

Abstract
Genome rearrangements are mediators of evolution and disease. Such rearrangements are frequently bounded by transfer RNAs (tRNAs), transposable elements, and other repeated elements, suggesting a functional role for these elements in creating or repairing breakpoints. Though not well explored, there is evidence that origins of replication also colocalize with breakpoints. To investigate a potential correlation between breakpoints and origins, we analyzed evolutionary breakpoints defined between Saccharomyces cerevisiae and Kluyveromyces waltii and S. cerevisiae and a hypothetical ancestor of both yeasts, as well as breakpoints reported in the experimental literature. We find that origins correlate strongly with both evolutionary breakpoints and those described in the literature. Specifically, we find that origins firing earlier in S phase are more strongly correlated with breakpoints than are later-firing origins. Despite origins being located in genomic regions also bearing tRNAs and Ty elements, the correlation we observe between origins and breakpoints appears to be independent of these genomic features. This study lays the groundwork for understanding the mechanisms by which origins of replication may impact genome architecture and disease.
Keywords: genomic rearrangements, tRNAs, transposable elements, S. cerevisiae, K. waltii, comparative genomics
Introduction
Genomes are fluid entities that undergo structural rearrangements producing phenotypic changes. Somatic chromosome rearrangements have long been associated with the development of cancers (Hanahan and Weinberg 2000; Pollack et al. 2002; Lahortiga et al. 2007; Smith et al. 2007; Weaver et al. 2007; Weir et al. 2007; Darai-Ramqvist et al. 2008), and more recently germ line rearrangements have been implicated in human variation (Sharp et al. 2006; Kidd et al. 2008) and disease (Inoue and Lupski 2002; Stankiewicz and Lupski 2002). On an evolutionary timescale, chromosome rearrangements play a defining role in the divergence of species (Pevzner and Tesler 2003; Kellis et al. 2004; Clark et al. 2007; Kehrer-Sawatzki and Cooper 2007; Darling et al. 2008). Given the role of genome rearrangements in evolution and disease, it is imperative to understand how and where genome breakage and rearrangements occur.
Genome rearrangements were originally thought to occur at nonspecific and independent sites (Nadeau and Taylor 1984). It was discovered, however, that breakpoints must be reused in order to transform the mouse gene order into the human gene order (Pevzner and Tesler 2003; Peng et al. 2006). In yeast, it was found that breakpoints in a checkpoint-deficient mutant occur at genetically determined locations (Cha and Kleckner 2002). With further data from cancer and infertility studies (Cohen et al. 1996; Sankoff et al. 2002), the random-breakage model lost favor and was replaced by the fragile-site model, wherein specific regions in the genome are thought to act as hot spots for genome rearrangements. The fragile-site model readily leads to the conjecture that specific genomic sequences, functional elements, or structures increase the propensity of a genomic region either to produce or misrepair a break, ultimately creating a rearrangement.
Numerous studies in yeast systems have mapped rearrangement breakpoints close to transfer RNA (tRNA) genes, transposable elements, and their long terminal repeats (LTRs). After exposing diploid Saccharomyces cerevisiae cells to ionizing radiation under conditions favoring homologous recombination (HR), Argueso et al. (2008) found that almost all chromosome aberrations were bordered by Ty elements. Dunham et al. (2002) analyzed S. cerevisiae strains evolved under glucose-limiting conditions and mapped rearrangements in six evolved strains to at least one Ty, solo LTR, or tRNA. In mutants with low levels of either alpha (Lemoine et al. 2005) or delta (Lemoine et al. 2008) DNA polymerase, deletion and translocation events were localized to Tys and LTRs. Kellis et al. (2003) identified 20 inversions among S. paradoxus, S. mikatae, and S. bayanus relative to S. cerevisiae, all of which were flanked by tRNAs, and seven of which are translocation events between transposable elements.
What biological mechanism in breakpoint formation is implied by the proximity of observed breakpoints to tRNAs, Tys, and LTRs? Breakpoint formation is the product of two distinct cellular events: 1) a double-stranded DNA (dsDNA) break and 2) nonconservative repair of that break. Consequently, the site of the initial break may not be the same location where the break is ultimately repaired. Hence, untangling in which step in breakpoint formation a proximal sequence element participated can be difficult. Separation of the processes of breakage and repair has been emphasized by work in which a genomic break is induced and the ensuing repair is observed. VanHulle et al. (2007) demonstrated that following an induced double-strand break in sister chromatids, repair can occur via recombination of nonallelic Ty elements located 30 kb from the break site.
Thus, the proximity of tRNAs, transposable elements, and other repeats to a repaired break site does not exclude the possibility that additional, uncharacterized nearby elements may have played a role in either the initial breakage or its ultimate repair. For example, although a breakpoint may map to a tRNA gene, Ty, or solo LTR, these elements may have served only as sites of recombination following resection from an independent breakage event located at some distance from the repair event. It is intriguing, then, that the literature on genome architecture, experimental evolution, and repair also contains examples of origins of replication associated with breakpoints. In a system generating spontaneous duplications, Payen et al. (2008) note that three of the 18 breakpoints they map are located at autonomously replicating sequence (ARS) elements. In analyzing genomic rearrangements between S. cerevisiae and S. pastorianus, Dunn and Sherlock (2008) observe that origins are often found in close proximity to these breakpoints. In another recent work (Hwang et al. 2008), translocations in a strain defective in sister chromatid recombination (smc6-9) were mapped to LTRs, tRNAs, and ARS elements. On reexamining the works of Dunham et al. (2002), Kellis et al. (2003), Lemoine et al. (2005, 2008), and Argueso et al. (2008), we note that ARS elements are found in the vicinity of many of the breakpoints reported therein.
As comprehensive well-defined origin lists have only become available in the past few years, it has been difficult to formally include replication origins in the analyses of breakpoints. Now, however, with the advent of high-resolution origin data sets with origin-firing times annotated (Feng et al. 2006; Nieduszynski et al. 2007; McCune et al. 2008) it is possible to test if the casually observed colocalization of origins and breakpoints is significant. To comprehensively ask if replication origins act as fragile genomic sites, here we explore on a genome-wide scale correlations between S. cerevisiae origins and genomic breakpoints generated over evolutionary time and those breakpoints described in the S. cerevisiae experimental evolution and repair literature. Although we cannot know the set of all origins in the ancestral yeasts giving rise to S. cerevisiae, we can determine if extant S. cerevisiae origins are present at regions created by genome rearrangements and thus may have survived a rearrangement event or arisen subsequently. On the other hand in analyzing breakpoints experimentally produced in S. cerevisiae, we can ask if origin locations correlate with sites of genome rearrangements, although we cannot know if the origin survives after the break is repaired. Using evolutionary and experimentally generated breakpoints, the work presented here establishes an association between origins of replication and genome rearrangement sites before and after rearrangement events. Although an association between origins and breakpoints has been reported anecdotally, this study is the first systematic, genome-scale examination for such an association, and this work highlights the possibility that a potential contributor to genome plasticity has hitherto been overlooked.
Methods
Yeast Genomes
The S. cerevisiae, Kluyveromyces waltii, and Ancestor genomes used in this work are summarized in supplementary table S1 (Supplementary Material online). The Kellis S. cerevisiae gene content used is described in Kellis et al. (2004), and the S. cerevisiae gene content for the Wolfe data set was taken from Byrne and Wolfe (2005). Chromosomal coordinates for the S. cerevisiae genes in the Wolfe set were referenced from the Saccharomyces Genome Database (SGD; December 2007; Cherry et al. 1998; http://www.yeastgenome.org/). The K. waltii genome used in both the Kellis and Wolfe data set was derived from Kellis et al. (2004). To get chromosomal coordinates for all K. waltii genes, the K. waltii genome was assembled by concatenating the contigs in the order described by Kellis et al. (2004) with an “N” representing a gap in the sequence (see the supplementary section Resources and Datasets [Supplementary Material online] for a discussion on the minimal impact of these gaps on our analyses). Twelve contigs showing significant overlap were joined to create six longer contigs. Kluyveromyces waltii open reading frames (ORFs) annotated by Kellis et al. (2004) and belonging to assembled chromosomes were Blasted against the assembled K. waltii genome to produce chromosomal coordinates for these ORFs. Saccharomyces cerevisiae–K. waltii homolog matches for the Kellis data set were taken from Kellis et al. (2004). The top matching K. waltii ORF for a given S. cerevisiae gene was selected. Wolfe S. cerevisiae–K. waltii homolog matches were taken from Byrne and Wolfe (2005). The Wolfe Ancestor gene content and the S. cerevisiae–Ancestor and the K. waltii–Ancestor matches were taken from Yeast Gene Order Browser (YGOB; Version 2.0, PNAS 2007; http://wolfe.gen.tcd.ie/ygob/). For further details, the assembled genome, and the list of genes used in this study see the supplementary section Resources and Datasets (Supplementary Material online).
Genomic Features
The list of genomic features correlated with breakpoints in this study is shown in supplementary table S2 (Supplementary Material online). Saccharomyces cerevisiae genomic features were taken from SGD in December 2007 (http://www.yeastgenome.org/). Spo11 hot spots were taken from Borde et al. (2004). The ARSs in OriDB (Nieduszynski et al. 2007) were downloaded from OriDB in January 2008. These ARSs are categorized in OriDB as “confirmed,” “likely,” and “dubious” according to the extent of experimental proof that exists for that ARS. A list of 411 high-confidence ARSs (hcARSs) was created from the confirmed OriDB ARSs supplemented with additional ARSs found by single-stranded DNA (ssDNA) origin mapping in a rad53 strain using a high-density microarray (Feng W and Brewer B, unpublished data) that mapped nearly identical origins to those described in Feng et al. (2006) but with greater resolution. Origins known to fire in an observable proportion of cells in a population, McCune origins, were taken from McCune et al. (2008). These origins were previously annotated as early or late firing according to two different metrics: Rad53 checkpoint–mediated regulation (previously “unchecked” or “checked”) and dependence on Clb5 (previously CDR [Clb5 Dependent Region] values 0–3). Here we have termed early-firing origins as Rad53 unregulated (previously “unchecked”) or not Clb5 dependent (non-CDR, CDR = 0) and late-firing origins as Rad53 regulated (previously “checked”) and Clb5 dependent (CDR, CDR value >0). Any origins represented by a single coordinate were given a 1-kb resolution. Kluyveromyces waltii centromeres were taken from Kellis et al. (2004) and K. waltii tRNAs were predicted using tRNAscan-SE (Lowe and Eddy 1997). For further details and the list of all genomic features used, see the supplementary section Resources and Datasets (Supplementary Material online).
Defining Evolutionary Breakpoints
Breakpoints between S. cerevisiae and K. waltii were found by analyzing intergenic regions in S. cerevisiae that are bounded by genes having K. waltii homologs (fig. 1A). The total set of S. cerevisiae genes as defined by a given data set was reduced to the set of S. cerevisiae genes having K. waltii homologs (supplementary table S1, Supplementary Material online). Intergenic regions in this reduced S. cerevisiae genome were defined between adjacent nonoverlapping genes. Overlapping genes were excluded because they were found to be enriched for SGD 2004 genome annotation errors and dubious genes (supplementary table S3, Supplementary Material online). To determine if an intergenic region contained a breakpoint, the K. waltii homologs of the flanking genes were interrogated. Breakpoints were defined according to the definitions in fig. 1A. If the K. waltii homologs of the flanking S. cerevisiae genes were located on different chromosomes in the K. waltii genome, an interchromosomal break was called. If the K. waltii homologs were from the same K. waltii chromosome but more than 20 genes apart on the K. waltii chromosome, an intrachromosomal break was called. The cutoff of 20 genes is the same cutoff as that used by Byrne and Wolfe (2005). Furthermore, a histogram of the number of K. waltii genes between K. waltii homologs flanking a noninterchromosomal breakpoint in S. cerevisiae showed a long narrow tail with a 20-gene cutoff being conservative (supplementary fig. S1, Supplementary Material online). Tandem duplications were defined when the S. cerevisiae genes flanking the intergenic region were homologous to the same K. waltii gene. These tandem duplications were manually confirmed. Six of these tandem duplications were the products of the 2004 SGD (used to define S. cerevisiae genes in Kellis et al. 2004) calling two genes that have since been merged into one. Tandem expansions beyond a simple duplication (three or more tandem genes deriving from the same ancestral gene) appear in the data as consecutive tandem duplications. To approximately define inversions and other local gene rearrangement events, two consecutive intergenic regions were considered. As a prerequisite, K. waltii genes were ordered and given a number representing their placement from left to right along the native K. waltii chromosome. Then the order of the K. waltii homologs flanking the two S. cerevisiae intergenic regions was compared. Gene order changes seen were assigned to the second intergenic region. If, for example, the K. waltii homologs were found to be increasing in order in the first intergenic region but decreasing in the second, a gene order change was noted in the second intergenic region (see fig. 1A). If an intergenic region was determined to contain a breakpoint, the breakpoint was defined as the entire intergenic region. Breakpoints between S. cerevisiae–Ancestor and K. waltii–Ancestor were defined identically.
FIG. 1.—

(A) A cartoon illustration of breakpoints mapped between genes in S. cerevisiae with K. waltii homologs. The red arrows represent S. cerevisiae genes ordered as they appear in the S. cerevisiae chromosome. The colored arrows below represent homologs in K. waltii. These genes are colored according to which chromosome they belong to in K. waltii and have been numbered according to how the genes are arranged on a given K. waltii chromosome. The black boxes represent S. cerevisiae intergenic regions that contain breakpoints as defined through comparison with K. waltii. An intergenic region between two S. cerevisiae genes with K. waltii homologs belonging to different K. waltii chromosomes is defined as an interchromosomal breakpoint (type I in the figure). An S. cerevisiae intergenic region with K. waltii homologs from the same chromosome but more than 20 genes apart is called an intrachromosomal breakpoint (II). A tandem duplication (III) is called if the K. waltii homologs flanking the S. cerevisiae intergenic region are the same. If two consecutive intergenic regions do not otherwise have a breakpoint, the order of the K. waltii genes is checked. If the K. waltii homologs flanking the first S. cerevisiae intergenic region are found to be increasing/decreasing while they are decreasing/increasing in the second intergenic region, then a gene order change (IV) is called in the second intergenic region. See the text for breakpoints not detected. (B) The locations of breakpoints (Kellis data set defined), tRNAs, Ty/LTRs, and hcARSs along S. cerevisiae chromosome V are shown as black bars. The width of the black vertical bars reflects the size of the intergenic region to which a breakpoint maps or the physical size of the feature. (C) Minimal end point measure of the distance between breakpoints and a feature. Distances are measured between the ends of the breakpoint and the feature such that the shortest distance is measured. If the breakpoint and feature overlap (the hcARS at 258 kb is an example), a distance of 0 is used (see inset for distances in this example).
We checked that our breakpoint-finding algorithm was in agreement with previously mapped breakpoints between S. cerevisiae and K. waltii by manually comparing a subset of our breakpoints with those mapped by Kellis et al. (2004) and those shown in the YGOB (version PNAS 2007) (Byrne and Wolfe 2005). Kellis et al. (2004) define 353 syntenic blocks as clusters of genes that are less than 20 genes apart and contain at least three genes. These blocks demarcate gross rearrangements and contain within them many smaller-scale genome rearrangements. We confirmed that our algorithm successfully identified breakpoints at the boundaries of each of the syntenic blocks called by Kellis et al. (2004) on S. cerevisiae chromosomes X and XII and, in addition, also identified many other smaller-scale rearrangements within each block. In YGOB, we scanned through S. cerevisiae chromosomes X and XII and observed that all 119 of the breakpoints we call on these chromosomes are clearly marked by the browser. Differences between our breakpoints and those in YGOB occur where one species has a gene the other species does not (captured by YGOB), a breakpoint is called by YGOB but corresponds to a junction between supercontigs in our assembly (and therefore is not seen as a break), and sites where a K. waltii gene was not part of the assembled K. waltii genome (called by YGOB but ignored in our analysis because the genomic location of the K. waltii gene is unknown).
Literature Breakpoints
Breakpoints were curated from the S. cerevisiae breakage, repair, and experimental evolution literature. The list of papers used is shown in supplementary table S4 (Supplementary Material online). These papers include experimental evolution under nutrient limitation, stress, or a mutagen, or experimental evolution in a genome lacking a gene; gross chromosomal rearrangement and HO-generated break and repair assays; analysis of checkpoint or repair mutants; genomic comparison of S. cerevisiae to another strain or to another sensu stricto species; and the location of horizontally transferred genes. Overall, 442 breakpoints from 29 papers were curated. Inclusion criteria were as follows: A breakpoint had to be mapped, had to be unique to a given experimental setup or strain in a given paper, and had to represent a physical breakage or copy number alteration. The mapping resolution of the breakpoint was taken into account when recording the coordinates of the breakpoint. For gene resolution of breakpoints, as in comparative genomic hybridization papers, the breakpoints were named as the coordinates of the genes on the extremes of the copy number–altered region. For example, if genes 13–19 were duplicated, breaks were named at genes 13 and 19. To equalize resolution across experimental works, if the break had been delineated to a more specific region, the break was extended to be either the entire genic or intergenic region. For example, if a break was found at a Ty element, then the break was extended to include the entire intergenic region to which the Ty element belongs. Dubious genes in the intergenic region were not used to define the intergenic region boundaries. Six literature breakpoints that exceeded 17 kb were discarded.
The list of papers was broken into two sets: evolutionarily generated breakpoints and experimentally generated breakpoints. The evolutionarily generated breakpoints from seven papers (147 breakpoints) were mapped among sensu stricto yeast. The remaining 22 papers (295 breakpoints) were those in which the authors generated the breakpoints and were known to have both the ancestor and derived strains. For further details and the list of all literature breakpoints used see supplementary table S4 (Supplementary Material online) and the supplementary section Resources and Datasets (Supplementary Material online).
Enrichment Analysis
Each S. cerevisiae chromosome was broken into 5-kb bins for a total of 2,423 bins across the entire genome; bins did not bridge separate chromosomes (see supplementary fig. S2 [Supplementary Material online] for the choice of 5-kb bins). Breakpoints and genomic features were assigned to bins corresponding to where their midpoints fell. For each bin, the presence or absence of a breakpoint or feature was recorded. Then for each feature, the total number of bins with that feature and a breakpoint was determined. To test for enrichment of breakpoints and each feature, a hypergeometric distribution was assumed (phyper function in the statistics package R). P values <0.05 were considered as evidence of a correlation and P values <0.05 after a Bonferroni correction were considered strongly significant.
Distance Method
The S. cerevisiae genome was scanned for breakpoints. When a breakpoint was encountered, the distance to the nearest genomic feature of a particular class was recorded. Two methods of measuring distances between breakpoints and features were used. The midpoint method measures the distance from the midpoint of the breakpoint to the midpoint of the feature. The minimal end point method gives a distance of zero to features falling at least partially within a breakpoint (fig. 1C). For features falling outside of a breakpoint, the method uses the distance between the end point of the feature and the end point of the breakpoint. Breakpoints and intergenic regions over a cutoff were excluded from the latter method. The cutoff was used so that the few very large breakpoints would not give a distance of zero to features falling within these large sections of the genome. For the S. cerevisiae–K. waltii and S. cerevisiae–Ancestor analysis, a cutoff of 17 kb was used, thus excluding breaks in a few telomeric regions and in the ribosomal DNA (rDNA) locus. This cutoff was selected by observing that all or all but one of the intergenic regions over 17 kb contained breakpoints, whereas below 17 kb only a subset of the intergenic regions had breakpoints (supplementary fig. S3, Supplementary Material online). For the K. waltii–Ancestor analysis a cutoff of 14 kb was applied. The modified total number of intergenic regions and breakpoints is shown in supplementary table S5 (Supplementary Material online).
Simulation
To determine the significance of the distance measures between breakpoints and features, the null distribution of distances from breakpoints to features was found by randomizing breakpoints through a circular permutation algorithm (fig. 2A). Specifically, S. cerevisiae chromosomes were randomly arranged in a circle. Then breakpoints were shifted around the circle of chromosomes a random number of intergenic regions. Literature breakpoints were similarly treated except that they were shifted a random number of base pairs because these breakpoints are not constrained to intergenic regions. Based on preliminary simulation results, the number of breakpoints bearing a feature within 1 kb was further studied (fig. 2B). In all, 10,000 simulations were run, and in each simulation the number of breakpoints with a feature within 1 kb was recorded. From the resulting distribution, a P value was obtained by summing the number of simulations during which the number of breakpoints with a feature within 1 kb was equal to or greater than observed in the real data (fig. 2C). P values <0.05 were considered as evidence of a correlation, and P < 0.05 after a Bonferroni correction were considered strongly significant.
FIG. 2.—

The simulation method. (A) To randomly place breakpoints in the genome, breakpoints (b-238, b-40, etc.) were shifted a random number of intergenic regions over a randomly ordered set of concatenated chromosomes. In this example, break b-238 moves from chromosome V to chromosome XIV. (B) With the set of real and simulated breakpoints, the distance to the nearest genomic feature was found. In this example the distances to the nearest hcARS were plotted for one simulated set of Wolfe breakpoints using minimal end point measures. We noted that for ARSs/origins, tRNAs, Tys, and LTRs, the number of breakpoints with a genomic feature within 1 kb was greater for the real set of breakpoints than for the random set of breakpoints. (C) For 10,000 simulations, the number of breakpoints with a feature within 1 kb was counted. This distribution was used to obtain a significance value for the number of real breakpoints with the feature within 1 kb (dark-blue line). The significance value was found by summing the number of simulations where the number of breakpoints with the feature within 1 kb was greater than or equal to the real data (dark-blue line). The distribution and real data are shown for Wolfe breakpoints and hcARSs using the minimal end point measures. The data for this example produce a P value of 0.0002. The complete data for all genomic features are shown in supplementary table S7 (Supplementary Material online).
Logistic Regression Analysis
The genome was divided into 5-kb bins, and each bin was scored as 0 if it did not contain a breakpoint or 1 if it did. Concurrently, the number of each type of feature was tallied for each bin. Regression analysis on a full interaction model (all features multiplied together) was performed using the logit (identical results were obtained using the probit). In a logit model, the unobserved probability of breakage is set as logistically distributed, whereas the probit model assumes a normal distribution. The hcARS set was used for this analysis as this set contains the greatest number of potential origins. In R, the full model with all genomic features and breakpoints from the Kellis data is written as, Logitfull <-glm(breakpoint ∼ centromere × telomere × telomereic_repeat × snoRNA × snRNA × ribosomal_gene × Spo11_hot spot × tRNA × Ty × LTR × hcARS, family = binomial(link = “logit”), data = KellisRegressionData). The multiplication symbol represents both the additive and interactive effects of two terms (e.g., tRNA × hcARS = tRNA + hcARS + tRNA:hcARS, where the last term is the interaction term). Terms were sequentially removed and/or converted to additive and interaction terms. With each simplification of the model, the new model was compared with the previous model using a likelihood ratio test with a 0.05 cutoff. If the new model was not found to be significantly different from the previous model, the new simplified model was accepted. Hence, a fully reduced model is one in which every remaining term contributes significantly to the prediction of the data; any further eliminations of terms significantly degrades the fit to the data. A total of 1,000 bootstrap simulations were performed to obtain typical confidence intervals for the model's coefficients.
Algorithms were written in perl, and statistical analyses were performed in R. All perl scripts are available upon request.
Results
Defining Evolutionary Breakpoints between S. cerevisiae and K. waltii
To generate an extensive genome-wide list of evolutionary breakpoints in S. cerevisiae, we compared the genomes of S. cerevisiae and K. waltii (also known as Lachancea waltii). The yeasts S. cerevisiae and K. waltii are related by an ancestor that underwent a whole-genome duplication event in the lineage ultimately producing S. cerevisiae (Kellis et al. 2004). Comparison of the S. cerevisiae and K. waltii genomes reveals that the 100- to 150-My divergence of these species is characterized by massive gene loss and by extensive chromosomal rearrangements, including inversions, translocations, and smaller duplications (Kellis et al. 2004). By comparing the locations of these rearrangement breakpoints with extant S. cerevisiae replication origins, we can ask if there is any evidence of origins correlating with the production or resolution of genomic breaks over evolutionary time.
We defined breakpoints between the S. cerevisiae and K. waltii genomes by looking for interruptions in synteny. Only those S. cerevisiae genes for which a K. waltii homolog could be identified were used to establish synteny. By aligning K. waltii homologs onto the S. cerevisiae genome, we identified four specific types of genome rearrangement breakpoints (fig. 1A): I) interchromosomal breakpoints—S. cerevisiae intergenic regions flanked by K. waltii homologs found on two different K. waltii chromosomes; II) intrachromosomal breakpoints—S. cerevisiae intergenic regions where the adjacent genes and their K. waltii homologs are on the same K. waltii chromosome but more than 20 genes apart; III) tandem duplications—S. cerevisiae intergenic regions where the two flanking S. cerevisiae genes match the same K. waltii homolog; and IV) gene order changes—S. cerevisiae intergenic regions where there is an inversion of gene order within a region of synteny (e.g., in fig. 1A S. cerevisiae genes 6 and 7 correlate to K. waltii genes 90 and 99, respectively; however, S. cerevisiae genes 7 and 8 correspond to K. waltii genes 99 and 91, respectively, reflecting a change in gene order with respect to the K. waltii chromosome).
Our method to define breakpoints between these species introduces a few important limitations. First, not all types of breakpoints are captured. Because of the requirement that each S. cerevisiae gene must have a K. waltii homolog, deletions and exogenous gene insertions were not identified. The homolog-matching algorithm we created also could not detect single-gene inversions. Second, our ability to match the S. cerevisiae and K. waltii genomes is dependent on being able to assign homologs in the two species. These homologs are the only information that allows us to define blocks of synteny and locations of breakpoints. To mitigate against potential inaccuracies in any one homolog assignment between these genomes, we applied our breakpoint definitions to two sets of matched S. cerevisiae–K. waltii genomes, the Kellis and Wolfe data sets (Kellis et al. 2004; Byrne and Wolfe 2005; see supplementary table S1 [Supplementary Material online] and Methods). Third, once a homology assignment was made, any information regarding how completely the homologs matched each other was lost. For example, gene fragments are not distinguished from highly conserved genes. As a result, breakpoints cannot, by definition, occur within a gene; our list of breakpoints therefore is limited to intergenic regions. Fourth, S. cerevisiae intergenic regions are defined as sequences lying between S. cerevisiae genes that have K. waltii homologs. It follows then that some intergenic regions can be quite long if a stretch of S. cerevisiae genes without K. waltii homologs is encountered (supplementary fig. S3, Supplementary Material online). Such long stretches are found near the S. cerevisiae telomeres and the rDNA locus. Last, by our methods, each single-gene translocation results in two breakpoints, creating breakpoint clusters. Despite the limitations of our methods, the rearrangement breakpoints we identified between S. cerevisiae and K. waltii are in good agreement with those previously mapped by Kellis et al. (2004) and those indicated on the YGOB (Byrne and Wolfe 2005; see Methods).
We applied our breakpoint definitions to these two data sets, yielding a total of 1,152 breakpoints for the Kellis data set and 718 breakpoints for the Wolfe data set (supplementary table S5, Supplementary Material online). The greater number of breakpoints in the Kellis data set reflects the differences in how homologs were assigned in the two data sets. First, the Wolfe homolog assignment did not allow for tandem duplications. Second, the Kellis data set included dynamic regions (e.g., subtelomeric) in the S. cerevisiae genome. Third, the Kellis K. waltii homologs were not manually selected to maintain synteny between S. cerevisiae and K. waltii. Fourth, the Wolfe data set manually selected the best homolog assignments and enforces a 1:2 mapping between K. waltii and S. cerevisiae. The Kellis data set, on the other hand, uses the best K. waltii match for each S. cerevisiae gene and allows for greater than a 1:2 mappings between K. waltii and S. cerevisiae. In total there are 75 K. waltii genes with a greater than a 1:2 mapping, with the greatest mapping being 1:18 (see the supplementary section Resources and Datasets [Supplementary Material online] for the complete homolog assignments between S. cerevisiae and K. waltii for the two data sets). By using two homolog data sets to find S. cerevisiae–K. waltii breakpoints, we have identified breakpoints more inclusively in the Kellis data set and more conservatively in the Wolfe data set.
Origins Are Correlated with S. cerevisiae–K. waltii Evolutionary Breakpoints Using an Enrichment Test
To investigate whether a correlation exists between the location of evolutionary breakpoints and origins of replication in S. cerevisiae, we began with an enrichment test. We broke the S. cerevisiae genome into 5-kb bins and scored each bin for the presence or absence of a breakpoint so that clusters of breakpoints in a bin would be treated as a single event. The same procedure was repeated for genomic features (tRNAs, origins, see supplementary table S2, Supplementary Material online). For each feature, we tallied the number of bins containing that feature and a breakpoint. Significance values were determined by comparing the correlations observed in the real data with correlations that would be expected if breakpoints were randomly placed into bins. As we have restricted each bin to having only a single breakpoint, we are effectively sampling bins in the genome without replacement, and thus a hypergeometric distribution gives the null distribution of the number of bins expected to have a breakpoint and a feature.
Our enrichment results (table 1 and supplementary table S6, Supplementary Material online) corroborate the previously noted correlation between evolutionary breakpoints and S. cerevisiae tRNAs for the Kellis and Wolfe data sets (Fischer et al. 2000; Kellis et al. 2003; Dietrich et al. 2004; Hughes and Friedman 2004; Kellis et al. 2004; Garfinkel 2005; Liti and Louis 2005; Dujon 2006) even after a Bonferroni correction for multiple testing. Though not significant in the Kellis data set after a Bonferroni correction, we also found that evolutionary breakpoints correlated with Ty LTRs in the Wolfe data set. In contrast, we did not find correlations between breakpoints and ribosomal protein genes, small nucleolar RNAs, small nuclear RNAs, and Spo11 hot spots (supplementary table S6, Supplementary Material online), features with no anticipated association with breakpoints. These results gave us confidence that the enrichment analysis and our definition of breakpoints were consistent with previous analyses.
Table 1.
Enrichment and Simulation Analysis Using Minimal End Point and Midpoint Measures for Wolfe S. cerevisiae–K. waltii breakpoints and Selected Genomic Features
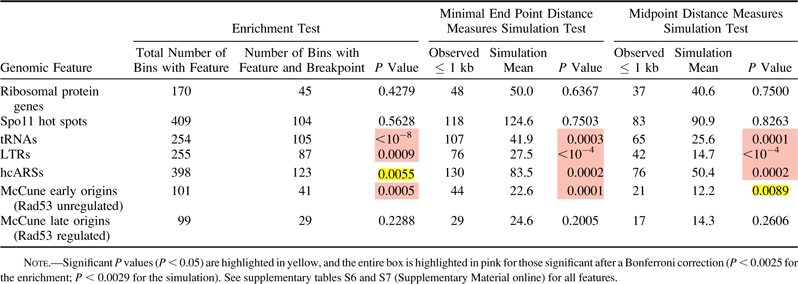 |
We applied these same methods to origins. Significant results (table 1 and supplementary table S6, Supplementary Material online) were found for hcARSs compiled for this work as well as confirmed ARSs in OriDB (Nieduszynski et al. 2007) (see supplementary table S2 [Supplementary Material online] and Methods). These results were significant after a Bonferroni correction in only the Kellis data set. Because ARSs do not necessarily act as replication origins in the chromosomal context, we then tested a newly published list of origins distinguished by their ability to fire in a significant proportion of cells in a population (McCune et al. 2008). As a group, these origins were significant though not after a Bonferroni correction for both breakpoint sets (supplementary table S6, Supplementary Material online). However, after separating the McCune origins into early- and late-firing origins as defined by Rad53 checkpoint–mediated regulation (Feng et al. 2006; see Methods), we discovered that early-firing origins are significantly correlated to breakpoints in both data sets even after a Bonferroni correction (table 1 and supplementary table S6, Supplementary Material online). Origins with earlier firing times are less likely to be passively replicated by a nearby origin and may be regulated differently than later-firing origins (Feng et al. 2006; McCune et al. 2008). These results suggest that origins are overrepresented at sites that have experienced a rearrangement event and that the physical firing of origins or the regulation of early-firing origins is associated with rearrangements.
Breakpoints Are Correlated with Origins That Map within 1 kb
Although the findings from the enrichment analysis agree with previous analyses, it assumed a theoretical null distribution describing how randomly placed breaks associate with features. Moreover in breaking the genome into 5-kb bins, contiguity between features in adjacent bins is lost. To better analyze the potential correlation between evolutionary breakpoints and origins of replication without assuming a theoretical null distribution and without the use of bins, we designed an alternative analysis method. For each type of genomic feature, this method obtains the distance from a breakpoint to the nearest feature of that type (i.e., the distance from a breakpoint to the nearest tRNA, the distance from a breakpoint to the nearest origin, etc.). A permutation scheme then estimates the probability of the observed distance between breakpoints and features occurring if breakpoints were randomly located in the genome.
Distances between breakpoints and features were found by measuring the minimal distance between the end points or the distance between midpoints of the breakpoint and feature (see fig. 1C and Methods). To calculate significance values for the distances obtained, the distances of breakpoints to features were compared with those measured using a set of random breakpoints. As local clusters of breakpoints exist around single-gene translocations and highly dynamic regions, we randomized breakpoints by employing a shifting algorithm to maintain the genic spacing between breakpoints. As described in fig. 2A and Methods, the S. cerevisiae chromosomes were randomly ordered and concatenated in silico. Breakpoints were shifted a random number of intergenic regions over the concatenated chromosomes. This randomization method maintains the genic spacing between breakpoints and ensures that breakpoints are only located in intergenic regions following the permutations.
As a preliminary analysis, we compared the distances measured for the real set of breakpoints and for one simulated set of breakpoints (fig. 2B). For both data sets, there were more tRNAs, Tys, LTRs, and origins within 1 kb of a breakpoint in the real data than in the one simulated set, indicating that there may be more breakpoints with these features within 1 kb than expected by chance. To see if there were indeed more breakpoints near tRNAs, Tys, LTRs, or origins than expected, we created 10,000 sets of simulated breakpoints. For each simulation, we counted the number of breakpoints with a given feature within 1 kb and determined the significance of this value (fig. 2C and Methods).
In agreement with previous work (Fischer et al. 2000; Kellis et al. 2003; Dietrich et al. 2004; Hughes and Friedman 2004; Kellis et al. 2004; Garfinkel 2005; Liti and Louis 2005; Dujon 2006), we found values significant after a Bonferroni correction for Tys, LTRs, and tRNAs in both data sets (fig. 3, table 1, and supplementary table S7, Supplementary Material online). As in the enrichment test, we found S. cerevisiae–K. waltii breakpoints to be highly correlated with ARSs in all four combinations of data sets and breakpoint–feature distance measures. Furthermore, we again discovered that early-firing origins are very strongly correlated with breakpoints even after a Bonferroni correction in three of the four analyses (see supplementary table S7 [Supplementary Material online] and Methods). Therefore, using two different methods of analysis—enrichment and the distance methods—on two different S. cerevisiae–K. waltii homolog sets, we corroborate previous work linking tRNAs, Tys, and LTRs to breakpoints, and we find evidence that origins of replication, specifically early-firing origins, are correlated with evolutionary breakpoints.
FIG. 3.—

The correlative significance of Wolfe S. cerevisiae–K. waltii breakpoints and selected genomic features as determined by minimal end point distance measures and simulation analysis. The early and late origins are McCune origins defined by Rad53 checkpoint–mediated regulation. See supplementary table S7 (Supplementary Material online) for all genomic features. The histograms show the number of simulations (y axis) in which a particular number of breakpoints (x axis) was within 1 kb of a genomic feature. The red vertical line shows the number of breakpoints within 1 kb of a genomic feature observed in the real data. The P values were derived by summing the number of simulations in which the number of breakpoints with a genomic feature within 1 kb was equal to or greater than the observed data (red line) and dividing this number by the total number of simulations performed (10,000).
Saccharomyces cerevisiae–Ancestor Evolutionary Breakpoints Are Also Correlated with Origins
The S. cerevisiae–K. waltii breakpoints we have considered thus far represent events occurring on two separate evolutionary paths: the lineage leading to S. cerevisiae and the lineage producing K. waltii. Ideally, we would like to consider the breakpoints in each lineage separately. To do so would require knowledge of the genome of the yeast ancestral to both S. cerevisiae and K. waltii. Although such an actual ancestral genome is unknown, we can approximate its gene content using the inferred ancestral genome constructed by Gordon et al. (2009).
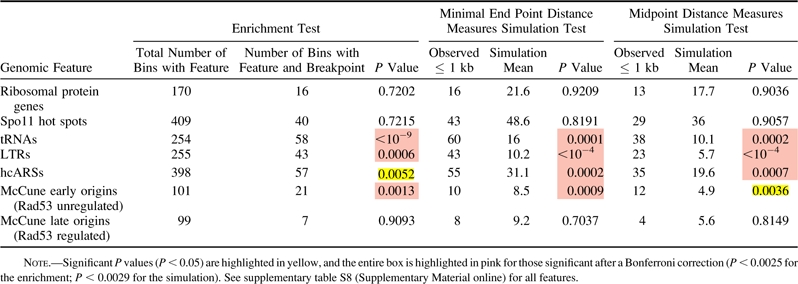
Using the same methods as before, we mapped S. cerevisiae–Ancestor and K. waltii–Ancestor breakpoints (supplementary table S5, Supplementary Material online). The enrichment and distance analyses revealed the same correlations for S. cerevisiae–Ancestor breakpoints as seen for S. cerevisiae–K. waltii breakpoints (table 2 and supplementary table S8, Supplementary Material online) and are in agreement with a recent analysis of S. cerevisiae–Ancestor breakpoints (Gordon et al. 2009) (see Discussion). To perform the parallel analysis on the K. waltii–Ancestor breakpoints, we would need K. waltii genomic features. Unfortunately, experimental work to uncover genomic features in K. waltii remains to be completed. We were able to predict, though, the location of tRNAs in the K. waltii genome using tRNAscan-SE (Lowe and Eddy 1997). Performing enrichment and distance measures tests on the K. waltii tRNAs showed that tRNAs are correlated with K. waltii–Ancestor breakpoints (supplementary table S9, Supplementary Material online). Therefore, in separating the S. cerevisiae and K. waltii lineages, we find further evidence that tRNAs, Tys, LTRs, and origins are correlated with evolutionary breakpoints along the S. cerevisiae lineage, and we find evidence that tRNAs are associated with breakpoints produced in the K. waltii lineage.
Table 2.
Enrichment and Simulation Analysis Using Minimal End Point and Midpoint Measures for Wolfe S. cerevisiae–Ancestor Breakpoints and Selected Genomic Features
 |
Recent Evolutionary Breakpoints Described in the Literature Are Correlated with Origins
Breakpoints between S. cerevisiae and K. waltii or the Ancestor have occurred over a 150-My period during which time tRNAs, transposable elements, and origins have likely not remained in conserved locations. We therefore wished to test whether the same correlations would be found using breakpoints defined over a shorter evolutionary distance and mapped by different methods. To this end, we curated breakpoints from the literature on genome arrangements in the sensu stricto yeast and used the locations of horizontally transferred genes in S. cerevisiae. Doing so provided us with 147 breakpoints from seven papers (supplementary table S4, Supplementary Material online).
As before, we applied the enrichment and distance analyses to these evolutionarily generated literature breakpoints. Unlike our defined evolutionary breakpoints, literature breakpoints are not confined to intergenic regions. Therefore, to simulate literature breakpoints, we used a shifting technique as before but shifted by a random number of base pairs rather than by intergenic regions over the concatenated chromosomes.
In analyzing the 147 evolutionary literature breakpoints, the enrichment and distance analyses produced highly significant correlations for tRNAs and LTRs and for origins of replication (supplementary table S10, Supplementary Material online). Furthermore, the correlation of early origins, rather than late origins, with breakpoints was again observed. Therefore, in addition to finding correlations between S. cerevisiae origins and evolutionary breakpoints we defined using the S. cerevisiae, K. waltii, and Ancestor genomes, we also find a significant correlation between origins and more recent breakpoints described in the literature.
Origins Exist at Sites That Experience Breakage in the Laboratory Setting
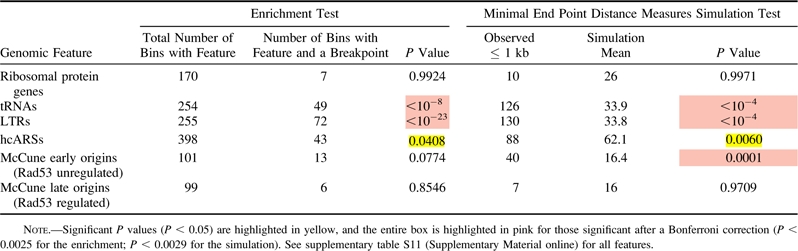
Thus far we have established a correlation between breakpoints generated over evolutionary timescales and extant S. cerevisiae replication origins as well as with tRNAs, Tys, and LTRs. This correlation cannot, however, untangle cause and effect—whether an origin was present prior to the rearrangement and thus may have promoted the rearrangement, or whether an origin arose at the breakpoint subsequent to and perhaps promoted by the rearrangement event. Although we cannot ask if origins existed at evolutionary break sites prior to rearrangements, we can examine breakpoints generated in systems where origin location is known prior to the break event. Accordingly, we analyzed the subset of literature breakpoints wherein the authors were known to have both the original and derived strains producing the published breakpoint (supplementary table S4, Supplementary Material online). As origin location is believed to be largely conserved among the sensu stricto species (Nieduszynski et al. 2006), we included breakpoints from studies using S. cerevisiae or sensu stricto yeast. Using this list of recent, experimentally observed breakpoints, we again found correlations, though not as highly significant across all analyses, of breakpoints with tRNAs, Tys, LTRs, and early-firing replication origins (fig. 4, table 3, and supplementary table S11, Supplementary Material online). Therefore, we have evidence that origins are found at rearrangement sites prior to breakage, consistent with the idea that origins may contribute to genome rearrangement events.
FIG. 4.—

The correlative significance of experimental breakpoints and selected genomic features as determined by minimal end point distance measures and simulation analysis. See supplementary table S11 (Supplementary Material online) for all genomic features. See the legend in fig. 3 for further details.
Table 3.
Enrichment and Simulation Analysis Using Minimal End Point Measures for Experimentally Derived Breakpoints from the Literature with Selected Genomic Features
 |
Evolutionary Breakpoints near Origins Are Also Experimental Breakpoints
By analyzing both experimentally and evolutionarily generated breakpoints, we have discovered that S. cerevisiae origins are present both at sites that break under experimental conditions and at sites that are the product of an evolutionary rearrangement. Is it then the case that origins are fragile sites in the genome that are repeatedly used as breakpoints over evolutionary time and in experimental systems? To address this question, we first asked if the evolutionary and experimental breakpoints overlap. We found that about half of the experimentally generated breakpoints are within 1 kb of an evolutionary breakpoint, although the collocation of these two types of breakpoints is not significant (supplementary table S12, Supplementary Material online). Next we asked if the specific subset of evolutionary breakpoints that is associated with origins correlates with the experimental breakpoints. For comparison the same procedure was repeated for tRNAs and LTRs. We found that, like tRNA- and LTR-associated evolutionary breakpoints, early-firing origin-associated evolutionary breaks defined by both data sets correlate significantly after a Bonferroni correction with the experimentally generated breakpoints (supplementary table S12, Supplementary Material online), and evolutionary breakpoints associated with hcARSs correlate strongly after a Bonferroni correction in one data set. Hence, we have evidence that, like tRNAs and Tys, early-firing replication origins are associated with fragile genomic sites.
tRNAs, Tys, LTRs, and Origins Are Clustered Together throughout the Genome
We have observed correlations of both evolutionary and experimental breakpoints with replication origins. These individual correlations, however, could be indirect due to clustering of features. For instance, replication origins may be indirectly correlated with breakpoints by being directly correlated with tRNAs, which are in turn directly correlated to breakpoints. Dunn and Sherlock (2008), for example, note that breakpoints mapped between S. cerevisiae and S. pastorianus are frequently found where tRNAs, Ty/LTRs, and origins are clustered together. The proximity of Tys and LTRs to tRNAs has been well documented (Kim et al. 1998; Bolton and Boeke 2003; Bachman et al. 2004; Garfinkel 2005). As well, a correlation between potential replication origins and tRNA/Tys has been reported previously (Wyrick et al. 2001; Gordon et al. 2009). We confirmed all the above correlations by performing enrichment tests among tRNAs, Tys, LTRs, and origins (supplementary table S13, Supplementary Material online). In this analysis, we found that specifically early-firing origins are highly correlated with tRNAs and LTRs.
This correlation suggests either that origins and tRNAs/LTRs are located together in the genome at places that subsequently become fragile sites or that genome breakage and subsequent rearrangements result in origins and tRNAs/LTRs coming together. We distinguished between these two possibilities by first removing hcARSs, tRNAs, Tys, and LTRs within 5 kb (by end point measures) of a breakpoint, thereby leaving only hcARSs, tRNAs, Tys, and LTRs that are not associated with breakpoints. In repeating the enrichment analysis, the correlation among hcARSs, tRNAs, Tys, and LTRs persisted (data not shown). Although it is possible that some of the hcARS-tRNA-Ty/LTR clusters have not yet experienced a break or are incapable of breaking for another reason, these results suggest that their collocations are independent of breakpoint formation.
Origins Increase the Chance of Breakage in the Genome Independent of Proximity to tRNAs, Tys, and LTRs
Because origins, tRNAs, and Ty/LTRs are clustered throughout the genome, how do we interpret correlations between these features and breakpoints? One possibility is that the presence of a breakpoint results from the presence of only one of the three types of features, whereas the other two types of features are clustered with the first feature for an independent reason. A second possibility is that breakpoint formation requires two or more features acting codependently. Lastly, the likelihood of a genomic region experiencing a rearrangement may increase by the additive and independent effects of each feature.
We distinguished between these possibilities by employing regression analysis on the S. cerevisiae–K. waltii breakpoints. We did not have enough experimental breakpoints to perform regression analysis on those breakpoints. For the Kellis and Wolfe data sets and the hcARSs, we derived the simplest model that predicts the presence of a breakpoint as well as does a model with all genomic features dependent on each other (see Methods). The simplest model for both data sets was a completely additive model in which tRNAs and hcARSs independently increase the likelihood of a breakpoint being present (supplementary table S14, Supplementary Material online). The lack of LTRs in the model may result from the close overlap of tRNAs with Tys/LTRs. This model thereby suggests that tRNAs and origins directly and independently contribute to breakpoint formation.
As a further reduced model lacking origins was a significantly poorer predictor of breakpoint location, the regression analysis predicts that there should be a population of origins unassociated with nearby tRNAs and Tys/LTRs that are, nevertheless, correlated with breakpoints. To test the conclusions of the regression analysis, we removed all hcARSs, tRNAs, and Tys/LTRs that have another different kind of feature within 5 kb measured by minimal end point measures. This distance measurement is stricter than the 5-kb bins used for regression analysis. After removing features, there remained 250 hcARSs, 34 tRNAs, 12 Tys, and 23 LTRs, representing loss of about half the hcARSs and about a 10-fold reduction in the number of tRNAs, Tys, and LTRs. Using the minimal end point distance analysis, we obtained significant P values for the Kellis-defined S. cerevisiae–K. waltii breakpoints associated with isolated hcARSs, whereas Wolfe breakpoints were only weakly correlated with isolated origins (Kellis: P value = 0.0007; Wolfe: P value = 0.0454). To test the validity of this analysis, we repeated the analysis for isolated tRNAs and LTRs. Due to the substantial overlap of tRNAs and Ty/LTRs, we treated these features as one group so that only half of tRNAs and Ty/LTRs were lost when we removed clusters of features. In doing so we obtained significant correlations between the evolutionary breakpoints and tRNAs or LTRs (P values <0.0005) for both data sets. The results here are in agreement with the regression results. They agree with an overlap of tRNAs with Tys/LTRs, and, more importantly, they indicate that origins of replication are fragile sites independent of being associated with tRNAs, Tys, and LTRs.
Discussion
Genome rearrangements alter the genome in ways that are neutral, beneficial, or detrimental to the organism. These rearrangements are the result of genome breakage followed by a repair process that alters the gene order in the genome. In an effort to understand cis-acting factors that may contribute to such rearrangements, we examined evolutionary breaks in synteny defined between S. cerevisiae and another yeast species (K. waltii, an inferred ancestor, or a more closely related species, each compared pairwise with S. cerevisiae) as well as breaks generated under laboratory conditions in S. cerevisiae—“experimental breaks.” With both sets of breakpoints, we found associations with previously known features—tRNAs, Tys, and LTRs—and we also identified origins of replication as an additional element associated with genome fragility. Specifically, we found a significant correlation between breakpoints and sequences observed to initiate replication not only in plasmid maintenance assays but also in their native chromosomal locations and with generally early-firing times in S phase. Not surprisingly, given the known colocalization of breakpoints with tRNAs, Tys, and LTRs, we also noted a colocalization of origins with these elements. However, we found no evidence that the correlation between origins and S. cerevisiae–K. waltii breakpoints depends on one of these other features.
Recently, using a simulation that was similar to our enrichment analysis, Gordon et al. (2009) also reported correlations among tRNAs, origins, and breakpoints mapped between S. cerevisiae and a hypothetical ancestor of K. waltii and S. cerevisiae, constructed from more species than the ancestor used in our study. As in our study, though both tRNAs and origins correlate significantly with S. cerevisiae–Ancestor breakpoints, the correlation between tRNAs and breakpoints appeared stronger than that for origins and breakpoints. Likewise, they found a strong correlation between tRNAs and origins themselves, although they did not attempt to unravel whether tRNAs and origins act independently, synergistically, or codependently with each other in their association with breakpoints. They additionally analyzed genes specifically gained in the lineage leading to S. cerevisiae and found these sites to be correlated with tRNAs and origins despite finding that gene gain sites are not correlated with breakpoints. As we did not consider gene gains in either S. cerevisiae or K. waltii, this result of Gordon et al. (2009) suggests that we have been conservative in estimating the number of genome-altering events associated with origins.
It is formally possible that origins arise or are inserted at sites of breakage. A recent report using human cell lines suggests that there is overreplication of 200-bp regions containing replication origins (Gomez and Antequera 2008). There is also evidence that nuclear fragments of DNA can repair dsDNA breaks by microhomology-mediated nonhomologous end joining (NHEJ) (Moore and Haber 1996; Ricchetti et al. 1999; Yu and Gabriel 1999). Hence, if fragments of origin sequence are present in the nucleus, they may have the potential to participate in microhomology-mediated NHEJ. This mechanism would result in origins migrating to break sites rather than existing prior to breakage, thereby leading to origins being found at sites that are the product of an evolutionary rearrangement. We observed in our analysis of experimentally generated breakpoints, however, that origins exist at sites that come to experience breakage. Therefore, our work suggests that origins play a role in creating breaks or misrepairing them, ultimately producing genome rearrangements.
Rearrangements in the genome are likely only observable when a threshold probability of breakage is exceeded and a nonconservative repair process is used. It is possible that while tRNAs, LTRs, and origins make individual contributions to producing rearrangements, the additive effects of these features together greatly increases the likelihood of a rearrangement event occurring. This notion may explain why it is frequently observed that rearrangements occur at sites containing a combination of these three features.
How might replication origins increase the propensity of a region to experience breakage? Do origins participate in the initial break or the subsequent repair? Although these questions have yet to be experimentally studied, we can review the breakpoint and repair literature to assess possible mechanisms of origin involvement in breakpoint formation. In considering breakage, is the origin sequence prone to breakage? Yeast origins of replication are composed of an approximately 200-bp sequence with a degenerate 17-bp AT-rich consensus sequence present in a nucleosome-free region (Nieduszynski et al. 2006). The AT-rich origin consensus sequence or its presence in a nucleosome-free region could make origins particularly vulnerable to DNA breakage. It is possible that the AT content of origins promotes local DNA unwinding, increasing the chance of breakage in origin sequences. However, because origins in S. cerevisiae are thought to be bound throughout the cell cycle by origin recognition complex (ORC) proteins (Diffley et al. 1994; Liang and Stillman 1997), any instability incurred by AT content or the lack of nucleosomes is likely to be transient.
Although the ORC at replication origins may protect AT- rich DNA, it may also be a source of instability. Protein complexes bound at specific sites throughout the genome present obstacles to the replication fork (Ivessa et al. 2003). For example, one mechanism by which tRNAs are thought to generate genomic instability is by bound RNA polymerase III inferring with the replication machinery, thereby causing fork pausing and breakage (Deshpande and Newlon 1996; Ivessa et al. 2003). One prediction of this idea is that origins bound by ORC but passively replicated by a fork emanating from a nearby origin would experience fork pausing and thus should appear to be correlated with breakpoints. If not replicated by a nearby origin, these passively replicated origins would fire late in S phase. Therefore, under this hypothesis, we would expect that later-firing origins would be correlated with breakpoints. We found, however, the converse to be the case—that early-firing origins are correlated with breakpoints, whereas later-firing origins are not. Thus, our data do not corroborate the notion of instability being generated by passive replication through an unfired origin.
Are there steps during DNA replication that might be inherently destabilizing? During replication of the lagging strand and processing of Okazaki fragments, ssDNA is produced (Garg and Burgers 2005). In general, any disruption of replication progression produces an accumulation of ssDNA believed to be mainly on the lagging strand (Sogo et al. 2002). As ssDNA is chemically less stable than dsDNA (Lindahl 1993), it may be more prone to nicking or forming secondary structures that are substrates for repair. ssDNA formation as currently understood, however, is unlikely to produce the observed correlations; unless there is notably more ssDNA exposed directly around origins or if ssDNA around origins persists longer, ssDNA generated during replication should lead to instability throughout the genome and not to hot spots around origins.
Alternatively, origins may not be more prone to breakage but may cause or be involved in an error-prone repair pathway. Several lines of reasoning are consistent with this hypothesis. First, with around 400 origins in the yeast genome, could the origin consensus sequence act as a repeated element capable of HR? As there are over 10,000 matches to the consensus sequence in the yeast genome (Nieduszynski et al. 2006), it seems improbable that the correlation between origins and breakpoints would be observed if these short repeats were functioning as sites of recombination. A correlation could be observed if an event during replication promoted recombination at origins. Though poorly understood, replication initiation has been associated with HR-independent recombination in S. cerevisiae (Lopes et al. 2003) and HR-dependent recombination in Saccharomyces pombe cells (Segurado et al. 2002). These recombination events are believed to help establish sister chromatid cohesion. Perhaps these events could lead to nonallelic recombination around origins and produce origin-proximal rearrangements.
Second, Payen et al. (2008) showed that whereas duplications produced through an HR-dependent mechanism could be interchromosomal or intrachromosomal events, break events repaired through an HR-independent mechanism produced smaller segmental duplications that were almost exclusively intrachromosomal. Though not conclusive, in analyzing rearrangement size, we found a better correlation of replication origins with small-scale rather than with large-scale rearrangements (data not shown). Following this reasoning, we would predict that origins contribute to breakpoint formation via an HR-independent mechanism.
Third, phosphorylation of Sae2 by Cdc28/Cdk1 has been shown to be involved in regulating the switch from error-prone NHEJ repair, which predominates in G1, to error-free HR in S phase (Huertas et al. 2008). Perhaps this switch is not complete early in S phase. Thus, any damage generated as replication forks emanate from early origins could undergo error-prone repair, leading to breakpoints proximal to early-firing origins, whereas damage generated at late-firing origins would be more likely to undergo error-free repair.
Another recent work also has suggested a link between early-firing origins and genome instability. Frum et al. (2008) observed replication pausing near newly replicated origins in early S phase in human fibroblasts. Separately, Caldwell et al. (2008) demonstrated that early episomal origins contribute to signaling the S phase DNA damage checkpoint and that such signaling is dependent on forks encountering a replication pause. Is it just a coincidence that only early origins are correlated with tRNAs (and Tys/LTRs) (supplementary table S13, Supplementary Material online), which have been shown to cause replication fork pausing (Deshpande and Newlon 1996; Voineagu et al. 2008)? This coincidence raises some questions on the genomic organization of origins, tRNAs, and associated Tys/LTRs. For example, are early-firing origins with a proximal tRNA selected for via increased S phase DNA damage checkpoint signaling? Or do proximal tRNAs impact origin-firing time by some manner?
Relevant to this discussion is whether origin location is conserved. Among hemiascomycetes, origins have only been completely mapped in S. cerevisiae. Though there is evidence suggesting that origins show synteny over the sensu stricto species (Yang et al. 1999; Nieduszynski et al. 2006), whether synteny of origins is extended to K. waltii is uncertain. Unlike other genomic features, origins show an exceptional level of redundancy to the point where an S. cerevisiae chromosome lacking all known origins segregates normally 97% of the time (Dershowitz et al. 2007). Therefore, the interpretation of the correlation between evolutionarily defined breakpoints and extant S. cerevisiae origins is not straightforward; it can only be known that origins now exist at sites that underwent a genome rearrangement. It is only in studying the recently generated experimental breakpoints that we can directly show that origins are located at sites that experience rearrangements. A cursory look at tRNA conservation between S. cerevisiae and K. waltii reveals that about 50% of tRNAs are syntenically conserved between these species. These tRNAs are located both within and at the ends of syntenic blocks. We infer then that tRNAs are capable of participating in as well as surviving breakage and repair events. Experimental work is called for to verify the role of origins in genome rearrangements and to determine how origins are affected following a rearrangement. Investigating the effect of origins on genome stability and the fate of origins after a break event may reveal new mechanisms in genome evolution and further our understanding of genome architecture.
Supplementary Material
Supplementary figs. S1–S3, tables S1–S14, and section Resources and Datasets are available at Genome Biology and Evolution online (http://www.oxfordjournals.org/our_journals/gbe/)
Funding
This work was supported by the National Institute of General Medical Sciences (grant number 18926 to B.J.B. and M.K.R.; Genetics Predoctoral Training Program to S.C.D).
Supplementary Material
Acknowledgments
We are grateful to Josh Akey for his guidance on the statistical methods, computational resources, and for comments on the manuscript. We thank Ken Wolfe for answering our questions regarding the Ancestor. We thank Maitreya Dunham and Kim Lindstrom for helpful discussions and critical comments on the manuscript.
References
- Argueso JL, et al. Double-strand breaks associated with repetitive DNA can reshape the genome. Proc Natl Acad Sci USA. 2008;105:11845–11850. doi: 10.1073/pnas.0804529105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachman N, Eby Y, Boeke JD. Local definition of Ty1 target preference by long terminal repeats and clustered tRNA genes. Genome Res. 2004;14:1232–1247. doi: 10.1101/gr.2052904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolton EC, Boeke JD. Transcriptional interactions between yeast tRNA genes, flanking genes and Ty elements: a genomic point of view. Genome Res. 2003;13:254–263. doi: 10.1101/gr.612203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borde V, et al. Association of Mre11p with double-strand break sites during yeast meiosis. Mol Cell. 2004;13:389–401. doi: 10.1016/s1097-2765(04)00034-6. [DOI] [PubMed] [Google Scholar]
- Byrne KP, Wolfe KH. The Yeast Gene Order Browser: combining curated homology and syntenic context reveals gene fate in polyploid species. Genome Res. 2005;15:1456–1461. doi: 10.1101/gr.3672305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caldwell JM, et al. Orchestration of the S-phase and DNA damage checkpoint pathways by replication forks from early origins. J Cell Biol. 2008;180:1073–1086. doi: 10.1083/jcb.200706009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cha RS, Kleckner N. ATR homolog Mec1 promotes fork progression, thus averting breaks in replication slow zones. Science. 2002;297:602–606. doi: 10.1126/science.1071398. [DOI] [PubMed] [Google Scholar]
- Cherry JM, et al. SGD: Saccharomyces Genome Database. Nucleic Acids Res. 1998;26:73–79. doi: 10.1093/nar/26.1.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark AG, et al. Evolution of genes and genomes on the Drosophila phylogeny. Nature. 2007;450:203–218. doi: 10.1038/nature06341. [DOI] [PubMed] [Google Scholar]
- Cohen O, et al. Cartographic study: breakpoints in 1574 families carrying human reciprocal translocations. Hum Genet. 1996;97:659–667. doi: 10.1007/BF02281879. [DOI] [PubMed] [Google Scholar]
- Darai-Ramqvist E, et al. Segmental duplications and evolutionary plasticity at tumor chromosome break-prone regions. Genome Res. 2008;18:370–379. doi: 10.1101/gr.7010208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darling AE, Miklos I, Ragan MA. Dynamics of genome rearrangement in bacterial populations. PLoS Genet. 2008;4:e1000128. doi: 10.1371/journal.pgen.1000128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dershowitz A, et al. Linear derivatives of Saccharomyces cerevisiae chromosome III can be maintained in the absence of autonomously replicating sequence elements. Mol Cell Biol. 2007;27:4652–4663. doi: 10.1128/MCB.01246-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshpande AM, Newlon CS. DNA replication fork pause sites dependent on transcription. Science. 1996;272:1030–1033. doi: 10.1126/science.272.5264.1030. [DOI] [PubMed] [Google Scholar]
- Dietrich FS, et al. The Ashbya gossypii genome as a tool for mapping the ancient Saccharomyces cerevisiae genome. Science. 2004;304:304–307. doi: 10.1126/science.1095781. [DOI] [PubMed] [Google Scholar]
- Diffley JF, Cocker JH, Dowell SJ, Rowley A. Two steps in the assembly of complexes at yeast replication origins in vivo. Cell. 1994;78:303–316. doi: 10.1016/0092-8674(94)90299-2. [DOI] [PubMed] [Google Scholar]
- Dujon B. Yeasts illustrate the molecular mechanisms of eukaryotic genome evolution. Trends Genet. 2006;22:375–387. doi: 10.1016/j.tig.2006.05.007. [DOI] [PubMed] [Google Scholar]
- Dunham MJ, et al. Characteristic genome rearrangements in experimental evolution of Saccharomyces cerevisiae. Proc Natl Acad Sci USA. 2002;99:16144–16149. doi: 10.1073/pnas.242624799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn B, Sherlock G. Reconstruction of the genome origins and evolution of the hybrid lager yeast Saccharomyces pastorianus. Genome Res. 2008;18:1610–1623. doi: 10.1101/gr.076075.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng W, et al. Genomic mapping of single-stranded DNA in hydroxyurea-challenged yeasts identifies origins of replication. Nat Cell Biol. 2006;8:148–155. doi: 10.1038/ncb1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer G, James SA, Roberts IN, Oliver SG, Louis EJ. Chromosomal evolution in Saccharomyces. Nature. 2000;405:451–454. doi: 10.1038/35013058. [DOI] [PubMed] [Google Scholar]
- Frum RA, Chastain PD, 2nd, Qu P, Cohen SM, Kaufman DG. DNA replication in early S phase pauses near newly activated origins. Cell Cycle. 2008;7:1440–1448. doi: 10.4161/cc.7.10.5879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garfinkel DJ. Genome evolution mediated by Ty elements in Saccharomyces. Cytogenet Genome Res. 2005;110:63–69. doi: 10.1159/000084939. [DOI] [PubMed] [Google Scholar]
- Garg P, Burgers PM. DNA polymerases that propagate the eukaryotic DNA replication fork. Crit Rev Biochem Mol Biol. 2005;40:115–128. doi: 10.1080/10409230590935433. [DOI] [PubMed] [Google Scholar]
- Gomez M, Antequera F. Overreplication of short DNA regions during S phase in human cells. Genes Dev. 2008;22:375–385. doi: 10.1101/gad.445608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon JL, Byrne KP, Wolfe KH. Additions, losses, and rearrangements on the evolutionary route from a reconstructed ancestor to the modern Saccharomyces cerevisiae genome. PLoS Genet. 2009;5:e1000485. doi: 10.1371/journal.pgen.1000485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- Huertas P, Cortes-Ledesma F, Sartori AA, Aguilera A, Jackson SP. CDK targets Sae2 to control DNA-end resection and homologous recombination. Nature. 2008;455:689–692. doi: 10.1038/nature07215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes AL, Friedman R. Transposable element distribution in the yeast genome reflects a role in repeated genomic rearrangement events on an evolutionary time scale. Genetica. 2004;121:181–185. doi: 10.1023/b:gene.0000040383.51611.e3. [DOI] [PubMed] [Google Scholar]
- Hwang JY, et al. Smc5-Smc6 complex suppresses gross chromosomal rearrangements mediated by break-induced replications. DNA Repair (Amst) 2008;7:1426–1436. doi: 10.1016/j.dnarep.2008.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue K, Lupski JR. Molecular mechanisms for genomic disorders. Annu Rev Genomics Hum Genet. 2002;3:199–242. doi: 10.1146/annurev.genom.3.032802.120023. [DOI] [PubMed] [Google Scholar]
- Ivessa AS, et al. The Saccharomyces cerevisiae helicase Rrm3p facilitates replication past nonhistone protein-DNA complexes. Mol Cell. 2003;12:1525–1536. doi: 10.1016/s1097-2765(03)00456-8. [DOI] [PubMed] [Google Scholar]
- Kehrer-Sawatzki H, Cooper DN. Understanding the recent evolution of the human genome: insights from human-chimpanzee genome comparisons. Hum Mutat. 2007;28:99–130. doi: 10.1002/humu.20420. [DOI] [PubMed] [Google Scholar]
- Kellis M, Birren BW, Lander ES. Proof and evolutionary analysis of ancient genome duplication in the yeast Saccharomyces cerevisiae. Nature. 2004;428:617–624. doi: 10.1038/nature02424. [DOI] [PubMed] [Google Scholar]
- Kellis M, Patterson N, Endrizzi M, Birren B, Lander ES. Sequencing and comparison of yeast species to identify genes and regulatory elements. Nature. 2003;423:241–254. doi: 10.1038/nature01644. [DOI] [PubMed] [Google Scholar]
- Kidd JM, et al. Mapping and sequencing of structural variation from eight human genomes. Nature. 2008;453:56–64. doi: 10.1038/nature06862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JM, Vanguri S, Boeke JD, Gabriel A, Voytas DF. Transposable elements and genome organization: a comprehensive survey of retrotransposons revealed by the complete Saccharomyces cerevisiae genome sequence. Genome Res. 1998;8:464–478. doi: 10.1101/gr.8.5.464. [DOI] [PubMed] [Google Scholar]
- Lahortiga I, et al. Duplication of the MYB oncogene in T cell acute lymphoblastic leukemia. Nat Genet. 2007;39:593–595. doi: 10.1038/ng2025. [DOI] [PubMed] [Google Scholar]
- Lemoine FJ, Degtyareva NP, Kokoska RJ, Petes TD. Reduced levels of DNA polymerase delta induce chromosome fragile site instability in yeast. Mol Cell Biol. 2008;28:5359–5368. doi: 10.1128/MCB.02084-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemoine FJ, Degtyareva NP, Lobachev K, Petes TD. Chromosomal translocations in yeast induced by low levels of DNA polymerase a model for chromosome fragile sites. Cell. 2005;120:587–598. doi: 10.1016/j.cell.2004.12.039. [DOI] [PubMed] [Google Scholar]
- Liang C, Stillman B. Persistent initiation of DNA replication and chromatin-bound MCM proteins during the cell cycle in cdc6 mutants. Genes Dev. 1997;11:3375–3386. doi: 10.1101/gad.11.24.3375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindahl T. Instability and decay of the primary structure of DNA. Nature. 1993;362:709–715. doi: 10.1038/362709a0. [DOI] [PubMed] [Google Scholar]
- Liti G, Louis EJ. Yeast evolution and comparative genomics. Annu Rev Microbiol. 2005;59:135–153. doi: 10.1146/annurev.micro.59.030804.121400. [DOI] [PubMed] [Google Scholar]
- Lopes M, Cotta-Ramusino C, Liberi G, Foiani M. Branch migrating sister chromatid junctions form at replication origins through Rad51/Rad52-independent mechanisms. Mol Cell. 2003;12:1499–1510. doi: 10.1016/s1097-2765(03)00473-8. [DOI] [PubMed] [Google Scholar]
- Lowe TM, Eddy SR. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25:955–964. doi: 10.1093/nar/25.5.955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCune HJ, et al. The temporal program of chromosome replication: genomewide replication in clb5[Delta] Saccharomyces cerevisiae. Genetics. 2008;180:1833–1847. doi: 10.1534/genetics.108.094359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore JK, Haber JE. Capture of retrotransposon DNA at the sites of chromosomal double-strand breaks. Nature. 1996;383:644–646. doi: 10.1038/383644a0. [DOI] [PubMed] [Google Scholar]
- Nadeau JH, Taylor BA. Lengths of chromosomal segments conserved since divergence of man and mouse. Proc Natl Acad Sci USA. 1984;81:814–818. doi: 10.1073/pnas.81.3.814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieduszynski CA, Hiraga S, Ak P, Benham CJ, Donaldson AD. OriDB: a DNA replication origin database. Nucleic Acids Res. 2007;35:D40–D46. doi: 10.1093/nar/gkl758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieduszynski CA, Knox Y, Donaldson AD. Genome-wide identification of replication origins in yeast by comparative genomics. Genes Dev. 2006;20:1874–1879. doi: 10.1101/gad.385306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payen C, Koszul R, Dujon B, Fischer G. Segmental duplications arise from Pol32-dependent repair of broken forks through two alternative replication-based mechanisms. PLoS Genet. 2008;4:e1000175. doi: 10.1371/journal.pgen.1000175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng Q, Pevzner PA, Tesler G. The fragile breakage versus random breakage models of chromosome evolution. PLoS Comput Biol. 2006;2:e14. doi: 10.1371/journal.pcbi.0020014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pevzner P, Tesler G. Genome rearrangements in mammalian evolution: lessons from human and mouse genomes. Genome Res. 2003;13:37–45. doi: 10.1101/gr.757503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollack JR, et al. Microarray analysis reveals a major direct role of DNA copy number alteration in the transcriptional program of human breast tumors. Proc Natl Acad Sci USA. 2002;99:12963–12968. doi: 10.1073/pnas.162471999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricchetti M, Fairhead C, Dujon B. Mitochondrial DNA repairs double-strand breaks in yeast chromosomes. Nature. 1999;402:96–100. doi: 10.1038/47076. [DOI] [PubMed] [Google Scholar]
- Sankoff D, Deneault M, Turbis P, Allen C. Chromosomal distributions of breakpoints in cancer, infertility, and evolution. Theor Popul Biol. 2002;61:497–501. doi: 10.1006/tpbi.2002.1599. [DOI] [PubMed] [Google Scholar]
- Segurado M, Gomez M, Antequera F. Increased recombination intermediates and homologous integration hot spots at DNA replication origins. Mol Cell. 2002;10:907–916. doi: 10.1016/s1097-2765(02)00684-6. [DOI] [PubMed] [Google Scholar]
- Sharp AJ, Cheng Z, Eichler EE. Structural variation of the human genome. Annu Rev Genomics Hum Genet. 2006;7:407–442. doi: 10.1146/annurev.genom.7.080505.115618. [DOI] [PubMed] [Google Scholar]
- Smith DI, McAvoy S, Zhu Y, Perez DS. Large common fragile site genes and cancer. Semin Cancer Biol. 2007;17:31–41. doi: 10.1016/j.semcancer.2006.10.003. [DOI] [PubMed] [Google Scholar]
- Sogo JM, Lopes M, Foiani M. Fork reversal and ssDNA accumulation at stalled replication forks owing to checkpoint defects. Science. 2002;297:599–602. doi: 10.1126/science.1074023. [DOI] [PubMed] [Google Scholar]
- Stankiewicz P, Lupski JR. Genome architecture, rearrangements and genomic disorders. Trends Genet. 2002;18:74–82. doi: 10.1016/s0168-9525(02)02592-1. [DOI] [PubMed] [Google Scholar]
- VanHulle K, et al. Inverted DNA repeats channel repair of distant double-strand breaks into chromatid fusions and chromosomal rearrangements. Mol Cell Biol. 2007;27:2601–2614. doi: 10.1128/MCB.01740-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voineagu I, Narayanan V, Lobachev KS, Mirkin SM. Replication stalling at unstable inverted repeats: interplay between DNA hairpins and fork stabilizing proteins. Proc Natl Acad Sci USA. 2008;105:9936–9941. doi: 10.1073/pnas.0804510105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver BA, Silk AD, Montagna C, Verdier-Pinard P, Cleveland DW. Aneuploidy acts both oncogenically and as a tumor suppressor. Cancer Cell. 2007;11:25–36. doi: 10.1016/j.ccr.2006.12.003. [DOI] [PubMed] [Google Scholar]
- Weir BA, et al. Characterizing the cancer genome in lung adenocarcinoma. Nature. 2007;450:893–898. doi: 10.1038/nature06358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyrick JJ, et al. Genome-wide distribution of ORC and MCM proteins in S. cerevisiae: high-resolution mapping of replication origins. Science. 2001;294:2357–2360. doi: 10.1126/science.1066101. [DOI] [PubMed] [Google Scholar]
- Yang C, Theis JF, Newlon CS. Conservation of ARS elements and chromosomal DNA replication origins on chromosomes III of Saccharomyces cerevisiae and S. carlsbergensis. Genetics. 1999;152:933–941. doi: 10.1093/genetics/152.3.933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu X, Gabriel A. Patching broken chromosomes with extranuclear cellular DNA. Mol Cell. 1999;4:873–881. doi: 10.1016/s1097-2765(00)80397-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.