D with and without two peptide ligands: GHRP binding to the “b” site disrupts its nearby calcium-binding site. Biochemistry. 2002;41(40):12124–12132. doi: 10.1021/bi0261894. [DOI] [PubMed] [Google Scholar]
- 24.Petrie TA, Capadona JR, Reyes CD, Garcia AJ. Integrin specificity and enhanced cellular activities associated with surfaces presenting a recombinant fibronectin fragment compared to RGD supports. Biomaterials. 2006;27(31):5459–5470. doi: 10.1016/j.biomaterials.2006.06.027. [DOI] [PubMed] [Google Scholar]
- 25.Martino MM, Mochizuki M, Rothenfluh DA, Rempel SA, Hubbell JA, Barker TH. Controlling integrin specificity and stem cell differentiation in 2D and 3D environments through regulation of fibronectin domain stability. Biomaterials. 2009;30(6):1089–1097. doi: 10.1016/j.biomaterials.2008.10.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Morton TA, Myszka DG, Chaiken IM. Interpreting complex binding kinetics from optical biosensors: a comparison of analysis by linearization, the integrated rate equation, and numerical integration. Anal Biochem. 1995;227(1):176–185. doi: 10.1006/abio.1995.1268. [DOI] [PubMed] [Google Scholar]
- 27.Leahy DJ, Aukhil I, Erickson HP. 2.0 A crystal structure of a four-domain segment of human fibronectin encompassing the RGD loop and synergy region. Cell. 1996;84(1):155–164. doi: 10.1016/s0092-8674(00)81002-8. [DOI] [PubMed] [Google Scholar]
- 28.Geer CB, Tripathy A, Schoenfisch MH, Lord ST, Gorkun OV. Role of ‘B-b’ knob-hole interactions in fibrin binding to adsorbed fibrinogen. J Thromb Haemost. 2007;5(12):2344–2351. doi: 10.1111/j.1538-7836.2007.02774.x. [DOI] [PubMed] [Google Scholar]
- 29.Weisel JW, Nagaswami C. Computer modeling of fibrin polymerization kinetics correlated with electron microscope and turbidity observations: clot structure and assembly are kinetically controlled. Biophys J. 1992;63(1):111–128. doi: 10.1016/S0006-3495(92)81594-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Laudano AP, Doolittle RF. Synthetic peptide derivatives that bind to fibrinogen and prevent the polymerization of fibrin monomers. Proc Natl Acad Sci U S A. 1978;75(7):3085–3089. doi: 10.1073/pnas.75.7.3085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Laudano AP, Doolittle RF. Influence of calcium ion on the binding of fibrin amino terminal peptides to fibrinogen. Science. 1981;212(4493):457–459. doi: 10.1126/science.7209542. [DOI] [PubMed] [Google Scholar]
- 32.Litvinov RI, Gorkun OV, Owen SF, Shuman H, Weisel JW. Polymerization of fibrin: specificity, strength, and stability of knob-hole interactions studied at the single-molecule level. Blood. 2005;106(9):2944–2951. doi: 10.1182/blood-2005-05-2039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Litvinov RI, Gorkun OV, Galanakis DK, Yakovlev S, Medved L, Shuman H, et al. Polymerization of fibrin: Direct observation and quantification of individual B:b knob-hole interactions. Blood. 2007;109(1):130–138. doi: 10.1182/blood-2006-07-033910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Everse SJ, Spraggon G, Veerapandian L, Riley M, Doolittle RF. Crystal structure of fragment double-D from human fibrin with two different bound ligands. Biochemistry. 1998;37(24):8637–8642. doi: 10.1021/bi9804129. [DOI] [PubMed] [Google Scholar]
- 35.Weisel JW. Fibrin assembly. Lateral aggregation and the role of the two pairs of fibrinopeptides. Biophys J. 1986;50(6):1079–1093. doi: 10.1016/S0006-3495(86)83552-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Blomback B, Bark N. Fibrinopeptides and fibrin gel structure. Biophys Chem. 2004;112(23):147–151. doi: 10.1016/j.bpc.2004.07.013. [DOI] [PubMed] [Google Scholar]
- 37.Taylor SJ, McDonald JW, 3rd, Sakiyama-Elbert SE. Controlled release of neurotrophin-3 from fibrin gels for spinal cord injury. J Control Release. 2004;98(2):281–294. doi: 10.1016/j.jconrel.2004.05.003. [DOI] [PubMed] [Google Scholar]