

Abstract
Motivation: Shotgun sequencing generates large numbers of short DNA reads from either an isolated organism or, in the case of metagenomics projects, from the aggregate genome of a microbial community. These reads are then assembled based on overlapping sequences into larger, contiguous sequences (contigs). The feasibility of assembly and the coverage achieved (reads per nucleotide or distinct sequence of nucleotides) depend on several factors: the number of reads sequenced, the read length and the relative abundances of their source genomes in the microbial community. A low coverage suggests that most of the genomic DNA in the sample has not been sequenced, but it is often difficult to estimate either the extent of the uncaptured diversity or the amount of additional sequencing that would be most efficacious. In this work, we regard a metagenome as a population of DNA fragments (bins), each of which may be covered by one or more reads. We employ a gamma distribution to model this bin population due to its flexibility and ease of use. When a gamma approximation can be found that adequately fits the data, we may estimate the number of bins that were not sequenced and that could potentially be revealed by additional sequencing. We evaluated the performance of this model using simulated metagenomes and demonstrate its applicability on three recent metagenomic datasets.
Contact: sean.d.hooper@genpat.uu.se
Supplementary information: Supplementary data are available at Bioinformatics online.
1 INTRODUCTION
Shotgun sequencing is the cornerstone of modern genomics. An entire genome is sheared into short fragments, which are then amplified and sequenced. The large number of short sequences produced, termed reads, are assembled into longer sequences (contigs) based on their overlaps, potentially providing the DNA sequence of an entire genome. Initially, sequencing projects focused on microbes that could be isolated and cultured. This ensured that the sequenced DNA had been obtained from a clonal isolate and that all of the reads originated from one genome. A few years ago, however, the focus of microbial genomics shifted to sequencing DNA recovered directly from environmental samples, thus sequencing the aggregate genomes of entire communities, or metagenomes (Breitbart et al., 2002; Stein et al., 1996). Since then, shotgun sequencing methods have been employed in numerous large-scale metagenomics projects (e.g. Tringe et al., 2005; Venter et al., 2004), resulting in a veritable explosion of data—and a need for more sophisticated methods of analysis (Raes et al., 2007).
In a typical metagenomics project, a portion of the DNA sampled from an environment is sequenced to yield a known number of reads with a known average read length. Each of these reads may represent a section of a genome, and in some cases several reads may represent the same section. In this work, we divide genomes in the metagenome into bins, each corresponding to the average read length, and try to estimate the frequency of these bins by the number of reads that are assigned to them.
Following assembly of the reads into contigs, one can calculate the number of bins that the contig represents, and thereby the number of reads per bin. This generates a coverage spectrum (or bin spectrum) that embodies the observed number of bins containing various numbers of reads in the sample. However, the observed bins are but a portion of the total DNA in the sample. It is often desirable to know what fraction of the total genetic diversity in the sample has been analyzed, i.e. how many more potential bins exist for which there are currently no reads. This estimate could, in turn, suggest how much additional DNA sequencing would be necessary in order to capture a desired proportion of the sampled DNA for purposes such as functional comparisons between metagenomes or mining for novel functions. Approximating answers to these questions necessitates being able to model the observed bin spectrum.
In many fields of science, it is desirable to gain an estimate of the number of unobservables in a collection of data. In ecology, the estimation of macrobiological species richness also requires that the unobservable species be estimated. Chao and co-workers (Chao, 1984; Chao et al., 1992) studied the lower bounds of the number of unobservables by non-parametric measures and by using mixed Poisson distributions (Chao and Bunge, 2002). Analogously, for metagenomes, 16S rDNA can be used as markers of species, which can be estimated using compound Poisson models (Quince et al., 2008) who also estimate the total amount of DNA by assuming that all genomes have the same number of genes. However, this assumption may hold better in the case of viromes, where genome sizes are more or less equal. Angly and coworkers used this assumption to good effect when estimating the species richnesses from the full DNA sample (Angly et al., 2005).
In demographics and casualty statistics, there is perhaps a longer history still. In this case, the unobservables may represent children per mother (Brass, 1958), automobile insurance claims or accidents (Dropkin, 1959; Simon, 1961). However, it is more difficult to establish whether DNA sequences are from the same source than to determine whether children have the same mother.
In this work, rather than estimating species abundances and richness, we will forego species markers and study the distribution of DNA in the form of bins. This could provide valuable insights into the functional complexity of a metagenome, i.e. the full genetic diversity, and would enjoy far greater sample sizes for estimations, thus increasing the accuracy of estimations. Also, we will apply concepts from statistical fields to metagenomics, in order to evaluate their ability to quickly and accurately estimate the parameters of the bin abundance and also the number of unobservable bins. In particular, the Brass estimators (Brass, 1958) prove to be not only easily accessible but also accurate. Hopefully, this could complement the existing estimators such as the Chao1 and ACE estimators.
We report here our exploration of the effect of bin abundances on the distribution of reads per bin of sampled DNA. We test our conclusions against simulated assemblies and find a high degree of accuracy. Lastly, we illustrate the applicability of our approach using datasets from three metagenomics projects. We find that two of them were successful, and we discuss possible reasons why the third set could not be estimated.
2 METHODS
2.1 Poisson occupancy models
A genome g of length gL can be divided into non-overlapping bins of length L, where L is the average read length. During the sequencing process, the sequences of bins sampled from g can be determined, and are referred to as reads. Each bin can be covered by zero or more reads, and a read may only belong to one bin. This process of assigning reads to bins is easiest to visualize as a stochastic process; let us assume that we pick one bin at random from g and sequence it, resulting in a read. Thereafter, we pick another bin from g and record the read. There is a possibility that the read will fully or partially overlap the original bin. This would therefore result in an assembly of the reads into a contig. Usually, the output from an assembly is in the form of reads and contigs, but from this information, we could calculate how many bins the contig represents, and also how the reads are distributed on the bins (see Supplementary Material). This process of picking bins from g can be expressed as a Poisson process, and is related to the coverage per nucleotide (Lander and Waterman, 1988; Wendl, 2006). In this case, the expected number of reads per bin is RL/gL, i.e. the number of reads times the probability of picking the bin. If we denote the number of reads per bin as k, and the expected number of reads per bin in genome g as λg, we can therefore express the distribution of k as
| (1) |
When there is only one genome in the sample, i.e. the number of genomes G = 1, all bins are expected to have more or less the same expected number of reads. However, when working with actual genomes, various factors can cause us to observe unequal occupancy frequencies. Paralogous sequences, for instance, can be virtually identical, thus artificially inflating the number of reads observed for such bins. If there were two paralogous bins with identical nucleotide sequences, we would observe one bin with twice the expected number of reads. A similar multiplication effect can result when repetitive DNA, such as transposons or prophages, blurs the distinction between bins. However, these effects are often of little consequence in most sequencing projects of isolated organisms (G = 1) given a high average coverage.
When the sample includes more than one genome (G> 1), we cannot assume the same λg for all bins from all genomes. The expected number of reads per bin will depend on the relative abundance of the source genomes in the sample (Fig. 1) so that the expected number of reads per bin for bins in genome i (assuming that the effects of repeated identical sequences is negligible) is 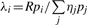 , where R is the total number of reads, pi is the relative proportion of genome i in the metagenome and ηj is the number of bins in genome j. Therefore, a more general form of Equation (1) is needed—a mixed Poisson distribution for all values of λg, where λg is itself distributed according to a secondary distribution.
, where R is the total number of reads, pi is the relative proportion of genome i in the metagenome and ηj is the number of bins in genome j. Therefore, a more general form of Equation (1) is needed—a mixed Poisson distribution for all values of λg, where λg is itself distributed according to a secondary distribution.
| (2) |
Fig. 1.

The effect of community complexity on the expected number of reads per bin (λi) for each bin i. In a simple community with a single genome, λ is approximately equal for all bins. Here, the observed bin spectrum (number of reads per bin) follows a Poisson distribution. However, in metagenomic samples from complex communities, bins will be drawn from different genomes that are present in varying abundances. Therefore, the value of λ is not the same for all bins. If λ follows a gamma distribution, then the bin spectrum will follow a negative binomial distribution and can be modeled.
When G = 1, f(λg) is a constant; when G > 1, it varies with a secondary probability distribution that reflects the relative abundance of the source genomes. Here, α and β are parameters that determine the scale and shape of the secondary distribution. Many different distributions can be used for this purpose. Commonly used distributions are the exponential, lognormal and gamma. In this work, we chose to focus on the gamma distribution as it is general enough to describe both exponentially decaying communities (i.e. those with a large variation in bin abundances) and those which are more Gaussian (i.e. those where contig abundances are less varied), although we also compare our results to a Poisson-lognormal distribution (Izsák, 2007). Thus
| (3) |
An additional advantage of using the gamma distribution is that the resultant compound Poisson-Gamma distribution is itself well-studied and understood as the negative binomial distribution. It has often been used to model other over-dispersed Poisson distributions such as the distribution of bacteria in homogeneous mediums (el-Shaarawi, 1985) and insurance claims (Dropkin, 1959; Simon, 1961). Here we employ it to approximate the distribution of reads per bin, i.e. the bin spectrum. Following this approach, Equation (2) can be rewritten as
| (4) |
where q = 1 − p, r = α and 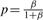 .
.
When analyzing a sample where G > 1, we typically want to estimate the bins abundance and the total number of bins, including those with no corresponding reads. Since we cannot observe the number of bins with k = 0 reads, the distribution of f(k|λg) is null-truncated, i.e. the number of bins with zero reads is missing. Correcting for this yields
| (5) |
since P(k = 0) = pr in Equation (4).
2.2 Estimating parameters
If x1, x2,…, xN are observations of the reads per bin, we can attempt to estimate the parameters r and p. The Maximum Likelihood (ML) estimates for r and p are therefore
These equations are cumbersome and can only be solved numerically. A less exact but easier way is to study the moments of the null-truncated negative binomial using the Brass estimates (Brass, 1958) to exploit the ratio of n1/N, where n1 is the number of bins with k = 1 reads:
| (6) |
Here,  is the mean of the reads per observed bin, and var(x) is the variance. These estimates are reasonably accurate (see Supplementary Material for the variances of the Brass estimators) and can also be used as initial values for ML estimates. With p* and r* estimated through Equations (6) or ML estimates, we can also estimate the number of bins which were not occupied by a read: N* = N + N0, where N is the number of observed bins, N0 is the number of bins with zero reads, and N* is the estimated total number of bins. From Equations (5), we recall that N0 = N* pr, i.e. the number of unobserved bins is N* times the probability of k = 0. The variance of N0, given our estimates of r and p, is therefore var(N0) = N* pr(1 − pr).
is the mean of the reads per observed bin, and var(x) is the variance. These estimates are reasonably accurate (see Supplementary Material for the variances of the Brass estimators) and can also be used as initial values for ML estimates. With p* and r* estimated through Equations (6) or ML estimates, we can also estimate the number of bins which were not occupied by a read: N* = N + N0, where N is the number of observed bins, N0 is the number of bins with zero reads, and N* is the estimated total number of bins. From Equations (5), we recall that N0 = N* pr, i.e. the number of unobserved bins is N* times the probability of k = 0. The variance of N0, given our estimates of r and p, is therefore var(N0) = N* pr(1 − pr).
2.3 Evaluating resequencing efforts
Often, the first study of a metagenome is exploratory in nature. Following the initial foray into the DNA in a sample, the potential for follow-up projects is often evaluated. In this case, it may be of interest to adjust the scale of the sequencing to capture a larger degree of variation. Since the number of unobservable bins is N0 = N* pr, we can study the effect of varying the function pr, which is the probability of a bin having zero reads in the negative binomial distribution. We cannot solve both p and r in this equation, but if we tentatively consider r to determine the shape of the underlying gamma distribution (since α = r) and therefore be immutable, we can form an estimate p(c0), which would be the value required to capture a certain percent of the bins in the sample. The general equation for estimating p(c0) is
| (7) |
where c0 is the proportion of bins covered by zero reads. Since the expected average number of reads per bin for all bins x1,…, xN in the set is
| (8) |
we can calculate the new average reads per bin for all bins in the set by replacing p with p(c0).
However, it is also likely that as coverage increases, the shape parameter r will also change. Thus, the estimate may also change, which would suggest that a new, more accurate estimate could be made.
2.4 Removal of high-occupancy contigs
This method of forming a negative binomial approximation for the coverage spectrum is robust when bins have few reads, but is more sensitive to fluctuations in the number of bins with very many reads. Such situations can occur in metagenomic datasets due to the presence of a highly dominant species in an environment, unintentional inclusion of phage, or the introduction of an allochthonous contaminant during processing. These sometimes artificially over-abundant genomes or viromes result in one or more highly covered bins, while the rest of the sample is represented by bins with significantly lower coverages.
When using a gamma distribution to model genome abundance, high-occupancy bins often carry a very low probability of occurrence, which causes the approximation to fail. Our method of handling this situation is to ignore the high-occupancy bins and the taxa they represent, then attempt to model the remaining population as a gamma distribution. This approach minimizes two particular difficulties. First, when a metagenome contains a very dominant species, this skews the estimates of how much additional material one would expect to find by further sequencing, i.e. the total number of genomes in the metagenome. Second, the effects of contamination or unusual assembly protocols on the gamma approximation are minor when they affect low-occupancy bins, but they can be quite dramatic when they produce high-occupancy bins. Thus, it can be advantageous to remove high-occupancy bins.
The negative binomial approximation is unable to model the coverage spectrum if r or p are estimated to be negative or if r approaches infinity. When r → ∞, the negative binomial approaches a Poisson distribution (i.e. the variance and mean are equal), suggesting that all genomes occur more or less at an equal frequency. Conversely, when r → 0, the distribution is heavily skewed towards the k = 0 value. Here the approximation may fail because the dispersion (variance divided by the mean) of the distribution is too large to be accurately modeled by the negative binomial. This situation often arises when high-occupancy bins are present. Thus, we want to examine the portion of data that lies between these two conditions, i.e. where the dispersion is low enough to model with a negative binomial but still larger than the value where the negative binomial approaches the Poisson distribution.
This approach of removing or partitioning the spectrum has also been used by Chao and coworkers [described in Chao and Bunge (2002)], where a suitable cutoff is determined by recalculating the goodness of fit to the observed data. Analogously, we can suggest a series of gamma distributions that can be used to approximate the bin abundances.
3 RESULTS
3.1 Simulated single genomes
When all reads are derived from an isolate organism, the expected number of reads per bin (λ) is constant. If there are no major contributions from paralogs or high-copy number sequences, then the distribution of reads per bin will follow a Poisson distribution, i.e. Po(λ). Recall that the negative binomial approximation approaches the Poisson distribution when r → ∞, i.e. when the mean and variance of the coverage spectrum are equal. Here, no estimates of α and β are possible, since these values cannot be defined. We can still estimate λ from the mean of the estimated distribution, which in our terms is λ = r(1 − p)/p. To study such a situation, we simulated in silico a genome of 1000 bins (including bins with no reads) according to Po(λ = 1) 1000 times. Each of these bins would therefore be covered by on average one read, which suggests that ∼37% of bins would not be observed [Equation (1), P(k = 0)]. The resultant average estimates were 1007.2 ± 71 for N* (the total number of bins) and 0.9987 ± 0.08 for λ. Thus, although observing only ∼63% of bins, the number of unobservables was accurately estimated. However, α and β were estimated as either some arbitrary high numbers (>100) or, due to the structure of Equation (5), as negative values, simply because there is no distribution of λ—it is a scalar value. Since the distribution is approximately Poisson, the Lander–Waterman equations should be used instead (Lander and Waterman, 1988).
3.2 Simulated metagenomic datasets
We simulated two sets of metagenomes, with 10 000 and 20 000 bins, respectively (see Supplementary Tables S1 and S2) with varying degrees of coverage. Simulations were performed 100 times for each combination of the negative binomial parameters r and p, which represent the bin abundance. We note that when r is low and p is high, the proportion of bins occupied by a read decreases, and estimates of the parameters lose precision. This is particularly evident in Supplementary Table S1 when r = 0.5 and p ≥ 0.7 (average read per bin = 0.21). At this low degree of reads per bin, it is unlikely that any estimate will be very accurate. In Supplementary Table S2, this effect is compensated somewhat by the higher number of bins, but still fails at (r = 0.5, p = 0.8, average number of reads per bin 0.125).
We also note a slight (2–3%) overestimation of the total number of bins (column 1, Supplementary Tables S1). This should be taken into consideration when estimating the bin abundance in metagenomes, and could possibly be compensated for. This overestimation decreases to roughly 1% with larger values of N (see Supplementary Table S2), suggesting that the accuracy of the model increases with larger datasets, even though the total occupancy may still be low. Notably, sampling of most complex environments is likely to yield a number of bins that is considerably greater than the N values used in our simulations. In summary, the larger and more complex the environmental sample, the more accurate the estimates made by this method. It is noteworthy to point out that estimates are good despite a very low bin count. This is a consequence of assuming a smooth and perfect bin abundance, and in a real world situation, the bin abundance will be less smooth even if the underlying bin abundance follows a gamma distribution.
3.3 Simulated de novo assembly
The previous simulations describe a perfect world, where populations are perfectly complex and assembly is fully accurate. However, the assembly step in real situations is error-prone and affects the final estimates of the total amount of DNA. To study these effects, we simulated metagenomes of 30 and 50 organisms with gamma-like abundances using MetaSim (Richter et al., 2008), while varying the number of reads drawn from the metagenome. In all cases, we drew a random selection of organisms from the full set of available taxa, excluding plasmids. We subsequently assigned each organism a relative abundance drawn from a gamma distribution. In the 30 organism case, the total amount of DNA is roughly 105 Mbps. From this dataset, 50 000 and 100 000 reads of 1000 bp were drawn and assembled using MIRA [used in e.g. Chevreux et al. (2004)], repeated twice and estimated directly without any bin removal. The estimated amount of total DNA was slightly lower than the true value in the 100 000 read case; 84 and 85 Mbps. The underestimation is likely due to misassemblies of unrelated sequences into larger contigs, which therefore inflates the reads per bins. One example of this is an assembly of Thermus thermophilus and Flavobacterium sp. sequences into a contig. Added to this is the effect of lower coverage which further complicates the estimates. In the 50 000 reads case, the estimates are 66 and 67 Mbps, reflecting the lower coverage. In the 50 organism case with 100 000 reads, we again see an underestimation of the real amount of DNA. We observe 85 Mbps in both replicates, compared to roughly 130 MBp. This is consistent with the observations from the 30 organism case, considering that the total coverage is lower relative to the 100 000 read simulation of 30 organisms. Thus, it can be concluded that the addition of an assembly step will affect the final estimate, depending on how strict or how liberal the assembler is. In this case, numbers are underestimated but still reasonable. Naturally, the cross-assembly also affects the estimates of the underlying gamma distribution, but may still serve as initial approximations.
3.4 Real metagenomes
3.4.1 Goodness of fit
To evaluate how well the approximated bin spectrum fits to the observed, we calculated the χ2-score for the number of bins with one to five reads. The critical χ2-score at four degrees of freedom is 9.448 (95% confidence). If the goodness of fit is higher than the critical value, we can reject the assumption that the underlying bin abundance can be approximated by a gamma distribution.
3.4.2 Lake sediment formate community
This study employed stable isotope probing combined with metagenomic analysis to characterize the ecological roles of microbes in sediment from Lake Washington (Kalyuzhnaya et al., 2008). Five samples of the sediment microbial community were exposed to different13C- labeled single-carbon compounds that are used as a carbon source by various methylotrophs. The sequence of the labeled DNA from each sample was then determined by whole genome shotgun sequencing. Here, we study the sample corresponding to13C-labeled formate. Considering reads of at least 500 bp, we observed 22 741 bins with an average read per bin of 1.2. The contig spectrum was then transformed into a bin spectrum (see Supplementary Material).
We can quickly find a good fit (Fig. 2A) at χ2 = 3.7 after removing only two bins of six reads each. This fit suggests that we cannot reject the hypothesis that the underlying bin abundance is gamma distributed. Furthermore, the lognormal distribution is rejected at χ2 = 10.82.
Fig. 2.

(a) Blue curve: estimated log bin spectrum for the Lake Washington formate dataset. Red stars: the log number of observed bins. Note that the observed value at zero reads per contig is zero. The χ2-score for this fit is 3.7; we cannot reject the assumption that the bin abundance is gamma-like. (b) Blue curve: observed and estimated log bin abundance distribution for the termite hindgut dataset. The χ2-value is 1.0.
The Brass estimates are r = 0.43 (variance 0.054) and p = 0.88 (variance 3.4 × 10−4) The best gamma estimates for this dataset are therefore α = 0.43 and β = 7.2. This suggests that there may be roughly 395 460 additional bins to discover, corresponding to 277 Mbp given an average read length of 700 bp. For comparison, the Chao1 estimator of the number of bins is 142 359, which of course is the lower bound of bins.
3.4.3 Lake sediment methylotrophic community
Another Lake Washington sample focused on the methylotrophic community by radiolabeling methylamine. This set was more difficult to fit, mostly due to a relatively high number of bins with more than five reads. In particular, we found more bins with six reads than bins with five reads, which is difficult to model using a mixed Poisson model. The best fit we could find was at χ2 = 59, which is much higher than the critical value. Thus we must reject the hypothesis that the underlying bin abundance follows a gamma distribution. We must furthermore reject the lognormal distribution, since the χ2-value was even higher at 196. This dataset was included as a demonstration that the methods described in this work will not always work; natural systems are sometimes too complex to be adequately approximated. Since we could not fit the dataset, we will not attempt to predict the amount of remaining DNA or the parameters of the underlying bin abundance.
3.4.4 Termite hindgut
The termite hindgut project (Warnecke et al., 2007) studied the role of bacterial symbionts in cellulose and xylan degradation in the termite hindgut. Samples from 165 individuals of a Nasutitermes species were pooled and sequenced. This dataset was of low coverage; the average read per observed bin was 1.26.
The best approximation is found after removing two bins of 10 and 11 reads each, resulting in a χ2-value of s = 1.0 (Fig. 2B). The Brass estimates are r = 1.5 (variance 0.048) and p = 0.85 (variance 1.3 × 10−4). The estimated gamma parameters are therefore α = 1.5 and β = 5.54. The χ2-value of the Poisson-lognormal is 7.1. Therefore, the gamma distribution is the best approximation, but we cannot reject the lognormal bin abundance either. Based on the gamma approximation, we suggest that there are 144 730 additional bins in the sample which where not covered by a read, corresponding to an additional 100 MBps assuming a read length of 700 bp. The Chao1 estimator suggests a lower bound of 126 670 bins, and the Poisson-lognormal suggests 116 903 bins.
3.5 Additional sequencing
For the formate and termite datasets, we can form non-rejectable Poisson-gamma approximations. Therefore, we can also attempt to estimate the effect of additional sequencing. For the termite set, which is a good fit for the PGD, the negative binomial parameters are r = 0.54 and p = 0.72. To capture 90% of this sample, we set c0 = 0.1 so that 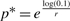 [see Equation (7)]. With the estimated value p* = 0.014, we can then calculate the average number of reads per bin of the project that captures 90% of the sample:
[see Equation (7)]. With the estimated value p* = 0.014, we can then calculate the average number of reads per bin of the project that captures 90% of the sample: 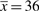 [Equation (8)]. This is quite a substantial increase in sequencing compared to the observed
[Equation (8)]. This is quite a substantial increase in sequencing compared to the observed 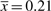 . This is not surprising considering that much of the DNA is present at very small frequencies, and a lot of effort would be spent trying to catch most of the rare bins. In terms of reads, the termite dataset has a total of ∼52 000 reads. The required number of reads to capture 90% would be roughly 180 times higher. If our goal is more moderate, i.e. to capture 50% of the bins in the set, we estimate
. This is not surprising considering that much of the DNA is present at very small frequencies, and a lot of effort would be spent trying to catch most of the rare bins. In terms of reads, the termite dataset has a total of ∼52 000 reads. The required number of reads to capture 90% would be roughly 180 times higher. If our goal is more moderate, i.e. to capture 50% of the bins in the set, we estimate 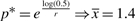 . Thus, the additional effort required to increase the coverage to 50% of bins is quite small compared to the effort needed to cover 90% of bins. For the termite set, we would need roughly seven times more reads.
. Thus, the additional effort required to increase the coverage to 50% of bins is quite small compared to the effort needed to cover 90% of bins. For the termite set, we would need roughly seven times more reads.
For the Lake Washington formate set, we estimated p = 0.88 and r = 0.43. The equivalent average reads per bin required to cover 90% of bins is considerable: 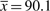 and for 50%
and for 50% 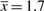 , which should be compared to the estimated
, which should be compared to the estimated 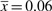 . The formate dataset has 25 000 reads. To cover 90% of the set, we need ∼1300 times more reads, and ∼30 times more reads to cover 50%.
. The formate dataset has 25 000 reads. To cover 90% of the set, we need ∼1300 times more reads, and ∼30 times more reads to cover 50%.
4 DISCUSSION
4.1 Applicability
The compound Poisson model for bin occupancy that we present here assumes that the assignment of reads to bins is mechanistic and can be described by a Poisson process, and the expected number of reads per bin depends on the abundance of the genome to which the bin belongs. We model the bin abundance distribution as a gamma distribution. However, as illustrated by the methylotroph set, there is no law of nature that requires the abundances to be so distributed, nor is a gamma distribution a consequence of large numbers of organisms. Nonetheless, the gamma distribution is flexible enough to accommodate a variety of patterns of relative abundances. Indeed, for all three real datasets, the gamma seems more appropriate than the lognormal. An additional reason to focus on the gamma distribution is the relative ease of handling as compared to many other compound Poisson distributions.
The methodology in this work is not novel; it has been used extensively in applied statistics, especially in the field of casualty actuarial science. Our contribution is to demonstrate how the work of Brass (1958), Dropkin (1959), Simon (1961), and others is relevant and applicable to metagenomics today.
The estimates of the parameters α and β are highly accurate for simulated datasets where the contig abundance follows a continuous gamma distribution, and reasonably accurate when lower numbers of species are drawn randomly from a gamma distribution. Thus, even imperfect abundances can be captured, given enough reads. With real metagenomic datasets, however, estimates are slightly less accurate, for several reasons. One major factor is the noise inherent in sequencing and assembly. For instance, cross-hybridization may produce incorrect read assignments and chimeric assemblies, thus leading to an inaccurate number of reads per bin. Secondly, highly similar or multi-copy sequences, such as transposons and phages, can result in a few bins with very high coverage. These occurrences are difficult to model, since the probability of such a high-coverage bin arising by pure chance is very low. Another important factor is the quality of assembly; as we have seen, a too greedy assembly will result in an underestimation of the total amount of DNA, just as an overly cautious assembly will inflate the estimate. Finally, the available data may be difficult to assess since some sequencing projects employ techniques that interfere with our subsequent analysis. For example, while assembling reads onto previously sequenced genomes of other organisms has its advantages, it does not help estimations of bin abundances. Likewise, some metagenomics projects filter the organisms in the environmental sample prior to sequencing based on for instance cell size (Venter et al., 2004) or metabolism (Kalyuzhnaya et al., 2008). Estimates based on such data would therefore reflect only this subset of organisms.
In some cases, a metagenome consists of many low abundance organisms and assembly is minimal. Frequently, assembly is not accurate (Mavromatis et al., 2007), which could result in an inaccurate estimation of bin abundances. For most analyses it is preferable not to attempt to assemble the sequences in such occasions and follow a more ‘gene centric’ type of analysis, for instance the Hypersaline mat and Soil projects (Kunin et al., 2008; Tringe et al., 2005). However, since we have no assembly and therefore no bin spectrum, so we cannot use this data for the prediction of the bin abundance. Indeed, the goals of these projects were to catalog protein functions in a metagenome, not to study the bin abundances.
Our methodology enables us to estimate bin abundance, but generally not species abundance. In order to directly relate the two, one must assume that all species have approximately the same genome size. This assumption may be warranted when studying species where all genome sizes are roughly equal, such as viral communities (Angly et al., 2005), but not for microbial metagenomes. Here, genome sizes vary significantly, and a species with a larger genome would contribute more bins to the sample per genome copy.
In some cases, the species abundance can be estimated by various phylogenetic binning procedures (Dalevi et al., 2006; Heath and Pati, 2007), but these methods will only be effective for dominant species with high coverage. For the large majority of cases, rare species will have only a small contribution to the bin spectrum. Thus these species, which may represent a large proportion of the species richness, cannot be phylogenetically binned.
Another approach for estimating the species abundance, or more often to catalog the species that are present in a metagenome, is to focus on a subset of sequences which are assumed to be species-specific signatures, such as the 16S ribosomal DNA (16S rDNA) (Quince et al., 2008; Schloss and Handelsman, 2005; Tringe et al., 2005). This subset can be handled in much the same way as a full metagenome, but with the distinction that one is selectively looking at only ∼1/1000th of the DNA sample. While 16S rDNA more accurately identifies unique species, the probability of observing such a signature sequence is much lower than the probability of observing any random sequence from the same genome. Thus rare species are more prone to go undetected. If we focus on DNA abundance on the other hand, each organism will contribute its full genome to the sample, thereby reducing small-sample effects.
4.2 Pitfalls of sequencing methodology
This, or any, model of assigning reads to bins requires that the model is reasonable. In the case where each read and bins are subject to the same mechanism and the bin abundance can be approximated by a gamma distribution, it will yield meaningful results. However, since the goals of a sequencing project may be more focused on an inventory of functions in the sample, or which use approaches where reads may be subject to varying mechanisms, there may be special considerations as to why this model will not work.
With the introduction of new high throughput sequencing technologies such as 454 pyrosequencing and Illumina, sequencing of metagenomic datasets is increasingly performed with more than one method. It is common to use any combination of Sanger, 454 and Illumina for a project, since they all yield nucleotide sequences. Furthermore, different versions of each platform result in different size reads. Assembly of such hybrid datasets results in contigs comprised of reads of varying length, which must be taken into account when calculating the number of reads per bin. This renders the initial assumption of equal read length invalid, and the process cannot be easily be modeled without a detailed knowledge of the contributions from each technology.
Furthermore, different sequencing technologies have their own biases. For instance, Sanger technology is known not to be able to sequence regions with strong secondary structures, while 454 may fail to accurately sequence regions with homopolymeric repeats. Illumina has biases in the base composition of the sequences and chimeric sequences (Quail et al., 2008). It is not uncommon for all methods to find an appreciable proportion of reads to be duplicates and cause an uneven distribution of read coverage across the targeted sequencing regions. As a consequence, these unfavorable features result in difficulties in under-represented genome regions, particularly when the sequences are from genomes with base compositions at the extremes of high or low G+C content. Conversely, duplications can result in artificially more highly occupied contigs.
It is also known that the quality of sequence is lower towards the end of the read. Furthermore, extraneous sequences from the vectors are frequently included. When these reads are used for assembly of isolate genomes, we expect that the errors will be eliminated by the large coverage of the sequencing. In the metagenomic datasets, however, the luxury of high coverage is rarely enjoyed. Using reads with errors or contamination can result in false positive assemblies, which will artificially increase the occupation of bins and thereby complicate the modeling.
Finally, some sequencing projects may employ novel assembly strategies that are tailored to the project at hand. While this is expected and sometimes necessary for the specific project, the resulting bin spectrum may not be suitable for studies such as described in this work. For instance, the metagenomes of the intestinal microbiota of two human subjects were sequenced (Vaishampayan et al., submitted for publication), and the coverage of the genomes of organisms present in both metagenomes was artificially increased by combining the reads from both subjects into one assembly. That consolidated assembly was later separated into subject-specific assemblies, each one retaining only the reads obtained from that subject and replacing any reads from the other with N's (undefined nucleotides). This artificial enrichment of reads per bin resulted in a considerable underestimation of the total DNA in the sample.
Although the modeling of a metagenome may seem daunting, given the nature of the technology, we should not be discouraged from attempting to seek a deeper theoretical understanding of what may be the most significant development in genomics in recent years, namely the sampling of DNA from mixed, complex communities. Here, the Brass estimates may provide a quick and easy indication of the underlying population of genetic material, and could be used in conjunction with other estimators, for instance the Chao1 estimator of the lower bound of species/bin richness.
Supplementary Material
ACKNOWLEDGEMENTS
The authors wish to thank R. Izsák for the Poisson-lognormal estimate software and his constructive input, A. Lapidus for her insights into assembly, M.P. Francino for early access to the human gut data, S. Tringe for her expertise in metagenome projects and M. Youle for critical reading of this manuscript.
Funding: US Department of Energy's Office of Science, Biological and Environmental Research Program and by the University of California, Lawrence Berkeley National Laboratory under contract No. DE-AC02-05CH11231, Lawrence Livermore National Laboratory under Contract No. DE-AC52-07NA27344, and Los Alamos National Laboratory under contract No. DE-AC02-06NA25396.
Conflict of Interest: none declared.
REFERENCES
- Angly F, et al. PHACCS, an online tool for estimating the structure and diversity of uncultured viral communities using metagenomic information. BMC Bioinformatics. 2005;6:41. doi: 10.1186/1471-2105-6-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brass W. Simplified methods of fitting the truncated negative binomial distribution. Biometrika. 1958;45:9. [Google Scholar]
- Breitbart M, et al. Genomic analysis of uncultured marine viral communities. Proc. Natl. Acad. USA. 2002;99:14250–14255. doi: 10.1073/pnas.202488399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao A. Nonparametric estimation of the number of classes in a population. Scand. J. Statist. 1984;11:5. [Google Scholar]
- Chao A, Bunge J. Estimating the number of species in a stochastic abundance model. Biometrics. 2002;58:531–539. doi: 10.1111/j.0006-341x.2002.00531.x. [DOI] [PubMed] [Google Scholar]
- Chao A, et al. Estimating population size for capture-recapture data when capture probabilities vary by time and individual animal. Biometrics. 1992;48:201–216. [PubMed] [Google Scholar]
- Chevreux B, et al. Using the miraEST assembler for reliable and automated mRNA transcript assembly and SNP detection in sequenced ESTs. Genome Res. 2004;14:1147–1159. doi: 10.1101/gr.1917404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalevi D, et al. Bayesian classifiers for detecting HGT using fixed and variable order markov models of genomic signatures. Bioinformatics. 2006;22:517–522. doi: 10.1093/bioinformatics/btk029. [DOI] [PubMed] [Google Scholar]
- Dropkin LB. Some considerations on automobile rating systems utilizing individual driving records. Proc. Casualty Actuarial Soc. 1959;XLVI:11. [Google Scholar]
- el-Shaarawi AH. Some goodness-of-fit methods for the Poisson plus added zeros distribution. Appl. Environ. Microbiol. 1985;49:1304–1306. doi: 10.1128/aem.49.5.1304-1306.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heath LS, Pati A. Genomic signatures in de Bruijn chains. WABI. 2007:216–227. [Google Scholar]
- Izsák R. Maximum likelihood fitting of the Poisson lognormal distribution. Environ. Ecol. Stat. 2007;15:23. [Google Scholar]
- Kalyuzhnaya MG, et al. High-resolution metagenomics targets specific functional types in complex microbial communities. Nature Biotechnol. 2008;26:1029–1034. doi: 10.1038/nbt.1488. [DOI] [PubMed] [Google Scholar]
- Kunin V, et al. Millimeter-scale genetic gradients and community-level molecular convergence in a hypersaline microbial mat. Mol. Systems Biol. 2008;4:198. doi: 10.1038/msb.2008.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lander ES, Waterman MS. Genomic mapping by fingerprinting random clones: a mathematical analysis. Genomics. 1988;2:231–239. doi: 10.1016/0888-7543(88)90007-9. [DOI] [PubMed] [Google Scholar]
- Mavromatis K, et al. Use of simulated data sets to evaluate the fidelity of metagenomic processing methods. Nature Methods. 2007;4:495–500. doi: 10.1038/nmeth1043. [DOI] [PubMed] [Google Scholar]
- Quail MA, et al. A large genome center's improvements to the Illumina sequencing system. Nature Methods. 2008;5:1005–1010. doi: 10.1038/nmeth.1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quince C, et al. The rational exploration of microbial diversity. ISME J. 2008;2:997–1006. doi: 10.1038/ismej.2008.69. [DOI] [PubMed] [Google Scholar]
- Raes J, et al. Get the most out of your metagenome: computational analysis of environmental sequence data. Curr. Opin. Microbiol. 2007;10:490–498. doi: 10.1016/j.mib.2007.09.001. [DOI] [PubMed] [Google Scholar]
- Richter DC, et al. MetaSim: a sequencing simulator for genomics and metagenomics. PLoS ONE. 2008;3:e3373. doi: 10.1371/journal.pone.0003373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schloss PD, Handelsman J. Introducing DOTUR, a computer program for defining operational taxonomic units and estimating species richness. Appl. Environ. Microbiol. 2005;71:1501–1506. doi: 10.1128/AEM.71.3.1501-1506.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon LJ. Fitting negative binomial distributions by the method of maximum likelihood. Proc. Casualty Actuarial Soc. 1961;XLVIII:8. [Google Scholar]
- Stein JL, et al. Characterization of uncultivated prokaryotes: isolation and analysis of a 40-kilobase-pair genome fragment from a planktonic marine archaeon. J. Bacteriol. 1996;178:591–599. doi: 10.1128/jb.178.3.591-599.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tringe SG, et al. Comparative metagenomics of microbial communities. Science. 2005;308:554–557. doi: 10.1126/science.1107851. [DOI] [PubMed] [Google Scholar]
- Venter JC, et al. Environmental genome shotgun sequencing of the Sargasso Sea. Science. 2004;304:66–74. doi: 10.1126/science.1093857. [DOI] [PubMed] [Google Scholar]
- Warnecke F, et al. Metagenomic and functional analysis of hindgut microbiota of a wood-feeding higher termite. Nature. 2007;450:560–565. doi: 10.1038/nature06269. [DOI] [PubMed] [Google Scholar]
- Wendl M. Occupancy modeling of coverage distribution for whole genome shotgun DNA sequencing. Bull. Math. Biol. 2006;68:179–196. doi: 10.1007/s11538-005-9021-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.