

Abstract
Born-type electrostatic continuum methods have been an indispensable ingredient in a variety of implicit-solvent methods that reduce computational effort by orders of magnitude compared to explicit-solvent MD simulations and thus enable treatment using larger systems and/or longer times. An analysis of the limitations and failures of the Born approaches serves as a guide for fundamental improvements without diminishing the importance of prior works. One of the major limitations of the Born theory is the lack of a liquidlike description of the response of solvent dipoles to the electrostatic field of the solute and the changes therein, a feature contained in the continuum Langevin-Debye (LD) model applied here to investigate how Coulombic interactions depend on the location of charges relative to the protein/water boundary. This physically more realistic LD model is applied to study the stability of salt bridges. When compared head to head using the same (independently measurable) physical parameters (radii, dielectric constants, etc.), the LD model is in good agreement with observations, whereas the Born model is grossly in error. Our calculations also suggest that a salt bridge on the protein's surface can be stabilizing when the charge separation is ≤4 Å.
Introduction
In principle, the most accurate description of molecular structure and dynamics in a solvated system requires the explicit retention of all charged groups, especially the solvent molecules. However, despite the continual increases in computational speed, capacity, and number of algorithms for all-atom explicit-solvent simulations of biomolecular systems, there are still important systems (e.g., ribosomes) and processes that are far too large in size and/or require excessively long computational times for treatment with explicit-solvent methods. Therefore, these systems and processes can only be described using reduced representations, such as those provided by implicit-solvent approaches in which the solvent is replaced by a continuum model. Despite the diversity among the continuum models used for this purpose, the indications are that these models can attain accuracy comparable to that of the explicit-solvent models when the correct physics is included (1). Moreover, the computational speed of implicit-solvent models, which are orders of magnitude greater than that of explicit-solvent models, makes it possible to use them to explore a wider range of conditions, parameter space, mutants, etc., to enhance the scope and implications obtainable from explicit-solvent simulations.
Electrostatic modeling figures predominantly in implicit-solvent continuum descriptions for a wide range of applications. Foremost among electrostatic continuum schemes are those based on the Born model or its equivalent in terms of the solution of the Poisson or Poisson-Boltzmann (PB) equation. Although these Born-type methods have been enormously useful, when used with independently measured parameters (ionic radii, dielectric constants, etc.), they suffer from some well-known limitations in treating, for instance, hydration energies of multivalent ions (2–6), calculating the absolute electrostatic solvation and transfer free energies between hydrophobic and hydrophilic environments (7), and estimating the absolute pKa shifts for protein residues (8–10). Attempts to remedy these difficulties within Born-type models include the empirical adjustment of atom radii to values deviating from more direct experimental measures to reproduce ion solvation energies (11–14) and thereby describe the major influences of the liquidlike electrostatic relaxation of the solvent, the strong orientation in the first solvent shell surrounding a charged or partially charged atom and/or the use of different model-dependent solvent dielectric constants for calculating different properties (9). However, the above parameter-jiggling approaches not only obscure the true physical description, but also fail for many molecular systems (and properties) that are dissimilar to the training compounds (and properties) (15–22). For example, when the Born radii are chosen to fit hydration energies, the Born calculations of transfer energies and pKa shifts grossly overestimate experiments (8,10). One possible reason for this failure may arise from oversimplifying the description of the liquidlike dielectric response of the solvent by introducing a single (discordant) effective radius (11), because a highly oriented first solvent layer affects the electric field, thereby tending to orient solvent molecules in the second layer, which further affects the field and orients the molecules in the next layer, and so on.
The venerable Langevin-Debye (LD) model has been developed to describe the nonlinear liquidlike electrostatic influences of solvent reorientation, an effect included in the catchall term “dielectric saturation” (23,24). After the inclusion of “corrections” by Onsager and Kirkwood (25,26), the LD model requires input of information (or its equivalent) concerning the bulk static and optical dielectric constants of the solvent and the dipole moment of the solvent in the gas and liquid phases. This information about the solvent is more detailed than that provided by Born-type models, which depend only on the bulk static dielectric constant of the solvent. Indeed, recent works by Sandberg and Mehler and their co-workers demonstrate that the LD model can be used to explain the hydration energy of multivalent ions (2,27). Recently, we described how its use leads to an ∼50% reduction of computed electrostatic transfer energies and pKa shifts, which may help to eliminate the gross overestimation predicted by Born-type models when using otherwise measured, realistic atomic radii and protein dielectric constants (7).
The numerical solution of the LD model produces a distance-dependent dielectric function. Various analytical alternatives to this numerical solution have been proposed, and applied, to describe electrostatic interactions in molecular dynamics simulations of biological macromolecules (28,29). For instance, the generalized Born model adopts a universal distance-dependent screening function for electrostatic pair interactions (11). However, our previous work uses the LD model to demonstrate that this single universal screening function for electrostatic pair interactions must, at least, be replaced by two universal screening functions, one for charges of like sign and the other for a pair of charges of opposite signs (10). Here, we extend the analysis of the LD model to study the dependence of electrostatic energy and screening functions on the location of a pair of ions within and/or near a solvated spherical protein (15). In addition to our goal of providing insight toward improving generalized Born approaches for protein systems, our work enables a quick, qualitative initial estimation of how the location dependence of dielectric reorientation/saturation affects the stability of salt bridges in proteins, thus generally succeeding in establishing the correct physics and providing motivation for a full quantitative extension to treat the stability with lengthy all-atom molecular dynamics simulations. Moreover, our results indicate that very short (≤4-Å) salt bridges on the protein's surface can stabilize folded protein structures.
Theory
LD model and numerical solutions
The LD model describes the electrostatics for a set of solvated charges in terms of a coupled set of equations for the external electric field, E; the electric displacement, D; the polarization, P; and the local field, F, inside a microscopically small sphere called a Lorentz sphere,
| (1) |
| (2) |
| (3) |
where β = 1/kT, k is Boltzmann's constant, T is the absolute temperature, ɛ0 is the permittivity of the vacuum, α0 and μ are the electric polarizability and the magnitude of permanent dipole moment of the solvent molecules, respectively, n is the number density of the solvent molecules, Cμ and g are the Onsager and Kirkwood correction factors, respectively, and L(x) = coth(x) − 1/x is the Langevin function. The electric polarizability, α0, and permanent dipole moment, μ, of the solvent molecules are estimated from the experimental bulk static and optical dielectric constants.
The numerical solution of Eqs. 1–3 makes it possible to express the electric field, E(r), as a function of D(r) at the spatial position r. Since Eqs. 1–3. imply that E(r) and D(r) are collinear, the relative permittivity, ɛ(r), at r is defined as the ratio of the magnitudes D(r) and E(r) and is calculated as a function of the known electric displacement,
| (4) |
where the shape of the function, f, depends on the physical properties of the solvent, including the dipole moment, μ, the polarizability, α0, and the number density, n, and therefore differs for charges in, for instance, proteins or water. Generally, ɛ(r) varies between the optical dielectric constant, 1.77 at short distances, r, from a charge and (for large r) the bulk static dielectric constant, which is ∼4 inside proteins (30) and 78.5 in water. The shapes of ɛ(r) for a single ion in a protein and in water are presented in our previous work (7). Many continuum model calculations of electrostatic interactions often artificially adjust the dielectric constant inside the protein to 15–20 to reproduce experimental data despite the fact that this value significantly exceeds a reasonable estimate of ∼2.5–4 for the actual dielectric constant (8,9,31). However, our previous article suggests that this disagreement between the experimental and “theoretical” dielectric constant arises at least partially from the attempts to model dielectric saturation, solvent electrostatic relaxation, and protein conformational relaxation as confined mostly to nearest neighbors and thus adequately described as having “fudged” atomic radii. In contrast, the transfer experiments can be correctly described using the LD model and can otherwise measure physically reasonable parameters (7). Fig. 4 A of our recent work (7) shows the LD prediction of an increase in the effective dielectric constant from the center to the surface of globular proteins. Moreover, this result agrees with the two-dielectric constant, concentric-sphere protein model postulated by Simonson and Brooks (32) as describing the origin of the high effective dielectric constants of proteins due to structural relaxation of charged residues.
Figure 4.
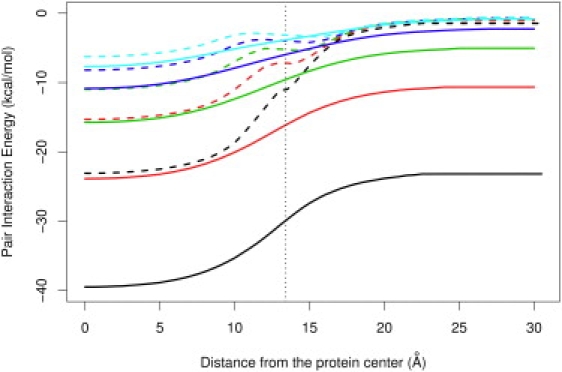
Electrostatic interaction energy for a pair of opposite unit charges as calculated from the LD (solid lines) and Born models (dashed lines). The ion pair separations are 3 Å (black), 4 Å (red), 5 Å (green), 6 Å (blue), and 7 Å (cyan).
A salt bridge is modeled here as a dipole, a pair of ions separated by a fixed distance, L, that is situated at various positions, h, with respect to a solvated spherical protein (Fig. 1). The electric displacement, D(r), for such a system with a pair of interacting ions is simply given by
| (5) |
where e is the elementary charge, R1 and R2 designate the positions of the two ions, and q1 and q2 are their charges.
Figure 1.
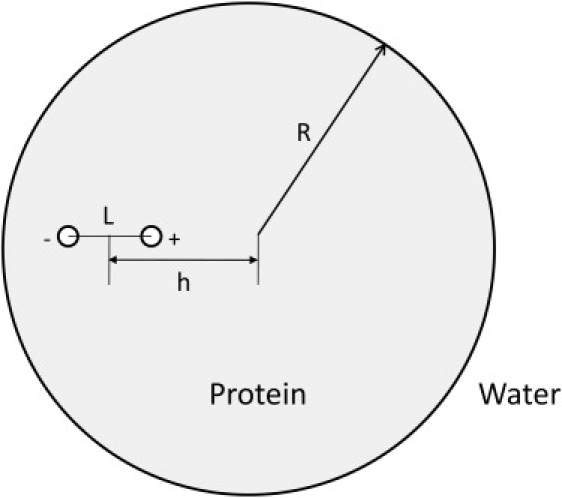
Geometrical description of the model biological system. A spherical protein of radius R is placed in water. A pair of oppositely charged ions, separated by a distance L, is placed along a radial line with the center of the dipole situated at a distance h from the center of the protein.
Electrostatic solvation energy, interaction energy, and screening function
The total electrostatic energy is determined from the integral,
| (6) |
where V represents integration over all space excluding the interior of the ions and f is the relative permittivity as a function of the electric displacement D(r) as obtained numerically from Eq. 4. Referring to the geometry of our model of an ion pair in Fig. 1, the protein-phase dielectric function, fprot, is introduced into the integrand of Eq. 6. when the position r lies inside the spherical protein, and the water function, fwater, is used otherwise. f is set to unity when calculating the total electrostatic energy of the dipole in vacuum. Equation 5 gives D for a two-charge system, whereas D = qe/(4πr2) applies for a single charge, with r the distance from the center of the ion.
The solvation energy of the dipole is defined as the difference in electrostatic energy between the solvated dipole in the target system and the dipole in vacuum,
| (7) |
where Wsolv is the electrostatic solvation energy, is the total electrostatic energy of a dipole placed in our model system of a spherical protein in water, and is the total energy of the dipole in a vacuum. The electrostatic pair-interaction energy, Wint, is the difference between the total energy of the two-charge system and the sum for two independent single-charge systems,
| (8) |
where V is the same as in Eq. 6, and V1 and V2 represent the full space excluding the interior of ions 1 and 2, respectively. D is the electric displacement of a two-charge system (see Eq. 6), and D1 and D2 are the corresponding values for the single-charge systems.
Setting the relative permittivity, f (Eq. 8), to unity throughout space yields the interaction energy for a pair of charges in vacuum as
| (9) |
The pair screening function, fS, is defined as the ratio of the interaction energies in vacuum and in the target biological system as
| (10) |
where h is the distance between the center of the dipole and the center of the spherical protein, L is the length of the dipole, R is the radius of the spherical protein, and q1 and q2 are the charges and a1 and a2 are the radii of the two ions. The integrals in Eqs. 6–9 can be reduced to two-dimensional integrations by adopting confocal ellipsoidal coordinates to enable their evaluation numerically.
Methods
All the parameters required to generate the numerical solution of the LD model for proteins and water are described in our prior work (7). The same radii and dielectric constants are used in calculations with the Born and LD models to compare them on an equal footing and to expose the general physics. The target system depicted in Fig. 1 describes a spherical protein in water with a dipole of fixed length, L, that is situated at locations ranging from a distant position in water to the interior of the protein. To reduce the numerical integration to two dimensions, the center of the spherical protein and the two ions are always taken as collinear. In addition, the radii of the ions are set to 1.4 Å, except when otherwise stated. The radius of gyration of a globular protein is calculated from the formula Rg = 2.83 × N0.34 (33), where N is the number of residues in the protein. Most of the calculations below set the protein radius to 13.4 Å, which equals the radius of gyration of a 100-residue globular protein. Similar calculations have been repeated for 200- and 300-residue proteins with radii equal to 16.9 Å and 19.5 Å, respectively, and the data are available on request. No qualitative changes are found.
Results and Discussion
The solvation energy of dipoles
Fig. 2 A presents the calculated solvation energy of a dipole with fixed length (L = 3 Å) as a function of the distance, h, of the center of the dipole from the protein center using the LD and Born models. By neglecting dielectric saturation and liquidlike solvent electrostatic relaxation, the Born model greatly overestimates the absolute solvation energy of a dipole, even in pure water.
Figure 2.
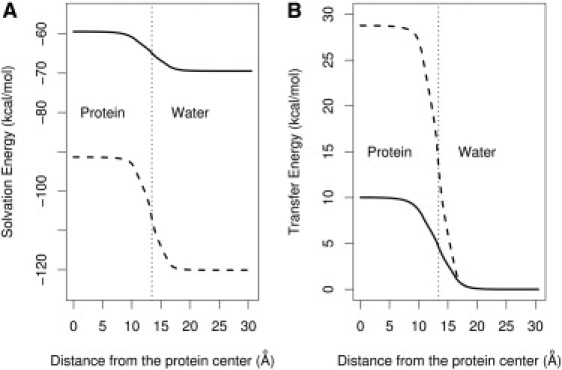
Comparison of solvation and transfer free energies between the LD model (solid line) and the Born model (dashed line). (A) The electrostatic solvation energy calculated for a dipole composed of two unit charges of opposite sign and with a fixed length of 3 Å placed at various positions in the protein-water system. (B) The transfer free energy required to move the dipole from water into the interior of the protein, calculated by subtracting the baseline from the two curves in A. The protein is spherical, with a radius of 13.4 Å, which corresponds to the radius of gyration of a 100-residue globular protein. The dotted vertical line denotes the position of the boundary between the spherical protein and water.
As a test of our model, the solvation energy of amides is predicted with the LD model using the partial charges and atomic radii directly from the Amber94 force field. The amide group is simply represented as two dipoles (C=O and NH), and their correlations are neglected. The sum of the calculated solvation energies for the two dipoles is −12.07 kcal/mol, which is our estimate of the solvation energy of amides. Our result compares well with the experimental solvation energy of −10 kcal/mol (34) and −11.75 kcal/mol calculated by Baldwin and his colleagues (34) using the DELPHI program (13), a PB solver program which uses an optimized set of atomic radii to match the solvation free energy of some small model compounds. Although independently measured parameters are used in our estimation, the energy is as good as in the DELPHI calculation, with parameters that depart from independent measurements of the same physical quantity. In contrast, the Born model predicts this solvation energy as −16.6 kcal/mol, using the same set of independently measured atomic radii.
The transfer free energy,
| (11) |
which is calculated by subtracting the solvation energy in pure water from the total solvation energy in the presence of the protein, displays the existence of even larger errors in the Born model estimations. As shown in Fig. 2 B, the Born model estimate for the energetic barrier for burying an interacting pair of ions is as high as 30 kcal/mol. In contrast, the LD model, by properly describing the long-range consequence of liquidlike electrostatic relaxation and dielectric saturation, reduces this counterintuitive high barrier by two-thirds to 10 kcal/mol, a more reasonable desolvation energy barrier, considering that such dipoles have been observed inside proteins.
The huge overestimation of the transfer free energy by the Born model is in accord with our prior (7) calculation of the solvation energy and electrostatic transfer free energy for burying a single charge inside a spherical protein. Although both calculations indicate the presence of large errors in the Born model, the overestimation of the energetic barrier in that model is more severe for dipole burial (∼3 times) than for burial of a single charge (∼2 times).
To analyze the origins of the difference between the two models, the transfer free energy is decomposed into three terms,
| (12) |
where ΔEtrans (h) is the transfer free energy calculated by subtracting the solvation energy for pure water from the solvation energy for the dipole centered at h, and ΔEint (h) = Wint (h) - Wint (∞) is the pair-interaction energy between the two ions relative to the value in pure water. The decomposition of the transfer free energy for the LD and Born models is contrasted in Fig. S1 of the Supporting Material. Both models predict that the electrostatic pair interactions contribute to stabilizing the burial of a dipole, presumably due to the low dielectric environment inside proteins. Although the Born model overestimates the absolute scale of the electrostatic interactions, the magnitude of overestimation is marginal compared with the errors incurred for the self-energy. However, the sum of the pair-interaction and solvation energies of the individual charges produces a larger error in the solvation energy obtained from the Born model.
Stability of salt bridges
Electrostatic interactions were initially believed to be important in stabilizing protein folding until Kauzmann proposed the dominant role of hydrophobic interactions (35). In a 1985 review, Dill also raised doubts concerning the significance of electrostatic interactions, because only ∼10% of the residues in globular proteins have charged side chains (36). Since Fersht's first estimate for the strength of a salt bridge (37), the question of whether a salt bridge stabilizes proteins has remained controversial (see reviews in Pace et al. (38) and Bosshard et al. (39)), because, despite a favorable interaction energy between the oppositely charged ion pairs, the desolvation penalty is highly unfavorable when the charged groups are moved during the folding process to a location in the interior or on the surface of a globular protein, where solvent accessibility is limited compared with the free accessibility in the solvent. pH titration and mutational analysis are experimental methods applied most frequently to evaluate the contribution of salt bridges to protein stability (39). Unfortunately, pH titrations can never achieve a state in which both opposite charges become neutral, so the titration experiments are unable to decouple the net contribution of the salt bridges from the desolvation energy of the individual charges. In a similar way, mutational analysis usually introduces energy changes from different side-chain packing and van der Waals interactions. Nevertheless, both charges can be neutralized simultaneously, and therefore, mutational analysis experiments probably provide a better approach to the analysis of salt-bridge energetics. Unfortunately, opposite conclusions can be drawn when the mutations introduce side groups that differ in length (40), which indicates that none of the above experimental methods can accurately estimate the net contribution of a salt bridge to protein stability (39). Recently, NMR measurements of pKa shifts have been used frequently to estimate the contribution of ion pairs to protein stability. However, most pKa values are measured by directly varying the pH (similar to pH titrations), and even for the small set of pKa values measured indirectly, no definitive conclusion can be drawn as to whether salt bridges stabilize or destabilize proteins (38). Although computations, in principle, can decouple the solvation and pair-interaction energies to estimate the net contribution of salt bridges, most computational treatments employ a Born-like continuum model, because calculations with microscopic models usually suffer from insufficient sampling and convergence problems. Consequently, the desolvation term is frequently overestimated, and some computational studies predict that salt bridges destabilize folded protein structures (41). Kumar and Nussinov systematically analyze the salt bridges in 36 high-resolution protein structures using continuum electrostatic calculation and discover large variations among the contributions from different ion pairs, although more salt bridges are evaluated as stabilizing than as destabilizing (42). Despite the above controversies, both statistical analyses and experimental works indicate that salt bridges increase the thermodynamic stability of folded proteins. Significantly more ion pairs are found on the surface of hyperthermophilic proteins than on their mesophilic counterparts (38,43). Through numerous mutational analyses of a thermophilic cold-shock protein, Pace successfully identifies a pair of oppositely charged surface residues that contribute significantly to thermal stability (44). By similar mutational analysis on a surface salt bridge of ubiquitin, Makhatadze and his colleagues draw the same conclusion: surface salt bridges are stabilizing (45). Their later series of studies also indicates that complex salt bridges, the electrostatic network among a group of charged residues, stabilize proteins more significantly due to the cooperativity between single ion pairs (46).
The contribution of a salt bridge to the free energy of protein folding can be calculated theoretically from the electrostatic energy difference between a pair of ions buried in a protein and two separate noninteracting ions solvated in water. This energy difference is estimated here using a two-step thermodynamics pathway in which a salt bridge is first formed in pure water and then moved to the interior of a spherical protein. Consequently, the total electrostatic free energy for this process is
| (13) |
where again h is the distance between the center of the dipole and the protein center, Wint(∞) is the interaction energy for a pair of ions in pure water, is the energy required to transfer a pair of ions from pure water to the new position h relative to the center of the protein, and the Born and LD models are contrasted on an equal footing using the same parameters for ion radii and static dielectric constants.
The calculated interaction energy values for a pair of oppositely charged ions with a separation of L = 3 Å in pure water are −23.20 kcal/mol and −1.47 kcal/mol using the LD and Born models, respectively. As discussed in our previous work (10), the Born model grossly overestimates the screening by water and therefore underestimates the magnitude of the interaction energy for an ion pair. The curve in Fig. 3 A, for the free energy to form a salt bridge in the protein/water system as a function the relative position h is obtained by displacing Fig. 2 B by the interaction energies in pure water. Despite the positive desolvation energy barrier, and neglecting the corrections discussed below, the LD model predicts that this pair of opposite charges will stabilize the protein by >10 kcal/mol regardless of whether the charges lie on the surface or in the interior of the protein. In contrast, due to both the overestimation of the solvation barrier and the underestimation of the electrostatic interaction energy, the Born model predicts that the salt bridge will be highly unfavorable (>10 kcal/mol) even when it lies on the surface of a protein. The apparently unreasonably large stability (10 kcal/mol) calculated above should be reduced in actual proteins, since the radii of charged groups in protein residues typically exceed the illustrative value of 1.4 Å adopted here. However, the sign of the above calculation still holds for calculations with realistic radii, indicating the positive contribution of very short salt bridges to protein stability. In summary, the LD model agrees (whereas the Born model disagrees) with the findings of Pace and Makhatadze and their respective co-workers that ion pairs on the protein surface can improve the stability of proteins (44–46).
Figure 3.
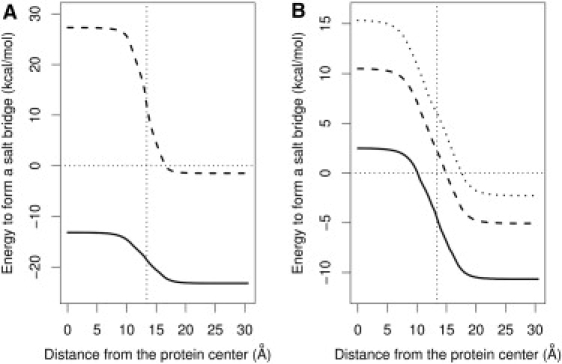
Free energy of formation of a salt bridge between a pair of oppositely charged unit charges in the protein/water system. (A) The energy for a dipole with an intercharge separation of 3 Å as calculated using the LD (solid line) and Born models (dashed line). (B) The energy profiles for ion pairs at larger intercharge separations for the LD model: 4 Å (solid line), 5 Å (dashed line); and 6 Å (dotted line). The vertical thin dotted lines in both graphs represent the location of the boundary between the protein and water. The horizontal thin dotted lines represent an energy of 0 kcal/mol, so the points beneath this line correspond to situations that stabilize protein folding.
Fig. 3 B presents the computed free-energy profiles for ion pairs with larger separations (4–6 Å). The weaker pair-interaction energies at larger separations cannot balance the desolvation energy barrier, especially when the ion pair resides at the center of the protein. However, at an ion separation of 4 Å, the overall energy required to form a salt bridge on the protein surface is still favorable. The relatively favorable energy of short salt bridges (≤4 Å) on the surface or in the interior of proteins explains the observation that there are significantly more opposite charges than like charges in charge-charge separations of ≤4 Å in protein structures (47).
To simplify the calculation, the above calculation assumes that the dipole in the initial hydrated state (outside the protein) is completely solvent-accessible. However, the initial state in a real folding process is the unfolded state, a polypeptide with high flexibility, in which the charged side chains may be in contact with other residues and thus not be as well solvated as is a single isolated dipole. Correcting for this approximation further elevates the energy of the initial state and lowers the overall free-energy stabilization of salt-bridge formation. Hence, the salt bridge should give more stability than what is calculated here. Although the representation of the charged residues comprising the salt bridges as pure ions is an oversimplification (because the residues delocalize the charge and therefore possibly generate weaker fields), the qualitative comparison of salt bridges between the Born and LD models is still robust, because the same ion charges and radii are used throughout our calculations.
Despite the neglect of atomistic molecular details, our simple calculation for the stability of salt bridges is fast and can be completed within a CPU hour. On the other hand, Warshel and colleagues, in an attempt to describe electrostatic relaxation and dielectric saturation effects inside proteins, calculate the electrostatic free energy in molecular simulations using a model with a finite lattice of Langevin dipoles (9,31). However, because only the linear response of the dipoles is evaluated, their work fails to describe accurately dielectric saturation in water. In addition, the molecular simulations in their study require significantly more computer time than our Debye model calculations.
The pair-interaction energy and screening function
Fig. 4 depicts the pair-interaction energy between two unlike charges as a function of the relative position between the dipole and the boundary between the protein and water. Although both the LD and Born models predict a favorable interaction, the Born model yields a severe underestimation, especially for ion pairs at short distances. In addition, all curves calculated by the Born model display artifactual kinks at the protein/water boundary. The differences between the LD and Born models gradually decrease when the separation distance of the ion pairs increases, possibly because the weakened electric field between the more distantly separated ion pairs results in diminished dielectric saturation effects, and the errors introduced by the Born model are thus smaller.
In Fig. 5, the screening functions (effective dielectric) between ion pairs at various separations show a similar tendency of the Born model to heavily overestimate screening by both protein and water. Even the twofold overestimation by the Born model of screening inside proteins can still greatly influence the calculation of the interaction energy (Fig. 4). When the fixed dipole is transferred from the center of a protein into water (Fig. 5, solid curves), the screening function increases gradually from very low values to the highest achievable values in pure water, as described in our previous work (10). In addition, the screening functions increase for longer dipoles because the dielectric saturation is weaker due to a weaker electric field.
Figure 5.
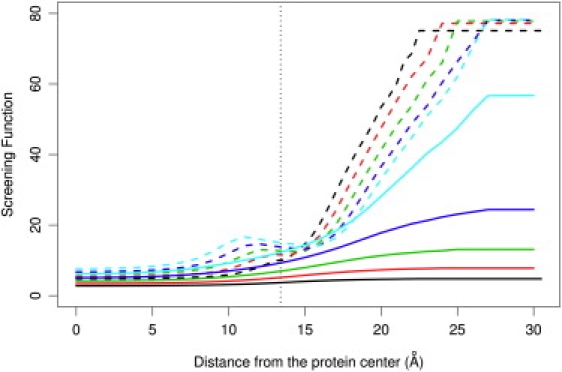
Screening function for a pair of opposite unit charges as calculated from the LD (solid lines) and Born models (dashed lines). The ion pair separations are 3 Å (black), 4 Å (red), 5 Å (green), 6 Å (blue), and 7 Å (cyan).
In our previous article, two universal equations were formulated to describe the screening function for a pair of charges in pure water, given the charges and radii of the ions (10). The work described here begins the process of devising formulas for the electrostatic pair-screening function in a protein/water system that will be useful for implicit-solvent molecular dynamics simulations for proteins. Indeed, an LD-based dielectric screening function applied in implicit-solvent molecular dynamics simulations of RNA molecules showed that LD treatment of the dielectric screening can stabilize the RNA molecules (H. Gong, E. Haddadian, T. R. Sosnick, and K. F. Freed, unpublished).
Supporting Material
A figure is available at http://www.biophysj.org/biophysj/supplemental/S0006-3495(09)01673-7.
Supporting Material
Acknowledgments
We thank Tobin Sosnick, Aaron Dinner, Steve Berry, George Rose, and Bertrand García-Moreno for helpful suggestions and comments.
This research was supported by the Chinese Grant 985 Phase II, by the Tsinghua-Yue-Yuen Medical Sciences Fund, and by National Institutes of Health grants GM081642 and GM57880.
Contributor Information
Haipeng Gong, Email: hgong@tsinghua.edu.cn.
Karl F. Freed, Email: freed@uchicago.edu.
References
- 1.Cramer C.J., Truhlar D.G. Implicit solvation models: equilibria, structure, spectra, and dynamics. Chem. Rev. 1999;99:2161–2200. doi: 10.1021/cr960149m. [DOI] [PubMed] [Google Scholar]
- 2.Sandberg L., Edholm O. Nonlinear response effects in continuum models of the hydration of ions. J. Chem. Phys. 2002;116:2936–2944. [Google Scholar]
- 3.Bucher M., Porter T.L. Analysis of the Born model for hydration of ions. J. Phys. Chem. 1986;90:3406–3411. [Google Scholar]
- 4.Kakitani T., Mataga N. On the possibility of dielectric saturation in molecular systems. Chem. Phys. Lett. 1986;124:437–441. [Google Scholar]
- 5.Sandberg L., Edholm O. A fast and simple method to calculate protonation states in proteins. Proteins. 1999;36:474–483. doi: 10.1002/(sici)1097-0134(19990901)36:4<474::aid-prot12>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- 6.Sandberg L., Edholm O. Calculated solvation free energies of amino acids in a dipolar approximation. J. Phys. Chem. B. 2001;105:273–281. [Google Scholar]
- 7.Gong H., Hocky G.M., Freed K.F. Influence of nonlinear electrostatics on transfer energies between liquid phases: charge burial is far less expensive than Born model. Proc. Natl. Acad. Sci. USA. 2008;105:11146–11151. doi: 10.1073/pnas.0804506105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dwyer J.J., Gittis A.G., García-Moreno E B. High apparent dielectric constants in the interior of a protein reflect water penetration. Biophys. J. 2000;79:1610–1620. doi: 10.1016/S0006-3495(00)76411-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Warshel A., Sharma P.K., Parson W.W. Modeling electrostatic effects in proteins. Biochim. Biophys. Acta. 2006;1764:1647–1676. doi: 10.1016/j.bbapap.2006.08.007. [DOI] [PubMed] [Google Scholar]
- 10.Gong H., Freed K.F. Langevin-Debye model for nonlinear electrostatic screening of solvated ions. Phys. Rev. Lett. 2009;102:057603. doi: 10.1103/PhysRevLett.102.057603. [DOI] [PubMed] [Google Scholar]
- 11.Still W.C., Tempczyk A., Hendrickson T. Semianalytical treatment of solvation for molecular mechanics and dynamics. J. Am. Chem. Soc. 1990;112:6127–6129. [Google Scholar]
- 12.Onufriev A., Case D.A., Bashford D. Effective Born radii in the generalized Born approximation: the importance of being perfect. J. Comput. Chem. 2002;23:1297–1304. doi: 10.1002/jcc.10126. [DOI] [PubMed] [Google Scholar]
- 13.Rocchia W., Alexov E., Honig B. Extending the applicability of the nonlinear Poisson-Boltzmann equation: multiple dielectric constants and multivalent ions. J. Phys. Chem. B. 2001;105:6507–6514. [Google Scholar]
- 14.Zhou B., Agarwal M., Wong C.F. Variable atomic radii for continuum-solvent electrostatics calculation. J. Chem. Phys. 2008;129:014509. doi: 10.1063/1.2949821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mallik B., Masunov A., Lazaridis T. Distance and exposure dependent effective dielectric function. J. Comput. Chem. 2002;23:1090–1099. doi: 10.1002/jcc.10104. [DOI] [PubMed] [Google Scholar]
- 16.Baumketner A., Shea J.E. Kinetics of the coil-to-helix transition on a rough energy landscape. Phys. Rev. E Stat. Nonlin. Soft Matter Phys. 2003;68:051901. doi: 10.1103/PhysRevE.68.051901. [DOI] [PubMed] [Google Scholar]
- 17.Hummer G., Garcia A.E., Garde S. Helix nucleation kinetics from molecular simulations in explicit solvent. Proteins. 2001;42:77–84. [PubMed] [Google Scholar]
- 18.Pak Y., Kim E., Jang S. Misfolded free energy surface of a peptide with αββ motif (1PSV) using the generalized Born solvation model. J. Chem. Phys. 2004;121:9184–9185. doi: 10.1063/1.1804159. [DOI] [PubMed] [Google Scholar]
- 19.Bursulaya B.D., Brooks C.L. Comparative study of the folding free energy landscape of a three-stranded β-sheet protein with explicit and implicit solvent models. J. Phys. Chem. B. 2000;104:12378–12383. [Google Scholar]
- 20.Nymeyer H., García A.E. Simulation of the folding equilibrium of α-helical peptides: a comparison of the generalized Born approximation with explicit solvent. Proc. Natl. Acad. Sci. USA. 2003;100:13934–13939. doi: 10.1073/pnas.2232868100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhou R., Berne B.J. Can a continuum solvent model reproduce the free energy landscape of a β-hairpin folding in water? Proc. Natl. Acad. Sci. USA. 2002;99:12777–12782. doi: 10.1073/pnas.142430099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhou R. Free energy landscape of protein folding in water: explicit vs. implicit solvent. Proteins. 2003;53:148–161. doi: 10.1002/prot.10483. [DOI] [PubMed] [Google Scholar]
- 23.Debye P., Pauling L. The inter-ionic attraction theory of ionized solutes. IV. The influence of variation of dielectric constant on the limiting law for small concentrations. J. Am. Chem. Soc. 1925;47:2129–2134. [Google Scholar]
- 24.Debye P.J.W. The Chemical Catalog Company; New York: 1929. Polar Molecules. [Google Scholar]
- 25.Onsager L. Electric moments of molecules in liquids. J. Am. Chem. Soc. 1936;58:1486–1493. [Google Scholar]
- 26.Kirkwood J.G. The dielectric polarization of polar liquids. J. Chem. Phys. 1939;7:911–919. [Google Scholar]
- 27.Hassan S.A., Mehler E.L. A critical analysis of continuum electrostatics: the screened Coulomb potential-implicit solvent model and the study of the alanine dipeptide and discrimination of misfolded structures of proteins. Proteins. 2002;47:45–61. doi: 10.1002/prot.10059. [DOI] [PubMed] [Google Scholar]
- 28.Ramstein J., Lavery R. Energetic coupling between DNA bending and base pair opening. Proc. Natl. Acad. Sci. USA. 1988;85:7231–7235. doi: 10.1073/pnas.85.19.7231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Min-yi S., Karl F.F. All-atom fast protein folding simulations: the villin headpiece. Proteins. 2002;49:439–445. doi: 10.1002/prot.10230. [DOI] [PubMed] [Google Scholar]
- 30.Gilson M.K., Honig B.H. The dielectric constant of a folded protein. Biopolymers. 1986;25:2097–2119. doi: 10.1002/bip.360251106. [DOI] [PubMed] [Google Scholar]
- 31.Schutz C.N., Warshel A. What are the dielectric constants of proteins and how to validate electrostatic models? Proteins. 2001;44:400–417. doi: 10.1002/prot.1106. [DOI] [PubMed] [Google Scholar]
- 32.Simonson T., Brooks C.L. Charge screening and the dielectric constant of proteins: insights from molecular dynamics. J. Am. Chem. Soc. 1996;118:8452–8458. [Google Scholar]
- 33.Gong H., Fleming P.J., Rose G.D. Building native protein conformation from highly approximate backbone torsion angles. Proc. Natl. Acad. Sci. USA. 2005;102:16227–16232. doi: 10.1073/pnas.0508415102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Avbelj F., Luo P., Baldwin R.L. Energetics of the interaction between water and the helical peptide group and its role in determining helix propensities. Proc. Natl. Acad. Sci. USA. 2000;97:10786–10791. doi: 10.1073/pnas.200343197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kauzmann W. Some factors in the interpretation of protein denaturation. Adv. Protein Chem. 1959;14:1–63. doi: 10.1016/s0065-3233(08)60608-7. [DOI] [PubMed] [Google Scholar]
- 36.Dill K.A. Theory for the folding and stability of globular proteins. Biochemistry. 1985;24:1501–1509. doi: 10.1021/bi00327a032. [DOI] [PubMed] [Google Scholar]
- 37.Fersht A.R. Conformational equilibria and the salt bridge in chymotrypsin. Cold Spring Harb. Symp. Quant. Biol. 1971;36:71–73. doi: 10.1101/sqb.1972.036.01.012. [DOI] [PubMed] [Google Scholar]
- 38.Pace C.N., Grimsley G.R., Scholtz J.M. Protein ionizable groups: pK values and their contribution to protein stability and solubility. J. Chem. Phys. 2009;284:13285–13289. doi: 10.1074/jbc.R800080200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bosshard H.R., Marti D.N., Jelesarov I. Protein stabilization by salt bridges: concepts, experimental approaches and clarification of some misunderstandings. J. Mol. Recognit. 2004;17:1–16. doi: 10.1002/jmr.657. [DOI] [PubMed] [Google Scholar]
- 40.Waldburger C.D., Schildbach J.F., Sauer R.T. Are buried salt bridges important for protein stability and conformational specificity? Nat. Struct. Mol. Biol. 1995;2:122–128. doi: 10.1038/nsb0295-122. [DOI] [PubMed] [Google Scholar]
- 41.Hendsch Z.S., Tidor B. Do salt bridges stabilize proteins? A continuum electrostatic analysis. Protein Sci. 1994;3:211–226. doi: 10.1002/pro.5560030206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kumar S., Nussinov R. Salt bridge stability in monomeric proteins. J. Mol. Biol. 1999;293:1241–1255. doi: 10.1006/jmbi.1999.3218. [DOI] [PubMed] [Google Scholar]
- 43.Karshikoff A., Ladenstein R. Ion pairs and the thermotolerance of proteins from hyperthermophiles: a “traffic rule” for hot roads. Trends Biochem. Sci. 2001;26:550–556. doi: 10.1016/s0968-0004(01)01918-1. [DOI] [PubMed] [Google Scholar]
- 44.Pace C.N. Single surface stabilizer. Nat. Struct. Biol. 2000;7:345–346. doi: 10.1038/75100. [DOI] [PubMed] [Google Scholar]
- 45.Makhatadze G.I., Loladze V.V., Thomas S.T. Contribution of surface salt bridges to protein stability: guidelines for protein engineering. J. Mol. Biol. 2003;327:1135–1148. doi: 10.1016/s0022-2836(03)00233-x. [DOI] [PubMed] [Google Scholar]
- 46.Gvritishvili A.G., Gribenko A.V., Makhatadze G.I. Cooperativity of complex salt bridges. Protein Sci. 2008;17:1285–1290. doi: 10.1110/ps.034975.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Barlow D.J., Thornton J.M. Ion-pairs in proteins. J. Mol. Biol. 1983;168:867–885. doi: 10.1016/s0022-2836(83)80079-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.