

Abstract
Osmolytes are small molecules that play a central role in cellular homeostasis and the stress response by maintaining protein thermodynamic stability at controlled levels. The underlying physical chemistry that describes how different osmolytes impact folding free energy is well understood, however little is known about their influence on other crucial aspects of protein behavior, such as native-state conformational changes. Here we investigate this issue with the Hsp90 molecular chaperone, a large dimeric protein that populates a complex conformational equilibrium. Using small angle X-ray scattering we observe dramatic osmolyte-dependent structural changes within the native ensemble. The degree to which different osmolytes affect the Hsp90 conformation strongly correlates with thermodynamic metrics of their influence on stability. This observation suggests that the well-established osmolyte principles that govern stability also apply to large-scale conformational changes, a proposition that is corroborated by structure-based fitting of the scattering data, surface area comparisons and m-value analysis. This approach shows how osmolytes affect a highly cooperative open/closed structural transition between two conformations that differ by a domain-domain interaction. Hsp90 adopts an additional ligand-specific conformation in the presence of ATP and we find that osmolytes do not significantly affect this conformational change. Together, these results extend the scope of osmolytes by suggesting that they can maintain protein conformational heterogeneity at controlled levels using similar underlying principles that allow them to maintain protein stability; however the relative impact of osmolytes on different structural states can vary significantly.
Keywords: Hsp90, osmolyte, small angle x-ray scattering, conformational change
Introduction
Osmolytes are small molecules that help maintain protein homeostasis under harsh environments and stress conditions. While their primary function involves cell volume regulation,1 many osmolytes have additionally been selected for their potent influence on protein folding and stability. Numerous studies have demonstrated that protecting osmolytes, such as trimethylamine N-oxide (TMAO), can force thermodynamically unstable proteins to fold2 and stabilize folded proteins to withstand harsh conditions such as high temperature and denaturant concentrations. This favorable folding property of TMAO operates in an additive and independent manner with urea,3–5 allowing counteraction of urea's destabilizing effects on protein stability in urea-rich cells.6 Indeed, urea and TMAO are often found in a 2:1 molar ratio in marine cartilaginous fish, which in vitro has been shown to result in minimally changed stability.6,7 Despite their importance to cellular homeostasis, the influence of osmolytes on crucial aspects of protein behavior, such as native state conformation, is much less understood than their influence on folding.
The underlying physical chemistry behind the strong ability of osmolytes to affect protein stability is strikingly simple. Systematic transfer free energy measurements have demonstrated that urea and strongly protecting osmolytes predominantly affect stability via their influence on the protein backbone, with sidechains making only a minimal contribution.8–10 This simple experimental observation demonstrates that the osmolyte mechanism is universal (i.e. largely independent of primary sequence), which clearly highlights their general utility in combating denaturing stresses. Since most backbone groups in folded proteins are not solvent accessible, osmolytes primarily affect stability via changes in the free energy of the unfolded state. Protecting osmolytes, such as TMAO, betaine, and sarcosine interact unfavorably with the unfolded state providing relative stability to the folded state.11,12 Conversely the denaturant urea can make hydrogen-bonds to the protein backbone13 that stabilize the unfolded state relative to purely aqueous solvation. This physical mechanism is consistent with the strong correlation between folding induced changes in surface area and the slope of free energy change with increasing urea concentration (m-value14).
These experimental results have significantly advanced the understanding of osmolyte effects on protein structure and stability. For example, the Tanford transfer model, which utilizes transfer free energies to predict folding m-values, is in strong agreement with the numerous proteins for which experimental data are available.8,9 In this approach, evaluation of the numbers and kinds of groups that become newly exposed upon unfolding can be used with experimentally determined group transfer free energies to predict the degree to which an osmolyte will affect protein stability.8,15 These transfer free energies have also been used to appropriately weight conformations sampled in simulations, subsequently predicting a wide variety of experimentally measured behaviors of protein L and cold shock protein.16 Finally, the transfer free energy values themselves have been found to correlate with osmolyte fractional polar surface area, and a simple energetic model for osmolyte/backbone interactions based on this observation has strong predictive power for their influence on protein stability.17
The general nature of the osmolyte mechanism suggests that they can affect other protein transformations besides folding. One possibility is osmolyte-driven changes in native-state conformation, which, like protein folding, can bury a significant surface area. Such effects would be important in sorting out the influence of counteracting osmolytes (such as urea and TMAO) on protein function. Many proteins exist in an ensemble of native conformations that can be regulated by ligand binding or post-translational modifications. Furthermore, the spatial organization of multidomain proteins is central to the efficiency of many multistep enzymatic reactions. It is likely that the conformational equilibria of these delicately balanced systems can be influenced by osmolytes.
Here we investigate the influence of osmolytes on native-state conformation by utilizing the Hsp90 molecular chaperone, a large dimeric protein that populates a wide range of distinct states (Fig. 1). The Hsp90 monomer is composed of three domains. The N-terminal domain (NTD; Figure 1, light green) contains an ATP binding site, for which binding causes a reorientation at the NTD-middle domain (MD, light purple) and MD-C-terminal domain (CTD, aqua) interfaces resulting in closure via N-terminal dimerization. The CTD is a permanent dimerization site. In the open state, each domain contributes exposed hydrophobic surface postulated as important for substrate protein binding18. Closure to the ATP state buries a significant fraction of this surface area,19 suggesting that the Hsp90 conformation is directly coupled to substrate interaction.
Figure 1.
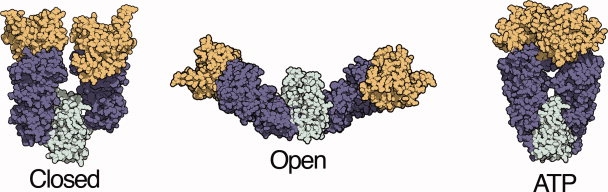
Distinct conformational states of the Hsp90 molecular chaperone. Previous crystallography, electron microscopy, and small angle x-ray scattering (SAXS) measurements have determined a multi-state conformational equilibrium for the Hsp90 homolog from E. coli, HtpG.20–22 The relative population of these conformations can be determined from SAXS measurements and structure-based fitting. The ATP state was modeled from a crystal structure of a yeast Hsp90 homolog.19,20 The closed state is similar to a crystal structure of the Hsp90 homolog from the ER, Grp94, and has been referred to as a Grp94-like state.
Hsp90 plays a unique role as a chaperone in that it not only facilitates folding but also enables certain classes of substrates, such as kinases and nuclear receptors, to become activated in the native state.23 Some of the signaling pathways that are regulated by Hsp90 are overactive in cancerous cells and accordingly inhibitors are being actively pursued as potential therapeutics.24 Understanding and manipulating the Hsp90 conformational equilibrium will be critical in efforts to elucidate its substrate interaction mechanism. Small angle X-ray scattering (SAXS) is a powerful tool for dissecting this conformational equilibrium, revealing that the open, closed, and ATP states coexist in an equilibrium that can be shifted by the binding of ATP (or the non-hydrolysable analog AMPPNP) and also by pH for the bacterial homolog (HtpG).20–22 The three states populated by HtpG (Fig. 1) have distinct scattering curves, and structure-based fitting can determine their relative populations. These characteristics make Hsp90 an ideal system for examining which conformational states are favored by different osmolytes, the extent to which osmolytes can shift a conformational equilibrium, and the interplay between ligand-induced and osmolyte-induced conformational changes.
Results
The Hsp90 from E. coli (HtpG) populates three conformations that can be clearly resolved by SAXS. The influence of osmolytes on these states was investigated by measurements of this chaperone in the presence of 1M each of the osmolyte solutions (TMAO, betaine, sarcosine, glycerol, or urea). Other well studied osmolytes such as sucrose, trehalose, and sorbitol show substantial background scattering that complicates their analysis (data not shown) and are not included. The scattering intensity was measured between Q values (4πsinθ /λ where 2θ is the scattering angle) of 0.01 to 0.3 Å−1, radially averaged and then buffer subtracted. The resulting data were transformed to an interatomic distance distribution, P(r), by Fourier transform. In the absence of osmolytes [Fig. 2(A), red curve], the distance distribution for HtpG at pH 7.5 agrees with previous studies.22
Figure 2.
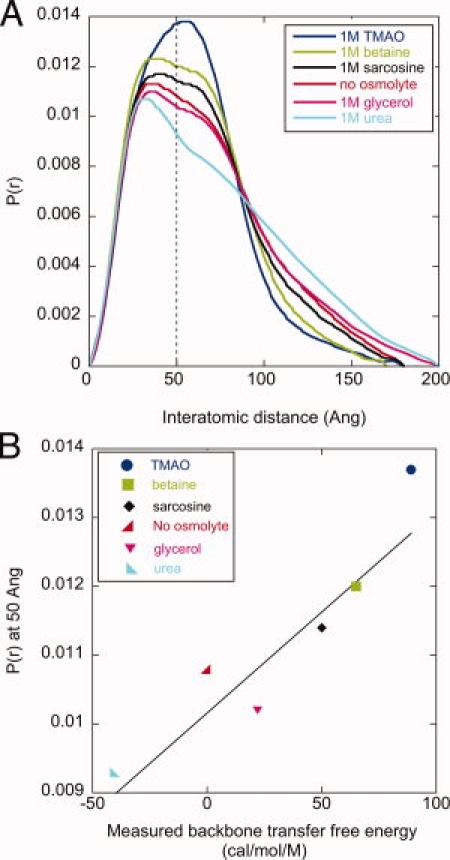
Osmolyte-dependent conformational changes in HtpG. A: The interatomic distance distribution of HtpG in the absence and presence of various 1M osmolyte solutions indicates dramatic, osmolyte-dependent conformational changes. P(r) values at 50 Å are highlighted by the dashed line. B: There is a strong correlation (R = 0.90) between the measured backbone transfer free energy of these osmolytes and their respective P(r) values at 50 Å. Buffer conditions: 50 mM TRIS pH 7.5, 50 mM KCl, 10 mM MgCl2, 1 mM DTT.
The addition of osmolytes produces striking structural changes that are evident in the distance distribution [Fig. 2(A)]. For the protecting osmolytes TMAO, betaine and sarcosine there is a noticeable contraction toward smaller distances, whereas for the denaturant urea there is a clear expansion. Glycerol, a weakly stabilizing osmolyte, has a minimal influence on the distance distribution. The differing influence of these osmolytes can be rank ordered by their respective P(r) values at a reference distance, such as 50 Å [dashed line, Fig. 2(A)]. This ordering: TMAO > betaine > sarcosine > glycerol > urea, is the same ranking for the degree to which these osmolytes affect protein stability, as discussed next.
The backbone transfer free energy of an osmolyte is the key metric that quantifies its influence on folding free energy.9,25 For example, the stabilizing osmolyte TMAO has a positive value (89 cal/mol/M) indicating an unfavorable backbone interaction (thus raising the free energy of the unfolded state and thereby providing relative stability to the native state), whereas the denaturant urea has a negative value (-41 cal/mol/M). These energetic values provide the dominant contribution of an osmolyte to protein stability. The rank ordering of these transfer free energies, TMAO (89 cal/mol/M) > betaine (65 cal/mol/M) > sarcosine (56 cal/mol/M) > glycerol (22 cal/mol/M) > urea (-41 cal/mol/M), is the same as their rank order influence on the P(r) value for HtpG. Indeed, we observe a strong correlation (R = 0.90) between the backbone transfer free energy values of each osmolyte and the P(r) value at 50 Å [Fig. 2(B)]. Since P(r) reflects the ratio of a conformational equilibrium, the quantity ln[P(r)] may be more appropriate for comparison against an energetic value. This comparison also yields a similarly high correlation coefficient (0.90, Supporting Information Fig. 1). This favorable correlation immediately suggests that the same physical principles that govern the osmolyte influence on protein folding also apply to large-scale conformational changes within the HtpG native ensemble.
The Hsp90 chaperone is highly conserved, with homologs in every kingdom of life.26 The domain structure is consistent among species, but the conformational ensemble in the absence of nucleotide is species-specific,21 with human and yeast Hsp90s favoring the open state, whereas the bacterial homolog (HtpG) populates a mixture of open and closed states. However, like HtpG, both yeast and human Hsp90 exhibit TMAO-induced contractions in their respective SAXS distance distributions (Supporting Information Fig. 2), indicating that the osmolyte effect is maintained within this chaperone family.
Osmolyte-driven conformation changes
Previous work with HtpG demonstrated that SAXS data can be fit to determine the relative populations of different conformers, which differ by only rigid body domain rearrangements.20 As expected for rigid body motion, the conformational changes induced by 1M TMAO are not associated with an increase in circular dichroism or fluorescence (data not shown), in contrast to large scale folding events. Indeed, similar to previous studies, the scattering data is well fit with a simple linear combination of the open and closed states. The computational method behind this procedure simply involves a linear least squares fit of additive combinations of precalculated P(r) curves for the different HtpG conformations (see Methods).
This two-state treatment results in an excellent agreement with the data. For example, the P(r) curves for HtpG in the absence of osmolytes and at 1M TMAO are shown with their respective fits in Figure 3. The fitting demonstrates that the open/closed equilibrium, 81/19%, is shifted to 34/66% in 1M TMAO. The visual agreement between the model and data is apparent, and is quantified by a low R-factor (similar to a crystallographic R-factor, see equation in Methods) of 2.1 and 2.2%, respectively.
Figure 3.
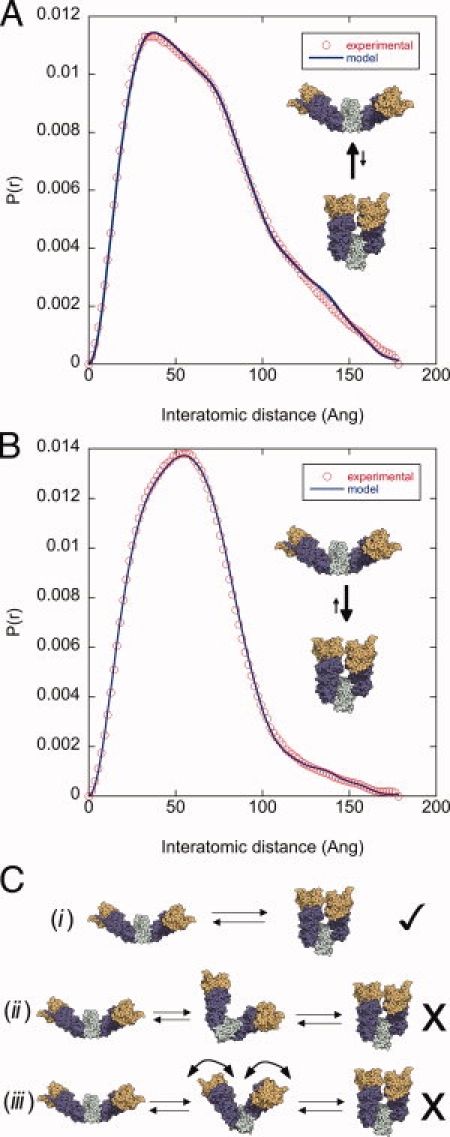
A cooperative, osmolyte-induced open/closed transition. In the absence of nucleotide, HtpG populates a two-state native equilibrium between the open and closed conformations. The SAXS data (open circles) of HtpG in the absence (A) and presence of 1M TMAO (B) can be fit very well by a two-state model (solid lines) with varying populations of the open and closed state. The fitting demonstrates that the open/closed equilibrium, 81/19%, is shifted to 34/66% in 1M TMAO. (C) Three models that differ greatly in their cooperativity can be distinguished by structure-based fitting. The most cooperative model (i) is the most appropriate for the scattering data, as discussed in Results.
This two-state model suggests that the open/closed transition is highly cooperative, with both monomers acting in concert despite the fact that neither the M-C nor the NTD interfaces are in contact (Fig. 1). This high cooperativity can be corroborated by investigating two alternative models [shown in Fig. 3(C)] that can be excluded by structure-based fitting. In the first (model ii), the individual monomers undergo independent transitions between the open and closed state, which would add a third mixed conformation into the equilibrium. In the absence of cooperativity, this mixed state would be maximally populated (50%) at an osmolyte concentration at the midpoint of the open/closed equilibrium. At 0.6M TMAO the open/closed equilibrium is 46.7/53.3% with an R-factor of 2.55%, providing an excellent opportunity to test this prediction. A three state fit indicates that a mixed state is either very minimally populated or not populated at all (open/closed/mixed is 53.1/46.1/0.8 with a minimally changed R-factor of 2.54%).
A second model with less cooperativity could involve a gradual shift to intermediate conformations that are between the open and closed states [Fig. 3(C), model iii], reminiscent of one-state protein folding models.27 This model implies that at intermediate TMAO concentrations, the conformational equilibrium would be better fit by a combination of the closed state and a new state that is more closed than the open state. We tested this prediction at 0.4M TMAO, where the open/closed equilibrium is 53/47% (R-factor of 2.3%). By fitting these data with the closed state and a state with rigid body freedom to explore intermediate conformations via different M-C opening angles (see Methods), we found that the optimal fit involved a state that is similar to the open state (Supporting Information Fig. 3).
Finally, the cooperativity is supported by an equivalent distribution of R-factors for the two-state fit at TMAO concentrations taken at regular intervals. These R-factors (which represent the quality of the two-state model) are: 2.2, 2.5, 2.3, 2.6, 2.6, 2.1% for TMAO concentrations of 0.0, 0.2, 0.4, 0.6, 0.8, 1.0M. This indicates that the two-state model is similarly appropriate over a wide range of the open/closed equilibrium.
Given that a two-state transition is an appropriate description of the osmolyte-induced conformational change, we can determine an open/closed equilibrium constant and subsequently its free energy. Since the open/closed equilibrium is controlled by a simple domain-domain interaction that buries a significant surface area, this should allow for a comparison with folding studies where a clear correlation has been observed between the folding-induced change in surface area and m-value (slope of the free energy change vs TMAO concentration). An equivalent conformational m-value can be determined from an osmolyte concentration series.
The TMAO titration shows a smooth change in the P(r) curves (Figure 4A) and subsequently the open/closed population (Figure 4B) and the associated free energy (inset). The linear extrapolation model28,29 predicts a linear relationship (with a slope of m) between RTln(K) and [TMAO], where K is the open/closed equilibrium constant. This concentration series shows an initially large slope of 2 kcal/mol/M at TMAO concentrations less than 0.4M, and above this concentration the slope reduces to 0.85 kcal/mol/M (inset, dashed lines). This change of m-value suggests larger surface area burial contributing to the conformational change at low TMAO concentrations. As discussed next, we can use the Tanford transfer model to suggest a structural interpretation of this behavior.
Figure 4.
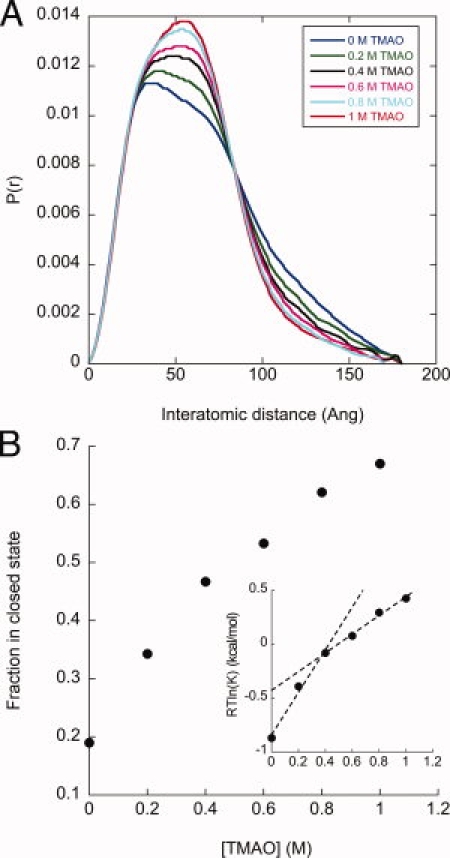
Determining m-values for the open/closed transition. (A) A titration of TMAO concentrations indicates a smooth shift in the P(r) distributions, reflecting a shift in the open/closed equilibrium. (B) The two-state fitting method was used to determine the population of the open and closed states, and define the equilibrium constant (K = [open]/[closed]). The inset plots the free energy of the open/closed equilibrium and is fit with two linear arms above and below 0.4M TMAO (dashed lines).
The open/closed conformational change involves rigid body rearrangements at the M-C interface (residue 500 marks the division between these domains). A predicted TMAO m-value associated with making/breaking this domain-domain interface can be determined from the Tanford transfer model, which scales the number of backbone and sidechain groups against the sum of experimentally measured transfer free energies associated with each group (details of this calculation method are given in other publications15). The predicted value for making/breaking the M-C interface is 0.7 kcal/mol/M, which is close to the limiting m-value at high TMAO concentrations (0.85 kcal/mol/M). This suggests that in the higher TMAO concentration regime the structural influence of TMAO is given by its role in forming the M-C domain interface. By contrast, at lower TMAO concentrations there are likely additional processes with significant surface area changes not reflected in the rigid rotation model. As discussed later, this type of variable m-value shows similarities to subglobal folding transitions that have been observed in osmolyte-induced protein folding/unfolding measurements.
Comparison of ligand-induced and osmolyte-induced conformational changes
The ATPase activity of Hsp90 is required for viability in eukaryotes30 and the binding of ATP is associated with an additional conformational change involving N-terminal dimerization (Fig. 1). This allows us to investigate the interplay between ligand- and osmolyte-induced conformational changes. The addition of a saturating concentration of 10 mM AMPPNP causes a significant change in conformation [compare Fig. 5(A) and Fig. 3(A)]. The two-state open/closed model no longer fits the data well (R-factor of 10%, Figure 5A dashed line), but a three-state open/closed/ATP fit results in good agreement (R-factor of 2.5%, solid line), similar to previous studies.22
Figure 5.
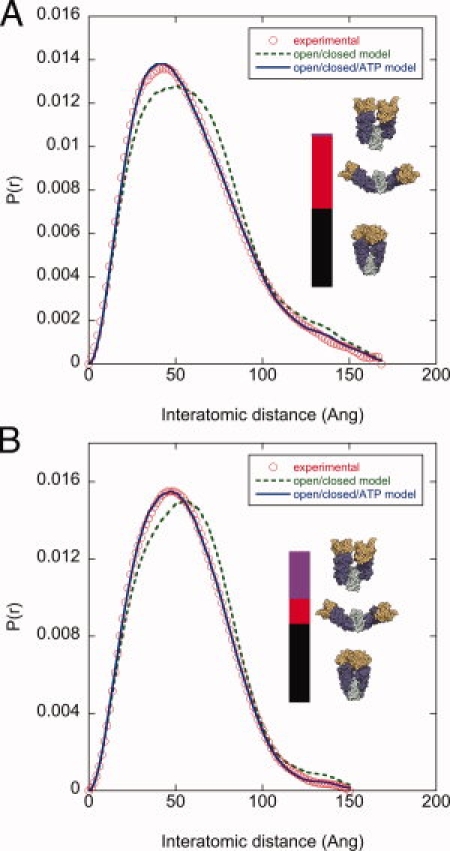
Comparison of TMAO- and nucleotide-induced conformational changes. (A) Addition of 10 mM of the non-hydrolysable ATP analog AMPPNP causes a significant population shift to the ATP state. The two-state model (open/closed, dashed line) does not fit the data, however a three state model (open/closed/ATP, solid line) fits well. The populations of the open/ATP states are depicted by the relative red/black bar heights. The negligible population of the closed state is indicated by the thin blue bar. (B) Subsequent addition of 1M TMAO does not substantially affect the population of the ATP state, but clearly affects the open/closed equilibrium (red/blue bar heights). Buffer conditions: 50 mM TRIS pH 7.5, 50 mM KCl, 10 mM MgCl2, 10 mM AMPPNP.
The addition of 10 mM AMPPNP and 1M TMAO shows this osmolyte affects the open/closed equilibrium but not the ATP population. The fitting (solid line, Figure 5B) indicates a similar population of the ATP state (57 versus 55% with and without 1M TMAO, respectively), but a dramatically different open/closed equilibrium (13/30% versus 45/0%). These changes are indicated by the relative height of the red/blue bars in Figure 5. To explore this effect over a broad range of conditions, we performed a concentration series of AMPPNP with and without TMAO, which demonstrates only modest TMAO-induced changes in the population of the ATP state [Supporting Information Fig. 4(A)]. Consistent with the lack of TMAO-induced changes in the ATP population, we measured no change in the rate of ATP hydrolysis by HtpG in 1M TMAO (data not shown).
The ATP state has a fully formed M-C interface and buries an additional 1900 Å2 via N-terminal dimerization, and yet the addition of TMAO alone does not populate this conformation. The observation that TMAO does not populate the more buried ATP state is possibly because of the complex, ligand-specific, reorganizations required to access this state. Indeed, ATP binding (i) restructures an N-terminal lid region that makes important dimer contacts, (ii) dramatically changes the organization with the middle domain via an interaction between an arginine (residue 336 in HtpG), and the ATP γ-phosphate, and (iii) is associated with the release of a β-strand which then makes cross monomer interactions that stabilize the N-terminal dimerization.18,19 These ligand-specific effects are unique to ATP, as ADP has been shown by SAXS to promote a conformational equilibrium similar to apo conditions. As expected then, in the presence of 1M TMAO the subsequent addition of ADP has no significant effect on the conformational equilibrium [Supporting Information Fig. 4(B)].
Discussion
Higher eukaryotes are enriched in genes composed of multiple domains. For these proteins, the native-state conformational ensemble is often critical to function and regulation. Hsp90 exemplifies the sensitivity of a native state conformation to solution conditions, demonstrating that the open/closed equilibrium can be significantly modulated in both directions by protecting/denaturing osmolytes [Fig. 2(A)]. This is the first example of a large osmolyte-induced conformational change in the native state. This effect is general within the Hsp90 family as we observe TMAO-induced structural contractions in Hsp90 homologs from E. coli, yeast, and human (Supporting Information Fig. 2). However, the structural states adopted by the yeast and human homologs may be different since the closed state has only been reported for HtpG and the Hsp90 homolog from the endoplasmic reticulum (Grp9431).
For HtpG, we observe a clear correlation between an osmolytes' measured backbone transfer free energy and its corresponding structural influence (Fig. 2). Given the dominance of the backbone transfer free energy in protein stability,8,15 this favorable correlation provides evidence that the underlying physical chemistry that governs stability can also be applied to these large conformational transitions. This connection between folding and large-scale conformational dynamics mirrors results for small-scale dynamics measured by hydrogen exchange, in which TMAO and urea have been demonstrated to have counteracting effects on amide exchange rates.32 The degree to which osmolytes affect different structural states can vary significantly however, as we observe a strong influence on the open/closed transition but a minimal influence on the ATP state population (Fig. 5).
We employed structure-based fitting of the scattering data [Fig. 3(A,B)], which indicates that the open/closed transition is remarkably co-operative with both monomers coupled despite their lack of direct contact through the M-C or NTD interfaces. Our analysis is unique in that plausible non-cooperative models can be directly excluded from structure-based fitting [Fig. 3(C)], whereas in folding other indirect arguments generally must be employed to assess cooperativity. These include (i) coincidence of CD and fluorescence, (ii) agreement between Van't Hoff and calorimetric enthalpy, and (iii) deviations from expected m-value (lower than expected values would indicate non-cooperative folding). One possible mechanism for the monomer-monomer coupling is an amphipathic helix pair (helix 21) that lies deep in the dimer cleft near the M-C interfaces. These helices, which show conformational heterogeneity in crystal structures, possibly make contacts that link the interface rearrangements on opposite monomers. Since these helices make important contacts in the proposed interaction site of the glucocorticoid receptor,33 this speculation suggests a coupling between substrate binding and Hsp90 conformation. Indeed, the closed state of HtpG has been shown to increase anti-aggregation activity for the citrate synthase model substrate.22
Given that TMAO induces a cooperative two-state conformational change, we can define the equilibrium constant, Gibbs free energy, and m-value associated with this change (Fig. 4). We find a biphasic m-value where at high TMAO concentrations the limiting value (0.85 kcal/mol/M) is close to the predicted value (0.7 kcal/mol/M) associated with making/breaking the M-C interface. At lower TMAO concentrations we observe a larger m-value (2 kcal/mol/M), suggesting additional folding/restructuring events, which remain undetected by SAXS, while significantly altering surface area exposure. Similar to this behavior, hydrogen exchange clearly demonstrates how subglobal folding/unfolding events can be induced at TMAO/urea concentrations outside of the threshold associated with global folding changes.34 This type of structural change can significantly alter the exposed surface area and contribute to nonlinear free energy dependence that has been observed with some proteins in the presence of GdmCl.35 Further studies will be required to determine the details of these TMAO-dependent surface changes in HtpG and their relative contribution to the m-value.
Materials and Methods
The expression and purification of the Hsp90 homologs from E. coli, yeast and human has been described previously.18,20,21,36 Small angle X-ray scattering was measured at the SIBYLS beamline (12.3.1) at the Advanced Light Source in Berkeley.37 Data was collected with 0.5, 2, and 5 second averaging times and the signal was circularly averaged from the detector. Each SAXS measurement was buffer subtracted with the appropriate osmolyte concentration. The raw scattering data, I(Q), was converted to an interatomic distance distribution with the GNOM program,38 using Dmax cut-off values that resulted in visually smooth tails in the P(r) distribution at high distance values. The backbone transfer free energy value for each osmolyte was compared with its respective P(r) value for HtpG at 50 Angstroms. These transfer free energy values are taken from previous studies,25 and represent an average of two independent experimental methods.
To determine the open/closed equilibrium from the HtpG scattering data, a two-state fit was calculated using the PRFIT program,22 and a similar three-state fit was employed by including an ATP state model of the crystal structure from the yeast Hsp90 homolog (2CG9). The R-factor that quantifies the quality of the fit is calculated similar to crystallographic analysis.
| (1) |
The rigid body modeling used to investigate intermediate conformations (Fig. 3, model iii) also uses the PRFIT program. The hinge point at the M-C domain interface was residue 500, as with previous studies,22 and was allowed to sample rotational motion along the x, y, and z axis rotations, starting with a course step size of 10° and then refining with 1° steps.
The open/closed m-value was determined by first estimating the M-C domain interface buried surface area. Since the rigid body model for the closed state does not include domain-packing details, we used a crystal structure of HtpG that has a formed M-C interface (2IOQ). The domain interface buried surface area was determined from calculating the buried area between the NM domain fragment (residues 1-500) and the C-terminal fragment (residues 501-624). An osmolyte specific m-value can be determined from the application of the Tanford transfer model to the structure and amino acid content of the M-C interface. This method scales the accessible surface area of the sidechain and backbone of residues by their measured transfer free energy values.15 The m-value for breaking the M-C interface was determined by taking the difference between the values for the entire monomer and the two fragments 1-500 and 501-624.
Acknowledgments
A special thanks to the Agard lab for many helpful discussions and to G. Hura for help with the SAXS data collection.
References
- 1.Yancey PH, Clark ME, Hand SC, Bowlus RD, Somero GN. Living with water stress: evolution of osmolyte systems. Science. 1982;217:1214–1222. doi: 10.1126/science.7112124. [DOI] [PubMed] [Google Scholar]
- 2.Baskakov I, Bolen DW. Forcing thermodynamically unfolded proteins to fold. J Biol Chem. 1998;273:4831–4834. doi: 10.1074/jbc.273.9.4831. [DOI] [PubMed] [Google Scholar]
- 3.Baskakov I, Wang A, Bolen DW. Trimethylamine-N-oxide counteracts urea effects on rabbit muscle lactate dehydrogenase function: A test of the counteraction hypothesis. Biophys J. 1998;74:2666–2673. doi: 10.1016/S0006-3495(98)77972-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Holthauzen LM, Bolen DW. Mixed osmolytes: the degree to which one osmolyte affects the protein stabilizing ability of another. Protein Sci. 2007;16:293–298. doi: 10.1110/ps.062610407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mello CC, Barrick D. Measuring the stability of partly folded proteins using TMAO. Protein Sci. 2003;12:1522–1529. doi: 10.1110/ps.0372903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang A, Bolen DW. A naturally occurring protective system in urea-rich cells: mechanism of osmolyte protection of proteins against urea denaturation. Biochemistry. 1997;36:9101–9108. doi: 10.1021/bi970247h. [DOI] [PubMed] [Google Scholar]
- 7.Lin TY, Timasheff SN. Why do some organisms use a urea-methylamine mixture as osmolyte? Thermodynamic compensation of urea and trimethylamine N-oxide interactions with protein. Biochemistry. 1994;33:12695–12701. doi: 10.1021/bi00208a021. [DOI] [PubMed] [Google Scholar]
- 8.Auton M, Bolen DW. Predicting the energetics of osmolyte-induced protein folding/unfolding. Proc Natl Acad Sci USA. 2005;102:15065–15068. doi: 10.1073/pnas.0507053102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Auton M, Holthauzen LM, Bolen DW. Anatomy of energetic changes accompanying urea-induced protein denaturation. Proc Natl Acad Sci USA. 2007;104:15317–15322. doi: 10.1073/pnas.0706251104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu Y, Bolen DW. The peptide backbone plays a dominant role in protein stabilization by naturally occurring osmolytes. Biochemistry. 1995;34:12884–12891. doi: 10.1021/bi00039a051. [DOI] [PubMed] [Google Scholar]
- 11.Qu Y, Bolen CL, Bolen DW. Osmolyte-driven contraction of a random coil protein. Proc Natl Acad Sci USA. 1998;95:9268–9273. doi: 10.1073/pnas.95.16.9268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bolen DW, Baskakov IV. The osmophobic effect: natural selection of a thermodynamic force in protein folding. J Mol Biol. 2001;310:955–963. doi: 10.1006/jmbi.2001.4819. [DOI] [PubMed] [Google Scholar]
- 13.Lim WK, Rosgen J, Englander SW. Urea, but not guanidinium, destabilizes proteins by forming hydrogen bonds to the peptide group. Proc Natl Acad Sci USA. 2009;106:2595–2600. doi: 10.1073/pnas.0812588106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Myers JK, Pace CN, Scholtz JM. Denaturant m values and heat capacity changes: relation to changes in accessible surface areas of protein unfolding. Protein Sci. 1995;4:2138–2148. doi: 10.1002/pro.5560041020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Auton M, Bolen DW. Application of the transfer model to understand how naturally occurring osmolytes affect protein stability. Methods Enzymol. 2007;428:397–418. doi: 10.1016/S0076-6879(07)28023-1. [DOI] [PubMed] [Google Scholar]
- 16.O'brien EP, Ziv G, Haran G, Brooks BR, Thirumalai D. Effects of denaturants and osmolytes on proteins are accurately predicted by the molecular transfer model. Proc Natl Acad Sci USA. 2008;105:13403–13408. doi: 10.1073/pnas.0802113105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Street TO, Bolen DW, Rose GD. A molecular mechanism for osmolyte-induced protein stability. Proc Natl Acad Sci USA. 2006;103:13997–14002. doi: 10.1073/pnas.0606236103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shiau AK, Harris SF, Southworth DR, Agard DA. Structural Analysis of E. coli hsp90 reveals dramatic nucleotide-dependent conformational rearrangements. Cell. 2006;127:329–340. doi: 10.1016/j.cell.2006.09.027. [DOI] [PubMed] [Google Scholar]
- 19.Ali MM, Roe SM, Vaughan CK, Meyer P, Panaretou B, Piper PW, Prodromou C, Pearl LH. Crystal structure of an Hsp90-nucleotide-p23/Sba1 closed chaperone complex. Nature. 2006;440:1013–1017. doi: 10.1038/nature04716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Krukenberg KA, Forster F, Rice LM, Sali A, Agard DA. Multiple conformations of E. coli Hsp90 in solution: insights into the conformational dynamics of Hsp90. Structure. 2008;16:755–765. doi: 10.1016/j.str.2008.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Southworth DR, Agard DA. Species-dependent ensembles of conserved conformational states define the Hsp90 chaperone ATPase cycle. Mol Cell. 2008;32:631–640. doi: 10.1016/j.molcel.2008.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Krukenberg KA, Southworth DR, Street TO, Agard DA. pH-Dependent Conformational Changes in Bacterial Hsp90 Reveal a Grp94-Like Conformation at pH 6 That Is Highly Active in Suppression of Citrate Synthase Aggregation. J Mol Biol. 2009;390:278–291. doi: 10.1016/j.jmb.2009.04.080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Young JC, Moarefi I, Hartl FU. Hsp90: a specialized but essential protein-folding tool. J Cell Biol. 2001;154:267–273. doi: 10.1083/jcb.200104079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pearl LH, Prodromou C, Workman P. The Hsp90 molecular chaperone: an open and shut case for treatment. Biochem J. 2008;410:439–453. doi: 10.1042/BJ20071640. [DOI] [PubMed] [Google Scholar]
- 25.Auton M, Bolen DW. Additive transfer free energies of the peptide backbone unit that are independent of the model compound and the choice of concentration scale. Biochemistry. 2004;43:1329–1342. doi: 10.1021/bi035908r. [DOI] [PubMed] [Google Scholar]
- 26.Chen B, Zhong D, Monteiro A. Comparative genomics and evolution of the HSP90 family of genes across all kingdoms of organisms. BMC Genomics. 2006;7:156. doi: 10.1186/1471-2164-7-156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sadqi M, Fushman D, Munoz V. Atom-by-atom analysis of global downhill protein folding. Nature. 2006;442:317–321. doi: 10.1038/nature04859. [DOI] [PubMed] [Google Scholar]
- 28.Greene RF, Pace CN. Urea and guanidine hydrochloride denaturation of ribonuclease, lysozyme, alpha-chymotrypsin, and beta-lactoglobulin. J Biol Chem. 1974;249:5388–5393. [PubMed] [Google Scholar]
- 29.Santoro MM, Bolen DW. Unfolding free energy changes determined by the linear extrapolation method. 1. Unfolding of phenylmethanesulfonyl alpha-chymotrypsin using different denaturants. Biochemistry. 1988;27:8063–8068. doi: 10.1021/bi00421a014. [DOI] [PubMed] [Google Scholar]
- 30.Obermann WM, Sondermann H, Russo AA, Pavletich NP, Hartl FU. In vivo function of Hsp90 is dependent on ATP binding and ATP hydrolysis. J Cell Biol. 1998;143:901–910. doi: 10.1083/jcb.143.4.901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dollins DE, Warren JJ, Immormino RM, Gewirth DT. Structures of GRP94-nucleotide complexes reveal mechanistic differences between the hsp90 chaperones. Mol Cell. 2007;28:41–56. doi: 10.1016/j.molcel.2007.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Qu Y, Bolen DW. Hydrogen exchange kinetics of RNase A and the urea:TMAO paradigm. Biochemistry. 2003;42:5837–5849. doi: 10.1021/bi0206457. [DOI] [PubMed] [Google Scholar]
- 33.Fang L, Ricketson D, Getubig L, Darimont B. Unliganded and hormone-bound glucocorticoid receptors interact with distinct hydrophobic sites in the Hsp90 C-terminal domain. Proc Natl Acad Sci USA. 2006;103:18487–18492. doi: 10.1073/pnas.0609163103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jaravine VA, Rathgeb-Szabo K, Alexandrescu AT. Microscopic stability of cold shock protein A examined by NMR native state hydrogen exchange as a function of urea and trimethylamine N-oxide. Protein Sci. 2000;9:290–301. doi: 10.1110/ps.9.2.290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ferreon AC, Bolen DW. Thermodynamics of denaturant-induced unfolding of a protein that exhibits variable two-state denaturation. Biochemistry. 2004;43:13357–13369. doi: 10.1021/bi048666j. [DOI] [PubMed] [Google Scholar]
- 36.Cunningham CN, Krukenberg KA, Agard DA. Intra- and intermonomer interactions are required to synergistically facilitate ATP hydrolysis in Hsp90. J Biol Chem. 2008;283:21170–21178. doi: 10.1074/jbc.M800046200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hura GL, Menon AL, Hammel M, Rambo RP, Poole Ii FL, Tsutakawa SE, Jenney FE, Jr, Classen S, Frankel KA, et al. Robust, high-throughput solution structural analyses by small angle X-ray scattering (SAXS) Nat Methods. 2009;6:606–612. doi: 10.1038/nmeth.1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Svergun D. Mathematical models in small-angle scattering data analysis. J Appl Cryst. 1991;24:485–492. [Google Scholar]