

Summary
Native disulfide bond formation is critical for the proper folding of many proteins. Recent studies using newly identified protein oxidants, folding catalysts, and mutant cells provide insight into the mechanism of oxidative protein folding in vivo. This insight promises new strategies for more efficient protein production.
Introduction
The formation of disulfide bonds in proteins requires a sufficiently oxidizing environment [1]. Eukaryotic cells contain compartments of widely varying reduction potential (E°′) [2]. Those proteins destined for secretion are co-translationally translocated into the oxidizing environment of the endoplasmic reticulum (ER), E°′= −0.18 V, where they can fold and acquire their native disulfide bonds. Oxidative folding in the ER of the yeast Saccharomyces cerevisiae requires two proteins: endoplasmic reticulum oxidoreductin 1 protein (Ero1p) and protein disulfide isomerase (PDI).
Ero1p is a 65-kDa, membrane-associated resident of the ER lumen that is essential for S. cerevisiae viability. In vivo, Ero1p oxidizes disulfide-containing proteins. Ero1p was identified using genetic screens for proteins that either, when overproduced, confer resistance to dithiothreitol (DTT, a small-molecule reductant) or, when mutated, cause sensitivity to DTT. Addition of the thiol oxidant diamide to the growth medium can complement an Ero1p deficiency, presumably by performing the oxidative function of the missing gene product. These results indicate that the essential function of Ero1p is to oxidize newly synthesized proteins [3,4].
PDI is a 57-kDa resident of the ER that is essential for S. cerevisiae viability. In vitro, PDI catalyzes three reactions: the oxidation of thiols and the reduction and isomerization of disulfides. The two PDI active sites consist of a CXXC motif with the sequence: Cys–Gly–His–Cys. Experiments using active-site variants of PDI indicate that its essential function is the isomerization of non-native disulfide bonds (Figures 1 and 2) [5,6].
Figure 1.

Primary pathway of native disulfide bond formation in the ER. PDI transfers oxidizing equivalents from Ero1p to reduced, unfolded proteins [9]••. Cells lacking Ero1p cannot oxidize newly synthesized proteins in the ER and are inviable. Non-native disulfide bonds must isomerize to the native state. Those proteins that do not attain the native state are degraded rather than secreted, and cells lacking a disulfide isomerase are inviable [5].
Figure 2.

Putative mechanism of disulfide bond isomerization [15]••. Isomerization begins with nucleophilic attack of a thiolate provided by the catalyst (such as PDI) on a non-native disulfide bond. The resulting, covalent substrate–catalyst complex contains a substrate thiolate and can perform intramolecular thiol–disulfide exchange reactions to form native disulfide bonds and, eventually, release of the catalyst (redox-inactive mechanism). The second thiol in the active site of the catalyst can act as a clock to ensure timely rearrangement of the substrate [16]. Those substrates that are slow to rearrange can be partially reduced (and subsequently reoxidized) by a dithiol catalyst (reduction–reoxidation mechanism). Those catalysts lacking a second thiol can become trapped in mixed disulfides with the substrate.
The CXXC active-site motif of PDI is also found in homologous thiol–disulfide oxidoreductases [7]. In bacteria, the pathway for native disulfide bond formation involves the CXXC-containing Dsb family of proteins (DsbA–D) and may be analogous to the eukaryotic pathway [8]. This review will focus on intriguing new insights into the mechanism of oxidative folding for eukaryotic secretory proteins and how these developments are being harnessed to guide the efficient production of these proteins.
Role of protein oxidants
Oxidative protein folding involves both the oxidation of thiols and the isomerization of non-native disulfide bonds. Recently, Kaiser and Frand [9]•• delineated how Ero1p and PDI accomplish the first step of this process (Figure 1). Ero1p and PDI form a disulfide-linked complex in vivo. Likewise, PDI and carboxypeptidase Y (CPY), a protein that contains five disulfide bonds, form mixed-disulfides. Reduced forms of both PDI and CPY accumulate in cells lacking functional Ero1p. Reduced CPY also accumulates in the ER of PDI-depleted cells. Thus, oxidizing equivalents flow from Ero1p to PDI to substrate proteins (Figure 1).
Apparently, only a small fraction of the PDI active sites are reduced in the ER [9]••. Yet, the dithiol form of PDI is required for catalysis of disulfide isomerization, its essential function (Figure 2). An E°′ of −0.18 V for the ER was found by measuring the concentrations of reduced and oxidized glutathione (GSH and GSSG) [2]. This value is in gratifying agreement with the in vitro optimum for the oxidative folding activity of PDI [7]. In a solution of E°′= −0.18 V, fully 50% of the PDI active sites should be reduced [6]. Either the ER is much more oxidizing than was believed previously or, in vivo, PDI is maintained out of equilibrium with its environment and away from its in vitro optimum. Determining the relative contributions of oxidase and isomerase activities during catalysis by PDI remains an interesting challenge.
Although the in vivo oxidase role of PDI has been underappreciated, this role is not essential. Even though oxidation of newly synthesized proteins is significantly impaired in the absence of PDI [9]••, a variant of PDI with CGHS active sites can complement a wild-type PDI deficiency. This variant cannot catalyze reduction or oxidation, but it is an efficient catalyst of disulfide isomerization (a redox-inactive process) [5,6]. Perhaps Ero1p can oxidize proteins directly and PDI is required for the proper folding of Ero1p, which contains 14 cysteine residues. Alternatively, Ero1p-independent oxidation pathways may exist in the ER.
A role for GSSG in the oxidation of proteins has been ruled out [10]•. GSSG has long been thought the primary source of protein oxidation, with the accumulation of GSSG being a result of its selective import into the ER [2]. Yet, GSSG can be formed in the ER from GSH by the action of Ero1p. Mutant strains of yeast unable to synthesize glutathione show no defect in protein disulfide formation. Indeed, knocking out glutathione synthesis restores viability to cells lacking functional Ero1p. Disulfide bonds form efficiently in the ER of these cells. Taken together, these results indicate that glutathione contributes net reducing equivalents to the ER and actually competes with protein thiols for oxidation. What is the role of glutathione in the ER? Most likely, it acts as a buffer against transient changes in oxidative stress [10]•. Perhaps it also helps to maintain the essential pool of reduced PDI.
If cells lacking Ero1p and glutathione can exhibit normal oxidation kinetics, then the ER must possess other oxidants. One candidate is sulfhydryl oxidase, an enzyme that catalyzes the oxidation of thiols by O2 [11]. Another putative electron acceptor is flavin-containing monooxygenase (FMO), an O2- and NADPH-dependent catalyst of thiol oxidation that is localized to the cytoplasmic face of the ER [12]. The appeal of both sulfhydryl oxidase and FMO as putative oxidants is the direct link they provide to the ultimate electron acceptor: O2.
Ero1p is a primary source of protein oxidation in the ER. How Ero1p becomes oxidized remains unknown (Figure 1). Ero1p performs a function analogous to that of the bacterial protein DsbB [8]. In Escherichia coli, the respiratory chain can oxidize DsbB and perhaps provide the ultimate source of protein oxidation [13,14]. Ero1p may possess an Fe–S cluster that accepts electrons from its active-site cysteine residues and transfers them to the respiratory chain, allowing further rounds of oxidation [3,4].
Importance of covalent catalysis
Although efficient oxidation is undoubtedly important, the isomerization of non-native disulfide bonds may often limit the folding rate of many proteins. Indeed, the essential in vivo function of PDI is to catalyze the unscrambling of disulfide bonds in other proteins [5,6]. The simplest mechanism for PDI-catalyzed disulfide isomerization is shown in Figure 2. A striking feature of this mechanism is that it requires only the provision of a reactive thiolate on the part of the catalyst.
Each CGHC active site of PDI possesses a high disulfide reduction potential (E°′= −0.18 V) and an often unprotonated thiol (pKa = 6.7). These properties can be combined to calculate that during the conditions of efficient in vitro oxidative folding (E°′ = −0.18 V, pH 7.0, and 30 °C), 33% of PDI active sites contain a thiolate [6]. To effect the mechanism shown in Figure 2, PDI can be pared down to a thiolate.
Support for this mechanism (Figure 2) comes from the activity of the dithiol: (±)-trans-1,2-bis(2-mercaptoacetamido)cyclohexane (BMC) [15]••. The physical properties of BMC are listed in Table 1 and are similar to those of PDI. Unlike PDI, BMC cannot bind to a protein substrate. Still, using only covalent interactions, BMC is able to catalyze native disulfide bond formation, both in vitro and in vivo.
Table 1. Properties of (±)-trans-1,2-bis(2-mercaptoacteamido)cyclohexane.
| Structure | 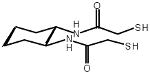 |
| Molecular Mass (Da) | 262 |
| E°′ (V) | −0.24 |
| pKa | 8.3; 9.9 |
| % Thiolate* | 0.05 |
Calculated for the conditions: E°′solution = −0.18 V; pH 7.0; 30 °C. Data from ref [15]••.
In vitro, BMC catalyzes the reactivation of scrambled ribonuclease (RNase) A, a substrate with 4 non-native disulfide bonds [15]••. In this assay, BMC and PDI produce similar yields of folded RNase A. GSH and a monothiol analogue of BMC each give lower yields than do the dithiols. In effect, the second thiol provides an intramolecular clock for substrate-induced thiol–disulfide exchange [16]. Those substrates that are slow to rearrange can be released through the formation of a disulfide bond within the catalyst. These partially reduced intermediates now have a second thiolate to induce additional disulfide rearrangements and can eventually be reoxidized by the catalyst. The contribution of this reduction–reoxidation mechanism is determined in part by the effective concentration (EC) of the active-site thiols [17,1]. The redox-inactive mechanism requires at least two productive thiol–disulfide exchanges within the substrate before catalyst release (Figure 2). A higher EC within the catalyst will decrease the contribution of the redox-inactive mechanism.
BMC added to the growth medium of Saccharomyces cerevisiae cells producing Schizosaccharomyces pombe acid phosphatase, which has 8 disulfides, causes more efficient protein folding in vivo and results in a 3-fold increase of secreted, active enzyme [15]••. This increase is equivalent to that achieved with co-overproduction of PDI (Table 2) [18]. The accordance of achievable yield between exogenously added BMC and endogenously produced PDI suggests a shared mechanism of action. Like PDI, BMC may function in vivo as a direct catalyst of disulfide isomerization. Still, BMC could act either to reduce a greater fraction of the basal PDI present in the ER, increasing its efficiency as an isomerase, or to oxidize acid phosphatase directly (after having been oxidized itself). Regardless, at optimum concentrations of BMC, a step other than native disulfide bond formation likely limits the secretion of acid phosphatase.
Table 2. Production of folded acid phosphatase in yeast*.
| Condition | Relative yield |
|---|---|
| PDI (basal) | 1 |
| PDI (basal) plus BMC (0.2 mg/mL) | 3 |
| PDI (15-fold overproduction) | 3 |
BMC may provide better methods for the production of disulfide-bonded proteins. The action of BMC in vivo may allow for increased yields of active protein simply through its addition to the growth medium. BMC-based redox buffers may also prove useful for the efficient in vitro folding of proteins from inclusion bodies.
Support for the biological relevance of the mechanism shown in Figure 2 comes from the observation by Molinari and Helenius of transient mixed disulfides between PDI and a newly synthesized viral glycoprotein in vivo [19]•. These complexes provide the first demonstration of a covalent interaction between PDI and a substrate in mammalian cells. Together with the observation of disulfide-linked complexes between PDI and CPY or Ero1p in yeast cells [9]••, these studies support an in vivo role for the mechanism of PDI catalysis derived from in vitro studies (Figure 2). It is not clear whether these complexes involve substrates undergoing oxidation or isomerization (or both).
Molinari and Helenius also find that PDI has substrate specificity in vivo [19]•. Although PDI was only observed in mixed disulfides with one of two disulfide-containing viral proteins, its homolog ERp57 was found linked to both substrates. A pancreas-specific homologue of PDI, but not PDI itself, has been found to specifically recognize peptides containing tyrosine or tryptophan residues [20]. The biological relevance of substrate specificity during PDI catalysis is not yet clear.
The in vitro folding pathway of multiply disulfide-bonded proteins is complex [21]. During the folding of reduced RNase A, PDI has little effect on the distribution of single-disulfide intermediates formed in glutathione [22] or DTT [23] redox buffers. The rate-limiting step during the in vitro folding of RNase A [23] and lysozyme [24] involves further oxidation of partially oxidized intermediates. PDI increases the rate of oxidative folding in vitro by isomerizing substrates that are neither fully oxidized nor fully reduced.
Structural basis for catalysis by PDI
The three-dimensional structure of PDI remains unknown. PDI consists of four domains with structural homology to the CXXC-containing oxidoreductase thioredoxin plus an acidic C-terminal domain [25]. The first and fourth domains contain the two active sites. Recent studies have identified an important role for the third domain in substrate binding [26,27]. The C-terminal domain is not required for catalytic activity [28].
Recent crystal structures of a PDI homolog composed solely of two catalytic domains from Pyrococcus furiosus [29] and of a dimeric E. coli thioredoxin active-site variant with enhanced isomerase activity [30] provide clues as to how the thioredoxin-like domains of PDI may interact. In both structures, the thioredoxin domains form a continuous β-sheet (though the interfaces occur at opposite edges of the β-sheet). This arrangement suggests that the thioredoxin domains of PDI may be colinear.
A crystal structure of the dimeric bacterial PDI homolog DsbC provides a slightly different model for the structure of PDI [31]•. The DsbC dimer contains two catalytic domains and two dimerization domains. This arrangement is reminiscent of the four thioredoxin-like domains in PDI. The dimer structure is V-shaped, with the two CXXC active sites pointing towards each other across a large, hydrophobic cleft. This cleft seems well-suited for the binding of unfolded proteins. The catalytic and dimerization domains in a DsbC monomer are connected by a hinged linker helix that allows for conformational changes associated with substrate recognition. Such flexibility has been implicated in catalysis by PDI [27], though it is not known if PDI possesses interdomain linkers similar to those in DsbC [32]. Knowledge of the structure of intact PDI will allow for a more detailed model of the mechanism by which PDI catalyzes native disulfide bond formation.
Importance of redox environment
Stable disulfide bonds rarely form in the cytoplasm [33]. The bacterial cytoplasm (E°′= −0.27 V) is normally even more reducing than its eukaryotic counterpart (E°′= −0.23 V) [2] and is not a good environment for the production of properly folded, multiply disulfide-bonded proteins. Of course, bacterial expression systems can produce large quantities of heterologous proteins. Unfortunately, insoluble aggregates of these proteins tend to form when they misfold, and the in vitro folding of these inclusion bodies can be difficult and time-consuming [34].
One approach to this problem has been to secrete disulfide-bonded proteins along with PDI to the more oxidizing periplasm in E. coli [35]. This approach can hit-or-miss, as rat PDI does not improve yields of human tissue plasminogen activator (tPA), a 17-disulfide protein, but yeast PDI increases the yield by 50%. In addition to problems of specificity, PDI activity may be hindered by the redox environment of the periplasm. The yield of active tPA obtained by coproduction of yeast PDI was lower than that obtained from cells overproducing DsbC [36], whose CXXC active site is maintained in the reduced state [8]. Perhaps the periplasmic environment is too oxidizing for efficient catalysis of disulfide isomerization by PDI. The ability of PDI to function in place of the periplasmic oxidant DsbA during the oxidative folding of E. coli alkaline phosphatase supports this idea [35].
In the E. coli periplasm, the prior overproduction of DsbC or DsbA (the bacterial analogue of Ero1p) doubles production of insulin-like growth factor-I (IGF-I) relative to the coproduction of the proteins [37]. The abundance of Dsb proteins in the periplasm during production of IGF-I leads, however, to increased aggregation rather than more efficient in vivo folding. Fortunately, IGF-I can be folded easily from inclusion bodies, and 8.5 g of IGF-I can be isolated from 1 L of cell culture. This high yield may be due to protection of translocated IGF-I from proteolysis or assistance in its translocation. Surprisingly, most of the DsbA in cells after its transient overproduction is found in the reduced form rather than the oxidized form that normally accumulates and is responsible for catalysis of protein oxidation in the periplasm [8]. The decrease in periplasmic folding efficiency when IGF-I is produced subsequent to DsbA or DsbC overproduction argues against improved thiol–disulfide exchange as a cause of the boost in yield [37].
Manipulation of the redox environment in the bacterial cytoplasm may allow for more efficient oxidative protein folding in vivo [38]••. Such a strategy has been made possible by the development of E. coli strains that grow normally despite having an oxidizing cytoplasm. These strains were isolated as suppressors of the slow-growing phenotype displayed by cells lacking the genes for thioredoxin reductase and glutathione synthesis. The cytoplasmic folding of four normally secreted, multiply disulfide-bonded proteins was more efficient in these strains than in wild-type cells. Thioredoxin, which normally acts as a cytoplasmic reductant, acts as a protein oxidant in these strains [39], consistent with its ability to act as an oxidant when exported to the periplasm [40,41].
The cytoplasmic folding of truncated tPA (vtPA), which has 9 disulfides, can be improved by coproduction of active-site thioredoxin variants with higher E°′. The optimum yield of vtPA in the cytoplasm is obtained with coproduction of DsbC in these mutant strains, which gives a 200-fold increase in folding efficiency relative to wild-type cells (Table 3). This result underscores the importance of maintaining a dithiol catalyst for the shuffling of non-native disulfide bonds (Figure 2). Surprisingly, the coproduction of PDI in the cytoplasm had little effect on the cytoplasmic folding of vtPA. The folding of vtPA in the cytoplasm of the mutant strains was more efficient than was its folding in the periplasm of wild-type cells (Table 3) [38]••. Oxidation may be slower in the cytoplasm than in the periplasm, decreasing the need for disulfide isomerization, and thus explaining the greater efficiency of cytoplasmic folding.
Table 3. Production of truncated tissue plasminogen activator in bacteria*.
| Condition | Relative yield |
|---|---|
| Wild-type cytoplasm | 0.1 |
| Oxidizing cytoplasm | 1 |
| Wild-type periplasm plus DsbC | 10 |
| Oxidizing cytoplasm plus DsbC | 21 |
Data from ref [38]••.
Modulation of the redox environment in the cytoplasm may be an effective strategy for the production of eukaryotic secretory proteins in bacteria. Folding efficiency in this system could be improved further by coproduction of a disulfide isomerase with active-site properties appropriate for the redox environment. The mechanism of disulfide bond isomerization (Figure 2) can guide the design and choice of this catalyst.
Conclusions
The recent past has provided many exciting advances in the understanding of native disulfide bond formation. Among these is a new-found appreciation of the role played by protein oxidants, such as Ero1p, in the ER. This protein most likely functions as the primary electron acceptor in the PDI-catalyzed oxidation of newly synthesized proteins. Further, the long-held hypothesis that glutathione is the primary oxidant in the ER has been disproved. The elaboration of the roles played by other protein oxidants in the ER and identification of the oxidant for Ero1p remain interesting challenges. The observation of covalent complexes between PDI and newly synthesized proteins supports the mechanism of PDI catalysis developed from in vitro studies. Catalysis of protein folding by BMC, a small-molecule mimic of PDI, reveals the most important properties (E°′ and pKa) of PDI. Small-molecule PDI mimics could be useful for the efficient preparation of secretory protein, both in vivo and in vitro. The structure of PDI, which would aid in the design of novel catalysts and provide a more detailed understanding of its catalytic mechanism, has remained elusive. The structure of DsbC, however, provides a hint of how the four thioredoxin-like domains of PDI might interact. Lastly, the development of E. coli strains with an oxidizing cytoplasm provides a new strategy for efficient in vivo native disulfide bond formation. The modulation of the redox environment in the eukaryotic cytoplasm or the ER, perhaps through altered levels of glutathione or Ero1p (or both), in addition to the provision of properly designed folding catalysts, may provide further improvements in efficient protein production. Such a strategy depends on a firm understanding of the mechanism by which native disulfide bonds form.
Acknowledgments
We thank E.A. Kersteen and T.A. Klink for helpful discussions and critical reading of the manuscript. K.J.W. was supported by a WARF predoctoral fellowship and Chemistry–Biology Interface Training Grant GM08505 (NIH). Work in the authors' laboratory was supported by grant BES-9604563 (NSF).
Abbreviations
- BMC
(±)-trans-1,2-bis(2-mercaptoacetamido)cyclohexane
- ER
endoplasmic reticulum
- Ero1p
endoplasmic reticulum oxidoreductin 1 protein
- GSH
reduced glutathione
- GSSG
oxidized glutathione
- PDI
protein disulfide isomerase
References and recommended reading
- 1.Raines RT. Nature's transitory covalent bond. Nature Struct Biol. 1997;4:424–427. doi: 10.1038/nsb0697-424. [DOI] [PubMed] [Google Scholar]
- 2.Hwang C, Sinskey AJ, Lodish HF. Oxidized redox state of glutathione in the endoplasmic reticulum. Science. 1992;257:1496–1502. doi: 10.1126/science.1523409. [DOI] [PubMed] [Google Scholar]
- 3.Frand AR, Kaiser CA. The ERO1 gene of yeast is required for oxidation of protein dithiols in the endoplasmic reticulum. Mol Cell. 1998;1:161–170. doi: 10.1016/s1097-2765(00)80017-9. [DOI] [PubMed] [Google Scholar]
- 4.Pollard MG, Travers KJ, Weissman JS. Ero1p: A novel and ubiquitous protein with an essential role in oxidative protein folding in the endoplasmic reticulum. Mol Cell. 1998;1:171–182. doi: 10.1016/s1097-2765(00)80018-0. [DOI] [PubMed] [Google Scholar]
- 5.Laboissière MCA, Sturley SL, Raines RT. The essential function of protein-disulfide isomerase is to unscramble non-native disulfide bonds. J Biol Chem. 1995;270:28006–28009. doi: 10.1074/jbc.270.47.28006. [DOI] [PubMed] [Google Scholar]
- 6.Chivers PT, Laboissière MCA, Raines RT. Protein disulfide isomerase: Cellular enzymology of the CXXC motif. In: Guzman NA, editor. Prolyl Hydroxylase, Protein Disulfide Isomerase, and Other Structurally-Related Proteins. Marcel Dekker; New York: 1998. pp. 487–505. [Google Scholar]
- 7.Gilbert HF. Thiol–disulfide exchange of divalent sulfur. In: Sinnott M, editor. Comprehensive Biological Catalysis: A Mechanistic Reference. Academic Press; San Diego: 1998. pp. 609–625. [Google Scholar]
- 8.Debarbieux L, Beckwith J. Electron avenue: Pathways of disulfide bond formation and isomerization. Cell. 1999;99:117–119. doi: 10.1016/s0092-8674(00)81642-6. [DOI] [PubMed] [Google Scholar]
- 9.Frand AR, Kaiser CA. Ero1p oxidizes protein disulfide isomerase in a pathway for disulfide bond formation in the endoplasmic reticulum. Mol Cell. 1999;4:469–477. doi: 10.1016/s1097-2765(00)80198-7. [DOI] [PubMed] [Google Scholar]; ••Redox-sensitive PDI-Ero1p and PDI-CPY complexes are trapped in vivo and identified by immunoprecipitation. With PDI expression turned off, reduced CPY and oxidized Ero1p accumulate in the ER. No complexes between Ero1p and CPY are found. In wild-type cells, only a small fraction of the free PDI molecules are modified by an alkylating agent. In cells lacking functional Ero1p, almost all of the free PDI is modified, indicating that Ero1p maintains PDI in a mostly oxidized state. The authors conclude that the predominant pathway of protein oxidation in the ER is driven by Ero1p and mediated by PDI.
- 10.Cuozzo JW, Kaiser CA. Competition between glutathione and protein thiols for disulphide-bond formation. Nature Cell Biol. 1999;1:130–135. doi: 10.1038/11047. [DOI] [PubMed] [Google Scholar]; •The production of GSSG in vivo is found to correlate with Ero1p activity. Growth of cells lacking functional Ero1p is restored by a knockout of glutathione biosynthesis, as is the oxidation of CPY. Induction of the unfolded protein response correlates with intracellular glutathione levels. The lack of glutathione causes an increase in sensitivity to exogenous addition of diamide, a thiol oxidant, indicating a role for reduced glutathione in protecting the ER against hyperoxidizing conditions.
- 11.Hoober KL, Sheasley SL, Gilbert HF, Thorpe C. Sulfhydryl oxidase from egg white: A facile catalyst for disulfide formation in proteins and peptides. J Biol Chem. 1999;274:22147–22150. doi: 10.1074/jbc.274.32.22147. [DOI] [PubMed] [Google Scholar]
- 12.Suh JK, Poulsen LL, Ziegler DM, Robertus JB. Yeast flavin-containing monooxygenase generates oxidizing equivalents that control protein folding in the endoplasmic reticulum. Proc Natl Acad Sci USA. 1999;96:2687–2691. doi: 10.1073/pnas.96.6.2687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bader M, Muse W, Ballou DP, Gassner C, Bardwell JCA. Oxidative protein folding is driven by the elctron transport system. Cell. 1999;98:217–227. doi: 10.1016/s0092-8674(00)81016-8. [DOI] [PubMed] [Google Scholar]
- 14.Kobayashi T, Ito K. Respiratory chain strongly oxidizes the CXXC motif of DsbB in the Escherichia coli disulfide bond formation pathway. EMBO J. 1999;18:1192–1198. doi: 10.1093/emboj/18.5.1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Woycechowsky KJ, Wittrup KD, Raines RT. A small-molecule catalyst of protein folding in vitro and in vivo. Chem Biol. 1999;6:871–879. doi: 10.1016/s1074-5521(00)80006-x. [DOI] [PubMed] [Google Scholar]; ••BMC, a small-molecule mimic of PDI, is synthesized and characterized. BMC increases the reactivation of scrambled RNase A in vitro. The yield of active RNase A is similar to that obtained with PDI and higher than that obtained with monothiol reagents. Further, addition of BMC to the growth medium of yeast cells overexpressing acid phosphatase causes an increase in the secretion of active enzyme.
- 16.Walker KW, Gilbert HF. Scanning and escape during protein-disulfide isomerase-assisted protein folding. J Biol Chem. 1997;272:8845–8848. doi: 10.1074/jbc.272.14.8845. [DOI] [PubMed] [Google Scholar]
- 17.Chivers PT, Prehoda KE, Raines RT. The CXXC motif: A rheostat in the active site. Biochemistry. 1997;36:4061–4066. doi: 10.1021/bi9628580. [DOI] [PubMed] [Google Scholar]
- 18.Robinson AS, Hines V, Wittrup KD. Protein disulfide isomerase overexpression increases secretion of foreign proteins in Saccharomyces cerevisiae. BioTechnology. 1994;12:381–384. doi: 10.1038/nbt0494-381. [DOI] [PubMed] [Google Scholar]
- 19.Molinari M, Helenius A. Glycoproteins form mixed disulfides with oxidoreductases during folding in living cells. Nature. 1999;402:90–93. doi: 10.1038/47062. [DOI] [PubMed] [Google Scholar]; •Mammalian cells are infected with a virus that causes all cellular protein synthesis to become dedicated to the production of viral proteins. These infected cells are pulse-labeled with radioactive sulfur, and the newly synthesized viral proteins are separated by two rounds of gel electrophoresis: the first under non-reducing conditions and the second in the presence of reductant. The two disulfide-containing viral proteins are found to form mixed disulfides with PDI and its homolog Erp57, but not with Erp72. The amount of these complexes present in the ER peaks 10 min after the pulse and then decreases steadily, illustrating the transient nature of these folding intermediates. Blocking proper glycosylation of the nascent proteins interferes with their oxidative folding.
- 20.Ruddock LW, Freedman RB, Klappa P. Specificity in substrate binding by protein folding catalysts: tyrosine and tryptophan residues are the recognition motifs for the binding of peptides to the pancreas-specific protein disulfide isomerase PDIp. Protein Sci. 2000;9:758–764. doi: 10.1110/ps.9.4.758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wedemeyer WJ, Welker E, Narayan M, Scheraga HA. Disulfide bonds and protein folding. Biochemistry. 2000;39:4207–4216. doi: 10.1021/bi992922o. [DOI] [PubMed] [Google Scholar]
- 22.Vinci F, Ruoppolo M, Pucci P, Freedman RB, Marino G. Early intermediates in the PDI-assisted folding of ribonuclease A. Protein Sci. 2000;9:525–535. doi: 10.1110/ps.9.3.525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shin HC, Scheraga HA. Effect of protein disulfide isomerase on the regeneration of bovine ribonuclease A with dithiothreitol. FEBS Lett. 1999;456:143–145. doi: 10.1016/s0014-5793(99)00946-1. [DOI] [PubMed] [Google Scholar]
- 24.van den Berg B, Chung EW, Robinson CV, Mateo PL, Dobson CM. The oxidative folding of hen lysozyme and its catalysis by protein disulfide isomerase. EMBO J. 1999;18:4794–4803. doi: 10.1093/emboj/18.17.4794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ferrari DM, Söling HD. The protein disulphide-isomerase family: Unravelling a string of folds. Biochem J. 1999;339:1–10. [PMC free article] [PubMed] [Google Scholar]
- 26.Klappa P, Ruddock LW, Darby NJ, Freedman RB. The b¢ domain provides the principal peptide-binding site of protein disulfide isomerase but all domains contribute to binding misfolded proteins. EMBO J. 1998;17:927–935. doi: 10.1093/emboj/17.4.927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cheung PY, Churchich JE. Recognition of protein substrates by protein-disulfide isomerase. J Biol Chem. 1999;274:32757–32761. doi: 10.1074/jbc.274.46.32757. [DOI] [PubMed] [Google Scholar]
- 28.Koivunen P, Pirneskoski A, Karvonen P, Ljung J, Helaakoski T, Notbohm H, Kivirikko KI. The acidic C-terminal domain of protein disulfide isomerase is not critical for the enzyme subunit function or for the chaperone or disulfide isomerase activities of the polypeptide. EMBO J. 1999;18:65–74. doi: 10.1093/emboj/18.1.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ren B, Tibbelin G, Pascale Dd, Rossi M, Bartolucci S, Ladenstein R. A protein disulfide oxidoreductase from the archaeon Pyrococcus furiosus contains two thioredoxin fold units. Nature Struct Biol. 1998;5:602–611. doi: 10.1038/862. [DOI] [PubMed] [Google Scholar]
- 30.Schultz LW, Chivers PT, Raines RT. The CXXC motif: Crystal structure of an active-site variant of Escherichia coli thioredoxin. Acta Cryst. 1999;D55:1533–1538. doi: 10.1107/s0907444999008756. [DOI] [PubMed] [Google Scholar]
- 31.McCarthy AA, Haebel PW, Törrönen A, Rybin V, Baker EN, Metcalf P. Crystal structure of the protein disulfide bond isomerase DsbC from Escherichia coli. Nature Struct Biol. 2000;7:196–199. doi: 10.1038/73295. [DOI] [PubMed] [Google Scholar]; •The crystal structure of E. coli DsbC is solved. This protein is a dimer with each monomer consisting of two domains. The catalytic domain has a thioredoxin-like fold and contains a CXXC active site. The dimerization domain has a 6-stranded anti-parallel β-sheet, with the last two strands exchanged between monomers. The domains within the monomer are connected by a hinged linker helix. The dimer has a large cleft that contains 46 hydrophobic or uncharged residues and comprises 45% of the total solvent-accessible surface area, suggesting a role for this cleft in binding unfolded proteins.
- 32.Darby NJ, van Straaten M, Penka E, Vincentelli R, Kemmink J. Identifying and characterizing a second structural domain of protein disulfide isomerase. FEBS Lett. 1999;448:167–172. doi: 10.1016/s0014-5793(99)00374-9. [DOI] [PubMed] [Google Scholar]
- 33.Locker JK, Griffiths G. An unconventional role for cytoplasmic disulfide bonds in vaccina virus proteins. J Cell Biol. 1999;144:267–279. doi: 10.1083/jcb.144.2.267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rudolph R, Lilie H. In vitro folding of inclusion body proteins. FASEB J. 1996;10:49–56. [PubMed] [Google Scholar]
- 35.Zhan X, Schwaller M, Gilbert HF, Georgiou G. Facilitating the formation of disulfide bonds in the Escherichia coli periplasm via coexpression of yeast protein disulfide isomerase. Biotechnol Prog. 1999;15:1033–1038. doi: 10.1021/bp990083r. [DOI] [PubMed] [Google Scholar]
- 36.Qiu J, Swartz JR, Georgiou G. Expression of human tissue-type plasminogen activator in Escherichia coli. Appl Environ Microbiol. 1998;64:4891–4896. doi: 10.1128/aem.64.12.4891-4896.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Joly JC, Leung WS, Swartz JR. Overexpression of Eschericjia coli oxidoreductases increases recombinant insulin-like growth factor-I accumulation. Proc Natl Acad Sci USA. 1998;95:2773–2777. doi: 10.1073/pnas.95.6.2773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bessette PH, Åslund F, Beckwith J, Georgiou G. Efficient folding of proteins with multiple disulfide bonds in the Escherichia coli cytoplasm. Proc Natl Acad Sci USA. 1999;96:13703–13708. doi: 10.1073/pnas.96.24.13703. [DOI] [PMC free article] [PubMed] [Google Scholar]; ••High yields of active E. coli alkaline phosphatase (2 disulfides), mouse urokinase (6 disulfides), truncated human tissue plasminogen activator (9 disulfides), and full-length human tissue plasminogen activator (16 disulfides) are obtained by cytoplasmic production in E. coli mutants that have an oxidizing cytoplasm. Coproduction of DsbC in the cytoplasm gives the highest yields.
- 39.Stewart EJ, Åslund F, Beckwith J. Disulfide bond formation in the Escherichia coli cytoplasm: An in vivo role reversal for the thioredoxins. EMBO J. 1998;17:5543–5550. doi: 10.1093/emboj/17.19.5543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Debarbieux L, Beckwith J. The reductive enzyme thioredoxin 1 acts as an oxidant when it is exported to the Escherichia coli periplasm. Proc Natl Acad Sci USA. 1998;95:10751–10756. doi: 10.1073/pnas.95.18.10751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jonda S, Huber-Wunderlich M, Glockshuber R, Mössner E. Complementation of DsbA deficiency with secreted thioredoxin variants reveals the crucial role of an efficient dithiol oxidant for catalyzed protein folding in the bacterial periplasm. EMBO J. 1999;18:3271–3281. doi: 10.1093/emboj/18.12.3271. [DOI] [PMC free article] [PubMed] [Google Scholar]