

Abstract
Solid tumor malignancies including breast, lung and prostate carcinomas are considered to be angiogenesis dependent. Tumor angiogenesis is often mediated by hypoxia secondary to tumor growth or by increased oncogenic signaling. Both mechanisms result in increased hypoxia-inducible factor-1 alpha (HIF-1α) signaling and its transcriptional target vascular endothelial growth factor (VEGF). Critical to HIF-1α signaling are post translational modifications including acetylation mediated by histone acetyltransferases (HATS) and deacetylation by histone deacetylases (HDACs). More recently, HDACs were shown to be up-regulated in response to hypoxia mediating increased HIF-1α signaling. HDAC inhibitors represent a new class of anti-cancer therapeutics which show great promise at inhibiting angiogenesis in pre-clinical animal models and early phase clinical trials. This review will discuss the role of HIF-1α and VEGF influence on tumor angiogenesis and how HDACs play a critical role in HIF-1α transcriptional activity. Furthermore it will also be discussed how targeting HDACs via their inhibition create new avenues in treating solid malignancies by increasing the activity of established and novel therapeutic applications.
Introduction
Angiogenesis describes the formation of new blood vessels from the existing vasculature and is required for the promotion of fundamental physiological processes including embryonic development, fertility and tissue repair [1]. While angiogenesis has strong implications in homeostasis, it also has the potential to promote tumor growth and metastasis [1, 2]. Within tumors, new blood vessel formation can occur by sprouting from pre-existing vasculature which maybe assisted by the recruitment of circulating cells such as bone marrow derived endothelial progenitor cells, macrophages and fibroblasts [3, 4]. These cells along with malignant cells are able to secrete pro-angiogenic factors including vascular endothelial growth factor (VEGF), which induce tumor blood vessel formation [5].
The transcription factor hypoxia-inducible factor 1 alpha (HIF-1α) regulates the expression of numerous genes involved in various cellular signaling pathways including angiogenesis via the increased expression of VEGF [6]. Over-expression of VEGF mediated by the stabilization of HIF-1α has been identified in multiple malignancies [6] and for this reason targeting the tumor vasculature via the inhibition of VEGF either directly or indirectly has become an attractive target in novel anti-cancer drug development.
This review will focus on the regulation of HIF-1α transcriptional activity by histone deacetylases (HDACs), the anti-angiogenic properties of HDAC inhibitors and their implications as anti-angiogenic agents in treating patients either as a monotherapy or in combination with other available chemotherapy agents.
1. HIF-1 and Angiogenesis
The HIF protein family of transcription factors consists of a constitutively expressed beta subunit HIF-1β whose mRNA and protein levels remain constant and are not regulated by oxygen levels [7] and three alpha subunits; HIF-1α, HIF-2α and HIF-3α which are tightly regulated by oxygen tension levels within a cell [8]. While HIF-2α and HIF-3α are expressed in selected tissues [9], HIF-1α is ubiquitously expressed in both human and mouse tissue and studies have revealed HIF-1α to be the primary executioner of general responses to hypoxia [10]. As part of this, HIF-1α is responsible for the expression of genes that facilitate survival and adaption of cells in both normoxia (normal O2 levels) and hypoxia (low O2 levels) conditions [10].
Under conditions of normoxia, post-translational modifications including the hydroxylation of proline residues and acetylation of a lysine residue within the oxygen-dependent degradation domain (ODDD) promotes HIF-1α interaction with von Hipple-Lindau (pVHL) ubiquitin E3 ligase complex. This occurs concurrently with the hydroxylation of an asparagine residue by the aparaginyl hydroxylase FIH-1 and inhibits the binding of transcriptional co-activators p300 and CBP to HIF-1α. These events result in polyubiquitination and the proteosomal degradation of HIF-1α [11–14]. In contrast, conditions of cellular hypoxia result in HIF-1α stabilized expression by remaining unhydroxylated. Stabilized HIF-1α escapes pVHL mediated degradation and is able to bind p300 and CBP where it translocates to the nucleus from the cytoplasm and heterodimerizes with HIF-1β to initiate transcription of its target genes [7, 15]. (Figure 1). Within the nucleus HIF-1α regulates gene expression of 2% of all human genes either directly or indirectly as shown by studies with endothelial cells using DNA microarrays. This response counter acts hypoxia by inducing multiple physiological responses including erythropoiesis and glycolysis (short term solutions) and angiogenesis (long term solution) [10].
Fig 1.

Schematic cartoon demonstrating the regulation of HIF-1α transcriptional activity. Under normoxic conditions (top row) HIF-1α is hydroxylated, acetylated and bound by the von Hipple-Lindau (pVHL) ubiquitin E3 ligase complex, resulting in polyubiquitination and the proteosomal degradation of HIF-1α. Under hypoxic conditions (bottom row) HIF-1α hydroxylation and acetylation are inhibited due to low oxygen, stabilizing HIF-1α. HIF-1α translocates to the nucleus to bind HIF-1β and recruit CBP/p300 resulting in gene transcription. Hypoxia also induces HDAC expression (middle row) which deacetylates HIF-1α either directly or indirectly to increase HIF-1α transcriptional activity. HDAC inhibition reverses the activity of HDACs resulting in the degradation of HIF-1α.
As mentioned previously, angiogenesis is a multistep and complex process which is necessary for homeostasis but also plays an integral role in solid tumor survival, progression and metastasis [1, 16].. Studies in transgenic mice have confirmed a significant role for HIF-1α in mediating vasculature maturity. Animals over-expressing just VEGF alone demonstrated hypervascularity with hyperpermeable skin [17], while animals with vessels induced with a transgene expressing a stable form of HIF-1α did not exhibit leaky vasculature [18].
2. HIF-1 and Cancer
Angiogenic growth factors, in particular VEGF, and enzymes involved in glucose metabolism exhibit increased expression in malignant tissue when compared to normal tissue. This is due largely to the overexpression of HIF-1α which is observed in various tumors including breast, prostate, brain, lung and head and neck [19, 20]. HIF-1α expression often increases in response to tumor cell proliferation and intratumoral hypoxia, as well as genetic alterations resulting in the activation of oncogenic signaling and inactivation of tumor suppressors [21] though the mechanisms behind this are yet to be fully elucidated. Thus far genetic models have shed some light on how HIF-1α expression maybe altered due to genetic instabilities including the loss of pVHL expression/function resulting in the activation of HIF-1α [22], loss of the tumor suppressors p53 [23] and/or loss of PTEN [24] expression resulting in the activation of PI3K/AKT/mTOR pathway signaling also resulting in the activation of HIF-1α [24, 25].
Other than loss of tumor suppressor gene or gain of oncogene expression, HIF-1α activation maybe induced by various cytokines and growth factors including insulin [26], insulin like growth factors [27], P42/44 mitogen activated kinase (MAPK) whose activation by tyrosine kinases amplifies the transcriptional response of HIF-1α [28]. In addition, the existence of alternate mechanisms to those mentioned above by which HIF-1α expression/activation can be increased have been identified which include the tumor microenviroment [29] and mutations within the ODDD domain of HIF-1α [30].
The over-expression of HIF-1α and VEGF has been reported in many solid tumors, and for this reason both have become attractive targets for anticancer treatment. Although targeting angiogenesis to treat cancer was first proposed in 1971 [31], it wasn’t until 33 years later that the first development and approval of the angiogenesis inhibitor bevacizumab proceeded [32]. The monoclonal antibody Bevacizumab inhibits VEGF and displayed anti-angiogenic and increased survival in colorectal and non small cell lung carcinoma patients when combined with conventional chemotherapy [33, 34]. Following these initial findings, two additional anti-angiogenic compounds, Sorafenib and Sunitinib were produced. Both compounds are small molecule receptor tyrosine kinase inhibitors, targeting multiple signaling pathways including VEGF receptors and platelet-derived growth factor (PDGF) receptors [35]. Sorafenib treatment of hepatocellular carcinoma and Sunitinib and Sorafenib treatment of renal cell carcinomas resulted in modest survival benefits [36–38], as well as toxic side effects [39]. Despite the promising success of these agents, targeting VEGF signaling appears to be insufficient to permanently inhibit tumor angiogenesis. Often tumors treated with anti-VEGF therapy develop resistance by selection of ‘hypoxia resistant’ cells or by activating alternate angiogenic pathways [40], thus suggesting that alternate therapy to target angiogenesis needs to be identified.
3. HDAC, HDAC inhibition and Angiogenesis
The primary role of HDACs is to oppose the activity of histone acetyltransferases (HATs). HDACs remove the acetyl groups from lysine residues of both histone and non-histone proteins [41]. HDACs can be divided into four classes: class I consists of HDAC 1, 2, 3 and 8 (nuclear localization), class II consists of HDAC 4, 5, 6, 7, 9 and 10 (cytoplasm and nuclear localization), class III consists of sirtuins (SIRT1–7) and class IV consists of HDAC 11 which exhibits features of both class I and II HDACs [42]. Class I, II and IV HDACs share homology in both structure and sequence and require a Zinc (Zn+) ion for their catalytic activity, whereas the class III sirtuins share no similarities in their structure or sequence with class I, II or IV HDACs and require a nicotamide adenine dinucleotide (NAD+) ion for their catalytic activity [41].
The regulation of HDAC function under hypoxic conditions as well as HDAC involvement in oxygen regulated gene expression and hypoxia-induced angiogenesis was first investigated and described seven years ago [14, 43]. Initial findings by Kim et al demonstrated that under hypoxic conditions, various cell lines in vitro (both malignant and primary) exhibited increased expression of HDAC1, HDAC2 and HDAC3 mRNA and protein [43]. Furthermore, experiments also showed over-expression of HDAC1 mediated the reduction of the p53 and pVHL expression. The suppression of these two tumor suppressor genes resulted in the over-expression of HIF-1α and VEGF, which was inversed by the use of the histone deacteylase inhibitor (HDACI) Trichostatin A (TSA) both in vitro and in vivo [43]. Mahon et al added to this by showing that the reduction in p53 and pVHL expression also resulted in reduction of factor inhibiting HIF-1α (FIH) allowing for the stimulation of angiogenesis in endothelial cells [14]. These initial reports demonstrated clearly that HDACs regulated HIF-1α activity indirectly under hypoxic conditions. Using biochemical, pharmalogical and genetic approaches Fath et al also showed that HIF-1α activity could also be negatively regulated indirectly by the induced acetylation of p300. This repression of HIF-1α transactivation activity was also independent of p53 and pVHL [44]. Another report also demonstrated that indirect regulation of HIF-1α could induce its degradation independent of pVHL and ubiquitin proteosomal degradation [11]. Via the inhibition of HDAC6 by HDACI, it was shown that hyperacetylation of Hsp90 resulted in the increased interaction and degradation of HIF-1α by Hsp70 [45] (Figure 1; Table 1).
Table 1.
Direct and Indirect influence of HDACs on HIF-1α activity
| Target Protein | HDAC | Direct/Indirect interaction with HIF-1α | Mechanism | Effect of HDAC inhibition |
|---|---|---|---|---|
| pVHL and p53 | HDAC1 | Indirect | Overexpression of HDAC1 reduced pVHL and p53 expression resulting in increased HIF-1α transcription | HDAC1 inhibition results in re-expression of pVHL and p 53 inducing HIF-1 a degradation |
| p300 | HDAC? | Indirect | Proposed HDAC deacetylation regulates binding of p300 to HIF-1α to induce HIF 1α transactivation | HDAC inhibition induces acetylation of p300 causing its disassociation and degradation of HIF-1α |
| Hsp90/Hsp70 axis | HDAC6 | Indirect | HDAC6 regulates Hsp90 function and its interaction with HIF-1α | HDAC6 inhibition results in hyperacetylation of Hsp90, accumulation of immature HIF-1α/Hsp70 complex and degradation of HIF-1α |
| HIF-1α | HDAC1 | Direct | HDAC1 binds the ODDD of HIF-1α to positive regulate HIF-1α stability and transactivation | HDAC1 inhibition results in the degradation and loss of HIF-1α transcriptional activity |
| HIF-1α | HDAC3 | Direct | HDAC3 binds the ODDD of HIF-1α to positive regulate HIF-1α stability and transactivation | HDAC3 inhibition results in the degradation and loss of HIF-1α transcriptional activity |
| HIF-1α | HDAC7 | Direct | HDAC7 co-translocates to the nucleus to increase HIF-1α transcriptional activity via the formation of a HIF-1α/HDAC7/p300 complex | No HDAC inhibition studies were performed |
| HIF-1α | HDAC4 | Direct | HDAC4 associates with HIF-1α to increase its stability and transcriptional activity | Pharmalogical inhibition and shRNA against HDAC4 resulted in decreased HIF-1α expression, transcriptional activity and proteosomal degradation independent of pVHL |
| HIF-1α | HDAC6 | Direct | HDAC6 associates with HIF-1α to increase its stability and transcriptional activity | Pharmalogical inhibition and shRNA against HDAC6 resulted in decreased HIF-1α expression, transcriptional activity and proteosomal degradation independent of pVHL |
Although there is conclusive data showing HDACs regulate HIF-1α activity through indirect mechanisms, it has also been observed that HDACs interact directly with HIF-1α to regulate its activity. HDAC1 and HDAC3 have been shown to directly regulate HIF-1α stability and transcriptional activity via interaction with the ODDD of HIF-1α [46], though previous work conducted in our laboratory contradicts this report. Treatment of PC3 and C2 cell lines with the class I specific HDACI MS275 (Pili laboratory; unpublished data) did not result in the loss of HIF-1α expression as shown by Kim et al [46]. One possible explanation for these differences could be that HIF-1α direct interaction with HDAC1 and HDAC3 maybe cell specific. Furthermore, HDAC7, a class II HDAC, has strong interaction with FIH-1, but under hypoxic conditions HDAC7 translocates from the cytoplasm to the nucleus to bind HIF-1α and increase its transcriptional activity [47]. While HDAC7 has a role in regulating angiogenesis in tumor cells [47], it also influences angiogenesis in primary endothelial cells. Using small interfering siRNAs, Mottet et al inhibited expression of both HDAC1 and HDAC7 in human umbilical vein endothelial cells (HUVECs) and indicated that HDAC7 was necessary for the assembly of endothelial cell in tube like structures in vitro [48]. In addition, the loss of HDAC7 expression resulted in morphological changes and decreased endothelial cell migration, concurrent with increased expression platelet derived growth factor (PDGF)-B and its β receptor (PDGF-β) [48]. Another study by Qian et al describes that HDAC4 and HDAC6 are vital for protein stability and transcriptional activity of HIF-1α. Using shRNA and pharmalogical inhibition targeting HDAC4 and HDAC6, it was shown that these class II HDACs induced HIF-1α protein stability via proteosome-dependent pathway in renal cell carcinoma cell lines devoid of pVHL [11] (Figure 1; Table 1).
The above mentioned studies convincingly show that the involvement of HDACs plays a vital role in hypoxic induced angiogenesis, and that targeting HDACs via their inhibition offers a new strategy in anti-cancer therapy through their ability to inhibit angiogenesis. To date numerous pre-clinical and clinical studies now exist which confirm a role for the anti-angiogenic activities of HDACI in the treatment of multiple tumors.
The anti-angiogenic properties of HDACI have been associated with the alteration of numerous pro- and anti-angiogenic genes [49]. Other than HIF-1α, VEGF and FGF (mentioned above), examples of genes which can be commonly down-regulated by HDACI are angiopoietin, tunica intima endothelial kinase 2 (TIE2), and endothelial nitric oxide synthase (eNOS) [50, 51]. In cancer cells HDACI have also been found to up-regulate p53, pVHL, thrombospondin 1 and anti-angiogenic activin A transcription [43, 52–54]. More specifically, valproic acid displayed anti-tumor effect by inhibiting the expression of VEGF and FGF in the colon carcinoma cell line Caco-2. Furthermore, the inhibition of VEGF and FGF was associated with the activation of the ubiquitin-proteosome degradation pathway [55]. Another example of anti-angiogenic activity by HDACI was displayed by the use of FK228, which inhibited hypoxia-induced angiogenesis. FK228 anti-angiogenic activities were a result of the transcriptional induction of pVHL and neurofibromin-2 (NF-2) conceited by the transcriptional repression of HIF-1α, VEGF and VEGF receptor [52, 56]. A more recent study identified novel genes involved in the negative regulation of endothelial cell growth and angiogenesis. Using microarray technology coupled with RNA interference for functional validation it was described that the genes clusterin, fibrillin 1 and quiescin Q6 potentiated tumor conditioned endothelial cell growth and angiogenesis [57]. Interestingly, the epigenetic silencing of these genes occurred as a result of promoter histone H3 deacetylation and loss of H3 lysine 4 methylation but did not involve DNA methylation of promoter CpG islands [57]. Each gene was re-expressed following treatment with either TSA or 5-aza-2′-deoxycytodine demonstrating that the silencing of these genes during angiogenesis occurs concurrent with promoter histone modifications and not promoter DNA hyper-methylation [57] (Table 2).
Table 2.
Pro- and anti-angiogenic genes altered by HDACI in both cancer and endothelial cells
| Gene | Target Cell | Activity on angiogensis | Effect on gene transcription by HDAC inhibition |
|---|---|---|---|
| p53 | Cancer | inhibits | Up-regulation |
| pVHL | Cancer | induces | Up-regulation |
| HIF-1α | Cancer | induces | Down-regulation |
| VEGF | Cancer | induces | Down-regulation |
| Activin A | Cancer | inhibits | Up-regulation |
| bFGF | Cancer | induces | Down-regulation |
| Thrombospondin 1 | Cancer | inhibits | Up-regulation |
| MMP-2 | Cancer | induces | Up-regulation |
| MMP-9 | Cancer | induces | Up-regulation |
| RECK | Cancer | inhibits | Up-regulation |
| FLT1 | Cancer | induces | Down-regulation |
| FLK1 | Cancer | induces | Down-regulation |
| Neurofibromin2 | Cancer | inhibits | Up-regulation |
| Ang1 | Cancer | induces | Down-regulation |
| VEGF receptor 1 | Endothelial | induces | Down-regulation |
| VEGF receptor 2 | Endothelial | induces | Down-regulation |
| Neuropilin-1 | Endothelial | induces | Down-regulation |
| Semaphoring III | Endothelial | inhibits | Up-regulation |
| Tie2 | Endothelial | induces | Down-regulation |
| Ang2 | Endothelial | induces | Down-regulation |
| eNOS | Endothelial | induces | Down-regulation |
| VEGFD | Endothelial | induces | Down-regulation |
| Clusterin | Endothelial | inhibits | Up-regulation |
| Fibrillin1 | Endothelial | inhibits | Up-regulation |
| Quiescin Q6 | Endothelial | inhibits | Up-regulation |
Adapted from Liu et al [49]
HDACI have also been noted to directly affect endothelial cells with the most prominent observation being the up-regulation p21WAF1/CIP1, which induces G1 cell cycle arrest and down-regulate the expression of survivin, an inhibitor of apoptosis in proliferating endothelial cells [50]. Qian et al also demonstrated that LAQ824 blocked pro-angiogenic tyrosine kinase receptors Tie 2 and Tie 2 ligand and angiopoietin 2 mRNA and protein [50]. Previous studies conducted by Deroanne et al also showed that TSA and vorinostat inhibit VEGF and VEGF receptors VEGFR1 and VEGFR2, as well as up-regulate the VEGF competitor protein, semaphorin II [58]. In addition the down regulation of eNOS in endothelial cells by valproic acid was shown to be important for HDACI mediated anti-angiogenic activity [59] (Table 2).
Preclinical in vivo models have also demonstrated mediation of angiogenic genes can determine the therapeutic benefit by HDACI. Experiments utilizing the chicken chorioallantoic membrane assay and matrigel plug assays in mice demonstrated that valproic acid inhibited angiogenesis in vivo, and it is hypothesized from coupled in vitro experiments to be mediated by decreased eNOS expression which was preceeded by HDAC inhibition [60]. Furthermore, mouse embryos from valproic acid treated mice displayed disturbed vessel formation [60]. More recently, Qian et al used LBH589 to pre-treat mice before injecting VEGF-A enriched matrigel plugs. Upon retrieval of the plugs it was observed that in vivo neo-vascularization was inhibited by LBH589 [61]. Also, LBH589 treatment of xenograft mice bearing PC3 (prostate carcinoma) tumors also showed significant growth delay. The anti-tumor effect was accounted for by LBH589 ability to inhibit angiogenesis as assessed by decreased CD31+ vessel structures when compared to vehicle treated mice. Interestingly, TUNEL staining revealed that there was no difference in apoptotic cell death in vehicle or LBH589 treated mice, determining that inhibition of angiogenesis and induction of apoptosis in vivo are two separate events [61].
While a large emphasis has been placed on the role of class I, II and IV HDACs in angiogenesis and tumorgenesis, more recent studies have been in effort to identify the function of class III HDACs in both events. Sirtuins have been demonstrated to regulate such targets as p53 [62], indicating that deregulated expression of sirtuins may influence both oncogenic and/or angiogenic signaling. Consistent with this possibility, sirtuin over expression has been documented in a wide range of tumors [63, 64]. Potente et al recently described that SIRT1 was critical for angiogenic signaling in response to ischemic stress [65]. Furthermore, loss of SIRT1 expression via RNAi-mediated gene silencing, pharmalogical inhibition or Cre-mediated excision of the floxed SIRT1 deacetylase domain, was demonstrated to prevent endothelial cell vascular-like sprout formation in vitro [62], suggesting that class III HDACs may create novel targets in treating cancers that rely on angiogensis for survival.
4. HDAC inhibitors in combination to target Angiogenesis
More over HDACI as monotherapy are displaying promising, although limited responses in the clinic and therefore their role in combination therapy may see HDACI reach their full potential as anti-cancer agents.
While HDACI are being combined with multiple anti-cancer agents (both novel and conventional) [41], only a few strategies targeting angiogenesis are currently under development that may show promise to clinical translation. As mentioned previously, the development and use of tyrosine kinase inhibitors (TKI) resulted in exciting results at first but disease relapses were common among patients and for this reason it has been investigated whether targeting HIF-1α and VEGF signaling by combining HDACI with TKI would enhance the anti-tumor effects of each compound. Research by Qian et al described that the TKI PTK787/ZK222584 only exhibited anti-angiogenesis effect on endothelial cells while the HDACI LAQ824 targeted both endothelial cells and tumor epithelial cells [50]. Combination treatment of both agents resulted in better efficacy of inhibiting in vitro and in vivo VEGF-mediated angiogenesis. Furthermore, LAQ824 inhibited the expression of angiogenic genes including angiopoietin-2, Tie-2 and survivin in endothelial cells and down regulated the expression of HIF-1α and VEGF in tumor cells [50]. Yu et al [66], recently described that combination of the multiple receptor tyrosine kinase inhibitor AEE788 with numerous HDACI (LBH589, LAQ824 and TSA) resulted in synergistic cytotoxicity in numerous solid and hematological cancer cell lines. AEE788 inhibition of mitogen activated protein kinase (MAPK) and Akt signaling enhanced HDACI-mediated apoptosis via the induction of ROS [66].
Tyrosine kinase signaling often results in the activation of survival pathways mediated by PI3K/Akt/mTOR signaling and for this reason has made this pathway attractive for therapy intervention. By using various techniques like pharmacological inhibition using LY294002 (targeting PI3K) and rapamycin (targeting mTOR) as well as biochemical methods including dominant negative expression of Akt, PI3K and PTEN it was observed that the induction of VEGF and HIF-1α was inhibited, linking the PI3K/Akt/mTOR pathway, HIF-1α and angiogensis [67]. Only recently though has the combination of mTOR inhibition with HDACI been evaluated [6]. Utilizing the capabilities of rapamycin and LBH589 to inhibit HIF-1α through different mechanisms, combination treatment demonstrated greater decrease in clonogenic potential as well as significantly lowering HIF-1α protein expression compared to single agents in PC3, C2 and HUVEC cell lines. In addition the combination of these agents resulted in significant inhibition of PC3 and C2 in vivo tumor growth and angiogenesis assessed by tumor weights and microvessel density [6].
The use of HDACI with demethylating agents has shown greater anti-tumor effect linked to the inhibition of angiogenesis. Maspin, a member of the serpin superfamily whose activity regulates such biological pathways including angiogenesis and metastasis was shown to have its expression silenced in oral cancer cell lines [68]. Upon treatment with the demethylating agent 5-aza-dC and/or the HDACI FR901228 the re-expression of maspin mRNA was observed [68]. Of interest the re-expression of maspin was not a result of the demethylation of CpG islands, indicating that histone post-translational modifications maybe the key mechanism behind maspin expression. Hellebrekers et al [69] experiments also demonstrated that HDACI and demethylating agents could inhibit immune escape of tumor conditioned endothelial cells in vitro and in vivo by the re-expression of intercellular adhesion molecule-1 (ICAM-1) restoring leukocyte-endothelial cell adhesion. Another study by Hellebrekers et al [57] mentioned previously in this review, discussed the silencing of novel genes which may mediate neo-angiogenesis in tumor conditioned endothelial cells, one of which was clusterin. An additional study carried out by Suuronen et al capitulates that clusterin mediates neo-angiogenesis. Treatment of the human cell line, retinal pigment epithelial cells (ARPE-19) with HDACI valproic acid or TSA and the demethylating agent 5-aza-2′-deoxycytidine resulted in increased clusterin mRNA and protein levels [70], further demonstrating that clusterin expression is epigentically regulated and may play a vital role in neo-angiogensis in a tumor setting.
5. Clinical application of HDACI targeting angiogenesis
While numerous clinical trial data has been published involving patients with various cancers being treated with HDACI, only few reports include correlative studies which investigate the mechanisms behind HDACI success in the clinic. Of the published data two recent reports on patients with refractory cutaneous T cell lymphoma (CTCL) indicate that the anti-angiogenic actions of HDACI may potentiate the clinical response noted in patients. In a phase II trial reported by Duvic et al refractory CTCL patients were treated with vorinostat [71]. Correlative studies performed on patient’s paired skin lesion samples pre-therapy compared with skin lesion samples collected 2 hours, 4 weeks and 8 weeks after vorinostat treatment revealed a significant decrease in the microvessel density in responding patients. Furthmore, decreased microvessel density was observed to be concurrent with an increase in thrombosponin-1 (TSP-1) expression in 67% of responding patients [71]. More recently, a report from a phase I clinical trial of refractory CTCL patients treated with panobinostat was published. In this small scale study of 10 patients, gene profiling from biopsy samples of 6 patients was conducted to further investigate possible mechanisms of HDACI anti-cancer activity. Interestingly, these studies revealed that a common set of 23 genes had significantly altered expression in all 6 patients independent of their clinical outcome, 4 of which were further validated by QRT-PCR [72]. Three from the 4 genes validated were identified as pro-angiogenic, including GUCY1A3, Angiopoietin-1 and COUPTF-II. All three genes were found to be highly expressed in baseline biopsies from patients and were all subsequently down regulated following panobinostat treatment [72].
Within our laboratory, pre-clinical studies suggest that targeting angiogensis with HDACI combined with other anti-angiogenic therapies targeting VEGF directly (eg: monoclonal antibodies or TKI) or indirectly via mTOR inhibition may prove more beneficial to patients, and thus clinical trails are being either conducted or planned to investigate this (Figure 2). The objective of a recently completed Phase I study by our group was to determine the safety, tolerability, and pharmacokinetic/pharmacodynamic profiles of the HDAC inhibitor vorinostat in combination with the VEGF inhibitor bevacizumab. Patients with measurable stage IV clear cell renal cell carcinoma and up to 2 prior regimens were eligible. Treatment consisted of vorinostat 200 mg orally twice daily x 2 weeks, and bevacizumab 15 mg/kg intravenously every 3 weeks. Each cycle was every 21 days. Western Blot analyses on peripheral-blood mononuclear cells (PBMCs) and platelet-rich plasma were performed. Eight male patients were enrolled, and 7 were evaluable; prior nephrectomy = 8; prior systemic therapy (receptor tyrosine kinase inhibitors) = 7. One of 7 patients experienced DLT with grade 4 thrombocytopenia. Other grade 3 toxicities (not DLTs) included anorexia, fatigue, hemothorax, and pulmonary embolism. The best responses in the 7 evaluable patients were stable disease. One patient with mixed response had stable disease greater than 18 months. Two other patients had stable disease for 5 and 6 months. Vorinostat mean Cmax concentration (Day1) and T1/2 were 1.264 ± 0.629 μM and 1.5 h, respectively (consistent with prior reports as single agent). Histones isolated from PBMCs showed transient but consistent post-therapy protein acetylation. Platelet- but not plasma-derived free VEGF was reduced by treatment. Decreased tumor metabolism and blood flow were observed by PET scan in two patients. The combination of vorinostat 200 mg PO BID with bevacizumab 15 mg/kg every 21 days is reasonably well tolerated. Tumor response and PD data suggest clinical and biological activity for this combination strategy. Furthermore, based on preclinical data generated in our lab we also designed a clinical trial of targeting HIF-1α by different mechanism of action. A Phase I/II clinical study will be conducted with the HDAC inhibitor LBH589 and the mTOR inhibitor RAD001 in patients with metastatic renal cell carcinoma who progress on anti VEGF therapies.
Fig 2.
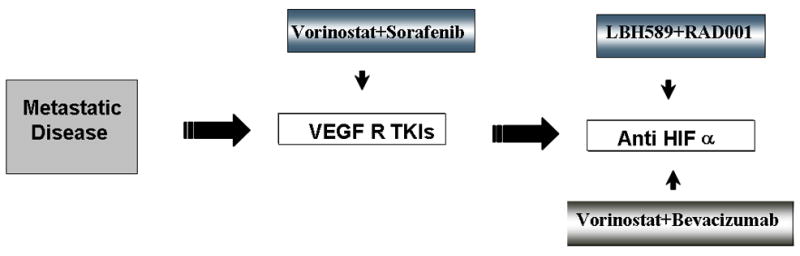
Schematic cartoon representing the strategies to treat solid tumor patients with metastatic disease. HDACI will be combined with TKI, monoclonal antibody or mTOR inhibitors.
6. Conclusion
As a better understanding of the pivotal role of angiogenesis in tumor biology is put together, it has been revealed that HIF-1α and its transcriptional product VEGF are key mediators of the response to tumor-induced hypoxia. For this reason targeting VEGF either directly or indirectly has become an attractive target of therapeutic approaches. Inhibiting VEGF specifically or the VEGF signaling pathway returned promising results though escape mechanisms counter acted the actions of the drugs, indicating the requirement for alternate therapies to be pursued. Currently, pre-clinical and clinical studies indicate that HDACI have positive effects on the expression of pro- and anti-angiogenic genes suggesting that the use of HDACI may potentiate the actions of current anti-VEGF therapies including bevacizumab and TKI as well as novel therapeutic approaches such as mTOR inhibition.
References
- 1.Carmeliet P. Angiogenesis in life, disease and medicine. Nature. 2005;438(7070):932–6. doi: 10.1038/nature04478. [DOI] [PubMed] [Google Scholar]
- 2.Avraamides CJ, Garmy-Susini B, Varner JA. Integrins in angiogenesis and lymphangiogenesis. Nat Rev Cancer. 2008;8(8):604–17. doi: 10.1038/nrc2353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Adams RH, Alitalo K. Molecular regulation of angiogenesis and lymphangiogenesis. Nat Rev Mol Cell Biol. 2007;8(6):464–78. doi: 10.1038/nrm2183. [DOI] [PubMed] [Google Scholar]
- 4.Kerbel RS. Tumor angiogenesis. N Engl J Med. 2008;358(19):2039–49. doi: 10.1056/NEJMra0706596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lin EY, Pollard JW. Tumor-associated macrophages press the angiogenic switch in breast cancer. Cancer Res. 2007;67(11):5064–6. doi: 10.1158/0008-5472.CAN-07-0912. [DOI] [PubMed] [Google Scholar]
- 6.Verheul HM, et al. Combination strategy targeting the hypoxia inducible factor-1 alpha with mammalian target of rapamycin and histone deacetylase inhibitors. Clin Cancer Res. 2008;14(11):3589–97. doi: 10.1158/1078-0432.CCR-07-4306. [DOI] [PubMed] [Google Scholar]
- 7.Kallio PJ, et al. Activation of hypoxia-inducible factor 1alpha: posttranscriptional regulation and conformational change by recruitment of the Arnt transcription factor. Proc Natl Acad Sci U S A. 1997;94(11):5667–72. doi: 10.1073/pnas.94.11.5667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Salceda S, Caro J. Hypoxia-inducible factor 1alpha (HIF-1alpha) protein is rapidly degraded by the ubiquitin-proteasome system under normoxic conditions. Its stabilization by hypoxia depends on redox-induced changes. J Biol Chem. 1997;272(36):22642–7. doi: 10.1074/jbc.272.36.22642. [DOI] [PubMed] [Google Scholar]
- 9.Ke Q, Costa M. Hypoxia-inducible factor-1 (HIF-1) Mol Pharmacol. 2006;70(5):1469–80. doi: 10.1124/mol.106.027029. [DOI] [PubMed] [Google Scholar]
- 10.Semenza GL. Hypoxia-inducible factor 1: master regulator of O2 homeostasis. Curr Opin Genet Dev. 1998;8(5):588–94. doi: 10.1016/s0959-437x(98)80016-6. [DOI] [PubMed] [Google Scholar]
- 11.Qian DZ, et al. Class II histone deacetylases are associated with VHL-independent regulation of hypoxia-inducible factor 1 alpha. Cancer Res. 2006;66(17):8814–21. doi: 10.1158/0008-5472.CAN-05-4598. [DOI] [PubMed] [Google Scholar]
- 12.Hewitson KS, et al. Hypoxia-inducible factor (HIF) asparagine hydroxylase is identical to factor inhibiting HIF (FIH) and is related to the cupin structural family. J Biol Chem. 2002;277(29):26351–5. doi: 10.1074/jbc.C200273200. [DOI] [PubMed] [Google Scholar]
- 13.Lando D, et al. FIH-1 is an asparaginyl hydroxylase enzyme that regulates the transcriptional activity of hypoxia-inducible factor. Genes Dev. 2002;16(12):1466–71. doi: 10.1101/gad.991402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mahon PC, Hirota K, Semenza GL. FIH-1: a novel protein that interacts with HIF-1alpha and VHL to mediate repression of HIF-1 transcriptional activity. Genes Dev. 2001;15(20):2675–86. doi: 10.1101/gad.924501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huang LE, et al. Activation of hypoxia-inducible transcription factor depends primarily upon redox-sensitive stabilization of its alpha subunit. J Biol Chem. 1996;271(50):32253–9. doi: 10.1074/jbc.271.50.32253. [DOI] [PubMed] [Google Scholar]
- 16.Zagzag D, et al. Expression of hypoxia-inducible factor 1alpha in brain tumors: association with angiogenesis, invasion, and progression. Cancer. 2000;88(11):2606–18. [PubMed] [Google Scholar]
- 17.Thurston G, et al. Leakage-resistant blood vessels in mice transgenically overexpressing angiopoietin-1. Science. 1999;286(5449):2511–4. doi: 10.1126/science.286.5449.2511. [DOI] [PubMed] [Google Scholar]
- 18.Elson DA, et al. Induction of hypervascularity without leakage or inflammation in transgenic mice overexpressing hypoxia-inducible factor-1alpha. Genes Dev. 2001;15(19):2520–32. doi: 10.1101/gad.914801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brahimi-Horn MC, Pouyssegur J. The hypoxia-inducible factor and tumor progression along the angiogenic pathway. Int Rev Cytol. 2005;242:157–213. doi: 10.1016/S0074-7696(04)42004-X. [DOI] [PubMed] [Google Scholar]
- 20.Quintero M, Mackenzie N, Brennan PA. Hypoxia-inducible factor 1 (HIF-1) in cancer. Eur J Surg Oncol. 2004;30(5):465–8. doi: 10.1016/j.ejso.2004.03.008. [DOI] [PubMed] [Google Scholar]
- 21.Kim W, Kaelin WG., Jr The von Hippel-Lindau tumor suppressor protein: new insights into oxygen sensing and cancer. Curr Opin Genet Dev. 2003;13(1):55–60. doi: 10.1016/s0959-437x(02)00010-2. [DOI] [PubMed] [Google Scholar]
- 22.Ivan M, Kaelin WG., Jr The von Hippel-Lindau tumor suppressor protein. Curr Opin Genet Dev. 2001;11(1):27–34. doi: 10.1016/s0959-437x(00)00152-0. [DOI] [PubMed] [Google Scholar]
- 23.An WG, et al. Stabilization of wild-type p53 by hypoxia-inducible factor 1alpha. Nature. 1998;392(6674):405–8. doi: 10.1038/32925. [DOI] [PubMed] [Google Scholar]
- 24.Zundel W, et al. Loss of PTEN facilitates HIF-1-mediated gene expression. Genes Dev. 2000;14(4):391–6. [PMC free article] [PubMed] [Google Scholar]
- 25.Stiehl DP, et al. Normoxic induction of the hypoxia-inducible factor 1alpha by insulin and interleukin-1beta involves the phosphatidylinositol 3-kinase pathway. FEBS Lett. 2002;512(1–3):157–62. doi: 10.1016/s0014-5793(02)02247-0. [DOI] [PubMed] [Google Scholar]
- 26.Zelzer E, et al. Insulin induces transcription of target genes through the hypoxia-inducible factor HIF-1alpha/ARNT. Embo J. 1998;17(17):5085–94. doi: 10.1093/emboj/17.17.5085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Feldser D, et al. Reciprocal positive regulation of hypoxia-inducible factor 1alpha and insulin-like growth factor 2. Cancer Res. 1999;59(16):3915–8. [PubMed] [Google Scholar]
- 28.Richard DE, et al. p42/p44 mitogen-activated protein kinases phosphorylate hypoxia-inducible factor 1alpha (HIF-1alpha) and enhance the transcriptional activity of HIF-1. J Biol Chem. 1999;274(46):32631–7. doi: 10.1074/jbc.274.46.32631. [DOI] [PubMed] [Google Scholar]
- 29.Weidemann A, Johnson RS. Biology of HIF-1alpha. Cell Death Differ. 2008;15(4):621–7. doi: 10.1038/cdd.2008.12. [DOI] [PubMed] [Google Scholar]
- 30.Mabjeesh NJ, Amir S. Hypoxia-inducible factor (HIF) in human tumorigenesis. Histol Histopathol. 2007;22(5):559–72. doi: 10.14670/HH-22.559. [DOI] [PubMed] [Google Scholar]
- 31.Folkman J. Tumor angiogenesis: therapeutic implications. N Engl J Med. 1971;285(21):1182–6. doi: 10.1056/NEJM197111182852108. [DOI] [PubMed] [Google Scholar]
- 32.Ferrara N, et al. Discovery and development of bevacizumab, an anti-VEGF antibody for treating cancer. Nat Rev Drug Discov. 2004;3(5):391–400. doi: 10.1038/nrd1381. [DOI] [PubMed] [Google Scholar]
- 33.Hurwitz H, et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med. 2004;350(23):2335–42. doi: 10.1056/NEJMoa032691. [DOI] [PubMed] [Google Scholar]
- 34.Sandler A, et al. Paclitaxel-carboplatin alone or with bevacizumab for non-small-cell lung cancer. N Engl J Med. 2006;355(24):2542–50. doi: 10.1056/NEJMoa061884. [DOI] [PubMed] [Google Scholar]
- 35.Faivre S, et al. Molecular basis for sunitinib efficacy and future clinical development. Nat Rev Drug Discov. 2007;6(9):734–45. doi: 10.1038/nrd2380. [DOI] [PubMed] [Google Scholar]
- 36.Escudier B, et al. Sorafenib in advanced clear-cell renal-cell carcinoma. N Engl J Med. 2007;356(2):125–34. doi: 10.1056/NEJMoa060655. [DOI] [PubMed] [Google Scholar]
- 37.Llovet JM, et al. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008;359(4):378–90. doi: 10.1056/NEJMoa0708857. [DOI] [PubMed] [Google Scholar]
- 38.Motzer RJ, et al. Activity of SU11248, a multitargeted inhibitor of vascular endothelial growth factor receptor and platelet-derived growth factor receptor, in patients with metastatic renal cell carcinoma. J Clin Oncol. 2006;24(1):16–24. doi: 10.1200/JCO.2005.02.2574. [DOI] [PubMed] [Google Scholar]
- 39.Verheul HM, Pinedo HM. Possible molecular mechanisms involved in the toxicity of angiogenesis inhibition. Nat Rev Cancer. 2007;7(6):475–85. doi: 10.1038/nrc2152. [DOI] [PubMed] [Google Scholar]
- 40.Kerbel R, Folkman J. Clinical translation of angiogenesis inhibitors. Nat Rev Cancer. 2002;2(10):727–39. doi: 10.1038/nrc905. [DOI] [PubMed] [Google Scholar]
- 41.Bolden JE, Peart MJ, Johnstone RW. Anticancer activities of histone deacetylase inhibitors. Nat Rev Drug Discov. 2006;5(9):769–84. doi: 10.1038/nrd2133. [DOI] [PubMed] [Google Scholar]
- 42.Glozak MA, Seto E. Histone deacetylases and cancer. Oncogene. 2007;26(37):5420–32. doi: 10.1038/sj.onc.1210610. [DOI] [PubMed] [Google Scholar]
- 43.Kim MS, et al. Histone deacetylases induce angiogenesis by negative regulation of tumor suppressor genes. Nat Med. 2001;7(4):437–43. doi: 10.1038/86507. [DOI] [PubMed] [Google Scholar]
- 44.Fath DM, et al. Histone deacetylase inhibitors repress the transactivation potential of hypoxia-inducible factors independently of direct acetylation of HIF-alpha. J Biol Chem. 2006;281(19):13612–9. doi: 10.1074/jbc.M600456200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kong X, et al. Histone deacetylase inhibitors induce VHL and ubiquitin-independent proteasomal degradation of hypoxia-inducible factor 1alpha. Mol Cell Biol. 2006;26(6):2019–28. doi: 10.1128/MCB.26.6.2019-2028.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kim SH, et al. Regulation of the HIF-1alpha stability by histone deacetylases. Oncol Rep. 2007;17(3):647–51. [PubMed] [Google Scholar]
- 47.Kato H, Tamamizu-Kato S, Shibasaki F. Histone deacetylase 7 associates with hypoxia-inducible factor 1alpha and increases transcriptional activity. J Biol Chem. 2004;279(40):41966–74. doi: 10.1074/jbc.M406320200. [DOI] [PubMed] [Google Scholar]
- 48.Mottet D, et al. Histone deacetylase 7 silencing alters endothelial cell migration, a key step in angiogenesis. Circ Res. 2007;101(12):1237–46. doi: 10.1161/CIRCRESAHA.107.149377. [DOI] [PubMed] [Google Scholar]
- 49.Liu T, et al. Histone deacetylase inhibitors: multifunctional anticancer agents. Cancer Treat Rev. 2006;32(3):157–65. doi: 10.1016/j.ctrv.2005.12.006. [DOI] [PubMed] [Google Scholar]
- 50.Qian DZ, et al. The histone deacetylase inhibitor NVP-LAQ824 inhibits angiogenesis and has a greater antitumor effect in combination with the vascular endothelial growth factor receptor tyrosine kinase inhibitor PTK787/ZK222584. Cancer Res. 2004;64(18):6626–34. doi: 10.1158/0008-5472.CAN-04-0540. [DOI] [PubMed] [Google Scholar]
- 51.Rossig L, et al. Inhibitors of histone deacetylation downregulate the expression of endothelial nitric oxide synthase and compromise endothelial cell function in vasorelaxation and angiogenesis. Circ Res. 2002;91(9):837–44. doi: 10.1161/01.res.0000037983.07158.b1. [DOI] [PubMed] [Google Scholar]
- 52.Kwon HJ, et al. Histone deacetylase inhibitor FK228 inhibits tumor angiogenesis. Int J Cancer. 2002;97(3):290–6. doi: 10.1002/ijc.1602. [DOI] [PubMed] [Google Scholar]
- 53.Sasakawa Y, et al. Antitumor efficacy of FK228, a novel histone deacetylase inhibitor, depends on the effect on expression of angiogenesis factors. Biochem Pharmacol. 2003;66(6):897–906. doi: 10.1016/s0006-2952(03)00411-8. [DOI] [PubMed] [Google Scholar]
- 54.Kang JH, et al. CCAAT box is required for the induction of human thrombospondin-1 gene by trichostatin A. J Cell Biochem. 2008;104(4):1192–203. doi: 10.1002/jcb.21697. [DOI] [PubMed] [Google Scholar]
- 55.Zgouras D, et al. Modulation of angiogenesis-related protein synthesis by valproic acid. Biochem Biophys Res Commun. 2004;316(3):693–7. doi: 10.1016/j.bbrc.2004.02.105. [DOI] [PubMed] [Google Scholar]
- 56.Mie Lee Y, et al. Inhibition of hypoxia-induced angiogenesis by FK228, a specific histone deacetylase inhibitor, via suppression of HIF-1alpha activity. Biochem Biophys Res Commun. 2003;300(1):241–6. doi: 10.1016/s0006-291x(02)02787-0. [DOI] [PubMed] [Google Scholar]
- 57.Hellebrekers DM, et al. Identification of epigenetically silenced genes in tumor endothelial cells. Cancer Res. 2007;67(9):4138–48. doi: 10.1158/0008-5472.CAN-06-3032. [DOI] [PubMed] [Google Scholar]
- 58.Deroanne CF, et al. Histone deacetylases inhibitors as anti-angiogenic agents altering vascular endothelial growth factor signaling. Oncogene. 2002;21(3):427–36. doi: 10.1038/sj.onc.1205108. [DOI] [PubMed] [Google Scholar]
- 59.Michaelis M, et al. Valproic acid and interferon-alpha synergistically inhibit neuroblastoma cell growth in vitro and in vivo. Int J Oncol. 2004;25(6):1795–9. [PubMed] [Google Scholar]
- 60.Michaelis M, et al. Valproic acid inhibits angiogenesis in vitro and in vivo. Mol Pharmacol. 2004;65(3):520–7. doi: 10.1124/mol.65.3.520. [DOI] [PubMed] [Google Scholar]
- 61.Qian DZ, et al. Targeting tumor angiogenesis with histone deacetylase inhibitors: the hydroxamic acid derivative LBH589. Clin Cancer Res. 2006;12(2):634–42. doi: 10.1158/1078-0432.CCR-05-1132. [DOI] [PubMed] [Google Scholar]
- 62.Potente M, Dimmeler S. Emerging roles of SIRT1 in vascular endothelial homeostasis. Cell Cycle. 2008;7(14):2117–22. doi: 10.4161/cc.7.14.6267. [DOI] [PubMed] [Google Scholar]
- 63.Saunders LR, Verdin E. Sirtuins: critical regulators at the crossroads between cancer and aging. Oncogene. 2007;26(37):5489–504. doi: 10.1038/sj.onc.1210616. [DOI] [PubMed] [Google Scholar]
- 64.Michan S, Sinclair D. Sirtuins in mammals: insights into their biological function. Biochem J. 2007;404(1):1–13. doi: 10.1042/BJ20070140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Potente M, et al. SIRT1 controls endothelial angiogenic functions during vascular growth. Genes Dev. 2007;21(20):2644–58. doi: 10.1101/gad.435107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yu C, et al. Abrogation of MAPK and Akt signaling by AEE788 synergistically potentiates histone deacetylase inhibitor-induced apoptosis through reactive oxygen species generation. Clin Cancer Res. 2007;13(4):1140–8. doi: 10.1158/1078-0432.CCR-06-1751. [DOI] [PubMed] [Google Scholar]
- 67.Zhong H, et al. Modulation of hypoxia-inducible factor 1alpha expression by the epidermal growth factor/phosphatidylinositol 3-kinase/PTEN/AKT/FRAP pathway in human prostate cancer cells: implications for tumor angiogenesis and therapeutics. Cancer Res. 2000;60(6):1541–5. [PubMed] [Google Scholar]
- 68.Murakami J, et al. Effects of demethylating agent 5-aza-2(′)-deoxycytidine and histone deacetylase inhibitor FR901228 on maspin gene expression in oral cancer cell lines. Oral Oncol. 2004;40(6):597–603. doi: 10.1016/j.oraloncology.2003.12.008. [DOI] [PubMed] [Google Scholar]
- 69.Hellebrekers DM, et al. Epigenetic regulation of tumor endothelial cell anergy: silencing of intercellular adhesion molecule-1 by histone modifications. Cancer Res. 2006;66(22):10770–7. doi: 10.1158/0008-5472.CAN-06-1609. [DOI] [PubMed] [Google Scholar]
- 70.Suuronen T, et al. Epigenetic regulation of clusterin/apolipoprotein J expression in retinal pigment epithelial cells. Biochem Biophys Res Commun. 2007;357(2):397–401. doi: 10.1016/j.bbrc.2007.03.135. [DOI] [PubMed] [Google Scholar]
- 71.Duvic M, et al. Phase 2 trial of oral vorinostat (suberoylanilide hydroxamic acid, SAHA) for refractory cutaneous T-cell lymphoma (CTCL) Blood. 2007;109(1):31–9. doi: 10.1182/blood-2006-06-025999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ellis L, et al. Histone deacetylase inhibitor panobinostat induces clinical responses with associated alterations in gene expression profiles in cutaneous T-cell lymphoma. Clin Cancer Res. 2008;14(14):4500–10. doi: 10.1158/1078-0432.CCR-07-4262. [DOI] [PubMed] [Google Scholar]