

Abstract
Phosphorylation of respiratory chain components has emerged as a mode of regulation of mitochondrial energy metabolism, but its mechanisms are still largely unexplored. A recently discovered intramitochondrial signalling pathway links CO2 generated by the Krebs cycle with the respiratory chain, through the action of a mitochondrial soluble adenylyl cyclase (mt-sAC). Cytochrome oxidase (COX), whose deficiency causes a number of fatal metabolic disorders, is a key mitochondrial enzyme activated by mt-sAC. We have now discovered that the mt-sAC pathway modulates mitochondrial biogenesis in a reactive oxygen species dependent manner, in cultured cells and in animals with COX deficiency. We show that upregulation of mt-sAC normalizes reactive oxygen species production and mitochondrial biogenesis, thereby restoring mitochondrial function. This is the first example of manipulation of a mitochondrial signalling pathway to achieve a direct positive modulation of COX, with clear implications for the development of novel approaches to treat mitochondrial diseases.
Keywords: mitochondria, sAC, cytochrome oxidase, mtDNA, ROS
INTRODUCTION
Reversible protein phosphorylation is a mechanism for short-term regulation of oxidative phosphorylation (OXPHOS) in mammalian mitochondria. A well-known example is the pyruvate dehydrogenase kinase (PDK)-dependent phosphorylation of subunit E1α of pyruvate dehydrogenase (Hansford, 1991; Linn et al, 1969). Amino acid phosphorylation has been identified in several other OXPHOS components, including complex I (Chen et al, 2004; De Rasmo et al, 2008; Papa et al, 1996; Piccoli et al, 2006; Sardanelli et al, 1995; Scacco et al, 2000; Signorile et al, 2002) and complex IV (cytochrome oxidase, COX) of the respiratory chain, where it modulates enzyme kinetics (Bender & Kadenbach, 2000; Helling et al, 2008; Kadenbach, 2003; Lee et al, 2005; Miyazaki et al, 2003).
Mitochondria contain all the components for OXPHOS protein phosphorylation, including different families of kinases and phosphatases (Horbinski & Chu, 2005; Lu et al, 2007; Pagliarini & Dixon, 2006). The presence of both protein kinase A (PKA) (Pagliarini & Dixon, 2006; Thomson, 2002) and A kinase-anchoring proteins (AKAPs) (Feliciello et al, 2005; Lewitt et al, 2001) has been demonstrated in mammalian mitochondria. In particular, the presence of PKA in the mitochondrial matrix has been shown with different approaches (Livigni et al, 2006; Prabu et al, 2006; Ryu et al, 2005; Schwoch et al, 1990). However, an important missing piece of the puzzle, which is currently being investigated, is the identity of the mitochondrial phosphatases needed to dephosphorylate PKA targets.
We have previously discovered that a soluble adenylyl cyclase (Acin-Perez et al, 2009; Chen et al, 2000; Zippin et al, 2003, 2001) (sAC) is the intra-mitochondrial source of cAMP that regulates PKA, and that stimulation of the mitochondrial sAC–cAMP–PKA (mt-sAC) signalling pathway increases mitochondrial respiration and ATP synthesis, while its inhibition decreases OXPHOS activity (Acin-Perez et al, 2009). This intramitochondrial signalling is necessary to stimulate COX activity, since extramitochondrial cAMP is ineffective (Acin-Perez et al, 2009). In addition, we have identified phosphodiesterase activity inside mitochondria, although the specific mitochondrial phosphodiesterase has not yet been identified.
Despite increasing evidence that OXPHOS proteins are subject to reversible phosphorylation, the physiopathological significance of these modifications and the underlying signalling pathways are still elusive. Virtually no information is available on the link between OXPHOS function, reactive oxygen species (ROS) production and mitochondrial cAMP signalling, in mitochondrial disorders, where the components of the respiratory chain are mutated or missing (Dimauro & Davidzon, 2005). OXPHOS protein phosphorylation may be one of the mechanisms whereby cells attempt to adapt to bioenergetic defects by optimizing enzyme activities and improving the balance between energy production and ROS generation (Kadenbach et al, 2009).
Here, we have investigated OXPHOS regulation by the mt-sAC pathway in genetic models of COX deficiency, which is one of the most common causes of inherited mitochondrial disorders (Shoubridge, 2001). We demonstrate that PKA and mt-sAC modulation induce ROS-mediated changes in the regulation of OXPHOS biogenesis that are different in COX deficient and wild-type (WT) cells. Strikingly, upregulating the expression of mitochondrial sAC (mt-sAC) in COX defective cells results in a dramatic amelioration of the biochemical defect, with improved COX activity, mitochondrial respiration and ability to grow under strictly aerobic conditions, and at the same time it restores normal regulation of OXPHOS biogenesis.
RESULTS
Differential modulation of OXPHOS by PKA in WT and respiratory chain mutant cells
We investigated the PKA-dependent modulation of OXPHOS in mtDNA mutant cybrid cells derived from the fusion of mtDNA-less human osteosarcoma cells with cytoplasts from a patient with a mitochondrial encephalopathy, who harboured a G6930A stop-codon mutation in the COXI gene (Bruno et al, 1999). This mutation in the homoplasmic state results in a complete loss of COX activity, whereas in the heteroplasmic state (i.e. a mixture of mutant and WT mtDNA) the severity of the defect depends on the proportion of mutant mtDNA (D'Aurelio et al, 2001). Heteroplasmic cybrids containing 75% mutant mtDNA (named CA75) have partially reduced COX activity (by 80%), cell respiration (by 45%) and ATP synthesis (by 65%) (Table 1). CA75 cells recapitulate the genetic and biochemical features of mtDNA diseases in patients, where mutations are often heteroplasmic and result in partial OXPHOS defects.
Table 1.
Mitochondrial functional parameters in COX deficient cells
| Cell line | Mutation | Whole cell respiration (fmol O2/min/cell) | TMPD/ascorbate respiration (fmol O2/min/cell) | COX activity (IU/mg protein) | CS activity (IU/mg protein) | ATP synthesis (nmol ATP/min/mg) |
|---|---|---|---|---|---|---|
| WT | 0 | 4.51 ± 0.319 (22) | 27.20 ± 3.67 (7) | 0.052 ± 0.016 (6) | 0.143 ± 0.025 (9) | 33.375 ± 7.558 (3) |
| CA75 | COXI (75%) | 2.535 ± 0.300*** (9) | 12.29 ± 1.04*** (7) | 0.010 ± 0.002*** (6) | 0.174 ± 0.005* (9) | 8.479 ± 0.607*** (3) |
| fWTHtert | 0 | 5.625 ± 0.332 (5) | n.d | 0.042 ± 0.007 (6) | n.d | 23.988 ± 0.146 (3) |
| fSCO2 Htert | SCO2 MT | 3.681 ± 0.131** (5) | n.d | 0.029 ± 0.005 ** (6) | n.d | 10.038 ± 0.238*** (3) |
Whole cell respiration, TMPD/ascorbate-dependent respiration, COX enzymatic activity, citrate synthase (CS) activity and ATP synthesis with pyruvate and malate as substrates measured in WT cybrids, heteroplasmic COXI mutant cybrids (CA75, 75% mutant mtDNA), WT human fibroblasts (fWT Htert) and Sco2 mutant human fibroblasts (fSco2 Htert). The number in brackets indicates the n for each experiment. n.d., not determined.
p < 0.01.
p < 0.001.
p < 0.0001.
8Br-cAMP, a membrane permeant PKA agonist, stimulated intact cell respiration (Fig 1A), ATP synthesis (Fig 1B), COX-dependent respiration with TMPD–ascorbate as substrates (Fig 1C) and COX enzymatic activity measured spectrophotometrically (data not shown). This stimulation was significantly stronger in CA75 than in WT cells. Conversely, the PKA inhibitor H89 (Chijiwa et al, 1990) decreased respiration (Fig 1A), ATP synthesis (Fig 1B) and COX activity (Fig 1C) significantly less in CA75 than in WT cells.
Figure 1. Abnormal PKA modulation of OXPHOS in COX defective cells.
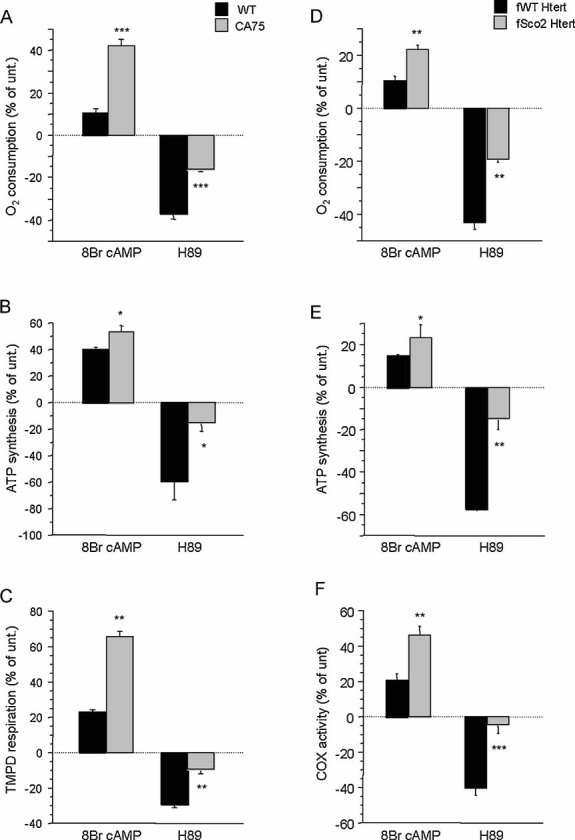
OXPHOS function was assessed in WT and CA75 cybrids incubated in the presence of 8Br-cAMP or H89.
- Mitochondrial respiration in intact cells (WT unt., n = 22; WT 8Br-cAMP, n = 9; WT H89, n = 9; CA75 unt., n = 9; CA75 8Br-cAMP, n = 5; CA75 H89, n = 5).
- ATP synthesis driven by pyruvate plus malate (n = 3 for all samples).
- TMPD/ascorbate driven respiration (n = 7 for all unt. samples, and n = 3 for all treated samples). OXPHOS function was also assessed in human WT and Sco2 mutant fibroblasts by:
- Intact cell respiration (n = 7),
- ATP synthesis (n = 6) and
- COX enzymatic activity (n = 6). For each cell line, changes induced by the treatments are indicated as a percentage of untreated controls (unt.). *, p < 0.01; **, p < 0.001; ***, p < 0.0001.
We then studied OXPHOS modulation by PKA in immortalized fibroblasts derived from a patient with mutant Sco2, a nuclear-encoded copper chaperone protein necessary for COX function (Papadopoulou et al, 1999; Salviati et al, 2002). Sco2 mutant fibroblasts (fSco2 Htert) had a partial deficiency in COX activity (by 25%), cell respiration (by 35%) and ATP synthesis (by 55%) (Table 1). fSco2 Htert cells showed an enhanced stimulation and blunted inhibition of cell respiration (Fig 1D), ATP synthesis (Fig 1E) and COX activity (Fig 1F) in response to 8Br-cAMP and H89, respectively, as compared to WT fibroblasts (fWT Htert).
To determine whether the different modulating effect of PKA was specific to COX mutants we studied three additional mtDNA mutations. The first (SUA63) contained a homoplasmic C3256T mutation in the tRNALeuUUR gene (Hao & Moraes, 1996), and had a severe decrease of both complex I and COX assembly (Fig 2A) and decreased respiration (by 50%). SUA63 cells responded to PKA modulation similarly to COXI and Sco2 mutants, with enhanced stimulation and blunted inhibition of OXPHOS by 8Br-cAMP and H89, respectively (Fig 2B), as compared to WT cells (SUB36, 0% mutant mtDNA). The second mutant was a murine cybrid line (E09) containing a homoplasmic missense COXI mutation resulting in a partial COX deficiency (Acin-Perez et al, 2003). Consistent with the results in human cells, E09 cells exhibited an increased COX stimulation by 8Br-cAMP and a blunted inhibition by H89, as compared to two WT mouse cybrid lines (Fig 2C). The third mutant was a 4 bp frame-shift deletion in the cytochrome b (Cyt b) gene (Rana et al, 2000). We tested two heteroplasmic Cyt b mutant cybrids, D4cytb 5.2 (75% mutant mtDNA) and D4cytb 3 (90% mutant mtDNA). These mutants, with an isolated defect of Complex III assembly (Fig 2D) and decreased respiration (by 15 and 25%, respectively), showed COX responses to PKA stimulation and inhibition similar to those in WT cybrids (cytbA4.4.1) (Fig 2E).
Figure 2. PKA modulation of OXPHOS in mtDNA mutant cells.
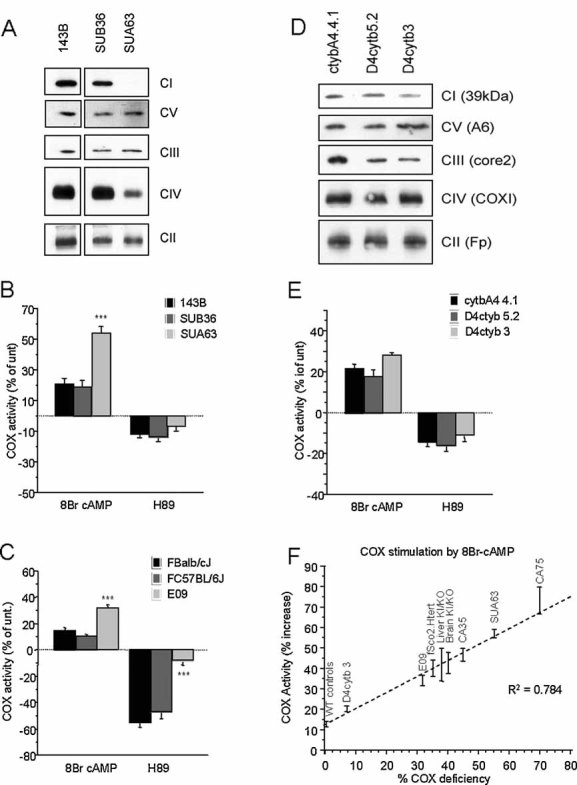
- BNGE Western blot of respiratory chain complexes in homoplasmic MELAS cybrids SUA63 and isogenic WT control cybrids SUB36.
- 8Br-cAMP and H89 modulation of COX activity in MELAS cybrids and controls (n = 7).
- 8Br-cAMP and H89 modulation of COX activity in mouse homoplasmic COXI mutant fibroblasts (E09) and WT controls (n = 4).
- BNGE Western blot of respiratory chain complexes in heteroplasmic Cyt b mutant cybrids (D4cytb 5.2 and D4cytb 3) and isogenic WT control cybrids.
- 8Br-cAMP and H89 modulation of COX activity in heteroplasmic Cyt b mutant cybrids and WT controls (n = 8). Values in (B), (C) and (E) are shown as percentage of untreated controls for each cell line. *, p < 0.01; **, p < 0.001; ***, p < 0.0001.
- Correlation plot between the percentage of residual COX activity and the percentage of COX stimulation upon 8Br-cAMP treatment, in the indicated COX deficient models. The percentages of COX stimulation by 8Br-cAMP is relative to untreated and is shown as the average ± SD of n = 5 determinations for each indicated COX deficient group. WT controls are averages (± SD) of non-COX deficient cells (143B, fWT Htert, SUB36, FBalb/cJ, FC57BL/6J, cytbA4 4.1, D4cytb 5.2) and tissues (brain WT and liver WT).
Taken together, these results indicate that PKA-mediated protein phosphorylation regulates COX, and that this regulation is different in WT and COX deficient cells. Basal protein phosphorylation is reduced in COX deficient cells, allowing for greater stimulation by 8Br-cAMP and lesser inhibition by H89. PKA modulation of OXPHOS is unchanged in respiratory chain defects that do not involve COX.
Differential modulation of OXPHOS by PKA in WT and COX deficient mice
The PKA-dependent modulation of OXPHOS was investigated in vivo, in a Sco2 KI/KO mouse model, where one allele harboured a E129K mutation, the mouse homologue of the human E140K mutation common to all patients with Sco2 defects (Papadopoulou et al, 1999), and the other a stop codon insertion. The Sco2 KI/KO mice have approximately a 35% reduction in COX activity in brain and liver and a 50% decrease in skeletal muscle (Schon et al, unpublished results). Similar to COX deficient cells, mitochondria from liver and brain of Sco2 KI/KO mice showed increased stimulation and blunted inhibition of ATP synthesis (Fig 3A,B) and COX activity (Fig 3C,D) by 8Br-cAMP and H89, respectively. Similar results were obtained in skeletal muscle (data not shown).
Figure 3. Abnormal PKA modulation of OXPHOS in COX defective mice.
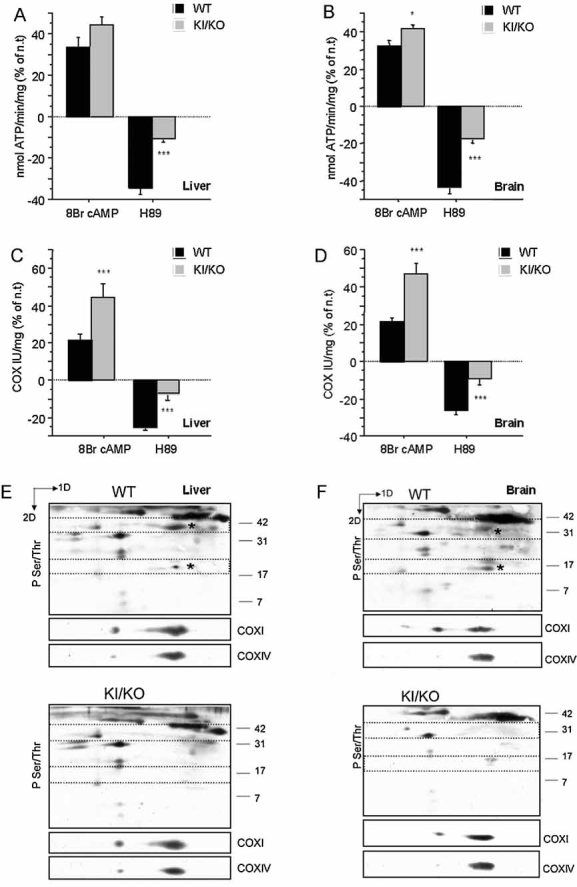
Liver and brain mitochondria from Sco2 KI/KO mice and WT control Sv/J mice were treated with 8Br-cAMP or H89. OXPHOS function was assessed by:
A, B. ATP synthesis (n = 6) and
C, D. COX enzymatic activity (n = 9). For each sample, changes induced by the treatments are indicated as a percentage of untreated controls (unt.). *, p < 0.01; ***, p < 0.0001.
E, F. Immunodetection of mitochondrial phospho-proteins in 2D-BNGE Western blot probed with anti-phospho Ser/Thr. The areas of the membrane delimited by dashed lines were probed with specific antibodies against COXI and COXIV subunits (shown below each panel) to demonstrate the identity of the phosphorylated proteins and to assess protein loading. The asterisks denote the putative phospho-COXI and phospho-COXIV in WT liver and brain mitochondria, which were undetectable in the KI/KO mitochondria. One representative Western blot of two independent experiments is shown.
The phosphorylation of COX subunits I and IV, which are targets of mitochondrial PKA (Acin-Perez et al, 2009), was investigated in Sco2 KI/KO mouse mitochondria by 2D blue native gel electrophoresis (2D-BNGE) and Western blot. In both liver and brain from Sco2 KI/KO mice, COXI and COXIV were less phosphorylated than in WT mitochondria (Fig 3E,F). The relative phosphorylation of each subunit was assessed by quantification of the intensities of the immuno-signal obtained with a phospho Ser/Thr antibody over the signal derived from the antibody against the total protein. In liver (n = 2), the phospho-COXI/COXI ratios were 0.05 in KI/KO and 0.60 in WT, and the phospho-COXIV/COXIV ratios were 0.01 and 0.3, respectively. In brain (n = 2), the phospho-COXI/COXI ratios were 0.01 in KI/KO and 0.35 in WT and the phospho-COXIV/COXIV ratios were 0.001 and 0.25, respectively.
These results indicate that in vivo COX deficiency results in decreased basal phosphorylation of COX subunits. COXI hypophosphorylation was also confirmed in the human fibroblast line fSco2 Htert (phospho-COXI/COXI ratio was 0.2 vs. 0.6 in fWT Htert, n = 2) and in the mouse fibroblast line E09 (phospho-COXI/COXI ratio: 0.3 vs. 0.6–0.7 in FBalb/cJ or FC57BL/6J, respectively, n = 2). Comparing all the systems together, we found that there was an inverse linear correlation (R2 0.782) between residual COX activity in COX-deficient mitochondria and the proportion of COX activity increased by 8Br-cAMP stimulation (Fig 2F). This correlation did not depend on whether the defect involved loss of COX subunits, such as in CA75, CA35, SUA63 and Sco2 mutants, or a loss of function with normal protein levels, such as in E09 cells. The result of this analysis indicates that COX regulation through PKA phosphorylation depends on COX activity and not on protein levels.
ROS and PGC1α mediate increased OXPHOS biogenesis in COXI mutant cybrids
When OXPHOS is impaired, cells need to develop strategies to compensate for the metabolic defect. Adaptive mechanisms include an increase in OXPHOS biogenesis, which is typically observed in muscle fibres harbouring certain mtDNA mutations (ragged red fibres). It has been proposed that ROS can behave as second messengers in the regulation of OXPHOS biogenesis (St-Pierre et al, 2006), and changes in OXPHOS biogenesis mediated by ROS production have been reported in cultured cells with mtDNA mutations (Moreno-Loshuertos et al, 2006). We hypothesized that decreased protein phosphorylation in COX deficient cells is a compensatory mechanism to enhance ROS production, and thus increase OXPHOS biogenesis. Indeed, ROS were elevated in heteroplasmic COXI mutants (Table 2). Consistently, the activity of citrate synthase (Table 1), a Krebs cycle enzyme and the mtDNA content (Table 2) were also increased. Furthermore, the expression of the cotranslational trans-activating factors PGC1α and NRF1 that control OXPHOS biogenesis (Scarpulla, 2008) was significantly increased in CA75 heteroplasmic COXI mutant cells (Table 2). Homoplasmic COXI mutants do not have increased ROS production, presumably because the electron flow is abolished, and thus did not upregulate OXPHOS biogenesis (Table 2).
Table 2.
ROS production and parameters of OXPHOS biogenesis in COXI mutant cybrids
| Cell line | Mutation load (%) | ROS production (F535 nm/max) | mtDNA/nDNA (% vs. 143B) | PGC1α/GAPDH (% vs. 143B) | NRF1/GAPDH (% vs. 143B) |
|---|---|---|---|---|---|
| 143B | 0 | 0.356 ± 0.078 (34) | 100 ± 9.79 (4) | 100 ± 12.41 (14) | 100 ±15.2 (12) |
| ctlB2 | 0 | 0.377 ± 0.041 (8) | n.d. | n.d. | n.d. |
| CA0 | 0 | 0.354 ± 0.026 (8) | 103.9 ± 7.57 (4) | n.d. | n.d. |
| CA35 | 35 | 0.560 ± 0.098** (18) | 174.33 ± 26.18** (6) | n.d. | n.d. |
| CA75 | 75 | 0.577 ± 0.104** (18) | 189.13 ± 13.55** (6) | 160.5 ± 29.52** (6) | 167.5 ± 11.64** (4) |
| CA100 | 100 | 0.383 ± 0.044 (8) | 90.26 ± 12.73 (4) | n.d. | n.d. |
ROS production in untreated cells is expressed as the ratio between the spontaneous DCFDA fluorescence (535 nm emission) and the maximal fluorescence elicited by exposure to H2O2 (300 µM). The mtDNA content determined by RT-PCR and normalized by nuclear 28S rRNA gene content is shown as percentage of 143B. PGC1α and NRF-1 mRNA levels determined by RT-PCR relative to GAPDH mRNA are shown as percentage of 143B. Three independent control cell lines were used: 143B parental osteosarcoma cells, ctlB2 human WT cybrids, CA0 WT cybrids isogenic for the CA COXI mutant cybrids. The number in brackets represents the n for each experiment. n.d., not determined.
p < 0.001.
8Br-cAMP decreased ROS production by stimulating COX and improving the efficacy of electron transfer, while H89 increased ROS production in WT cells, but significantly less in heteroplasmic COXI mutants (Fig 4A). This suggests that PKA blockage normally results in increased ROS, but mutant cells that have constitutively lower protein phosphorylation do not up-regulate ROS.
Figure 4. Effects of PKA modulation on ROS production and OXPHOS biogenesis.
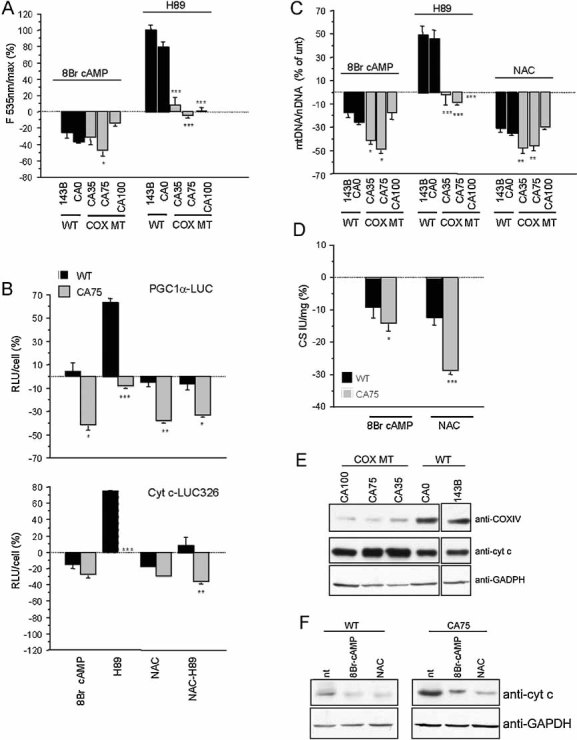
- WT and COXI mutant cybrids were incubated for 2 h in the presence of 8Br-cAMP or H89 and ROS production was measured by DCFDA fluorescence. Changes in fluorescence induced by the treatments are indicated as a percentage of untreated controls (unt.) (n = 9).
- LUC activity in PGC1α–LUC (upper panel) or Cyt c–LUC 326 (lower panel) transfected WT and CA75 cells, after 48 h treatment with 8Br-cAMP, H89, NAC or H89 plus NAC. Luminescence was normalized by the number of cells (RLU/cell ratio). Values are shown as a percentage of untreated controls for each cell line and represent the mean of three independent transfections, each measured in triplicates.
- MtDNA content after 48 h treatment with 8Br-cAMP, H89 or NAC (n = 7).
- Citrate synthase (CS) enzymatic activity after 48 h treatment with 8Br-cAMP or NAC (n = 6). In (C) and (D), values are shown as a percentage of untreated controls for each cell line. *, p < 0.01; **, p < 0.001; ***, p < 0.0001.
- Western blot for COXIV and Cyt c in COXI mutant cybrids and WT controls ran under the same conditions. Reprobing the blots with GAPDH confirms that same amount of proteins was loaded for all cell lines.
- Western blot for Cyt c in CA75 cybrids and WT controls after 48 h treatment with 8Br-cAMP and NAC. Panels (E) and (F) show representative Western blots from three independent experiments.
To investigate the role of PKA in OXPHOS biogenesis we used three different luciferase (LUC) reporter constructs. The first one expressed LUC under the control of the PGC1α promoter (Handschin et al, 2003); the second expressed LUC from the cytochrome c full promoter (Cyt c–LUC326, Xia et al, 1997), which is under the control of PGC1α through NRF2 (Scarpulla, 2008); the third had an incomplete Cyt c promoter, lacking SP1 and NRF2 binding sites (Cyt c–LUC66, Xia et al, 1997). Transfected cells were treated for 48 h with 8Br-cAMP, H89 or the antioxidant N-acetyl-cysteine (NAC). PGC1α–LUC (Fig 4B, upper panel) and Cyt c–LUC326 (Fig 4B, lower panel) expression decreased significantly in CA75 cells treated with 8Br-cAMP. Conversely, H89 significantly increased PGC1α–LUC expression in WT, but not in CA75 cells, confirming that OXPHOS biogenesis in the latter is poorly responsive to PKA inhibition. NAC had similar effects as 8Br-cAMP on the expression of the reporter genes in both WT and CA75 cells and suppressed the increase in LUC activity induced by H89 in WT cells, confirming that the changes in gene expression were mediated by ROS. Cyt c–LUC66 did not show any change in expression with any of the compounds (Fig S1 of Supporting Information). Changes in mtDNA content (Fig 4C) and citrate synthase activity (Fig 4D) paralleled those of PGC1α–LUC and Cyt c–LUC326.
In COXI mutant cells, Cyt c, which is under the control of PGC1α/NRF1, was strongly upregulated (Fig 4E). A 48-h treatment with 8Br-cAMP or NAC resulted in a decrease of Cyt c (Fig 4F), both in mutant and WT cells, indicating that chronic stimulation of PKA signalling affects OXPHOS biogenesis by decreasing ROS.
A 48 h treatment of WT cells with forskolin/IBMX, which increases cytosolic but not mitochondrial cAMP (Acin-Perez et al, 2009), did not affect ROS production, mtDNA content, cytrate synthase activity and the steady-state levels of Cyt c (Fig S2 of Supporting Information), indicating that OXPHOS biogenesis was not affected by cytoplasmic cAMP.
The effects of the chronic administration of 8Br-cAMP were recapitulated using a genetic approach, where an enzymatically active isoform of rat sAC (Hess et al, 2005) was expressed in 293T HEK cells (Fig S3A of Supporting Information), resulting in decreased mtDNA content (Fig S3B of Supporting Information), cytrate synthase activity (Fig S3C of Supporting Information), COXIV and Cyt c (Fig S3A of Supporting Information).
To assess the physiological consequences of OXPHOS biogenesis down-regulation we subjected cells to galactose medium, where cells depend on OXPHOS for ATP synthesis (Mattiazzi et al, 2004). In WT cells, the ratio between the doubling time in glucose and in galactose (glu/gal) was decreased by chronic treatment with 8Br-cAMP (Fig S4 of Supporting Information). Furthermore, 8Br-cAMP did not improve the inability of CA75 cells to replicate in galactose medium, because the decrease in OXPHOS biogenesis (Fig 4) offset the stimulation of COX activity.
Mitochondrially targeted sAC (mt-sAC) ameliorates the OXPHOS defect in COXI mutant cells
Treatment of COXI mutant cells with PKA analogues stimulates COX activity and OXPHOS (Fig 1), but it also inhibits mitochondrial biogenesis (Fig 4). Furthermore, chronic treatment with cAMP analogues is likely to have toxic extra-mitochondrial effects. For these reasons we undertook a genetic approach to constitutively increase cAMP selectively in mitochondria. Although a portion of sAC is localized in the mitochondrial matrix (Acin-Perez et al, 2009), it is not known yet which of the multiple splice variants of sAC is targeted to mitochondria and whether any specific sAC isoform is exclusively localized in mitochondria. Thus, to ensure its specific targeting to the mitochondrial matrix we generated a recombinant mitochondrial-sAC (mt-sAC) by adding a N-terminal cleavable presequence from the subunit c of human ATPase (Manfredi et al, 2002a) to the rat sAC isoform. We also appended a C-terminal HA-tag to distinguish mt-sAC from endogenous sAC (Fig 5A).
Figure 5. mt-sAC is targeted to mitochondria and is enzymatically active.

- Scheme of the sAC and the mitochondrially targeted sAC (mt-sAC) genetic constructs. P1 is the mitochondrial targeting peptide of the human P1 isoform of ATPase subunit c. Four amino acid of the mature subunit c (sub c) are included in the construct. The rat sAC gene is cloned downstream of the targeting peptide and contains an HA epitope tag.
- COS-1 cells expressing the empty vector (mock), sAC or mt-sAC were stained with Mitotracker (in red) and immuno-stained for HA (in green). In the merged images, colocalization of sAC and mitochondria is indicated by the overlap of the fluorescence (i.e. in yellow).
- sAC and mt-sAC distribution in subcellular fractions of transfected COS-1 cells. Total cell homogenate (T), mitochondrial fraction (M), cytosolic fraction (C). β-Actin and Tim23 are markers of cytosolic and mitochondrial proteins, respectively.
- cAMP levels in sAC and mt-sAC expressing cells measured in total cell homogenates (T) and in the mitochondria containing pellets (P), which result from plasma membrane permeabilization followed by centrifugation. ***, p < 0.0001.
In transiently transfected COS cells, mt-sAC was localized in mitochondria, as evidenced by colocalization with the mitochondrial dye Mitotracker Red, in contrast to sAC that was diffusely expressed (Fig 5B). Cell fractionation revealed that mt-sAC was in the mitochondrial fraction, except for a small proportion of unprocessed protein residing in the cytosol, while sAC was both in the cytosol and mitochondria (Fig 5C). After permeabilization of the plasma membrane, cAMP was measured in the pellet fraction (containing organelles) and in the total homogenate from transfected cells. Total cAMP levels were increased in both sAC and mt-sAC expressing cells, but in the latter cAMP was concentrated in the fraction containing mitochondria (Fig 5D).
After determining that the content of total and mitochondrial endogenous sAC was comparable in CA75 and WT cells (data not shown), we stably transfected them with mt-sAC and determined that cAMP was increased in the organellar fraction of mt-sAC expressing cells (Fig S5A of Supporting Information). The COXI mutation load in clones and mass cultures of transfected CA75 ranged between 62 and 87% (Fig 6A). The expression levels of mt-sAC varied among lines (Fig 6B), and COX activity was increased proportionally to mt-sAC expression (Fig 6C). CA75 lines with higher mt-sAC expression also had higher COX activity. For example, CA75 mt-sAC clones 1 and 2 had 76 and 72% mutant mtDNA, respectively; but clone 1 with higher mt-sAC expression also had higher COX activity. On the other hand, CA75 mt-sAC clone 3 with high mt-sAC expression had low COX activity due to a very high mutation load (89%).
Figure 6. mt-sAC expression in COXI mutant cell lines.
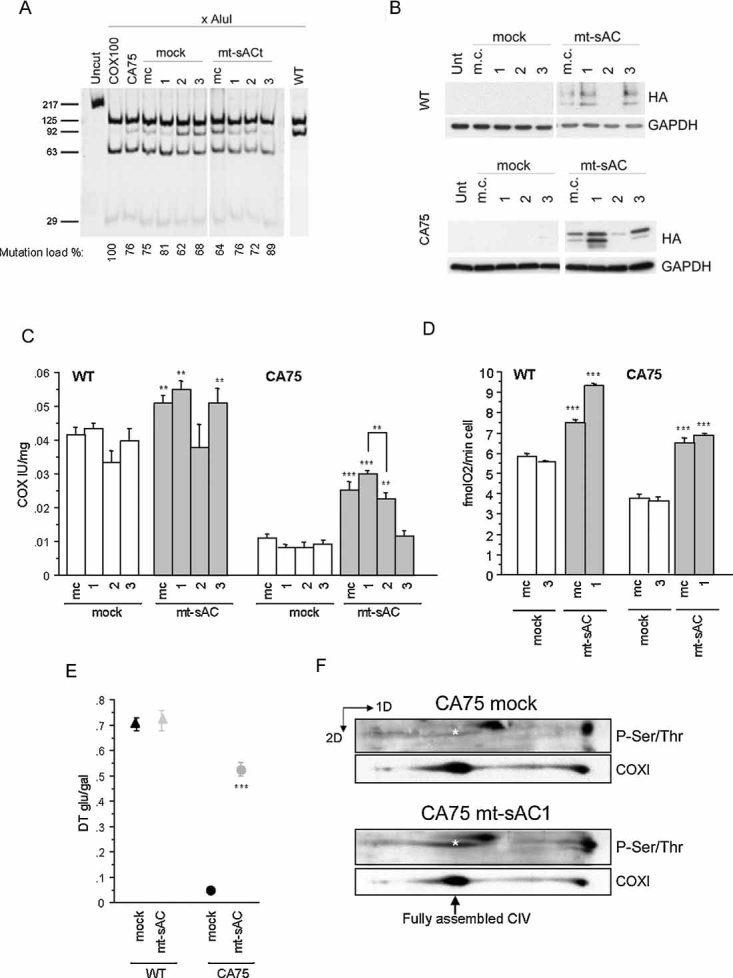
- COXI mutation load measured by last hot cycle PCR followed by AluI digestion in mass culture (m.c.) and individual clones of CA75 stably expressing mt-sAC. The level of heteroplasmy indicated as a percentage of mutant mtDNA is indicated below each lane, and represents the mean value of two independent determinations. WT lane originates from a different gel run in parallel with the CA75 samples.
- mt-sAC expression in total cell homogenates of WT and CA75 m.c. and clones ran under the same conditions determined by immunoblot with HA antibody. Reprobing the blots with GAPDH confirms that same amount of proteins was loaded for all cell lines.
- COX enzymatic activity in cell lysates of WT (on the left) or CA75 (on the right) expressing mt-sAC (n = 6).
- Intact cell respiration in WT (on the left) or CA75 (on the right) cells expressing mt-sAC (n = 6).
- The ratio of the doubling time in glucose medium and in galactose medium (DT glu/gal) in WT or CA75 cells expressing mt-sAC (n = 4). In panels, C–E, *, p < 0.01; **, p < 0.001; ***, p < 0.0001.
- 2D-BNGE of isolated mitochondria from mt-sAC expressing or mock transfected CA75 cells followed by successive immunodetection for phosphorylated proteins and COXI. The position of the fully assembled, monomeric, COX (CIV) is indicated by the arrow. The asterisk indicates the phosphorylated COXI, which is significantly lower in mock transfected than in mt-sAC expressing cells. The blots shown are representative of two independent experiments.
COX activity in mock transfected CA75 cells was relatively insensitive to the specific sAC inhibitor KH7 (Hess et al, 2005), consistent with down-regulation of COX PKA-dependent phosphorylation. However, KH7 completely abolished the increase in COX activity induced by mt-sAC expression in CA75 cells (Fig S5B of Supporting Information).
Cell respiration in CA75 mass cultures and clone 1 recovered to levels comparable to mock-transfected WT cells (Fig 6D). The functional amelioration of OXPHOS in CA75 cells expressing mt-sAC was further demonstrated by the improvement in the doubling time ratio glu/gal (Fig 6E). Of note, the growth in galactose did not change the mutation load in the mt-sAC CA75 cells (data not shown). At the proteomic level, the amount of phosphorylated COXI was increased in parallel with the improvement of OXPHOS function in mt-sAC expressing CA75 cells (Fig 6F).
The rescue of OXPHOS function induced by mt-sAC expression in CA75 cells correlated with a decrease in ROS production (Fig 7A), PGC1α (Fig 7B) and NRF1 (Fig 7C) mRNA, mtDNA (Fig 7D) and Cyt c content (Fig 7E), all of which reverted to normal levels. mt-sAC expression did not affect the levels of COXI and COXIV, which remained deficient (Fig 7E).
Figure 7. mt-sAC expression normalizes ROS-dependent OXPHOS biogenesis in COX deficient cells.
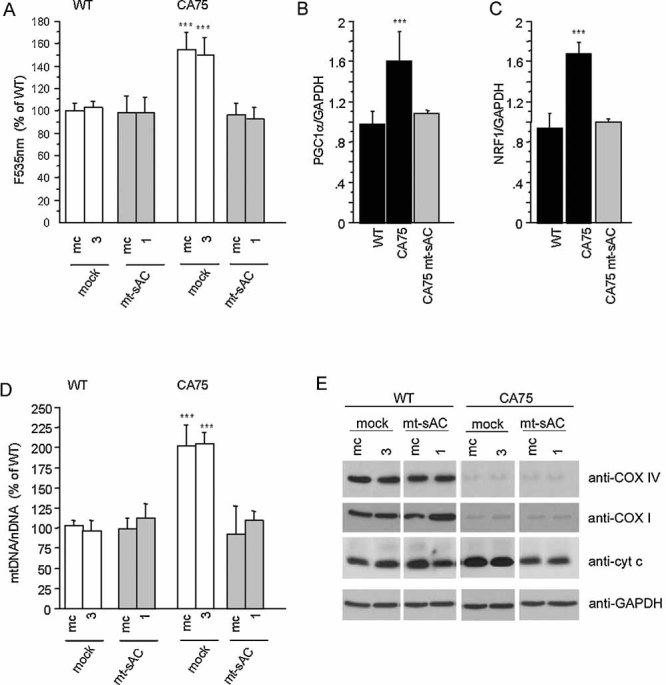
A. ROS production measured by DCFDA fluorescence in WT and CA75 mt-sAC expressing cells and mock-transfected controls. Values represent the percentage of DCFDA fluorescence relative to WT non-transfected cells (n = 15).
B, C. PGC1α and NRF-1 mRNA levels measured by RT-PCR and normalized by GAPDH mRNA levels, in non-transfected WT and CA75 cells and in CA75 mt-sAC expressing cells (n = 6).
D. mtDNA content in WT and CA75 mt-sAC expressing cells and mock-transfected controls. Values represent the percentage of mtDNA/nuclear DNA ratio relative to WT non-transfected cells (n = 9).
E. Western blot of total cell homogenates from WT and CA75 mt-sAC expressing cells and mock-transfected controls ran under the same conditions immunoprobed for Cyt c and for COX subunits I and IV. Reprobing the blots with GAPDH confirms that same amount of proteins was loaded for all cell lines. The blots shown are representative of two independent experiments.
Taken together these results indicate that the constitutive stimulation of cAMP production in mitochondria by expressing sAC targeted to the matrix triggers adaptation mechanisms in COX deficient cells that upregulate OXPHOS function and reduce the need for enhanced ROS-dependent OXPHOS biogenesis.
DISCUSSION
We have demonstrated that OXPHOS regulation by mitochondrial PKA is different in normal and COX defective cells and tissues, in humans and mice. OXPHOS activity in COX deficient systems is strongly stimulated and poorly inhibited by PKA agonists and antagonists, respectively, because there is a constitutive down-regulation of PKA-dependent protein phosphorylation. COX defective cells suppress phosphorylation of COX subunits and residual enzymatic activity, thereby increasing OXPHOS biogenesis via a ROS-mediated PGC1α/NRF1 pathway (Irrcher et al, 2009; St-Pierre et al, 2006). However, the ROS-mediated compensatory mechanism may come at a high price, because it enhances oxidative stress with potentially detrimental long-term effects. Interestingly, the up-regulation of ROS and OXPHOS biogenesis is only found in heteroplasmic, but not in homoplasmic, COXI mutants. The latter have a complete loss of COX activity, resulting in retrograde blockage of the pathways feeding reducing equivalents to the respiratory chain (Vives-Bauza et al, 2006), whereas a partial COX defect facilitates electron escape from the respiratory chain (Zuckerbraun et al, 2007). Undoubtedly, metabolic factors other than ROS may also contribute to the regulation of mitochondrial biogenesis in COX deficient cells. For example, changes in the AMP/ATP ratio could signal to PGC1α through AMP kinase (Bronner et al, 2004; Jager et al, 2007). Nevertheless, the observation that homoplasmic COXI mutant cells, which do not overproduce ROS, fail to upregulate mitochondrial biogenesis suggests that ROS may play an important role. The mechanism whereby COX deficient cells suppress COX phosphorylation, despite having all the components of the mt-sAC pathway in place, remains to be fully elucidated, but it is likely to result from reduced levels of CO2 produced by the Krebs cycle, which in mutant cells is slowed down by decreased utilization of NADH in the respiratory chain (Acin-Perez et al, 2009).
In COX deficient cells, OXPHOS can be ameliorated by stimulation of the PGC1α pathway of mitochondrial biogenesis. Increased expression of PGC1α by transgenic or pharmacological means resulted in a remarkable improvement of OXPHOS function and clinical phenotype in a mouse model of muscle COX deficiency (Wenz et al, 2008). The mt-sAC pathway opens a new window of opportunity to ameliorate mitochondrial defects. By genetic manipulation we stably upregulated mt-sAC and induced a long-term metabolic adaptation. mt-sAC expression dramatically improved mitochondrial function in COX deficient cells, which adapted to the sustained rise in mitochondrial cAMP by increasing OXPHOS activity. Therefore, activation of the mt-sAC pathway is an alternative approach to improve OXPHOS in COX deficient cells, without enhancing ROS production and OXPHOS biogenesis. In a therapeutic perspective, the two approaches, PGC1α stimulation and mt-sAC upregulation, could be complementary.
In line with our previous observation that COXI and COXIV are putative targets for the mt-sAC pathway (Acin-Perez et al, 2009), we find that these two subunits are hypophosphorylated in COX deficient mitochondria, suggesting that PKA-mediated phosphorylation of these two proteins is critical for enzymatic activity. Phosphorylation of various COX subunits has been recognized for over a decade (Bender & Kadenbach, 2000; Helling et al, 2008; Lee et al, 2005; Miyazaki et al, 2003; Samavati et al, 2008; Steenaart & Shore, 1997), although the physiological meaning of these phosphorylation events is still not completely understood. Cumulatively, COX subunits have more than 50 potential PKA target residues (Helling et al, 2008), but only a handful of targets have been identified so far. It is likely that in vivo multiple targets are simultaneously phosphorylated or dephosphorylated based on the needs for short-term reversible regulation of OXPHOS. So far, no specific phosphatases for any subunit of the respiratory chain have been described. Therefore, it is hard to predict the metabolic consequences of PKA modulation by analysing the effects of phosphorylation of individual amino acids in vitro. Extensive proteomic studies will be needed to determine under which conditions and at which residues COX is phosphorylated in vivo. Nevertheless, our results indicate that, in living cells, stimulation of COX phosphorylation by mitochondrial PKA results in increased enzymatic activity, which can be maintained over a prolonged period of time.
In summary, we have identified a novel pathway of OXPHOS regulation in healthy and COX deficient cells. We show that genetic manipulation of the mt-sAC pathway results in a resetting and optimization of the balance between energy metabolism, ROS production and OXPHOS biogenesis in COX deficient cells (summarized in the scheme of Fig 8). These findings provide new clues on the ways that mitochondria cope with metabolic defects and on potential pharmacologic interventions that target the mt-sAC pathway to improve mitochondrial function. It will be important to extend these observations to other mitochondrial disorders arising from genetic mutations of the mitochondrial or nuclear genome that affect the respiratory chain, as well as acquired mitochondrial defects that develop in the course of diverse neurodegenerative diseases (Kwong et al, 2006).
Figure 8. Schematic model of the proposed mitochondrial mt-sAC pathway of OXPHOS regulation in normal and COX deficient conditions.
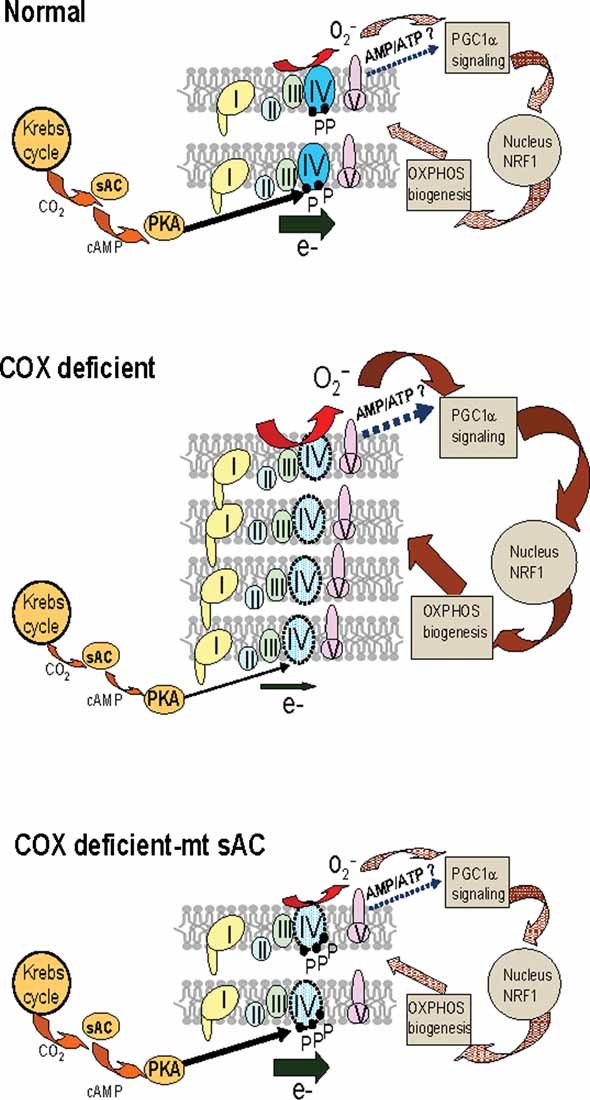
In normal mitochondria, the pace of electron (e−) flow through the respiratory chain complexes (I–IV) is finely regulated by the activity of complex IV (COX), which in turn is modulated by phosphorylation of its subunits (P, COX phospho-residues) by mitochondrial PKA. This regulation responds to the CO2 produced by the Krebs cycle through mitochondrial sAC. Electron flow is linked to ROS production (e.g. superoxide, O2.) and ATP synthesis, which in turn may affect the cellular AMP/ATP ratio. These, and other putative factors (indicated by ?), have signalling functions on the regulation of nuclear genes involved in mitochondrial biogenesis, such as PGC1α and NRF1. In COX deficient mitochondria with decreased electron flow, Krebs cycle activity is dampened and CO2 production reduced, resulting in hypophosphorylation of COX, increased ROS production and increased signalling for compensatory mitochondrial biogenesis through the PGC1α–NRF1 axis. Over-expression of mt-sAC in COX deficient cells increases COX phosphorylation by the sAC–cAMP–PKA pathway, thereby increasing electron flow and normalizing the signalling for OXPHOS biogenesis.
METHODS
Cell culture and pharmacological treatments
The previously described cybrids cell lines were derived from fusion of 143B osteosarcoma ρ0 cells with enucleated cells from patients harbouring mtDNA mutations (King & Attardi, 1989). Cybrids were grown in Dulbecco modified Eagle's medium (DMEM, Invitrogen) containing 4.5 g/l glucose and 1 mM pyruvate, supplemented with 10% foetal bovine serum (FBS, Invitrogen) and 50 µg/ml uridine (Sigma–Aldrich). COS-1 and 293T HEK cells were grown in the same conditions with the exclusion of uridine. Human fibroblasts were immortalized by expression of human telomerase as described (Lochmuller et al, 1999), and grown in the same conditions. For growth in galactose, cells were placed in medium without glucose and containing 4.5 g/l galactose (Sigma–Aldrich) plus 1 mM pyruvate and 10% dialyzed FBS. Cell replication rates in glucose and galactose medium were measured as described previously (Moreno-Loshuertos et al, 2006).
The concentrations of the compounds used on cultured cells for this study were as follows: 8Br-cAMP 1 mM (Sigma–Aldrich); H89 1 µM (Calbiochem); NAC 2.5 mM (Sigma–Aldrich); forskolin 10 µM (Sigma–Aldrich); 3-isobutyl-1-methylxanthine 50 µM (IBMX, Sigma–Aldrich), KH7 50 µM.
Generation of Sco2 KI/KO mice
To clone the murine Sco2 gene a genomic fragment (65d3) containing the Sco2 gene was isolated from a 129 Sv/J mouse BAC library (Genome System, Inc.). The isolated 5 kb fragment containing the entire Sco2 gene was subcloned into the KpnI site of pBluescript II KS+ and used to generate the targeting vector. To generate Sco2 knock out (KO) mice a 2 kb fragment composed of the gene encoding the neomycin phosphotransferase (NEO cassette) flanked by two 34-mer identical loxP sites (loxP–NEO–loxP) was inserted in the PacI site (converted from a HindIII), 30 bp downstream of the start codon of Sco2. To generate E129K mutant Sco2 knock in (KI) mice a single base pair mutation, which converted nucleotide 385 in the mouse Sco2 sequence from G to A, was introduced by in situ mutagenesis. The NEO cassette was inserted in the PacI site (converted from a SmaI site), 643 bp downstream of the stop codon of Sco2. To generate Sco2 KI/KO compound heterozygous mice the Sco2 homozygous KI mice were crossed with Sco2 heterozygous KO mice.
The paper explained
PROBLEM
Mitochondrial disorders are common genetic diseases of energy metabolism that affect one in every 5000 people. Impaired mitochondrial metabolism is also associated with aging and many neurodegenerative disorders. Deficiency of COX, a key mitochondrial respiratory chain enzyme, is a biochemical hallmark of mitochondrial disorders, either genetically determined or acquired in course of neurodegeneration and aging. No effective therapy is currently available for mitochondrial diseases.
RESULTS
The data here presented demonstrate that a recently discovered intra-mitochondrial signalling pathway, the mt-sAC pathway, is differentially regulated in healthy and COX-deficient mitochondria from humans and mice. COX deficient cells utilize the mt-sAC pathway for compensatory up-regulation of mitochondrial biogenesis through a free radical-mediated signalling to the nucleus. Importantly, genetic activation of the mt-sAC pathway stimulates COX phosphorylation, thereby enhancing its activity and correcting mitochondrial dysfunction in COX deficient cells.
IMPACT
This study defines the mt-sAC signalling pathway as a mechanism for metabolic adaptation to mitochondrial dysfunction and a novel potential target for therapeutic interventions to treat mitochondrial diseases.
Animal care
All experiments were performed according to the Institutional Animal Care and Use Committee-approved protocol at the Columbia University Medical Center (CUMC protocol number AAAA9997), which is consistent with the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Mitochondrial isolation from mouse tissue
Mouse liver and brain mitochondria were isolated as described (Fernandez-Vizarra et al, 2002). PKA activity was modulated by 8Br-cAMP (1 mM) and H89 (1 µM) in mitochondria and skeletal muscle homogenates as described elsewhere (Acin-Perez et al, 2009).
Cloning of sAC and mt-sAC
A C-terminal HA tag was added to the cDNA sequence from the rat sAC gene (a gift from Dr Levin and Dr Buck, Department of Pharmacology, Weill Medical College of Cornell University) by PCR amplification using appropriate primers (Methods of Supporting Information). The PCR product was digested with AgeI and NotI and cloned in the eukaryotic expression vector pTurbo-G418 (Evrogen-Axxora). To direct sAC to mitochondria (mt-sAC) the targeting signal of subunit c of ATPase (P1) (Manfredi et al, 2002b) plus four amino acids of the mature protein were cloned in frame at the N terminus of sAC using NheI and AgeI sites.
Cell transfection
Reporter constructs expressing LUC under the control of the complete Cyt c promoter (Cyt c–LUC 326) or an incomplete promoter (lacking NRF-1 and CREB binding sites, Cyt c–LUC 66) (Xia et al, 1997) and under the control of the PGC1α promoter (PGC1α–LUC) (Handschin et al, 2003) were used to transfect cells with FuGene6 (Roche). LUC activity was measured on lysates from 3 × 105 transfected cells using the Luciferase Assay System (Promega) in a luminometer (Optocomp I-MGM Electronics).
Stable mt-sAC cell lines were generated by transfection of 143B and CA75 cell lines followed by selection in 300 µg/ml G418 (Geneticin, Invitrogen) for 3 weeks.
Measurements of the mutation loads in COXI mutant clones was perform by last-hot cycle PCR followed by AluI digestion as described elsewhere (D'Aurelio et al, 2006).
sAC and mt-sAC localization
COS-1 cells transiently transfected with sAC or mt-sAC were plated on cover slips and stained with Mito-tracker Red (Invitrogen) followed by fixation in 4% paraformaldehide for immunocytochemistry using HA antibodies (Abcam). Transfected cells were analysed using a Zeiss LSM510 laser scanning confocal microscope (Carl Zeiss Microimaging, Inc., Thornwood, NY) equipped with a 63×/1.25 NA oil lens. A series of z-sections were taken spanning the thickness of the cell with intervals between sections set at 0.7 µm. Z-stack images were projected onto a single plane using the LSM Image Browser software (Carl Zeiss MicroImaging, Inc.). Digital magnification was 2× (total magnification was 126×).
For cell fractionation and intracellular localization of sAC or mt-sAC, 4 × 106 cells were transfected and mitochondria were isolated as described elsewhere (Birch-Machin & Turnbull, 2001). The total cell homogenate, the cytosolic fraction and the mitochondrial fraction were analysed by Western blot using HA antibodies.
Oxygen consumption and enzymatic assays
O2 consumption determinations in intact cells were carried out in an oxygraph equipped with a Clark electrode as previously described (Hofhaus et al, 1996). TMPD (1 mM) dependent respiration was assayed in intact cells after blocking the respiratory chain upstream of COX by addition of antimycin A (20 nM).
COX and cytrate synthase enzymatic activities were measured spectrophotometrically on isolated mitochondria (2–5 µg of protein) or in cell lysates (30–50 µg of protein) as described (Birch-Machin & Turnbull, 2001).
ATP synthesis in isolated mitochondria (15–25 µg of protein) or in cells permeabilized with digitonin (2 × 106 cells) was measured using a kinetic luminescence assay, as described (Vives-Bauza et al, 2007).
ROS production in intact cells
Cells were grown in glucose medium for 48 h in a 96-well plate and then incubated with the different compounds for 2 h. ROS were measured by 2′,7′-dichloroflourescein diacetate (H2-DCFDA; Invitrogen) fluorescence as previously described (Acin-Perez et al, 2009).
MtDNA content and mRNA levels by real time PCR
MtDNA content and mRNA levels of PGC1α, NRF-1 and GAPDH were measured with LightCycler-FastStart DNA Master SYBR Green I kit (Roche) or LightCycler® RNA Master SYBR Green I kit (Roche), respectively in a LightCycler capillary RT-PCR instrument (Roche) using appropriate primers (Methods of Supporting Information).
Western blot analyses
For denaturing SDS–PAGE 10–25 µg of protein from cell lysates, cytosolic or mitochondrial fractions was electrophoresed in a 12.5% acrylamide/bisacrylamide gel and electroblotted onto PVDF (BioRad) filters.
For 2D-BNGE, 50–75 µg of mitochondrial protein was applied on a 5–13% gradient BN gel followed by separation in a 12.5% denaturing gel (Schagger & von Jagow, 1991). After electrophoresis, proteins were electroblotted onto PVDF filters and sequentially probed with specific antibodies.
For protein detection the following antibodies were used: anti-NDUFA9 (Complex I), anti-Fp (Complex II), anti-core 2 (Complex III), anti-COXI and anti-COXIV (Invitrogen); anti-Cyt c (BD Pharmingen); anti-GAPDH and anti-HA (Abcam); anti-β-actin (Sigma–Aldrich); anti-Tim 23 and phospho Ser/Thr residues (BD Biosciences); anti-A6 (Complex V) (Rabbit polyclonal, Eric Schon, unpublished).
cAMP measurements
cAMP measurements were performed in transient or stable cell lines after subcellular fractionation. Fractions containing organelles (mitochondria) were collected after the cells were permeabilized with digitonin (50 µg/million cells) for 1 min and centrifuged at 2500 × g for 1 min. cAMP measurements were performed as described (Acin-Perez et al, 2009).
Statistical analyses
Comparisons between groups were made using one-way ANOVA. Pair wise comparisons were made by post-hoc Fisher PLSD test. Differences were considered statistically significant at p < 0.05. Data analyses were performed using the statistical program StatView. In all experiments, error bars indicate standard deviations (Adept Scientific, UK).
Author contributions
Rebeca Acin-Perez has designed the experiments together with Giovanni Manfredi, performed most of the work, and wrote the manuscript. Eric Salazar has performed the cAMP assays. Sonja Brosel has generated the immortalized human Sco2 fibroblasts. Hua Yang has generated the Sco2 KI/KO mice. Eric A. Schon is the principal investigator on the Sco2 work conducted in his laboratory and has made substantial intellectual contributions to this manuscript. Giovanni Manfredi is the laboratory head and has conceived and directed the project as well as written the manuscript together with Rebeca Acin-Perez.
Acknowledgments
We thank Dr José Antonio Enríquez (University of Zaragoza, Spain) for the mouse cybrids. Dr Lonny Levin and Dr Jochen Buck (Weill Medical College of Cornell University, New York) for the KH7 inhibitor, the rat sAC cDNA, the R21 antibody. Dr Carlos Moraes (University of Miami) and Dr Ireneaus de Coo (Erasmus Medical Center, Rotterdam, Netherlands) for the Cyt b cybrids. This work was supported by NIH (GM, EAS), the Muscular Dystrophy Association (GM, EAS), United Mitochondrial Disease Foundation (RA-P), Milstein Foundation, MSTP funding (ES) and The Marriot Foundation (EAS). All authors declare not to have any competing commercial interest in relation to this work.
Supporting information is available at EMBO Molecular Medicine online.
The authors declare that they have no conflict of interest.
For more information
United Mitochondrial Disease Foundation
Mitomap, a human mitochondrial genome database
Author's website
Giovanni Manfredi: http://www.med.cornell.edu/research/gmanfredi/index.html
Supplementary material
Detailed facts of importance to specialist readers are published as ”Supporting Information”. Such documents are peer-reviewed, but not copy-edited or typeset. They are made available as submitted by the authors.
References
- Acin-Perez R, Bayona-Bafaluy MP, Bueno M, Machicado C, Fernandez-Silva P, Perez-Martos A, Montoya J, Lopez-Perez MJ, Sancho J, Enriquez JA. An intragenic suppressor in the cytochrome c oxidase I gene of mouse mitochondrial DNA. Hum Mol Genet. 2003;12:329–339. doi: 10.1093/hmg/ddg021. [DOI] [PubMed] [Google Scholar]
- Acin-Perez R, Salazar E, Kamenetsky M, Buck J, Levin LR, Manfredi G. Cyclic AMP produced inside mitochondria regulates oxidative phosphorylation. Cell Metab. 2009;9:265–276. doi: 10.1016/j.cmet.2009.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bender E, Kadenbach B. The allosteric ATP-inhibition of cytochrome c oxidase activity is reversibly switched on by cAMP-dependent phosphorylation. FEBS Lett. 2000;466:130–134. doi: 10.1016/s0014-5793(99)01773-1. [DOI] [PubMed] [Google Scholar]
- Birch-Machin MA, Turnbull DM. Assaying mitochondrial respiratory complex activity in mitochondria isolated from human cells and tissues. Methods Cell Biol. 2001;65:97–117. doi: 10.1016/s0091-679x(01)65006-4. [DOI] [PubMed] [Google Scholar]
- Bronner M, Hertz R, Bar-Tana J. Kinase-independent transcriptional co-activation of peroxisome proliferator-activated receptor alpha by AMP-activated protein kinase. Biochem J. 2004;384:295–305. doi: 10.1042/BJ20040955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruno C, Martinuzzi A, Tang Y, Andreu AL, Pallotti F, Bonilla E, Shanske S, Fu J, Sue CM, Angelini C, et al. A stop-codon mutation in the human mtDNA cytochrome c oxidase I gene disrupts the functional structure of complex IV. Am J Hum Genet. 1999;65:611–620. doi: 10.1086/302546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen R, Fearnley IM, Peak-Chew SY, Walker JE. The phosphorylation of subunits of complex I from bovine heart mitochondria. J Biol Chem. 2004;279:26036–26045. doi: 10.1074/jbc.M402710200. [DOI] [PubMed] [Google Scholar]
- Chen Y, Cann MJ, Litvin TN, Iourgenko V, Sinclair ML, Levin LR, Buck J. Soluble adenylyl cyclase as an evolutionarily conserved bicarbonate sensor. Science. 2000;289:625–628. doi: 10.1126/science.289.5479.625. [DOI] [PubMed] [Google Scholar]
- Chijiwa T, Mishima A, Hagiwara M, Sano M, Hayashi K, Inoue T, Naito K, Toshioka T, Hidaka H. Inhibition of forskolin-induced neurite outgrowth and protein phosphorylation by a newly synthesized selective inhibitor of cyclic AMP-dependent protein kinase, N-[2-(p-bromocinnamylamino)ethyl]-5-isoquinolinesulfonamide (H-89), of PC12D pheochromocytoma cells. J Biol Chem. 1990;265:5267–5272. [PubMed] [Google Scholar]
- D'Aurelio M, Gajewski CD, Lenaz G, Manfredi G. Respiratory chain supercomplexes set the threshold for respiration defects in human mtDNA mutant cybrids. Hum Mol Genet. 2006;15:2157–2169. doi: 10.1093/hmg/ddl141. [DOI] [PubMed] [Google Scholar]
- D'Aurelio M, Pallotti F, Barrientos A, Gajewski CD, Kwong JQ, Bruno C, Beal MF, Manfredi G. In vivo regulation of oxidative phosphorylation in cells harboring a stop-codon mutation in mitochondrial DNA-encoded cytochrome c oxidase subunit I. J Biol Chem. 2001;276:46925–46932. doi: 10.1074/jbc.M106429200. [DOI] [PubMed] [Google Scholar]
- De Rasmo D, Panelli D, Sardanelli AM, Papa S. cAMP-dependent protein kinase regulates the mitochondrial import of the nuclear encoded NDUFS4 subunit of complex I. Cell Signal. 2008;20:989–997. doi: 10.1016/j.cellsig.2008.01.017. [DOI] [PubMed] [Google Scholar]
- Dimauro S, Davidzon G. Mitochondrial DNA and disease. Ann Med. 2005;37:222–232. doi: 10.1080/07853890510007368. [DOI] [PubMed] [Google Scholar]
- Feliciello A, Gottesman ME, Avvedimento EV. cAMP-PKA signaling to the mitochondria: protein scaffolds, mRNA and phosphatases. Cell Signal. 2005;17:279–287. doi: 10.1016/j.cellsig.2004.09.009. [DOI] [PubMed] [Google Scholar]
- Fernandez-Vizarra E, Lopez-Perez MJ, Enriquez JA. Isolation of biogenetically competent mitochondria from mammalian tissues and cultured cells. Methods. 2002;26:292–297. doi: 10.1016/S1046-2023(02)00034-8. [DOI] [PubMed] [Google Scholar]
- Handschin C, Rhee J, Lin J, Tarr PT, Spiegelman BM. An autoregulatory loop controls peroxisome proliferator-activated receptor gamma coactivator 1alpha expression in muscle. Proc Natl Acad Sci USA. 2003;100:7111–7116. doi: 10.1073/pnas.1232352100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansford RG. Dehydrogenase activation by Ca2+ in cells and tissues. J Bioenerg Biomembr. 1991;23:823–854. doi: 10.1007/BF00786004. [DOI] [PubMed] [Google Scholar]
- Hao H, Moraes CT. Functional and molecular mitochondrial abnormalities associated with a C –> T transition at position 3256 of the human mitochondrial genome. The effects of a pathogenic mitochondrial tRNA point mutation in organelle translation and RNA processing. J Biol Chem. 1996;271:2347–2352. doi: 10.1074/jbc.271.4.2347. [DOI] [PubMed] [Google Scholar]
- Helling S, Vogt S, Rhiel A, Ramzan R, Wen L, Marcus K, Kadenbach B. Phosphorylation and kinetics of mammalian cytochrome c oxidase. Mol Cell Proteomics. 2008;9:1717–1724. doi: 10.1074/mcp.M800137-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess KC, Jones BH, Marquez B, Chen Y, Ord TS, Kamenetsky M, Miyamoto C, Zippin JH, Kopf GS, Suarez SS, et al. The “soluble” adenylyl cyclase in sperm mediates multiple signaling events required for fertilization. Dev Cell. 2005;9:249–259. doi: 10.1016/j.devcel.2005.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofhaus G, Shakeley RM, Attardi G. Use of polarography to detect respiration defects in cell cultures. Methods Enzymol. 1996;264:476–483. doi: 10.1016/s0076-6879(96)64043-9. [DOI] [PubMed] [Google Scholar]
- Horbinski C, Chu CT. Kinase signaling cascades in the mitochondrion: a matter of life or death. Free Radic Biol Med. 2005;38:2–11. doi: 10.1016/j.freeradbiomed.2004.09.030. [DOI] [PubMed] [Google Scholar]
- Irrcher I, Ljubicic V, Hood DA. Interactions between ROS and AMP kinase activity in the regulation of PGC-1alpha transcription in skeletal muscle cells. Am J Physiol Cell Physiol. 2009;296:C116–C123. doi: 10.1152/ajpcell.00267.2007. [DOI] [PubMed] [Google Scholar]
- Jager S, Handschin C, St-Pierre J, Spiegelman BM. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1alpha. Proc Natl Acad Sci USA. 2007;104:12017–12022. doi: 10.1073/pnas.0705070104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadenbach B. Intrinsic and extrinsic uncoupling of oxidative phosphorylation. Biochim Biophys Acta. 2003;1604:77–94. doi: 10.1016/s0005-2728(03)00027-6. [DOI] [PubMed] [Google Scholar]
- Kadenbach B, Ramzan R, Vogt S. Degenerative diseases, oxidative stress and cytochrome c oxidase function. Trends Mol Med. 2009;15:139–147. doi: 10.1016/j.molmed.2009.02.004. [DOI] [PubMed] [Google Scholar]
- King MP, Attardi G. Human cells lacking mtDNA: repopulation with exogenous mitochondria by complementation. Science. 1989;246:500–503. doi: 10.1126/science.2814477. [DOI] [PubMed] [Google Scholar]
- Kwong JQ, Beal MF, Manfredi G. The role of mitochondria in inherited neurodegenerative diseases. J Neurochem. 2006;97:1659–1675. doi: 10.1111/j.1471-4159.2006.03990.x. [DOI] [PubMed] [Google Scholar]
- Lee I, Salomon AR, Ficarro S, Mathes I, Lottspeich F, Grossman LI, Huttemann M. cAMP-dependent tyrosine phosphorylation of subunit I inhibits cytochrome c oxidase activity. J Biol Chem. 2005;280:6094–6100. doi: 10.1074/jbc.M411335200. [DOI] [PubMed] [Google Scholar]
- Lewitt MS, Brismar K, Wang J, Wivall-Helleryd IL, Sindelar P, Gonzalez FJ, Bergman T, Bobek GA. Responses of insulin-like growth factor (IGF)-I and IGF-binding proteins to nutritional status in peroxisome proliferator-activated receptor-alpha knockout mice. Growth Horm IGF Res. 2001;11:303–313. doi: 10.1054/ghir.2001.0247. [DOI] [PubMed] [Google Scholar]
- Linn TC, Pettit FH, Reed LJ. Alpha-keto acid dehydrogenase complexes. X. Regulation of the activity of the pyruvate dehydrogenase complex from beef kidney mitochondria by phosphorylation and dephosphorylation. Proc Natl Acad Sci USA. 1969;62:234–241. doi: 10.1073/pnas.62.1.234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livigni A, Scorziello A, Agnese S, Adornetto A, Carlucci A, Garbi C, Castaldo I, Annunziato L, Avvedimento EV, Feliciello A. Mitochondrial AKAP121 links cAMP and src signaling to oxidative metabolism. Mol Biol Cell. 2006;17:263–271. doi: 10.1091/mbc.E05-09-0827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lochmuller H, Johns T, Shoubridge EA. Expression of the E6 and E7 genes of human papillomavirus (HPV16) extends the life span of human myoblasts. Exp Cell Res. 1999;248:186–193. doi: 10.1006/excr.1999.4407. [DOI] [PubMed] [Google Scholar]
- Lu G, Ren S, Korge P, Choi J, Dong Y, Weiss J, Koehler C, Chen JN, Wang Y. A novel mitochondrial matrix serine/threonine protein phosphatase regulates the mitochondria permeability transition pore and is essential for cellular survival and development. Genes Dev. 2007;21:784–796. doi: 10.1101/gad.1499107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manfredi G, Fu J, Ojaimi J, Sadlock JE, Kwong JQ, Guy J, Schon EA. Rescue of a deficiency in ATP synthesis by transfer of MTATP6, a mitochondrial DNA-encoded gene, to the nucleus. Nat Genet. 2002a;25:25. doi: 10.1038/ng851. [DOI] [PubMed] [Google Scholar]
- Manfredi G, Fu J, Ojaimi J, Sadlock JE, Kwong JQ, Guy J, Schon EA. Rescue of a deficiency in ATP synthesis by transfer of MTATP6, a mitochondrial DNA-encoded gene, to the nucleus. Nat Genet. 2002b;30:394–399. doi: 10.1038/ng851. [DOI] [PubMed] [Google Scholar]
- Mattiazzi M, Vijayvergiya C, Gajewski CD, DeVivo DC, Lenaz G, Wiedmann M, Manfredi G. The mtDNA T8993G (NARP) mutation results in an impairment of oxidative phosphorylation that can be improved by antioxidants. Hum Mol Genet. 2004;13:869–879. doi: 10.1093/hmg/ddh103. [DOI] [PubMed] [Google Scholar]
- Miyazaki T, Neff L, Tanaka S, Horne WC, Baron R. Regulation of cytochrome c oxidase activity by c-Src in osteoclasts. J Cell Biol. 2003;160:709–718. doi: 10.1083/jcb.200209098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno-Loshuertos R, Acin-Perez R, Fernandez-Silva P, Movilla N, Perez-Martos A, Rodriguez de Cordoba S, Gallardo ME, Enriquez JA. Differences in reactive oxygen species production explain the phenotypes associated with common mouse mitochondrial DNA variants. Nat Genet. 2006;38:1261–1268. doi: 10.1038/ng1897. [DOI] [PubMed] [Google Scholar]
- Pagliarini DJ, Dixon JE. Mitochondrial modulation: reversible phosphorylation takes center stage? Trends Biochem Sci. 2006;31:26–34. doi: 10.1016/j.tibs.2005.11.005. [DOI] [PubMed] [Google Scholar]
- Papa S, Sardanelli AM, Cocco T, Speranza F, Scacco SC, Technikova-Dobrova Z. The nuclear-encoded 18 kDa (IP) AQDQ subunit of bovine heart complex I is phosphorylated by the mitochondrial cAMP-dependent protein kinase. FEBS Lett. 1996;379:299–301. doi: 10.1016/0014-5793(95)01532-9. [DOI] [PubMed] [Google Scholar]
- Papadopoulou LC, Sue CM, Davidson MM, Tanji K, Nishino I, Sadlock JE, Krishna S, Walker W, Selby J, Glerum DM, et al. Fatal infantile cardioencephalomyopathy with COX deficiency and mutations in SCO2, a COX assembly gene. Nat Genet. 1999;23:333–337. doi: 10.1038/15513. [DOI] [PubMed] [Google Scholar]
- Piccoli C, Scacco S, Bellomo F, Signorile A, Iuso A, Boffoli D, Scrima R, Capitanio N, Papa S. cAMP controls oxygen metabolism in mammalian cells. FEBS Lett. 2006;580:4539–4543. doi: 10.1016/j.febslet.2006.06.085. [DOI] [PubMed] [Google Scholar]
- Prabu SK, Anandatheerthavarada HK, Raza H, Srinivasan S, Spear JF, Avadhani NG. Protein kinase A-mediated phosphorylation modulates cytochrome c oxidase function and augments hypoxia and myocardial ischemia-related injury. J Biol Chem. 2006;281:2061–2070. doi: 10.1074/jbc.M507741200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rana M, de Coo I, Diaz F, Smeets H, Moraes CT. An out-of-frame cytochrome b gene deletion from a patient with parkinsonism is associated with impaired complex III assembly and an increase in free radical production. Ann Neurol. 2000;48:774–781. [PubMed] [Google Scholar]
- Ryu H, Lee J, Impey S, Ratan RR, Ferrante RJ. Antioxidants modulate mitochondrial PKA and increase CREB binding to D-loop DNA of the mitochondrial genome in neurons. Proc Natl Acad Sci USA. 2005;102:13915–13920. doi: 10.1073/pnas.0502878102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salviati L, Hernandez-Rosa E, Walker WF, Sacconi S, DiMauro S, Schon EA, Davidson MM. Copper supplementation restores cytochrome c oxidase activity in cultured cells from patients with SCO2 mutations. Biochem J. 2002;363:321–327. doi: 10.1042/0264-6021:3630321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samavati L, Lee I, Mathes I, Lottspeich F, Huttemann M. TNFalpha inhibits oxidative phosphorylation through tyrosine phosphorylation at subunit I of cytochrome c oxidase. J Biol Chem. 2008;283:21134–21144. doi: 10.1074/jbc.M801954200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sardanelli AM, Technikova-Dobrova Z, Scacco SC, Speranza F, Papa S. Characterization of proteins phosphorylated by the cAMP-dependent protein kinase of bovine heart mitochondria. FEBS Lett. 1995;377:470–474. doi: 10.1016/0014-5793(95)01407-1. [DOI] [PubMed] [Google Scholar]
- Scacco S, Vergari R, Scarpulla RC, Technikova-Dobrova Z, Sardanelli A, Lambo R, Lorusso V, Papa S. cAMP-dependent phosphorylation of the nuclear encoded 18-kDa (IP) subunit of respiratory complex I and activation of the complex in serum-starved mouse fibroblast cultures. J Biol Chem. 2000;275:17578–17582. doi: 10.1074/jbc.M001174200. [DOI] [PubMed] [Google Scholar]
- Scarpulla RC. Transcriptional paradigms in mammalian mitochondrial biogenesis and function. Physiol Rev. 2008;88:611–638. doi: 10.1152/physrev.00025.2007. [DOI] [PubMed] [Google Scholar]
- Schagger H, von Jagow G. Blue native electrophoresis for isolation of membrane protein complexes in enzymatically active form. Anal Biochem. 1991;199:223–231. doi: 10.1016/0003-2697(91)90094-a. [DOI] [PubMed] [Google Scholar]
- Schwoch G, Trinczek B, Bode C. Localization of catalytic and regulatory subunits of cyclic AMP-dependent protein kinases in mitochondria from various rat tissues. Biochem J. 1990;270:181–188. doi: 10.1042/bj2700181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoubridge EA. Cytochrome c oxidase deficiency. Am J Med Genet. 2001;106:46–52. doi: 10.1002/ajmg.1378. [DOI] [PubMed] [Google Scholar]
- Signorile A, Sardanelli AM, Nuzzi R, Papa S. Serine (threonine) phosphatase(s) acting on cAMP-dependent phosphoproteins in mammalian mitochondria. FEBS Lett. 2002;512:91–94. doi: 10.1016/s0014-5793(02)02226-3. [DOI] [PubMed] [Google Scholar]
- St-Pierre J, Drori S, Uldry M, Silvaggi JM, Rhee J, Jager S, Handschin C, Zheng K, Lin J, Yang W, et al. Suppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivators. Cell. 2006;127:397–408. doi: 10.1016/j.cell.2006.09.024. [DOI] [PubMed] [Google Scholar]
- Steenaart NA, Shore GC. Mitochondrial cytochrome c oxidase subunit IV is phosphorylated by an endogenous kinase. FEBS Lett. 1997;415:294–298. doi: 10.1016/s0014-5793(97)01145-9. [DOI] [PubMed] [Google Scholar]
- Thomson M. Evidence of undiscovered cell regulatory mechanisms: phosphoproteins and protein kinases in mitochondria. Cell Mol Life Sci. 2002;59:213–219. doi: 10.1007/s00018-002-8417-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vives-Bauza C, Gonzalo R, Manfredi G, Garcia-Arumi E, Andreu AL. Enhanced ROS production and antioxidant defenses in cybrids harbouring mutations in mtDNA. Neurosci Lett. 2006;391:136–141. doi: 10.1016/j.neulet.2005.08.049. [DOI] [PubMed] [Google Scholar]
- Vives-Bauza C, Yang L, Manfredi G. Assay of mitochondrial ATP synthesis in animal cells and tissues. Methods Cell Biol. 2007;80:155–171. doi: 10.1016/S0091-679X(06)80007-5. [DOI] [PubMed] [Google Scholar]
- Wenz T, Diaz F, Spiegelman BM, Moraes CT. Activation of the PPAR/PGC-1alpha pathway prevents a bioenergetic deficit and effectively improves a mitochondrial myopathy phenotype. Cell Metab. 2008;8:249–256. doi: 10.1016/j.cmet.2008.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Xia Y, Buja LM, Scarpulla RC, McMillin JB. Electrical stimulation of neonatal cardiomyocytes results in the sequential activation of nuclear genes governing mitochondrial proliferation and differentiation. Proc Natl Acad Sci USA. 1997;94:11399–11404. doi: 10.1073/pnas.94.21.11399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zippin JH, Chen Y, Nahirney P, Kamenetsky M, Wuttke MS, Fischman DA, Levin LR, Buck J. Compartmentalization of bicarbonate-sensitive adenylyl cyclase in distinct signaling microdomains. FASEB J. 2003;17:82–84. doi: 10.1096/fj.02-0598fje. [DOI] [PubMed] [Google Scholar]
- Zippin JH, Levin LR, Buck J. CO(2)/HCO(3)(−)-responsive soluble adenylyl cyclase as a putative metabolic sensor. Trends Endocrinol Metab. 2001;12:366–370. doi: 10.1016/s1043-2760(01)00454-4. [DOI] [PubMed] [Google Scholar]
- Zuckerbraun BS, Chin BY, Bilban M, d'Avila JC, Rao J, Billiar TR, Otterbein LE. Carbon monoxide signals via inhibition of cytochrome c oxidase and generation of mitochondrial reactive oxygen species. FASEB J. 2007;21:1099–1106. doi: 10.1096/fj.06-6644com. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.