

Abstract
Hereditary diffuse gastric cancer (HDGC) is an autosomal dominant cancer susceptibility syndrome characterized by early-onset diffuse gastric cancer (DGC) and lobular breast cancer. E-cadherin (CDH1) heterozygous germline mutations and deletions are found in 40% of families. Independent of CDH1 alterations, most HDGC tumours display mislocalized or absent E-cadherin immunoexpression, therefore undetected defects at the CDH1 locus may still be involved. We aimed at determining whether CDH1 mutation-negative probands display germline CDH1 allele-specific expression (ASE) imbalance, using a single-nucleotide primer extension-based procedure and tried to uncover the underlying molecular defect. CDH1 ASE analysis was performed using three intragenic SNPs in RNA extracted from the blood of 21 cancer-free individuals and 22 HDGC probands (5 CDH1 mutation carriers and 17 CDH1 negative). Germline promoter methylation, deletions and haplotype-related susceptibility at the CDH1 locus were analysed. Both CDH1 alleles from cancer-free individuals displayed equivalent expression levels, whereas monoallelic CDH1 expression or high allelic expression imbalance (AI) was present in 80% of CDH1 mutant and 70.6% (n = 12) of CDH1-negative HDGC probands. Germline deletions and promoter hypermethylation were found in 25% of probands displaying high CDH1 AI. No particular haplotype was found to be associated with CDH1 high AI. Germline CDH1 AI is highly frequent among CDH1 mutation-negative probands but was not seen in cancer-free individuals. This implicates the CDH1 locus in the majority of mutation-negative HDGC families.
INTRODUCTION
Hereditary diffuse gastric cancer (HDGC) (OMIM No. 137215) is an autosomal dominant cancer-associated syndrome characterized by clustering of early-onset diffuse gastric cancer (DGC) (1) and lobular breast cancer (LBC) (2). Approximately 40% of HDGC families harbour heterozygous germline inactivating alterations of E-cadherin (CDH1) segregating with the disease (3,4). We have recently reported that HDGC is not only caused by CDH1 mutations but also by large deletions affecting the CDH1 locus (4). Although many additional high- and low-penetrance genes have been studied in HDGC, we and others failed to identify other germline genetic causes for cases that remain without molecular diagnosis.
DGC occurring in CDH1 germline mutation carriers displays abnormal or absent E-cadherin protein expression, due to the inactivation of the remaining wild-type allele through somatic promoter methylation, loss of heterozygozity or a second mutation (5–7). In our experience, tumours from families with clustering of DGC display similar morphological features and abnormal E-cadherin expression pattern, independent of harbouring germline CDH1 alterations (unpublished data). Therefore, we believe that other CDH1 germline genetic and epigenetic defects may be the cause of DGC clustering in families that remain genetically unexplained.
Recently, autosomal genes have been demonstrated to be the subject of random monoallelic inactivation (8). Yet, CDH1 was not one of those genes and was shown to be biallelically expressed in normal conditions (8). Approximately 10% of 4000 human autosomes analysed display random monoallelic expression a feature shared with imprinted genes or those encoded by the X-chromosome (8–10). Allelic expression imbalance (AI) for breast susceptibility genes BRCA2 and BRCA1 and for the colon cancer susceptibility gene APC, has also been associated as cancer-associated risk factors (11,12). More recently, germline AI of TGFBR1 was shown to confer increased risk of colorectal cancer, and two major haplotypes were predominantly found among cases displaying AI. Nevertheless, none of the previous reports identified the AI-causing mechanism (13,14). The measurement of CDH1 allele-specific expression (ASE) in the germline of CDH1-negative HDGC probands could be used to identify individuals with potential heterozygous germline genetic and epigenetic CDH1 abnormalities. Tan et al. (15) used common single-nucleotide polymorphisms within the CDH1 transcripts (cSNPs) to demonstrate the AI of CDH1 and other autosomal genes in a familial pancreatic cancer patient.
We studied whether patients with familial clustering of gastric cancer (GC) mainly of the diffuse type (HDGC) that tested negative for CDH1 germline alterations display germline CDH1 AI and attempted to identify the genetic abnormality underlying this phenomenon.
RESULTS
In this study, we aimed at investigating whether families with GC aggregation, namely HDGC families, that proved negative for CDH1 germline alterations display CDH1 AI in RNA derived from peripheral blood lymphocytes (PBLs). Highly polymorphic SNPs at the CDH1 mRNA (coding SNPs: rs1801552 and rs33964119, and 3′-UTR SNP rs1801026) were selected and used as allele discriminators for CDH1 AI determination.
Cancer-free individuals display equivalent germline RNA expression of CDH1 maternal and paternal alleles
Three SNPs were genotyped from PBLs’ RNA from 50 control cancer-free individuals to select a series of heterozygous individuals for ASE analysis. Twenty-one of the control cancer-free individuals were heterozygous at SNP rs1801552 (n = 14), rs33964119 (n = 1) and/or rs1801026 (n = 10), and their cDNAs used to determine the relative expression of CDH1 maternal and paternal alleles. In all cases, T and C alleles were identically represented (Fig. 1A), and a range of normalcy values was defined with an upper boundary for normal allelic expression ratio. The mean expression ratio in the cancer-free individuals’ germline RNA was 1.32 ± 0.14, and the ratio between alleles did not change significantly when a different SNP was used in the same sample (Fig. 1B). Moreover, these RNA results were similar to those obtained when using matched genomic DNA (gDNA) (Fig. 1C). The fact that the ratio remains equivalent independent of nucleic acid and SNPs used demonstrates that this assay is suitable for ASE quantitative measurement.
Figure 1.

CDH1 ASE analysis in cancer-free individuals. (A) CDH1 allelic expression ratio in cancer-free individuals. (B) ASE in RNA samples from four heterozygous individuals. (C) Allele-specific quantification in gDNA samples from four heterozygous individuals.
HDGC CDH1 germline mutation carriers display germline CDH1 AI
We applied the ASE quantification method, established for cancer-free individuals, in PBLs’ RNA from five HDGC probands shown elsewhere to be germline CDH1 mutation carriers (Table 1). Our aim was to understand whether CDH1 AI would reflect the presence of a CDH1 germline mutation.
Table 1.
Features of probands from GC families selected for ASE analysis
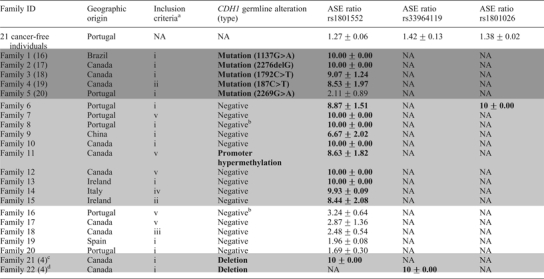 |
NA, not applicable; high A1 values and CDH1 alterations are highlighted in bold.
aSee Results section.
bMutation negative and not tested for large deletions.
cFamily 1.
dFamily 4 in reference (4).
After confirming that all five probands were constitutively heterozygous for SNP rs1801552 using polymerase chain reaction (PCR) sequencing, ASE analysis was conducted in germline RNA: 80% (4/5) of CDH1 mutation carriers showed high AI, which was not observed in gDNA using the same primer extension assay (Fig. 2A). One of the five mutation carriers lacked AI and displayed similar ASE mean ratios in RNA and DNA (data not shown). This specific patient carried a missense mutation, whereas the other four were carriers of truncating CDH1 mutations.
Figure 2.

CDH1 allele-specific quantification in CDH1-mutated and CDH1-negative probands. (A) ASE in CDH1 mutation carriers’ RNA and matched DNA. (B) Conventional boxplot (with whiskers) of allele expression ratio in cancer-free individuals (n = 21; IQR = 0.17; Q1 = 1.24; Q3 = 1.41), CDH1 mutation carriers (n = 5; IQR = 3.23; Q1 = 6.77; Q3 = 10) and CDH1 mutation-negative probands (n = 17; IQR = 7.02; Q1 = 2.96; Q3 = 9.98). P-values refer to Wilkoxon test (BC correction: ×3).
CDH1-negative probands display germline CDH1 AI
Of 27 CDH1 mutation-negative probands, 17 showed to be heterozygous for at least one of the SNPs (n = 16 for rs1801552, n = 1 for rs33964119 and n = 1 for rs1801026) and represented families with: (i) two or more GC cases with at least one DGC in first-degree relatives diagnosed before age 50 (n = 9); (ii) three or more GC in first-degree relatives with at least one DGC diagnosed at any age (n = 1); (iii) DGC before 45 years of age (n = 1); (iv) LBC before 45 years of age (n = 1); (v) clustering of GC cases of unknown histology (n = 5) (Table 1).
Fifteen out of 17 (88.2%) heterozygous CDH1-negative probands presented AI with mean ratios above the proposed normal values (Fig. 2B), whereas gDNA data showed equivalent representation of both alleles (Fig. 2C). In 70.6% (12/17) of patients with AI, the mean ratio was higher than 6, whereas the remaining demonstrated a more modest overrepresentation of one allele (Table 1). Complete reduction to monoallelic expression was observed in 41.2% (7/17) of cases (Table 1). The analysis of gDNA in these cases was repeated using a different DNA sample and the heterozygous state at the SNP position confirmed (data not shown). In four mutation-negative probands, we used a different single-base extension (SBE) primer to the same SNP to show that results were primer independent (Fig. 2C).
CDH1 germline promoter hypermethylation and germline large deletions occur in probands with germline CDH1 AI
To disclose putative epigenetic and structural mechanisms that could underlie the presence of high AI in the germline of CDH1-negative probands, we studied the CDH1 promoter methylation status as well as the presence of genomic rearrangements at the gene locus in PBLs’ DNA.
We studied germline CDH1 promoter hypermethylation in 12 probands with high CDH1 AI. A single proband from Family 11 displayed promoter hypermethylation at least in 11/19 (57.9%) CpG sites (Fig. 3A). Although not completing the HDGC criteria, this family has a history of at least two GC of unknown histology, and several cases of breast and colorectal cancer, which may also occur in the tumour spectrum of HDGC. Even so, all affected members in this family have deceased, and therefore a transgenerational study could not be performed. We had access to PBLs’ DNA from a probands’ brother and granddaughter (both non-affected by cancer so far) and observed that neither had CDH1 promoter hypermethylation.
Figure 3.

Germline CDH1 promoter hypermethylation analysis. (A) Partial electropherogram from Family 11 showing methylation at 9/19 CpG sites. (B) Schematic representation of CpG dinucleotides encompassed by the flanking PCR. Each line of circles represents an isolated allele; an open circle, a non-methylated CpG; and a black circle, a methylated CpG. (C) ARMs PCR scheme covering 13 CpG. The rs16260_A allele is methylated in at least eight CpG sites and partially methylated in three (circles with a black dot).
To understand whether CDH1 hypermethylation could be allele-specific as well as to determine the extent of methylation in CDH1 alleles of this case, 24 cloned alleles were sequenced (Fig. 3B). A significant proportion of alleles displayed variable allele-to-allele hypermethylation, supporting that germline allele-specific methylation could be occurring in this patient. To address this issue, we took advantage of patients’ heterozygozity at the common CDH1 promoter SNP rs16260 (−161C/A) to perform amplification refractory mutation system (ARMS)-based PCR (ARMS-PCR) and amplify each allele separately from bisulphite-converted DNA. With this approach, we showed that the methylated clones observed in Figure 3B are likely all derived from the −161A allele, since only this allele was hypermethylated (Fig. 3C). In parallel with the current study, a large series of CDH1 mutation-negative HDGC probands, which included 15/17 probands from the present CDH1-negative series, was screened for large genomic rearrangements at the CDH1 locus on chromosome 16 by MLPA and array CGH (4). By crossing our CDH1 AI data with the data arising from the rearrangement mapping, we verified that 2/12 (16.7%) probands with high CDH1 AI displayed large deletions at the CDH1 locus. One deletion (193.593 bp) was found in Family 21 (referred as Family 1 in our previous report) and encompassed the full sequence of CDH3, extending to position IVS2+57.595 in CDH1 (4). The other deletion found in Family 22 (referred as Family 4 in our previous report) was a 150 bp removal, encompassing the CDH1 TSS, with breakpoints located 125 bp upstream and 25 bp downstream of the TSS (4).
Our results show that 25% (3/12) of probands previously described as mutation negative display high CDH1 AI, which is likely generated by germline CDH1 methylation and large genomic rearrangements at the gene locus.
Germline CDH1 AI is not associated with a specific extended haplotype
A finding derived from the present ASE analysis was the exclusive underexpression of the rs1801552_C allele in all samples, independent of the level of AI detected as well as of the CDH1 germline alteration status, when PBLs’ RNA was compared with gDNA (Fig. 2A and C). This result was consistent when both flanking and SBE primers were changed (Fig. 2C, last panel), supporting the hypothesis of a rs1801552_C allele-associated downregulation, possibly reminiscent of an ancestral disease-associated haplotype and potentially associated with the occurrence of germline CDH1 alterations always in the C-allele. Supporting this idea, the haplotype reconstruction in Family 11 (displays high CDH1 AI and allele-specific CDH1 germline promoter methylation) showed that the hypermethylated rs16260_A CDH1 promoter allele was in phase with the underexpressed rs1801552_C allele.
To test this hypothesis, and assuming that, whatever the mechanism, downregulation of CDH1 was occurring always in the ancestral rs1801552_C-allele, we genotyped 12 SNPs, encompassing distinct haplotype blocks along CDH3 and CDH1 regions from chromosome 16 (Fig. 4A, Supplementary Material, Fig. S1) in 14 probands with high CDH1 AI (four with germline CDH1 mutation, one with germline CDH1 promoter methylation and nine without CDH1 alteration). Haplotypes were reconstructed using PHASE 2.02 software (16,17). HapMap haplotypes from a control population (n = 120) with a known phase were used to improve inference of probands’ haplotypes. The GOLD (graphical overview of linkage disequilibrium) application (18,19) shows, for our SNP data set, a common region of strong linkage disequilibrium (|D′| ≥ 0.84) between rs1801552, rs7203904 and rs2276330 (Supplementary Material, Fig. S2). However, the block structure observed in the HapMap control data (Supplementary Material, Figs S1 and S2A) for the region encompassing rs1801552, rs7203904 and rs2276330 was not maintained for the group of 14 probands with high CDH1 AI alone (Supplementary Material, Fig. S2B). Even when the nine probands with high CDH1 AI were analysed separately, similar results were obtained, suggesting that the proband series with high CDH1 AI is somewhat more diverse than the HapMap one (Supplementary Material, Fig. S2B1, 2C and 2A). Nevertheless, when we assessed the breakdown of rs1801552_T and rs1801552_C haplotypes (Fig. 4B—left and right panels, respectively) from probands and controls is made clear that the higher diversity of CDH1 high AI may be explained by rs1801552_C haplotypes alone. Moreover, the lack of a homogeneous region in the vicinity of rs1801552_C, common to all probands, refutes the hypothesis of high AI being associated with a specific extended haplotype.
Figure 4.

Haplotype analysis. (A) Adapted NCBI representation of 193 kb encompassing 12 coding and noncoding SNPs chosen along CDH3 and CDH1 loci. (B) Decay over distance of common haplotypes from rs1801552_T and rs1801552_C alleles. The most frequent haplotype is represented above the lines for each considered subgroup (HapMap, high CDH1 AI, germline mutant and methylated and CDH1 negative).
DISCUSSION
To date, ∼40% of families with well-defined clustering of DGC, described as HDGC, display CDH1 germline inactivation (4). A lower frequency of CDH1 alterations has also been described in isolated early-onset DGC patients, in families that do not fulfil the widely accepted clinical criteria and in families with clustering of early-onset LBC, without DGC involvement (3,20). Most reported germline CDH1 alterations lead to the production of truncated inactive peptides that lack normal function.
Given that a fair proportion of eligible families for germline CDH1 mutation screening lack such alterations despite displaying E-cadherin protein expression impairment in their tumours (data not shown), we propose that, in these cases, CDH1 is still compromised due to alterations that escape detection by conventional techniques.
Mutations leading to protein truncation constitute the majority of inactivating mutations in cancer susceptibility genes (3,21–24). Transcripts containing premature termination codons (PTCs), mainly as a result of frameshift and nonsense mutations, are eliminated by the nonsense-mediated mRNA decay (NMD) (25). Owing to the instability of PTC-containing transcripts, many mutations remained undetected by RNA-based techniques and only became discovered upon usage of DNA-based methods. Conversely, as the latter rely mostly on the amplification of individual exons with intronic flanking primers, large genomic deletions and changes in more distant noncoding regions were also missed, until recent technical improvements have been achieved (MLPA and arrayCGH) allowing their detection. Undiscovered alterations may therefore be revealed by techniques that permit individual assessment of paternal and maternal allelic expression. Without disregarding this interesting possibility, one has to consider that monoallelic expression with random choice between maternal and paternal alleles is no longer exclusive of imprinted and X-inactivated genes, as a group of autosomal gene families has been recently identified as a target of monoallelic downregulation (8).
In the last decade, at least three studies have been published re-enforcing the role of germline AI as a major risk factor for cancer development in sporadic and hereditary cohorts. These studies identified AI in BRCA1, BRCA2, APC and lately in TGFBR1, using well-selected cohorts of cancer patients and cancer-free control individuals (11–13). All these studies made use of different methodological approaches to analyse ASE in patients’ germline RNA; nevertheless, none described the molecular mechanism underlying the observed effect on RNA monoallelic downregulation.
In the present study, we established a reliable and straightforward assay to evaluate CDH1 ASE using SNPs within CDH1 mRNA. We demonstrated that CDH1 expression is biallelic in PBLs from heterozygous cancer-free individuals and that both CDH1 alleles express equivalent RNA levels, as mentioned previously (8). Moreover, the contribution of each allele was found to be independent from the SNP tested or SBE primers used (Fig. 1B and 2C). This result enabled us to set an upper artificial boundary for CDH1 normal expression ratio in cancer-free individuals. A valuable test of the approach reliability came from results obtained in four HDGC CDH1 germline-truncating mutation carriers, which showed that CDH1 AI in mutant probands was statistically higher than that of cancer-free individuals (P = 2.11e−03), as expected after PTC degradation by NMD (25,26). Similar results have been previously obtained for BRCA1, BRCA2 and APC (11,12).
Having established the assay, we had the proper tool to disclose whether this finely tuned biallelic expression was getting disrupted in our series of CDH1-negative probands. Our main concern was to select a series of patients that, although negative for CDH1 germline alterations, was, in all features, similar to any series previously used for CDH1 mutation screening. This selection was expected to increase the probability of finding a mechanism of disease, although it was highly dependent on RNA availability from probands’ PBLs. After the selection of 17 CDH1-negative probands, from different geographic backgrounds and fulfilling well-defined clinical criteria (see Results), we investigated the possibility of these probands presenting germline CDH1 AI. An impressive and unexpected proportion of these patients (70.6%) revealed AI ratios >6, with recurrent downregulation of the rs1801552_C allele. Coincidently, a recent study reporting ASE in a familial pancreatic cancer patient showed that the same rs1801552_C allele was underexpressed (15). Therefore, this allele was assumed as a preferential target for a germline downregulation mechanism so far unidentified.
Recent reports demonstrated that hMLH1 or hMSH2 monoallelic germline hypermethylation caused HNPCC and could be transmitted through generations (27,28). These studies established epimutations as the cause of a novel cancer susceptibility pattern of inheritance (27,28). Assuming that CDH1 could be the target of a similar epigenetic phenomenon, we have analysed CDH1 germline promoter hypermethylation in probands with high AI. The PBLs’ DNA extracted from the Family 11 proband displayed CDH1 hypermethylation with a striking pattern of clonal heterogeneity similar to that somatically found in sporadic carcinomas, cancer cell lines, and tumour foci from HDGC CDH1 germline mutation carriers, leading to CDH1 expression downregulation (7,29). We believe however that this proband harbours germline hypermethylation as the cloning experiment revealed that a large proportion of the sequences cloned were methylated. This result would not be expected if a small amount of cancer cells would be circulating in the blood, and is pointing to a germline defect rather than to somatic cancer-associated methylation reminiscent from cancer-circulating cells. The most interesting aspect in this case was that CpG methylation was occurring only at the rs16260_A allele, raising the hypothesis that allele-specific methylation is generating germline-specific downregulation of the rs1801552_C-containing allele. Further supporting this hypothesis is the fact that haplotype reconstruction by PHASE 2.02 software revealed that both alleles are indeed in phase. To assess CDH1 germline hypermethylation in a larger series, we extended the methylation analysis but failed to identify this phenomenon in 56 additional GC family probands (data not shown); therefore, we believe this is a rare mechanism in this setting, according to previously reported findings (30). Nevertheless, the overall frequency of CDH1 germline methylation (1.47%—1/68) herein identified does not differ from that reported for hMLH1 and hMSH2 in HNPCC-like probands (0.6 and 3.2%, respectively) (27,28).
We also found that 2/12 (16.7%) high CDH1 AI probands display large genomic deletions encompassing CDH1, so, in summary, we identified the potential mechanism underlying high AI in 25% (3/12) of the cases in this series, suggesting that AI most likely represents a marker for both germline genetic and epigenetic defects in CDH1.
Recently, germline heterozygous deletions of TACSTD1 3′-exons were found in 0.9% of HNPCC-suspected families, causing transcriptional read-through that resulted in hMSH2 downregulation (31,32). In the light of our CDH1 AI results, it is expected that the application of ASE analysis to hMSH2, in the aforementioned HNPCC families, would have spotted monoallelic expression of the gene. Conversely, it is also possible that a similar structural rearrangement, in the surroundings of CDH1 gene, could generate AI in some probands of our series.
Recently it was shown that TGFBR1 AI results in globally reduced expression of the gene, alters SMAD-mediated TGFβ signalling, is dominantly inherited, segregates in families and occurs in sporadic colorectal cancer (13). Two major TGFBR1 haplotypes were found predominant among TGFBR1 AI cases suggesting ancestral mutations; nevertheless, no causative germline change was identified (13).
As we failed to find a mechanism underlying the nine remaining cases displaying high CDH1 AI, and given that the rs1801552_C-containing allele was recurrently downregulated, we reasoned that this allele could be in linkage disequilibrium with a putative aetiological variant, either in CDH1 or in its proximity. The rs1125557_A allele (163+37235G>A) localized in CDH1 intron 2 was recently claimed as a susceptibility variant for sporadic DGC, mainly in homozygous AA individuals (33). This SNP was studied in our series and contrarily to what is reported, the percentage of AA individuals was similar between probands (28%) and controls (33%). Moreover, our quest for high CDH1 AI-related haplotypes generated no significant difference between haplotypic distribution in probands and in the HapMap reference/control group. As no haplotype is overrepresented within CDH1-negative probands, high CDH1 AI is most probably being caused by haplotype independent mechanisms.
Another interesting hypothesis arises from a study describing that two cis-regulatory FGFR2 SNPs are able to alter binding affinity for transcription factors Oct-1/Runx2 and C/EBPb (34), and that they synergize to augment FGFR2 expression, thereby increasing the propensity for tumour formation (34). Whether a similar phenomenon is occurring in our patients, and another SNP exists that synergizes with CDH1 rs1801552_C allele, still needs to be addressed.
A last hypothesis to cause CDH1 AI could be the existence of a trans-acting factor binding differentially to rs1801552_C and rs1801552_T alleles, as it was recently described for DEAF-1 (35). This protein was reported to allele-specific trans-regulate GDF5, through the differential binding to a GDF5 sequence encompassing a common SNP (rs143383). In the context of CDH1-negative probands, individuals carrying the common rs1801552_C allele would only be at risk of developing cancer in a background with abnormal expression of a trans-acting factor, with differential binding affinity for the rs1801552_C allele.
In summary, our results show that: (i) high CDH1 AI may be used as a marker for increased susceptibility to familial aggregation of GC, namely HDGC, as it occurs specifically in CDH1-truncating mutation carriers (100%) and CDH1-negative probands (70.6%), but not in cancer-free individuals; (ii) germline allele-specific CDH1 promoter methylation and deletion encompassing the CDH1 locus underlie high CDH1 AI in 25% of CDH1-negative probands; (iii) high CDH1 AI determination provides a simple, cost-effective and efficient tool to perceive indirectly changes of CDH1 expression that escape detection in gDNA-based screenings; and (iv) the remainder 75% of cases with high AI are not related to an extended susceptibility haplotype.
As AI detection does not vary upon the SNP used, any coding or even pre-mRNA SNP can be used to test ASE, and virtually every single individual can be analysed with this strategy. ASE quantification of gene transcripts opens the way to the detection of both trans- and cis-acting regulatory variations (35,36) and overcomes the difficulties resulting from non-genetic factors affecting mRNA levels and from mRNA instability, which can differ between samples.
In conclusion, we believe that most CDH1-negative probands, especially those presenting tumours with E-cadherin expression impairment, similar to that observed in CDH1 germline alteration carriers, do have a germline CDH1 expression defect caused by either direct or indirect mechanisms targeting the CDH1 genomic sequence.
MATERIALS AND METHODS
Patients and samples
The collection of clinical data from patients and families and PBL samples from patients and controls was approved by the appropriate Ethics Committee from each of the centres participating in this work: University of British Columbia, Vancouver, Canada; Institute of Molecular Pathology and Immunology of the University of Porto (IPATIMUP), Porto, Portugal; and Hospital Geral de Santo António, Porto, Portugal. Family history of cancer was obtained with informed consent.
Thirty-two probands from families displaying aggregation of GC mainly of diffuse histology (HDGC) and early onset isolated cases (aged <45 years) with either DGC or LBC were selected for the study. Twenty-seven of the 32 probands tested negative for CDH1 germline point mutations, whereas 5 have been described elsewhere to carry germline point mutations in the CDH1 gene (26,37–40). A series of 50 cancer-free individuals was used as controls.
For germline CDH1 hypermethylation analysis, we gathered 68 probands, including the 27 CDH1-negative probands from the aforementioned series and 41 other probands collected at IPATIMUP.
Nucleic acid preparation
gDNA was extracted from PBLs from patients and controls using a standard protocol. Total RNA was isolated from PBLs using the TriPure Isolation Agent (Roche Diagnostics, Basel, Switzerland) or the whole blood and bone marrow protocols—300 μl blood sample protocol from PURESCRIPT RNA Isolation Kit (GENTRA, MN, USA).
Approximately 300 ng of total RNA was used to synthesize first-strand cDNA with Superscript II Reverse Transcriptase and random hexamer primers (Invitrogen, CA, USA).
CDH1 SNP genotyping of cancer-free individuals and patients
Three CDH1 SNPs were genotyped in all patients and controls: two CDH1 coding SNPs rs1801552-2076C/T [minor allele frequency (MAF) T: 0.408 in HapMap phase II CEU panel from individuals with European ancestry] and rs33964119-2253C/T (MAF T: 0.045 in HapMap phase II European EGP CEPH panel); and a noncoding SNP rs1801026-g.*+54C>T located at the 3′-UTR before the polyadenylation signal (MAF T: 0.205 in HapMap phase II European EGP CEPH panel) by PCR using standard conditions. MAFs were obtained from the dbSNP home page at http://www.ncbi.nlm.nih.gov/projects/SNP/. PCR products were sequenced using BigDye Terminator v3.1 Cycle Sequencing Kits (Applied Biosystems, CA, USA) and run in an ABI Prism 3130 DNA automated sequencer (Applied Biosystems). Primer sequences and amplicon sizes are listed in Supplementary Material, Table S1.
ASE analysis
Primer extension analysis relies on the incorporation of a single dideoxynucleotide triphosphate (ddNTP) (complementary to the base of interest) that is selected to allow the differential extension of a primer annealed next to the polymorphic site (SBE primer). ASE products can be distinguished, and quantitatively measured, based on the presence of a specific fluorophore coupled to the incorporated ddNTP. PCR products encompassing the polymorphic site were used as templates for primer extension with fluorescently labelled dNTPs which are incorporated, causing chain termination in alleles having a C, G, A or T in this position. Purified PCR products were submitted to SNaPshot reaction using SNaPshot Multiplex Kit (Applied Biosystems) and an SBE-specific primer following manufacturer's instructions. Final products were analysed in a 310 Genetic Analyser (Applied Biosystems) using the size standard GeneScan 120 LIZ (Applied Biosystems). The peak area corresponding to each allele was quantified with the GeneScan results analyser software. The ratio of ddNTP incorporation by each allele was calculated by taking the ratio of the areas corresponding to T and C alleles. For samples presenting complete downregulation of one allele in germline RNA, a ratio value of 10 was attributed to characterize AI.
As a validation step, a PCR amplicon encompassing the same SNPs was generated from gDNA from patients and controls and submitted to the same analysis to demonstrate equivalent representation of each allele through equivalent ddNTP incorporation. Moreover, to exclude ASE bias mediated by a specific primer sequence, ASE results obtained from patients and control samples were confirmed using either different primer sequences for the primer extension assay or, whenever possible, another SNP. Primer sequences and amplicon sizes are listed in Supplementary Material, Table S2.
Promoter methylation analysis
DNA from probands’ PBLs belonging to 68 CDH1-negative families mainly displaying GC aggregation, originating from Canada (10), Ireland (2), Spain (1), Brazil (1), China (1), Italy (1) and Portugal (52), was screened for hypermethylation at the CpG island 3 of the CDH1 promoter. Approximately 50–100 ng of sodium bisulphite-modified DNA [EpiTect Bisulfite Kit (Qiagen, Hilden, Germany)—sodium bisulphite conversion of unmethylated cytosines in DNA from solutions with low concentrations of DNA] was used for PCR and sequencing to screen to screen for methylation at 19 CpG sites (position –56 bp to position +115 bp from the CDH1 TSS), with flanking primers not containing CpG sites. Samples displaying methylated CpG sites were resubmitted to an independent PCR that was cloned into pCR® 2.1-TOPO® vector (Invitrogen). Colony-PCR with Universal M13 primers was performed to determine the level and extent of CDH1 promoter methylation. Primer sequences and amplicon sizes are listed in the Supplementary Material, Table S3.
Amplification refractory mutation system-PCR
To analyse allele-specific CDH1 hypermethylation, heterozygozity at the common CDH1 promoter SNP rs16260 (−161C/A) was used to perform ARMS-PCR. Primers were designed to discriminate between templates which differed in a specific single nucleotide (41), thereby amplifying each allele separately from bisulphite-converted DNA. Briefly, primers flanking rs16260 SNP were used in a first-round PCR with 50 ng of bisulphite-converted DNA. Products were re-amplified with the same reverse primer and specific forward primers to the polymorphic A or C allele. Sequencing of each PCR product with the same reverse primer allowed analysis of 13 methylated CpG sites in independent alleles [the last three CpG sites had been previously analysed by cloning and sequencing (Fig. 3B and C)]. Primer sequences and amplicon sizes are listed in Supplementary Material, Table S4.
Haplotype analysis
Twelve coding and noncoding SNPs with heterozygous frequencies ranging from 0.217 to 0.648 (HapMap phase II CEU panel from individuals with European ancestry, http://www.ncbi.nlm.nih.gov/projects/SNP/) scattered along CDH1 and CDH3 loci and covering a region of approximately 193 kb (67.227.572–67.420.442) were genotyped in a multiplex PCR, according to manufacturer's instructions (QIAGEN Multiplex PCR Kit, Quiagen). SNP-specific primers for primer extension assay were designed to be used in a SNaPshot multiplex reaction by adding different size tags to each primer (SBE primers). Purified PCR products and SBE primers were used for primer extension assay according to the SNaPshot Multiplex Kit (Applied Biosystems). Selected SNPs, primers (PCR primers and SBEs) and respective amplicon sizes are listed in Supplementary Material, Table S5. The phased haplotypes from the CEU panel haplotypes from HapMap phase II database for European populations were used with our multilocus genotype data to statistically infer the probands’ haplotypes, using the software PHASE 2.02 (16,17). Linkage disequilibrium statistic D′ was calculated from the inferred haplotypes using the DNAsp program, version 5.0 (18).
Statistical analysis
R statistical program was used for the construction of a standard boxplot, plotted with whiskers and outliers. Owing to the non-normal distribution of the data, a non-parametric test, in particular the Wilcoxon rank sum test was used to calculate significance. The P-values obtained were further corrected using the Bonferroni correction due to the multiple testing performed.
SUPPLEMENTARY MATERIAL
FUNDING
This work was supported by The Portuguese Foundation for Science and Technology (FCT) (grant number PTDC/SAU-GMG/72168/2006; PhD grant numbers: SFRH/BD/41223/2007-HP, SFRH/BD/46462/2008-RC, SFRH/BD/44074/2008-JC, SFRH/BD/32984/2006-PO; and salary support from the programme Ciência 2007 to C.O.); and The Canadian Cancer Society/National Cancer Institute of Canada operating grant (grant number 018381).
Supplementary Material
ACKNOWLEDGEMENTS
We gratefully acknowledge patients, families and their caregivers for their willing participation in this project and who provided consent regarding the use of the information obtained from the study. We thank Luis Maia, Marta Novais and Teresa Coelho for providing biological material from cancer-free individuals.
Conflict of Interest statement. None declared.
REFERENCES
- 1.Guilford P., Hopkins J., Harraway J., McLeod M., McLeod N., Harawira P., Taite H., Scoular R., Miller A., Reeve A.E. E-cadherin germline mutations in familial gastric cancer. Nature. 1998;392:402–405. doi: 10.1038/32918. [DOI] [PubMed] [Google Scholar]
- 2.Pharoah P.D., Guilford P., Caldas C. International Gastric Cancer Linkage Consortium. Incidence of gastric cancer and breast cancer in CDH1 (E-cadherin) mutation carriers from hereditary diffuse gastric cancer families. Gastroenterology. 2001;121:1348–1353. doi: 10.1053/gast.2001.29611. [DOI] [PubMed] [Google Scholar]
- 3.Oliveira C., Seruca R., Carneiro F. Genetics, pathology, and clinics of familial gastric cancer. Int. J. Surg. Pathol. 2006;14:21–33. doi: 10.1177/106689690601400105. [DOI] [PubMed] [Google Scholar]
- 4.Oliveira C., Senz J., Kaurah P., Pinheiro H., Sanges R., Haegert A., Corso G., Schouten J., Fitzgerald R., Vogelsang H., et al. Germline CDH1 deletions in hereditary diffuse gastric cancer families. Hum. Mol. Genet. 2009;18:1545–1555. doi: 10.1093/hmg/ddp046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Grady W.M., Willis J., Guilford P.J., Dunbier A.K., Toro T.T., Lynch H., Wiesner G., Ferguson K., Eng C., Park J.G., et al. Methylation of the CDH1 promoter as the second genetic hit in hereditary diffuse gastric cancer. Nat. Genet. 2000;26:16–17. doi: 10.1038/79120. [DOI] [PubMed] [Google Scholar]
- 6.Barber M., Murrell A., Ito Y., Maia A.T., Hyland S., Oliveira C., Save V., Carneiro F., Paterson A.L., Grehan N., et al. Mechanisms and sequelae of E-cadherin silencing in hereditary diffuse gastric cancer. J. Pathol. 2008;216:295–306. doi: 10.1002/path.2426. [DOI] [PubMed] [Google Scholar]
- 7.Oliveira C., Sousa S., Pinheiro H., Karam R., Bordeira-Carriço R., Senz J., Kaurah P., Carvalho J., Pereira R., Gusmão L., et al. Quantification of epigenetic and genetic 2nd hits in CDH1 during hereditary diffuse gastric cancer syndrome progression. Gastroenterology. 2009;136:2137–2148. doi: 10.1053/j.gastro.2009.02.065. [DOI] [PubMed] [Google Scholar]
- 8.Gimelbrant A., Hutchinson J.N., Thompson B.R., Chess A. Widespread monoallelic expression on human autosomes. Science. 2007;318:1136–1140. doi: 10.1126/science.1148910. [DOI] [PubMed] [Google Scholar]
- 9.Reik W., Walter J. Genomic imprinting: parental influence on the genome. Nat. Rev. Genet. 2001;2:21–32. doi: 10.1038/35047554. [DOI] [PubMed] [Google Scholar]
- 10.Lyon M.F. X chromosomes and dosage compensation. Nature. 1986;320:313. doi: 10.1038/320313b0. [DOI] [PubMed] [Google Scholar]
- 11.Chen X., Weaver J., Bove B.A., Vanderveer L.A., Weil S.C., Miron A., Daly M.B., Godwin A.K. Allelic imbalance in BRCA1 and BRCA2 gene expression is associated with an increased breast cancer risk. Hum. Mol. Genet. 2008;17:1336–1348. doi: 10.1093/hmg/ddn022. [DOI] [PubMed] [Google Scholar]
- 12.Yan H., Dobbie Z., Gruber S.B., Markowitz S., Romans K., Giardiello F.M., Kinzler K.W., Vogelstein B. Small changes in expression affect predisposition to tumourigenesis. Nat. Genet. 2002;30:25–26. doi: 10.1038/ng799. [DOI] [PubMed] [Google Scholar]
- 13.Valle L., Serena-Acedo T., Liyanarachchi S., Hampel H., Comeras I., Li Z., Zeng Q., Zhang H.T., Pennison M.J., Sadim M. Germline allele-specific expression of TGFBR1 confers an increased risk of colorectal cancer. Science. 2008;321:1361–1365. doi: 10.1126/science.1159397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Castellví-Bel S., Castells A. Allele-specific expression as a new genetic susceptibility mechanism for colorectal cancer. Gastroenterology. 2009;136:2397–2399. doi: 10.1053/j.gastro.2009.04.035. [DOI] [PubMed] [Google Scholar]
- 15.Tan A.C., Fan J.B., Karikari C., Bibikova M., Garcia E.W., Zhou L., Barker D., Serre D., Feldmann G., Hruban R.H., et al. Allele-specific expression in the germline of patients with familial pancreatic cancer: an unbiased approach to cancer gene discovery. Cancer Biol. Ther. 2008;7:135–144. doi: 10.4161/cbt.7.1.5199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stephens M., Smith N.J., Donnelly P. A new statistical method for haplotype reconstruction from population data. Am. J. Hum. Genet. 2001;68:978–989. doi: 10.1086/319501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stephens M., Donnelly P. A comparison of bayesian methods for haplotype reconstruction from population genotype data. Am. J. Hum. Genet. 2003;73:1162–1169. doi: 10.1086/379378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Librado P., Rozas J. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics. 2009;25:1451–1452. doi: 10.1093/bioinformatics/btp187. [DOI] [PubMed] [Google Scholar]
- 19.Abecasis G.R., Cookson W.O. GOLD—graphical overview of linkage disequilibrium. Bioinformatics. 2000;16:182–183. doi: 10.1093/bioinformatics/16.2.182. [DOI] [PubMed] [Google Scholar]
- 20.Masciari S., Larsson N., Senz J., Boyd N., Kaurah P., Kandel M.J., Harris L.N., Pinheiro H.C., Troussard A., Miron P., et al. Germline E-cadherin mutations in familial lobular breast cancer. J. Med. Genet. 2007;44:726–731. doi: 10.1136/jmg.2007.051268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gayther S.A., Warren W., Mazoyer S., Russell P.A., Harrington P.A., Chiano M., Seal S., Hamoudi R., van Rensburg E.J., Dunning A.M., et al. Germline mutations of the BRCA1 gene in breast and ovarian cancer families provide evidence for a genotype–phenotype correlation. Nat. Genet. 1995;11:428–433. doi: 10.1038/ng1295-428. [DOI] [PubMed] [Google Scholar]
- 22.Gayther S.A., Mangion J., Russell P., Seal S., Barfoot R., Ponder B.A., Stratton M.R., Easton D. Variation of risks of breast and ovarian cancer associated with different germline mutations of the BRCA2 gene. Nat. Genet. 1997;15:103–105. doi: 10.1038/ng0197-103. [DOI] [PubMed] [Google Scholar]
- 23.Powell S.M., Petersen G.M., Krush A.J., Booker S., Jen J., Giardiello F.M., Hamilton S.R., Vogelstein B., Kinzler K.W. Molecular diagnosis of familial adenomatous polyposis. N. Engl. J. Med. 1993;329:1982–1987. doi: 10.1056/NEJM199312303292702. [DOI] [PubMed] [Google Scholar]
- 24.Peltomäki P., Vasen H.F. Mutations predisposing to hereditary nonpolyposis colorectal cancer: database and results of a collaborative study. The International Collaborative Group on Hereditary Nonpolyposis Colorectal Cancer. Gastroenterology. 1997;113:1146–1158. doi: 10.1053/gast.1997.v113.pm9322509. [DOI] [PubMed] [Google Scholar]
- 25.Frischmeyer P.A., Dietz H.C. Nonsense-mediated mRNA decay in health and disease. Hum. Mol. Genet. 1999;8:1893–1900. doi: 10.1093/hmg/8.10.1893. [DOI] [PubMed] [Google Scholar]
- 26.Karam R., Carvalho J., Bruno I., Graziadio C., Senz J., Huntsman D., Carneiro F., Seruca R., Wilkinson M.F., Oliveira C. The NMD mRNA surveillance pathway downregulates aberrant E-cadherin transcripts in gastric cancer cells and in CDH1 mutation carriers. Oncogene. 2008;27:4255–4260. doi: 10.1038/onc.2008.62. [DOI] [PubMed] [Google Scholar]
- 27.Hitchins M., Williams R., Cheong K., Halani N., Lin V.A., Packham D., Ku S., Buckle A., Hawkins N., Burn J., et al. MLH1 germline epimutations as a factor in hereditary nonpolyposis colorectal cancer. Gastroenterology. 2005;129:1392–1399. doi: 10.1053/j.gastro.2005.09.003. [DOI] [PubMed] [Google Scholar]
- 28.Chan T.L., Yuen S.T., Kong C.K., Chan Y.W., Chan A.S., Ng W.F., Tsui W.Y., Lo M.W., Tam W.Y., Li V.S., Leung S.Y. Heritable germline epimutation of MSH2 in a family with hereditary nonpolyposis colorectal cancer. Nat. Genet. 2006;38:1178–1183. doi: 10.1038/ng1866. [DOI] [PubMed] [Google Scholar]
- 29.Graff J.R., Gabrielson E., Fujii H., Baylin S.B., Herman J.G. Methylation patterns of the E-cadherin 5′ CpG island are unstable and reflect the dynamic, heterogeneous loss of E-cadherin expression during metastatic progression. J. Biol. Chem. 2000;275:2727–2732. doi: 10.1074/jbc.275.4.2727. [DOI] [PubMed] [Google Scholar]
- 30.Yamada H., Shinmura K., Goto M., Iwaizumi M., Konno H., Kataoka H., Yamada M., Ozawa T., Tsuneyoshi T., Tanioka F. Absence of germline mono-allelic promoter hypermethylation of the CDH1 gene in gastric cancer patients. Mol. Cancer. 2009;8:63. doi: 10.1186/1476-4598-8-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ligtenberg M.J., Kuiper R.P., Chan T.L., Goossens M., Hebeda K.M., Voorendt M., Lee T.Y., Bodmer D., Hoenselaar E., Hendriks-Cornelissen S.J., et al. Heritable somatic methylation and inactivation of MSH2 in families with Lynch syndrome due to deletion of the 3′ exons of TACSTD1. Nat. Genet. 2009;41:112–117. doi: 10.1038/ng.283. [DOI] [PubMed] [Google Scholar]
- 32.Niessen R.C., Hofstra R.M., Westers H., Ligtenberg M.J., Kooi K., Jager P.O., de Groote M.L., Dijkhuizen T., Olderode-Berends M.J., Hollema H., et al. Germline hypermethylation of MLH1 and EPCAM deletions are a frequent cause of Lynch syndrome. Genes Chrom. Cancer. 2009;48:737–744. doi: 10.1002/gcc.20678. [DOI] [PubMed] [Google Scholar]
- 33.Nasri S., More H., Graziano F., Ruzzo A., Wilson E., Dunbier A., McKinney C., Merriman T., Guilford P., Magnani M., Humar B. A novel diffuse gastric cancer susceptibility variant in E-cadherin (CDH1) intron 2: a case–control study in an Italian population. BMC Cancer. 2008;8:138. doi: 10.1186/1471-2407-8-138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Meyer K.B., Maia A.T., O'Reilly M., Teschendorff A.E., Chin S.F., Caldas C., Ponder B.A. Allele-specific up-regulation of FGFR2 increases susceptibility to breast cancer. PLoS Biol. 2008;6:e108. doi: 10.1371/journal.pbio.0060108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Egli R.J., Southam L., Wilkins J.M., Lorenzen I., Pombo-Suarez M., Gonzalez A., Carr A., Chapman K., Loughlin J. Functional analysis of the osteoarthritis susceptibility-associated GDF5 regulatory polymorphism. Arthritis Rheum. 2009;60:2055–2064. doi: 10.1002/art.24616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Milani L., Gupta M., Andersen M., Dhar S., Fryknäs M., Isaksson A., Larsson R., Syvänen A.C. Allelic imbalance in gene expression as a guide to cis-acting regulatory single nucleotide polymorphisms in cancer cells. Nucleic Acids Res. 2007;35:e34. doi: 10.1093/nar/gkl1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Suriano G., Yew S., Ferreira P., Senz J., Kaurah P., Ford J.M., Longacre T.A., Norton J.A., Chun N., Young S., et al. Characterization of a recurrent germ line mutation of the E-cadherin gene: implications for genetic testing and clinical management. Clin. Cancer Res. 2005;11:5401–5409. doi: 10.1158/1078-0432.CCR-05-0247. [DOI] [PubMed] [Google Scholar]
- 38.Humar B., Graziano F., Cascinu S., Catalano V., Ruzzo A.M., Magnani M., Toro T., Burchill T., Futschik M.E., Merriman T., Guilford P. Association of CDH1 haplotypes with susceptibility to sporadic diffuse gastric cancer. Oncogene. 2002;21:8192–8195. doi: 10.1038/sj.onc.1205921. [DOI] [PubMed] [Google Scholar]
- 39.Gayther S.A., Gorringe K.L., Ramus S.J., Huntsman D., Roviello F., Grehan N., Machado J.C., Pinto E., Seruca R., Halling K., et al. Identification of germ-line E-cadherin mutations in gastric cancer families of European origin. Cancer Res. 1998;58:4086–4089. [PubMed] [Google Scholar]
- 40.Simões-Correia J., Figueiredo J., Oliveira C., van Hengel J., Seruca R., van Roy F., Suriano G. Endoplasmic reticulum quality control: a new mechanism of E-cadherin regulation and its implication in cancer. Hum. Mol. Genet. 2008;17:3566–3576. doi: 10.1093/hmg/ddn249. [DOI] [PubMed] [Google Scholar]
- 41.Newton C.R., Heptinstall L.E., Summers C., Super M., Schwarz M., Anwar R., Graham A., Smith J.C., Markham A.F. Amplification refractory mutation system for prenatal diagnosis and carrier assessment in cystic fibrosis. Lancet. 1989;2:1481–1483. doi: 10.1016/s0140-6736(89)92931-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.