

Abstract
Glucocorticoids (GCs) are steroidal ligands for the GC receptor (GR), which can function as a ligand-activated transcription factor. These steroidal ligands and derivatives thereof are the first line of treatment in a vast array of inflammatory diseases. However, due to the general surge of side effects associated with long-term use of GCs and the potential problem of GC resistance in some patients, the scientific world continues to search for a better understanding of the GC-mediated antiinflammatory mechanisms.
The reversible phosphomodification of various mediators in the inflammatory process plays a key role in modulating and fine-tuning the sensitivity, longevity, and intensity of the inflammatory response. As such, the antiinflammatory GCs can modulate the activity and/or expression of various kinases and phosphatases, thus affecting the signaling efficacy toward the propagation of proinflammatory gene expression and proinflammatory gene mRNA stability. Conversely, phosphorylation of GR can affect GR ligand- and DNA-binding affinity, mobility, and cofactor recruitment, culminating in altered transactivation and transrepression capabilities of GR, and consequently leading to a modified antiinflammatory potential.
Recently, new roles for kinases and phosphatases have been described in GR-based antiinflammatory mechanisms. Moreover, kinase inhibitors have become increasingly important as antiinflammatory tools, not only for research but also for therapeutic purposes. In light of these developments, we aim to illuminate the integrated interplay between GR signaling and its correlating kinases and phosphatases in the context of the clinically important combat of inflammation, giving attention to implications on GC-mediated side effects and therapy resistance.
An integrated view of the interplay between glucocorticoid receptor signaling and its correlating kinases and phosphatases in the context of glucocorticoid-mediated inflammatory control, side-effects and resistance.
- I. Introduction
- A. Inflammation at a molecular level
- B. Glucocorticoid receptor-mediated signaling
- II. Phosphoregulation of the Glucocorticoid Receptor
- A. GR phosphorylation
- B. GR dephosphorylation
- C. Other posttranslational modifications of GR
- III. Kinases Targeted by Glucocorticoid Receptor-Mediated Signaling
- A. Mitogen-activated protein kinases (MAPKs)
- B. MAPK-activated protein kinases (MKs)
- C. Cyclin-dependent kinases (Cdks)
- D. IκB kinase α (IKKα)
- E. TANK-binding kinase 1 (TBK1)
- F. Other kinases
- IV. Phosphatases Targeted by Glucocorticoid Receptor-Mediated Signaling
- A. Dual specificity phosphatases (DUSPs)
- B. Other protein Y phosphatases
- C. Other phosphatases
- V. Kinase/Phosphatase Regulation in Glucocorticoid-Mediated Side Effects
- A. Skeleton and muscle effects
- B. Hyperglycemia and diabetes
- C. Other side effects
VI. Kinase/Phosphatase Regulation in Glucocorticoid Resistance
- VII. Future Perspectives in the Combat of Inflammation
- A. New glucocorticoid receptor ligands
- B. Combination therapies
- C. MicroRNA-specific modulation of GR
- D. Epigenetic approaches
VIII. Conclusions
I. Introduction
According to the World Health Organization (WHO; 2007 report), inflammation and inflammation-mediated illnesses are the biggest challenge in current medicine because 300 million people worldwide are estimated to suffer from asthma and 210 million people live with mild or severe chronic obstructive pulmonary disease (COPD), the latter leading up to 5% of global deaths. Furthermore, many people live uncomfortably with chronic inflammatory disorders, such as rheumatoid arthritis and inflammatory bowel disease. Moreover, the onset of cancer and cardiovascular diseases has also been linked to inflammation, claiming 13 and 30% of global deaths, respectively (WHO). As the costs of treating these disorders mount up and life comfort and expectancy are threatened, understanding and resolving inflammation is currently one of the main targets in science.
Today, glucocorticoid (GC)-based therapy is still the most commonly used treatment to combat chronic and acute inflammation. Since the discovery of the antiinflammatory properties of human cortisone in rheumatoid arthritis (1) and the cloning of the GC receptor (GR) (2), tremendous progress has been made in understanding how GCs inhibit inflammation: the molecular antiinflammatory mechanism of GCs consists of GR-mediated transactivation and transrepression mechanisms, the latter of which prominently features inhibition of nuclear factor-κB (NF-κB) activation and activity.
GCs have multiple physiological actions. As a consequence, a chronic exposure to pharmacological hormone doses becomes a problem in therapeutic settings, causing undesirable, yet on-target and thus GR-mediated, effects. The challenge is therefore not to develop more specific ligands for GR, but to change the spectrum of GR-mediated events and try to skew it more toward antiinflammatory pathways. This implies that selective (in terms of functionality) GR modulators could eliminate these adverse effects. Besides the undesirable effects, GC resistance, in which the patients do not respond to GCs, may also occur. Therefore, the mainstay of antiinflammatory research efforts is focused on further characterizing the antiinflammatory mechanisms of GCs in detail and developing new therapeutic strategies to fight inflammation with a better benefit-to-risk-ratio.
Protein kinases (afterward referred to as kinases) are enzymes that can rapidly and reversibly phosphorylate S, T, or Y residues of cellular proteins and as such affect their structure, function, location or metabolism. In turn, phosphatases function to revert the action of these kinases by dephosphorylating specific target residues (3). The GR itself is on the one hand subject to intense phosphoregulation, thus impacting its role in various antiinflammatory processes, and on the other hand this GR deploys and affects kinases and phosphatases as tools to implement its cellular antiinflammatory effects. In this review, we will focus on the above events, providing a contemporary view on the overall phosphomodulatory effects of and by the GR in the framework of inflammation. Additionally, the role of various phosphorylation events in the described GC-mediated side effects and the reported phenomenon of GC resistance will be discussed. Ultimately, we will discuss future therapeutic implications of phosphoregulation in the context of GR-based antiinflammatory strategies.
A. Inflammation at a molecular level
Inflammation is an initially advantageous response to intracellular damage or an extracellular challenger, provoking the activation of various proinflammatory mediators with the purpose to remove the damaging agent and to restore tissue structure and function. Physiologically, inflammation is locally marked by swelling, redness, pain, heat, and loss of function. Although inflammation can be a beneficial reaction, return to homeostasis is of the utmost importance to avoid the onset of an unfavorable chronic inflammation (4). The inflammatory reaction can be provoked by physical injury, tissue damage, or the invasion of alien pathogens. Alternatively, inflammation can also be provoked by an unwanted immune reaction of the body to its own proteins, i.e., an autoimmune reaction.
Biologically, inflammation progresses through different stages. First, local hyperemia is brought about by vasodilators. Ensuing inflammation is characterized by an exudation or leakage of plasma from the blood vessels into the inflamed tissue. This process is facilitated by an increased permeability of the endothelium and the augmented hydrostatic pressure in the blood capillaries. Next, cytokines and chemokines cause leukocytes (macrophages, neutrophils, etc.) to infiltrate the inflamed tissue to accommodate the phagocytosis of cellular debris and pathogens. Finally, fibroblasts proliferate to reinstate tissue structure. All cells involved in the inflammatory process can sense the environment, responding to various proinflammatory stimuli, and as a result initiate cytokine and chemokine cascades (5,6,7).
As such, the increase of inflammatory mediators, such as cytokines [TNFα (afterward referred to as TNF), IL-1β, IL-6, granulocyte monocyte-colony stimulating factor (GM-CSF), etc.], chemokines [IL-8; regulated upon activation, normal T-cell expressed and secreted (RANTES); melanoma growth-stimulating activity (Gro)-α; etc.], growth factors [fibroblast growth factor (FGF), epidermal growth factor (EGF), etc.], lipid-derived mediators (prostanoids, leukotrienes,), receptors [TNF receptor (TNFR), Toll-like receptor (TLR)], enzymes [inducible nitric oxide synthase (iNOS), cyclooxygenase-2 (COX-2), matrix metalloproteinases (MMPs), phospholipase A2, etc.), adhesion molecules [intercellular adhesion molecule-1 (ICAM1), vascular cell adhesion molecule-1 (VCAM1), E-selectin, P-selectin], and peptides (bradykinin, tachykinin, endothelin), is deemed pivotal for the propagation and progression of inflammation. Conversely, to control the course of the inflammatory process, modulatory and antiinflammatory cytokines such as IL-10, IL-4, and TGFβ are released (8).
On a molecular level, inflammatory diseases are mediated by stimulation via proinflammatory signals, like the bacterial lipopolysaccharide (LPS), viral factors, or the self-produced IL-1β or TNF. Binding of these agents to their respective receptors culminates predominantly in the activation of activator protein-1 (AP-1) and NF-κB, but also in the activation of other transcription factors. In turn, both NF-κB and AP-1 stimulate the expression of proinflammatory cytokines, chemokines, and adhesion molecules, thus propagating cellular inflammation (Fig. 1) (9,10,11,12).
Figure 1.

Inflammation at a molecular level: a simplified scheme. TNFR activation by TNF, IL1RI by IL1β, TLR3 by double-stranded RNA (dsRNA), TLR4 by bacterial LPS, and activation of other TLRs can signal via specific intermediary factors such as TRADD, TRAF2, RIP1, MEKK3, TAK1, TAB2/3, and NIK (for the TNFR), MyD88, IRAKs, TRAF6 and TAK1 (for IL1RI), Trif, RIP1, TRAF6 and TAK1 (for TLR3) and MyD88, Mal, Trif, Tram, RIP1, IRAKs, TRAF6 and TAK1 (for TLR4) to the MAPK pathway and to the activation and regulation of NF-κB and AP-1. Additionally, triggering TLR3 or TLR4 signaling cascades can instigate IKKε and TBK1 activation and subsequent IRF3-regulated gene transcription (10,11,12,29). TAB, TAK1-binding protein.
These activation pathways rely on a signaling cascade of intermediary factors and especially kinases. For instance, soluble TNF binds to a membrane-imbedded TNFR. Upon binding of TNF to TNFR1, the receptor homotrimerizes and recruits the TNFR-associated death domain (TRADD) protein to the cytoplasmic death domain of TNFR1 (13,14,15,16). Subsequently, the adhered protein complex is supplemented by receptor-interacting protein 1 (RIP1) and TNFR-associated factor 2 (TRAF2) (14,17,18,19). Ultimately, these adaptor proteins, specifically TGF-activated kinase 1 (TAK1), NF-κB-inducing kinase (NIK), or MAPK kinase kinase (MEKK3), phosphorylate the inhibitor of NF-κB (IκB) kinase (IKK) complex and trigger its dissociation (12,19,20,21,22). This process leads to the activation of the IKK-NF-κB pathway, critical for inducing tissue inflammation. Additionally, TNFR ligand binding results in the activation of ERK, p38, and c-Jun N-terminal kinase (JNK) MAPK via a multilayered kinase cascade (23,24,25). The MAPK family of protein kinases comprises ERK, JNK, and p38 MAPKs, of which the activation and function are regulated by upstream kinases and stress-related inducers (25). The multilayered activation cascade, enhancing the intracellular signal intensity and integrating various stimuli, is constructed bottom up by MAPKs, MAPK kinases (MAP2Ks or MKKs) and MAPK kinase kinases (MAP3Ks or MEKKs) (see Fig. 5). Preferentially, MAPKs target S/T protein residues, followed by a P (3,25). These MAPKs diverge the web of TNF-affected factors via multiple downstream kinase, cofactor, and transcription factor targets. Interestingly, the MAPK cascade is also involved in the posttranslational control of NF-κB and activation of AP-1 (26,27).
Figure 5.
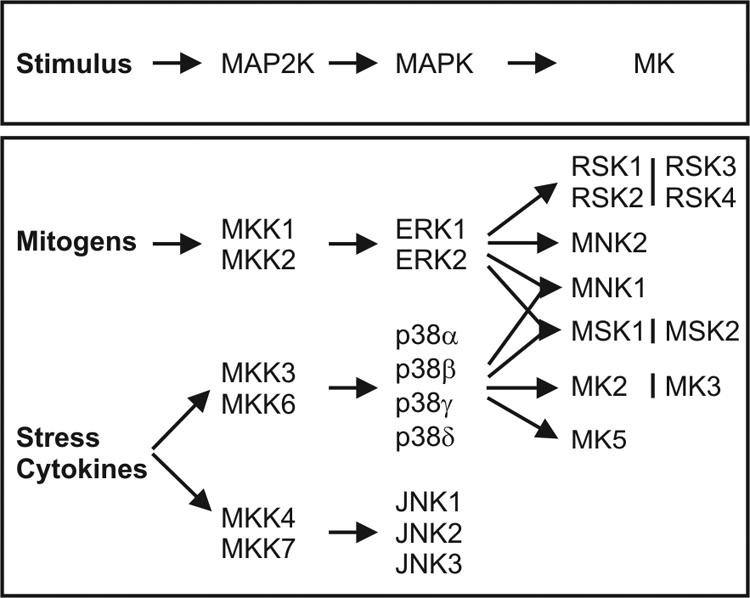
The layered MAPK signaling cascade. A schematic representation of the signaling cascades initiated by mitogens and stressors that lead to the activation of the MAP2Ks/MKKs, MAPKs, and ultimately the various MKs.
Alternatively, IL-1β signals via activation of the type I IL-1 receptor (IL1RI) and its associate intermediates myeloid differentiation primary response gene 88 (MyD88) and IL-1R-associated kinases (IRAK) 1, IRAK2, and IRAK4 and TAK1 toward activation of the MAPK pathway, AP-1, and NF-κB (11).
Lastly, various viral and bacterial factors can activate the related Toll-like receptors (TLR1-10). For example, TLR3 is activated by double-stranded RNA and TLR4 by the bacterial cell wall component LPS (28,29). TLR3 and TLR4 signal via TIR domain-containing adapter-inducing interferon (IFN) β (Trif) and MyD88, and via MyD88-adapter like protein (Mal) and translocating chain-associating membrane protein (Tram), respectively, to once again result in the activation and modulation of the MAPK pathway, AP-1 and NF-κB transcription factors (11,12,28,29). Alternatively, TLR3 and TLR4 can signal via TANK-binding kinase 1 (TBK1) and IKKε to activate IFN regulatory factor (IRF) 3 and to the subsequent transcription of type I IFN-inducible genes (30).
1. NF-κB
Because NF-κB is activated by a broad range of inflammatory and environmental stimuli, this transcription factor, which plays critical roles in both innate and adaptive immunity, serves as a biological sensor. Moreover, NF-κB is a pivotal regulator of inflammation because its activity level is raised in—and defective NF-κB signaling is associated with—an ever increasing list of inflammatory and pathological conditions (31,32,33,34,35,36).
a. NF-κB structure.
The transcription factor NF-κB comprises a family of five members: p65 (RelA), RelB, c-Rel, NF-κB1 (p50/p105), and NF-κB2 (p52/p100). All members are characterized by an N-terminal Rel-homology domain, which is required for homo- or heterodimerization, sequence-specific DNA binding, and nuclear translocation of NF-κB via its nuclear localization signal (NLS). Furthermore, RelA/p65, RelB, and c-Rel have C-terminal transactivation domains (TADs), whereas NF-κB1 and NF-κB2 are, respectively, p105 and p100 C-terminal ankyrin repeat-containing precursors (10), which are proteasomally processed to yield N-terminal products NF-κB p50 and p52, respectively. These mature species then heterodimerize with a TAD-containing NF-κB family member or with the TAD-bearing IκB protein B cell lymphoma-3 (Bcl-3) to form transcriptionally active complexes (37).
All NF-κB family members can form homo- or heterodimers directing the dimer to a specific set of target genes. Recent evidence suggests that alternative splicing of NF-κB components provides an additional way of controlling NF-κB signaling (38). NF-κB dimer function is specific because the ablation of a certain NF-κB family member cannot be compensated for (39). Because specific NF-κB dimer-promoter interaction is both context- and stimulus-dependent, the sequence of the NF-κB recognition site is not the sole determinant of NF-κB dimer-promoter interfacing (40,41,42,43). The main research target in inflammation is the prevalent heterodimer NF-κB p65-p50, of which p50 can increase DNA binding (44) and p65 confers transcriptional regulation (10).
b. NF-κB activation.
With regard to the NF-κB activation mechanism, multiple pathways have been described. The mechanism by which NF-κB is activated by proinflammatory TNF is commonly referred to as the canonical activation pathway (Fig. 1). In this canonical NF-κB activation pathway, the transcription factor NF-κB dimer p65-p50 is, in its resting state, held in the cytoplasm by an inhibitory IκB molecule, most commonly IκBα (10,45). IκB associates via its ankyrin repeats domain with NF-κB, which thus masks the NF-κB and IκB NLS motifs, consequently restricting inactive NF-κB to the cytoplasm. When cells are challenged with a proinflammatory signal, such as TNF, the IKK complex becomes activated. The activated IKK complex can phosphorylate IκBα on S32 and S36 or IκBβ on S19 and S23, leading to polyubiquitination of IκBα or IκBβ and ultimately culminating in IκB degradation by the 26S proteasome (10,46,47). Because NF-κB is released from its cytoplasmic constraint, NF-κB translocates into the nucleus, guided by its NLS, where it can bind specific genomic target sequences.
The IKK complex comprises the catalytically active subunits IKKα and IKKβ together with the scaffold regulatory subunit IKKγ [NF-κB essential modulator (NEMO)] (48). Additional, but most likely transient, components of the IKK complex are the chaperoning heat shock protein 90 (Hsp90), cell division cycle 37 protein (cdc37), and protein rich in amino acids E, L, K, and S (ELKS) (49,50,51). Hsp90 is necessary to allow the relocalization and thus activation of the IKK complex to the membrane-associated and activated TNFR1 complex, whereas cdc37 mediates the transient recruitment of Hsp90 to the IKK complex (49,50,52). Furthermore, ELKS is a necessary regulatory component of the IKK complex, serving as an auxiliary docking protein for IκBα (51). The IKK complex-mediated phosphorylation of IκB requires IKKβ and IKKγ, but does not necessitate IKKα, although IKKα can also phosphorylate IκB (47,53).
Secondly, the noncanonical pathway, initiated via B cell-activating factor (BAFF), lymphotoxin β, and other inducers, entails cytokine, among which TNF, and virus-initiated NIK activation (54,55). Activated NIK triggers IKKα homodimer activation, and subsequently S865, S869, and S871 of the p100 subunit in the p100-RelB complex becomes phosphorylated (56). This phosphorylation induces proteasomal processing to the p52 NF-κB subunit (57,58,59). The p52-RelB complex, consequently, translocates to the nucleus, where it targets specific gene promoters, such as IL-2 (60,61), for activation.
c. Posttranslational modifications of NF-κB.
Subsequent to the NF-κB activation process, NF-κB activity is substantially modulated by various posttranslational modifications: acetylation (62,63,64,65), SUMOylation (66), and phosphorylation (63,67), of which the last is best characterized so far. The intracellular control of NF-κB transactivation (duration and intensity) (64,68,69), subcellular localization (70), DNA-binding affinity (64,65,69,71), and NF-κB interaction with cofactors and IκBα (72,73,74) via various posttranslational modifications forms an intricate web of NF-κB management. Phosphorylation sites of NF-κB p65 are spread out over the Rel-homology domain and TAD1 and TAD2 (Fig. 4C). Although some of these phosphomodifications contribute to the transcriptional activity of NF-κB (73,75,76,77,78), others do not (79).
Figure 4.

Structure and posttranslational modifications of the GR. A, The reported phosphomodulated sites for the hGR, mGR, and rGR are depicted in relation to the known functional domains of this receptor. B, The reported posttranslational modifications for the hGR, exempt from the phosphorylated sites, are depicted in relation to the known functional domains of this receptor. C, The reported phosphomodulated sites for human NF-κB-p65 are depicted in relation to the known functional domains of this transcription factor. NTD, N-Terminal domain; HR, hinge region; P, phosphorylation site; h, human; m, murine; r, rat; aa, amino acids; SUMO, SUMOylation site; Ub, ubiquitination site; Ac, acetylation site; Rel-HD, Rel-homology domain; TA, transactivation.
In particular, the phosphorylation of NF-κB p65 S276 by p38 and ERK MAPK-activated mitogen- and stress-activated protein kinase 1 (MSK1) or protein kinase A (PKA) is pivotal for proper initiation of specific inflammatory gene expression (73,74,76,80,81,82,83,84). Phosphorylation of NF-κB p65 S276 facilitates association of p65 with the coactivators cAMP-responsive element-binding protein (CREB)-binding protein (CBP) and p300 (73,74,83,85) and the transcription elongation complex P-TEFb (positive transcription elongation factor b), consisting of cyclin-dependent kinase, Cdk9 and cyclin T1 (81). Furthermore, NF-κB p65 S276 phosphorylation can enhance the displacement of the inhibitory histone deacetylase 1 (HDAC1)-NF-κB p50 complex to derepress proinflammatory gene promoters (85). Therefore, NF-κB S276 phosphorylation is a crucial step in NF-κB driven promoter activation of specific gene targets (73,76,80,86). However, not all NF-κB-dependent genes require the phosphorylation of NF-κB S276 for their transcription. Whereas the transcription of ICAM, VCAM, Gro-β, IL-8, and IL-6 depend on the NF-κB p65 S276 phosphorylation, the NF-κB-mediated transcription of major histocompatibility complex-I (MHC-I), mangano-superoxide dismutase (MnSOD), and IκBα do not (73,81,86). Of note, NF-κB-mediated gene transcription independent of NF-κB S276 phosphorylation shows constitutive binding of RNA polymerase II (RNA pol II) (81). This differentiation suggests that a NF-κB phosphorylation code controls NF-κB-mediated transactivation of specific target genes (87), possibly defined by the architecture and topology of the target promoter (88). Consequently, the selective phosphorylation of S276 of NF-κB p65 may be responsible for some of the differential sensitivity of certain κB sites to repression by GCs. Because the phosphorylation of NF-κB p50 S337, which appears to be essential to NF-κB p50 DNA binding, is mediated by PKAc and possibly MSK1, these kinases are attributed an important role in the regulation of NF-κB p65-p50-dependent gene transcription (89).
Various kinases can phosphorylate NF-κB S536: IKKα, IKKβ, TBK1, IKKε, ribosomal S6 kinase 1 (RSK1), and glycogen synthase kinase (GSK) 3β (71,90,91,92,93,94,95,96). Also, the phosphorylation of NF-κB p65 S536 can contribute to the activity of NF-κB p65, most likely via facilitating the interaction between activated NF-κB and p300 (62,93). Additionally, NF-κB p65 S536 phosphorylation is reported to weaken the binding of NF-κB to IκB, thus prolonging the activity of NF-κB in the nucleus (71,93). Of note, acetylation of NF-κB of multiple lysine residues, probably by the histone acetyl transferase (HAT) activity of CBP/p300, is preceded by and requires NF-κB S276 and S536 phosphorylation (62).
A third NF-κB phosphorylation that can positively affect NF-κB activity is the protein kinase C (PKC) ζ-mediated phosphorylation of NF-κB p65 S311. Similar to NF-κB p65 S276 and S536 phosphorylation, the phosphorylation of NF-κB at S311 can enhance the interaction of NF-κB p65 with CBP. Moreover, NF-κB p65 S311 phosphorylation augments NF-κB recruitment to κB sites close to the promoter of the proinflammatory cytokine IL-6 (75).
Phosphorylation of NF-κB p65 T254 by an unknown kinase can enhance the NF-κB p65 activity via inducing the interaction of T254 and P255 of NF-κB p65 with the nuclear peptidyl-prolyl isomerase Pin1 [protein NIMA (never in mitosis gene a)-interacting]. This interaction results in the isomerization of the P residue, entailing a conformational change of NF-κB p65. Consequently, NF-κB p65’s binding affinity for IκBα is decreased. Furthermore, this modification stabilizes the NF-κB p65 protein and promotes the nuclear translocation of NF-κB p65 (97). Conversely, ubiquitination of NF-κB p65, inducing its proteolysis, is mediated by the E3-ubiquitin ligase suppressor of cytokine signaling (SOCS) 1. Because the binding sites for Pin1 and SOCS1 lie in close proximity, competition for binding might be possible (98). The proteasomal degradation of DNA-bound NF-κB p65 promotes transcriptional termination (99).
For the phosphorylation of NF-κB p65 S529 by casein kinase 2 (CK-2), it is still unclear whether this phosphorylation can affect NF-κB-mediated transcription (100,101). In contrast, phosphorylation of T505 via activation of checkpoint kinase 1 (Chk1) and ATM/Rad3-related (ATR) checkpoint kinase could decrease the activity of NF-κB via enhancing its interaction with HDAC1 (102,103,104). Even so, the IKKβ- or GSK3β-mediated phosphorylation of NF-κB p65 S468 negatively affects NF-κB activity (68,79), whereas IKKε-mediated phosphorylation of this same residue was associated with NF-κB transactivation (105).
Conversely, endogenous protein phosphatase (PP) 2A can associate with and dephosphorylate NF-κB p65 (106). Moreover, pharmacological blockage of PP2A leads to increased phosphorylation of NF-κB p65 (68,106). Additionally, association of PP4 has been linked to the activation of NF-κB via the dephosphorylation of T505 (102,107,108). Furthermore, it was suggested that a rapid dephosphorylation of NF-κB S536 could contribute to switching off NF-κB-dependent gene transcription (109).
d. NF-κB crosstalk.
NF-κB-mediated transcription can furthermore be coregulated by crosstalk of NF-κB with other transcription factors and association of NF-κB with various cofactors (110). Positive crosstalk of NF-κB-promoting proinflammatory gene transcription has been described for aryl hydrocarbon receptor, specificity protein 1 (Sp1), IRF, signal transducer and activator of transcription (STAT), activating transcription factor (ATF), CREB, and AP-1 (111,112,113,114,115,116,117,118). The binding of multiple distinct transcription factor complexes occurs in a highly dynamic manner (110,119). An example of negative crosstalk with NF-κB is shown by GR (120). Cofactors can either coactivate or corepress NF-κB-mediated gene transcription. Functionally, these cofactors can stimulate or repress the transcriptional activity of the enhanceosome (i.e., the multiprotein complex mediating promoter activation and gene transcription) (121) or alter the chromatin structure. Many coactivators, including p300, CBP, p300/CBP- associated factor (p/CAF), and steroid receptor coactivator 1 (SRC1) have a HAT domain, capable of acetylating histones but also other proteins (122,123,124,125,126,127). Interestingly, IKKα can phosphorylate CBP, increasing CBP activity and CBP binding to p65 (128). Conversely, corepressors often include HDAC activity (HDAC1, HDAC2, HDAC3) and can be directly or indirectly recruited to NF-κB-dependent gene promoters (85,129,130,131,132,133). Other NF-κB-dependent gene-associated corepressors are the NF-κB p65-p50-binding silencing mediator for retinoid and thyroid-hormone receptors (SMRT) and the NF-κB p50-binding nuclear corepressor (NCoR) (134,135).
e. NF-κB-targeted genes.
After NF-κB activation, nuclear NF-κB p65 can enhance gene expression of multiple proinflammatory genes via the occupation of a NF-κB-specific promoter recognition site (136). Because NF-κB can be considered a central regulator of proinflammatory gene transcription, NF-κB function is a highly dynamic signaling event, showing differential temporal expression profiles for different genes (40,137). Moreover, association of NF-κB with its specific recognition sites is transient, with a half-life of seconds, suggesting a dynamic regulation of enhanceosome composition and gene transcription constantly sensing the inflammatory status of its environment (119).
The canonical or classical NF-κB activation pathway controls among others leukocyte activation/chemotaxis, cellular metabolism, antigen processing, and negative regulation of the TNF signaling pathway (138,139). The activation of NF-κB results in gene transcription of cytokines (e.g., IL-6), chemokines (e.g., IL-8 and RANTES), adhesion molecules (e.g., E-selectin), enzymes (e.g., iNOS), and other inflammatory mediators (8). Furthermore, the cytokine TNF, but also IL-1β, can activate NF-κB, and these are thus mediators of a feedforward mechanism perpetuating inflammation.
However, it is essential to prevent the uncontrolled propagation of inflammation, which would cause systemic disorders. Therefore, NF-κB activation is also subject to an autoregulatory negative feedback loop. First, gene transcription of IκBα is stimulated by NF-κB activation. The newly synthesized IκBα replenishes the former proteasomally degraded IκBα levels and can bind to active NF-κB complexes in the nucleus, weakening NF-κB DNA binding and subsequently transporting NF-κB back to the cytoplasm (10,140,141). Furthermore, upregulation of the anti-apoptotic protein A20, the A20-binding inhibitor of NF-κB activation, ABIN-1, the TNFα-converting enzyme TACE, the tumor suppressor cylindromatosis (CYLD), the antiinflammatory cytokine IL-10, and the microRNA miR-146 ultimately results in a negative regulation of NF-κB activity (142,143,144,145,146,147,148,149,150,151,152,153).
2. AP-1
AP-1 is a transcription factor of general importance for many cellular processes in different organs, including inflammation. Among the target genes of AP-1 are important regulators of cell proliferation, differentiation, and apoptosis. The DNA binding of the AP-1 complex to 12-O-tetradecanoylphorbol-13-acetate (TPA)-response element (TRE) sequences is rapidly induced by growth factors, cytokines, and oncoproteins, which are implicated in the proliferation, survival, differentiation, and transformation of cells (154).
a. AP-1 structure.
Similar to NF-κB, AP-1 is a homo- or heterodimeric transcription factor complex that can be targeted to its regulatory sites in a sequence-specific manner (Fig. 1). The subunits of AP-1 are selected from the Jun (c-Jun, v-Jun, Jun B, and Jun D), Fos (c-Fos, Fos B, Fra-1, and Fra-2), activating transcription factor [ATF2, ATF3, B-ATF, Jun dimerization protein (JDP)-1, JDP-2], or MAF (MAFA, MAFB, c-MAF, NRL, MAFF, MAFG, and MAFK) families (154,155,156). The most predominantly occurring forms of AP-1 are Fos/Jun heterodimers, which show preferential binding to heptameric TREs when compared with ATF2/Jun homo- or heterodimers, which bind preferentially to octameric TREs and are strongly induced by the tumor promoter TPA (157). AP-1 proteins are known as basic leucine-zipper proteins because they dimerize through a leucine zipper motif and contain a basic domain for interaction with the DNA backbone. While the Fos proteins do not form homodimers but can heterodimerize with members of the Jun family, the Jun proteins can both homodimerize and heterodimerize with other Jun or Fos members to form transcriptionally active complexes (155,158). In addition to Fos proteins, Jun proteins can also heterodimerize efficiently with other AP-1 family members, such as the ATF family (159), and other basic zipper-containing transcription factors (156,160). Although members of the Jun and Fos families share a high degree of structural homology, the individual AP-1 dimers exert significant differences in their DNA-binding affinity and their capability to activate or suppress gene expression (155).
b. AP-1 activation and activity.
Regulation of net AP-1 activity can be achieved through changes in transcription of genes encoding AP-1 subunits, control of their mRNA stability, posttranslational processing, turnover of preexisting or newly synthesized AP-1 subunits, and specific interactions between AP-1 proteins and other transcription factors and cofactors.
Various stimuli, including physiological agents such as growth factors and cytokines, pharmacological compounds, such as anisomycin, phorbol esters and okadaic acid, and stressors such as UV radiation, hyperosmotic and heavy metal stress, rapidly elicit transcription of “immediate early” (IE) genes, such as those of the Fos and Jun families, by activation of MAPK cascades (161). These so-called “IE genes” are activated directly and require no new transcription or translation for their induction. The Fos and Jun proteins then activate and repress other genes, thereby producing secondary transcriptional reprogramming appropriate to specific stimuli. Because these proteins are differentially expressed and regulated in a cell- and stimulus-specific manner, every cell type produces a complex mixture of AP-1 dimers with subtly different functions (154,161). In most cases, enhanced expression of c-jun gene, protein, and function is not a solitary event but can be accompanied by an induction of transcription factors that are related to c-Jun (e.g., Jun B, Jun D), Fos family members (Fos, FosB, Fra-1/2), or ATF family members allowing the formation of functionally different heterodimers in a cell- and time-specific manner. Whereas some AP-1-regulated genes are preferentially induced by cJun-cFos dimers, others are mainly induced by Jun D-Fra-1 dimers. Fra-1 and Fra-2 promoters are activated by Jun-Fos dimers. The fos and jun genes are controlled by multiple upstream elements; for human c-fos, these include a cis-inducible element, a serum response element (SRE), a ternary complex factor site (TCF), an AP-1 site, an AP-1/CRE, a direct repeat, and a cAMP response element. The human c-jun promoter is controlled by the Jun2 AP-1 site, a footprint (FP), an NF-Jun site, two overlapping Sp1 sites, a CCAAT box, the Jun1 AP-1 site, a related to serum response factor site (RSRF), and in the 5′-untranslated region, two AP-2 sites and a weak AP-1 site (158,161,162). These regulatory elements are highly conserved between mouse and human.
AP-1 is induced by several external stimuli that increase MAPK activity. Expression of c-Fos is induced by TCFs, which are activated through phosphorylation by the ERK MAPKs.
IE gene expression of c-Jun may be achieved through ATF or c-Jun. Alternatively, c fos and myocyte enhancer factor 2 (MEF2), transcription factors can also induce c-jun expression in other contexts. The targeting of MAPKs to transcription factors controlling c-fos and c-jun gene expression has been very clearly established, and phosphorylation of these factors is further implicated in recruitment of coactivators such as p300/CBP and p/CAF, to these promoters (163,164).
c. Posttranslational regulation of AP-1.
Posttranslational activation of AP-1 is produced by translocation of a stimulus-dependent kinase, such as MAPKs or their effector kinases, which bind to and phosphorylate these factors, effecting transactivation and transcription of the associated gene (154,158). The mechanism of posttranslational control is most extensively documented in the case of mitogen- and cellular stress-induced hyperphosphorylation and, in particular, activation of Jun through the JNK cascade (165,166). Activated by a MAPK cascade, the JNKs translocate to the nucleus, where they phosphorylate Jun within its N-terminal TAD at S63 and S73 and thereby enhance its transactivation potential. The JNKs also phosphorylate and potentiate the activity of JunD and ATF2. Alternatively, p38 MAPK-dependent phosphorylation of c-Jun S63 and S73 has been demonstrated in response to a UV stimulus (167). Moreover, DNA-binding activity of c-Fos, FosB, and JunB were also dependent on the p38 protein kinase activity, whereas JunD, Fra-1, and Fra-2 were not affected. A complex network of signaling pathways that involves external signals for growth factors-Ras-Raf-MEK-ERK families also regulates AP-1 activity (168). Activated Ras or MEK1 primarily induces Fra-1 and c-Jun after N-terminal phosphorylation by JNKs. In contrast to c-Jun, JunB is not an efficient substrate for JNK. Furthermore, although JunD can be phosphorylated by JNK, its phosphorylation requires interaction with partners that provide a docking sequence (169). JunB and JunD are less potent collaborators of Ras in cell transformation than c-Jun, which correlates with their lower transcriptional activity. ERKs are persistently activated by growth factors and oncogenic Ras in tumors and are positive regulators of tumorigenesis (170). As such, they contribute substantially to the increased expression and activation of AP-1 members in many tumor types. Potential candidates for kinases that regulate Fos activity are the Fos-regulating kinase (FRK), RSK2, and p38 and ERK MAPK (170). When the AP-1 complexes are present in larger quantities, glycogen synthase kinase 3β (GSK3β), RSK2, casein kinase 2, cdc2, PKA, and PKC phosphorylate Fos and Jun proteins, thereby regulating their protein stability, DNA-binding activity, and the transactivating potential of the AP-1 family members (154,158).
d. AP-1 dimers and crosstalk.
The activities of AP-1 are partially modulated through the differential expression of its individual components, which determines their dimer composition (154,171), and partially through their specific context—cell type, response element sequences and organization, modification state, interaction with other regulatory factors, promoter sequence and organization, etc. (172). Whereas Jun, Fos, and FosB are often associated with a strong transactivation potential, JunB, JunD, Fra-1, and Fra-2 are usually found in a context in which only a weak transactivation potential is supported. Under specific circumstances, the latter might even act as repressors of AP-1 activity by competing for binding to AP-1 sites or by forming “inactive” heterodimers with Jun, Fos, or FosB. The crucial and determining aspects of “context” are not yet fully understood. To illustrate the importance, it was described that the composition of AP-1 regulatory complexes and the biological activities of the bound factors are dynamic and dependent on cell and response element contexts. Col3A is the response element in the collagenase-3 gene that confers activation by phorbol esters and repression by GCs in human U2OS osteosarcoma cells. The subunit composition and activity of AP-1, which binds ColA3, parallels the intracellular level of c-Fos, which is modulated by phorbol esters and GCs. A similar AP-1 site at the collagenase-1 gene, however, not inducible in U2OS cells, was not bound by AP-1, underscoring the importance of a context-dependent gene regulation (172).
The decision as to whether AP-1 is oncogenic or antioncogenic might depend on the antagonistic activity of different Jun proteins, but it is probably also influenced by tumor type, tumor stage, and the genetic background of tumors (158,168). For example, c-Jun-Fra-2, but not c-Jun-Fra-1 or c-Jun-c-Fos, inhibits the growth arrest of immortalized fibroblasts at confluence and under low-serum conditions. Using dimer-specific mutants of AP-1 proteins, in which manipulation of the leucine-zipper domain allows only specific dimers to form, it was demonstrated that the c-Jun-induced transformation program can be separated into two distinct pathways: c-Jun-ATF2 activity triggers growth factor independence, and c-Jun-c-Fos activity causes anchorage-independent growth (158). To fully elicit their oncogenic potential, most AP-1 components need the activity of “cooperating” oncoproteins, which often induce the expression of Jun and Fos proteins but also support AP-1-mediated cell transformation by posttranscriptional mechanisms (173,174). The main cooperating partner of AP-1 is the Ras pathway because cell transformation by activated Ras or MEK1 (MAPK kinase) induces AP-1 protein expression (168). The oncogenic cooperation of c-Jun with Ras and other oncoproteins, functioning upstream of Ras, requires N-terminal phosphorylation of c-Jun by JNKs.
B. Glucocorticoid receptor-mediated signaling
Inflammation can be controlled by the stress-induced release of GCs, mainly cortisol in humans. Besides being efficient in combating inflammation, GCs display pleiotropic effects in the regulation of protein, lipid, and carbohydrate metabolism; innate and adaptive immune systems; stress homeostatic regulation; reproductive processes; and growth and brain functions such as memory and behavior (175).
1. GR domains
The GR, which binds and mediates the signals of the GCs, belongs to the superfamily of nuclear receptors. This superfamily can be categorized according to ontogeny and function (176). In that respect, the GR is classified as NR3C1, most proximate to the mineralocorticoid receptor (MR). However, GRs and MRs nevertheless affect distinct target genes (177). The GR consists of an N-terminal domain, encompassing a first TAD [activation function 1 (AF-1)] responsible for transcriptional activation and association with certain basal transcription factors (2,178), a DNA-binding domain (DBD), in which the dimerization or D-loop within the two zinc fingers plays a role in GR dimerization and DNA-binding functions (178,179,180,181,182,183), and a C-terminal ligand-binding domain (LBD), containing a second TAD (AF-2) and also protein-binding sites (178,184,185,186,187,188,189). Interestingly, in addition to GR ligands, different GR/DNA-binding sequences can differentially affect GR conformation and regulatory activity; as such, DNA can be considered as a sequence-specific allosteric ligand of GR (190). Furthermore, the DBD can also account for GR-transcription factor association (191,192,193). In close proximity of the DBD and at the end of the LBD, two nuclear localization sites, the ligand-independent NL1 and the ligand-dependent NL2, have been described that direct the activated GR toward the nucleus (Fig. 2) (194,195).
Figure 2.

Structure and domain functions of the GR. NTD, N-Terminal domain; HR, hinge region; TF, transcription factor.
The GR can exist as multiple isoforms due to alternative splicing (GRα, GRβ, GRγ, GR-A, GR-P) and different translational start sites (GRα-A, GRα-B, GRα-C1, GRα-C2, GRα-C3, GRα-D1, GRα-D2, or GRα-D3). Both the N-terminal and C-terminal domain of GR can vary depending on the isotype, but the DBD most often remains constant. GRα, stretching to 777 amino acids in humans, is the most predominant, functional GR and currently the main research target. In contrast, GRβ cannot bind GCs and is not ubiquitously expressed. However, GRβ can act in a dominant-negative manner to suppress actions of GRα and is implicated in GC resistance (196,197). Although GRα is expressed throughout the body, the expression pattern of the isoforms can be restricted to certain cell types, possibly fine-tuning the GC-GR response in various tissues (196,198,199,200). Additionally, various function-altering polymorphisms have been defined for the human GR (hGR) (199,200,201). Membrane-associated and mitochondrial GR proteins have also been described (202,203,204,205,206). Alternatively, GCs can act via G protein-coupled receptors and its downstream cascades (207). The precise role of GC signaling via these receptors, however, still awaits further research. It is expected that GRα, being the most predominant GR species, will remain a prime focus of therapeutic attention for some time still.
In an uninduced state, GRs reside predominantly in the cell cytoplasm in association with a multimeric molecular chaperone complex, keeping the ligand-binding pocket receptive to high-affinity hormone binding and inactivating the NLS. This chaperone complex consists of several Hsps, such as Hsp90, Hsp70, and the Hsp90-binding protein p23, the hsp-organizing protein Hop, and tetratricopeptide repeat proteins that also bind Hsp90 such as FK506-binding protein (FKBP) 51, FKBP52, cyclophilin 40 (Cyp40), the C-terminus of Hsp70-interacting protein (CHIP), or the phosphatase PP5 (see Section II.B). However, in a single lysate, not all chaperone complexes are equally composed (186,208).
2. Triggering GR-mediated signaling
Once their cellular target is reached, GCs can cross the membrane because they are small hydrophobic molecules. Alternatively, natural GCs can enter the cell via the steroid hormone recognition and effector complex (209). GC binding to cytosolic GR instigates a conformational change in this receptor (185,210). The active conformational state of GR and its subsequent modifications allow GR to shed most of its chaperone complex, unmasking the NLS. These steroid-dependent changes allow GR to freely and rapidly move along cytoskeletal tracts to ultimately translocate into the nucleus (186). Subsequently, GR can give rise to positive or negative transcriptional effects (Fig. 3) (120,211,212) and rapid nontranscriptional effects (213). Together, these genomic and nongenomic pathways controlled by GRs culminate in a multilayered and fine-tuned control mechanism for gene regulation. In addition to the classic slow mode of GC action occurring between hours to days, increasing evidence is culminating for more rapid GC effects on cellular responses, taking place within minutes. Because these GC effects are too fast to be regulated at the transcriptional level, they are termed nongenomic, to distinguish them from the traditional genomic mode of GC action (214). Rapid GC effects may be transmitted by the GR or, more controversial, nongenomic GC activities might be mediated through nonspecific physicochemical interactions with the plasma membrane at high GC concentrations (215).
Figure 3.

Activation and nuclear actions of the GR. The unactivated, cytoplasmic GR is complexed with chaperone proteins. Binding of GCs to the GR instigates the nuclear translocation of GR. The binding of dimeric, activated GR onto GREs, DNA binding of GR in a concerted manner with another transcription factor (TF), or binding of GR onto a TF via a tethering mechanism can all result in GC-directed promoter activation. This transactivation results in the expression of metabolic gene products, associated with the occurrence of side effects, and to the expression of a number of antiinflammatory proteins. The antiinflammatory effects of GCs are predominantly mediated via interference of monomeric GR with the transactivation capacity of TFs, such as NF-κB, via a tethering mechanism. Otherwise, GC-activated GR can also negatively regulate gene transcription via competition for an overlapping binding site (competitive GRE) or via DNA-binding crosstalk with another TF (composite GRE), or else via the sequestration of a DNA-bound TF. Although the nature of these sequences is not well-defined, gene repression via direct binding of GR onto a so-called nGRE has also been reported.
Although uninduced GR is mainly found in the cytoplasm and GC-induced GR is largely nuclear, constitutive shuttling between nucleus and cytoplasm has been reported for both nonactivated and activated forms of GR (216). It is, however, the import or export rate that determines the location of the bulk of GR at any given time. Whereas importinα- or importinβ-based nuclear import of GR is mediated by the GR NLSs NL1 and NL2 (195,217,218), calreticulin-based and chromosome region maintenance 1 (CRM1)/exportin1-based mechanisms have been described to account for the nuclear export of liganded or unliganded GR (195,219,220,221). Moreover, the location of GR is codetermined by the recently discovered nuclear retention signal (NRS), which actively configures GR to the nucleus (222). Additionally, the DNA-binding ability of GR and ligand-specific conformational changes in GR have been previously linked with GR’s nuclear mobility and cellular location (223,224,225).
Once activated, several mechanisms are set in motion to discontinue the GR response. Although GR is assumed to be constitutively expressed under a vast array of physiological conditions, GR mRNA expression is subject to negative regulation by GCs (226,227,228). This might be explained by the presence of negative GC response element (nGRE), AP-1, NF-κB, and CREB regulatory motifs in the promoter of GR, all of which are negatively regulated by GCs (196). Alternatively, GCs can destabilize GR mRNA (229). Moreover, ligand-activated GR protein is degraded upon prolonged exposure to GCs by the proteasome-ubiquitin degradation pathway (230,231). However, mutation of the N-terminal GR PEST motif abrogates ligand-dependent downmodulation and consequently boosts GR-mediated transactivation (232). Lastly, it was shown that protein degradation of GR is linked to its export. Although hormone dissociation results in a rapid release of GRs from chromatin, unliganded GR is delayed in its export. Accelerated nuclear export of a nuclear export sequence-tagged GR chimera is associated with an increased rate of hormone-dependent down-regulation. The protracted rate of receptor nuclear export may be a way of increasing the efficiency of biological responses to secondary hormone challenges, via a limitation on receptor down-regulation and hormone desensitization (233).
3. Nuclear activity of the GR
a. GR transactivation and transrepression mechanisms.
Ligand binding of GCs to GR results in the nuclear translocation of GR where this receptor can act to modulate transactivation of typical GC response element (GRE)-containing or other promoters.
Ligand-activated, nuclear GR can stimulate the expression of certain genes via DNA binding of a dimerized GR. These GR homodimers bind in the major groove of DNA via their zinc finger DBD and target the imperfect palindrome of the consensus GRE (5′ GGT ACA nnn TGT TCT 3′) (234,235,236). This GRE can differ among promoters in sequence, copy number, and relative location in the promoter (in relation to the TATA box or other transcription factor-binding sites), regulating the specificity and magnitude of its response. GR may thus be modified in an allosteric manner by its response elements to generate a pattern of regulation that is appropriate to an individual gene (237,238). In DNA-binding GR-mediated transactivation research, either a simple GRE or concatamer GRE mouse mammary tumor virus (MMTV) reporter gene construct is often used, which GCs can transiently activate (239,240). However, various studies revealed that many known GC-inducible genes do not contain consensus GRE sites and do not require binding of dimerized GR. Some of these could be classified as promoters containing composite elements, in which GR collaborates with another transcription factor to enhance transcription in a cooperative manner (211,241,242,243,244,245). Essentially all genes have GR binding motifs reasonably close (at an average of 15 kb) to the transcription start site, however few of those are functional. Many elements lie very far (>50kb) from the start sites, and at present there are no simple ways to prove that a given element is controlling a given gene. Assignments are done essentially by conservation, demonstration of GR occupancy in vivo, and proximity (246). Lastly, tethering, i.e., direct binding of GR to DNA-bound transcription factors, has also been described to positively regulate DNA transcription upon GC administration (Fig. 3) (247,248,249).
Various transcription factors [e.g., Sp1, STAT1, STAT3, STAT5, CCAAT enhancer-binding protein (C/EBP), Ets, Egr-1, AP-2, AP-1, and NF-κB] can function in concerted array with GR to regulate and fine-tune transcription in a positive or negative manner (250,251,252). For example, ligand-activated GR can inhibit most, but not all, NF-κB-driven gene expression (211). Indeed, GR acts selectively to inhibit NF-κB at some, but not all, NF-κB sites. Notably, it inhibits NF-κB action at the IL-8 but not at the IκBα gene. NFκB at IL-8 is phosphorylated at Ser276 and recruits P-TEFb to promote elongation. GR represses IL-8, where transcription elongation depends on P-TEFb-mediated phosphorylation of pol II C-terminal domain (CTD) S2, by competing P-TEFb from p65 association. In contrast, GR fails to repress IκB gene expression, where CTD S2 phosphorylation proceeds without p65 recruitment of P-TEFb, and GR binding to p65 therefore poses no interference (253,254). The findings presented above represent a good example of a situation in which the context dependency is mechanistically understood.
Likewise, activation of the transcription factor NF-κB p65 can repress most, but not all, GR-transactivated gene promoters (255,256). The NF-κB-mediated inhibition of GR transactivation mechanisms is called “reciprocal repression” and is partially based on the mutual interaction of GR with NF-κB and partially on the cellular context.
Additionally, SRCs of the p160 family, such as SRC-1 and SRC-2, interacting with GR via its LBD LxxLL motif, and various cofactors for chromatin remodeling and histone modification [e.g., SRC-1 and TIF-2 associated modulatory protein (STAMP), CBP/p300, p/CAF, switching of yeast mating type/sucrose nonfermenting (SWI/SNF), and coactivator-associated arginine methyltransferase (CARM) 1] can regulate GC-mediated promoter activation (240,257,258,259,260,261). The nature of the GR ligand impacts GR cofactor binding. Although administration of the synthetic GC dexamethasone leads to GR SRC-1 binding, ligand binding of GR to the GR antagonist RU486 stimulates binding of GR to the corepressor NCoR (262). Furthermore, the requirement for certain cofactors and different GR interfaces can depend on the targeted gene promoter (244,263). Concerning histone modifications, a GC-induced phosphorylation of histone H3 (H3) S10 and acetylation of H3 K14 in MMTV promoter chromatin is associated with a transcriptionally active promoter (257).
The DNA binding of ligand-activated GR is, however, not a static phenomenon. Upon activation, GR rapidly assembles onto known GREs. The chaperones Hsp90 and p23 can localize to GR-bound GREs in a GC-inducible manner and promote disassembly of a functional GR-GRE transactivation complex (264). Furthermore, the GR transactivation regulatory complex has proven to turn over in an extremely dynamic manner, via a ligand- dependent “hit and run” mechanism (223,265,266). Combined with the release of GCs from GRs (264), these mechanisms continuously sense cellular stress hormones and thus allow an appropriate cellular response to varying GC levels.
b. GR transactivation and inflammation.
GC-mediated up-regulation of IκBα, GC-induced leucine zipper (GILZ), dual specificity phosphatase (DUSP) 1 (see Section IV.A), lipocortin-1/annexin A1, secretory leukocyte protease inhibitor SLPI, IL-10, the decoy IL-1 receptor type II, dexamethasone-induced Ras1 (Dexras1), downstream of tyrosine kinase 1 (Dok-1), Src-like adaptor protein (SLAP), p11/calpactin binding protein, thymosin β-4sulfoxide, Clara cell secretory 10-kDa protein (CC10), β-adrenergic receptors, SOCS1, and tristetraprolin (TTP) have all been suggested to be involved in the GC-mediated combat of inflammation (211,243,267,268,269,270). For example, because DNaseI footprinting studies in T47D/A1–2 cells demonstrated that regulatory factors bind to the IκB-α promoter after GC treatment, Deroo and Archer (271) proposed that GCs may be required for transcription factor binding and subsequent transactivation of the IκB-α promoter. Higher levels of IκB-α would then block the activation of NF-κB. GILZ, a classical GRE-driven target gene, also seems to mimic some aspects of GC action and inhibits inflammatory cytokine-induced COX-2 expression in bone marrow mesenchymal stem cells, via blockage of the nuclear translocation of NF-κB (272). Annexin A1, another GC-regulated target gene, is believed to exert its effects through the FPR receptor family of G protein-coupled receptors, of which the implication in the regulation of many inflammatory processes is increasingly being recognized (273). GCs transcriptionally stimulate the synthesis of TTP, a zinc finger protein capable of destabilizing several proinflammatory cytokine mRNAs by binding to adenylate uridylate-rich elements (AREs) within their 3′ untranslated regions, subsequently targeting them for degradation (see Section III.B.2) (274). The effect of (de)phosphorylation on the functionality of some of the gene products of the above-mentioned list or the effect of these proteins on other kinases/phosphatase signaling pathways is discussed further below.
The overall role and contribution of the different described GC-induced antiinflammatory proteins in the GC-mediated antiinflammatory mechanism remains somewhat controversial (211,275,276). In some studies, GCs can repress proinflammatory gene expression of IL-6, ICAM1, and COX-2 without the need for de novo protein synthesis (277,278), whereas in other studies urokinase plasminogen activator, COX-2, and IL-8 mRNA transcriptional repression by GCs have been found to partially rely on de novo protein synthesis (279,280,281,282,283,284). Nevertheless, although a body of evidence supports that the principal and initial antiinflammatory potential of GR may reside in its direct repression of proinflammatory gene expression (236,277,285,286), it is apparent that GR-mediated transactivation also plays a role. Moreover, the precise contributions of transrepression vs. transactivation mechanisms in the antiinflammatory actions of GR seem highly context-dependent and make matters even more complex (120). Hence, the idea that dissociating ligands would remain powerful antiinflammatories while modulating side effects is likely simplistic.
c. GR-mediated promoter inhibition.
Besides GR-mediated transactivation, ligand-activated GR can also act as a DNA-binding factor to repress specific gene transcription via composite response elements or competitive mechanisms (241,251,287,288,289,290,291) or can tether to another transcription factor to modulate transrepression of targeted genes via crosstalk with kinases, cofactors, or other promoter-bound transcription factors (Fig. 3) (211,212,251). In the latter mechanism, GR can bind and modulate DNA-bound transcription factors such as AP-1, NF-κB, STAT5, octamer-binding transcription factor 1 (Oct-1), CREB, Smad3, Smad6, Ets2, T-box expressed in T cells (T-bet), and GATA3 (211,212,251,292). Typically, the tethering GR repression of transcription factor activity is reflected in a reciprocal repression of GR transactivation by the very same transcription factors (192,251,255,277).
The transcription factors NF-κB and AP-1 are key to the propagation of inflammation and are also targets of GC-dependent repression of proinflammatory gene transcription (31). Moreover, chromatin immunoprecipitation analysis has shown that GR binds proximal to the NF-κB or AP-1 binding site in various promoters (254,293,294,295). The nuclear interaction of GR with the C-terminal activation domains of NF-κB p65 is pivotal to the GC-mediated repressive effect on NF-κB-regulated gene expression (192,255,277). Mechanistically, GR does not need to bind the target genes’ promoter DNA to infer inhibition of NF-κB and AP-1 function, yet mutation analysis revealed that a functional GR DBD is necessary for repression of AP-1- and NF-κB-regulated genes (192,193,256,296). This is explained by the finding that the GR DBD is involved in AP-1 tethering interactions (297). Even so, the GR LBD has been implicated in repression of NF-κB-regulated genes (298). GR-interacting protein 1 (GRIP1) was originally identified as a corepressor for GR during tethering to AP-1, explaining the LBD requirement for repression (297). Nonetheless, GR typically does not inhibit binding of AP-1 or NF-κB to its respective response element within the endogenous promoters (253,262,293,295,299). In addition, the LIM-domain protein thyroid receptor-interacting protein 6 (Trip6) is suggested to function as an essential intermediary interaction partner in the association of GR, and AP-1 or NF-κB, because knockdown of Trip6 or abolishing the interaction of Trip6 with GR abrogates GR-mediated transrepression (295,300).
Additionally, GCs can directly affect various histone modifications that combine into a so-called “histone code,” thus influencing chromatin accessibility and the associated gene transcription. Effects of GCs on histone phosphorylation will be discussed below. Furthermore, GCs can inhibit cellular TNF-induced histone H4 K8 and K12 acetylation via reducing the HAT activity of CBP. Moreover, GC administration can increase the expression of HDAC2, target HDAC2 to NF-κB CBP complexes, and target HDAC1 to the SP-A gene promoter (130,294,301). These histone-deacetylating events are associated with a halted transcription of NF-κB-driven genes (130,294,301). At the single promoter level, recent studies revealed a GC-mediated decline of histone H3 and H4 acetylation on the SP-A and IL-8 gene promoters, respectively, which was associated with a decreased transcription of these genes (294,302). Lastly, GCs instigate the dimethylation of H3 K9 at the SP-A promoter (294). This histone modification constitutes a transcription-repressive chromatin mark (303).
Because besides DNA-bound transcription factors and the basal transcription machinery, the activated NF-κB-driven gene promoters recruit various cofactors in a gene- and cell type-specific manner, specific research has aimed to unravel possible GC effects on the composition and modulation of this enhanceosome. First, it was proposed that GR competes with NF-κB or AP-1 for a limited amount of cofactors, such as the HAT CBP/p300 or SRC-1 (304). However, overexpression of these cofactors or mutation of the coactivator-interacting domains of GR or NF-κB did not lead to a reversal of the marked inhibition (295,305,306,307,308). Furthermore, GC administration did not affect NF-κB p65 association with CBP (307). Lastly, the hypothesis of involvement of GR-interacting cofactors was challenged via a mutation experiment. Although the mutation of E755A in the GR C terminus, abolishing the interaction of GR with LxxLL-containing cofactors, significantly decreased GR-mediated transactivation of a GRE-regulated reporter gene construct, this mutation did not alter GR-mediated transrepression of Gal4-p65 activity (309). Nevertheless, gradual overexpression of SRC-1 or SRC-2 combined with the comodulator STAMP results in a lower EC50 value and a higher fold repression for GR-mediated inhibition of AP-1-mediated reporter gene activity, in which EC50 is defined as the GC concentration required for a half maximal response (310). Involvement of SRC-2 in GR-mediated repression of AP-1- or NF-κB-dependent gene expression was also confirmed for the endogenous genes collagenase-3 and IL-8 (297). However, because most data on cofactor function are derived from overexpression experiments, physiological relevance of nuclear cofactor modulation of GR-induced transrepression is yet to be determined. Lastly, not only could cofactor complex assembly be modulated, but like NF-κB and GR, cofactors such as SRC-1, SRC-2, SRC-3, PGC-1, CBP, NCoR, and SMRT are themselves also subject to extensive posttranslational modulation such as phosphorylation, methylation, SUMOylation, ubiquitination, and acetylation. These modifications can affect cofactor-nuclear receptor binding, activity, localization, and half-life (311,312,313). However, the impact of GCs on these modulations or vice versa, how these modulations impact GC-mediated mechanisms, is currently not very well known.
II. Phosphoregulation of the Glucocorticoid Receptor
Activational control of GR can be imposed via a combinatorial mechanism involving ligand accessibility, GR concentration, subcellular localization, and also posttranslational modifications of GR. The activity of GR is affected by various modifications, among which are phosphorylation, acetylation, nitrosylation, redox regulation, ubiquitination, and SUMOylation (Fig. 4) (314,315). In this section, we will discuss the impact of phosphomodulation of GR on various aspects of GR functionality and signaling.
A. GR phosphorylation
Phosphorylation is the reversible covalent association of a phospho group on a protein. Phosphorylation is regulated by the balance between phosphorylating kinases and dephosphorylating phosphatases. This modification may affect GR hormone and DNA binding and subcellular localization, alter GR interactions and protein half-life, ultimately affecting transactivating and transrepressing capabilities of GRs. These phosphorylations of the GR are mediated by specifically targeted kinases.
In murine GR (mGR), researchers have identified eight phosphorylation sites: S122, S150, S212, S220, S234, S315, S412, and T159, most of which reside in the N-terminal domain (Fig. 4A) (316,317). The rat GR (rGR) phosphorylation sites correspond to those of mGR (Fig. 4A) (318). Conversely, in hGR, five Ser residues were characterized as phosphorylation targets: S113, S141, S203, S211, and S226, and recently S404 (319,320,321). These residues could be sequence matched to the mGR phosphorylation targets S122, S150, S212, S220, S234, and S412, respectively (Fig. 4A). Additionally, recent mass spectrometry analysis of hGR confirmed the phosphorylation of hGR S226, but also suggested a cell cycle- dependent potential phosphorylation of T8, S45, S134, S234, and S267 (322), of which only the latter four S residues present a conserved counterpart in the mGR and rGR. However, additional evidence to confirm in vivo phosphorylation of these sites is yet to be reported. Of note, all phosphorylation sites are located in the AF-1-containing N-terminal domain of GR.
Phosphorylation of hGR S203 and S211 can both be mediated by Cdk2/cyclin A kinase complexes, whereas Cdk2/cyclin E targets only hGR S203 (323). In support, murine embryonic fibroblast cells devoid of the Cdk inhibitor p27Kip1, which affects Cdk2 activity, show enhanced GR phosphorylation at the corresponding mGR S212 and S220 and an increase in GR transactivation potential (324). In general, phosphorylation of hGR S203 and S211 or their murine counterparts is associated with an enhanced transactivation of GRE-regulated promoters (323,325,326,327). However, the interaction of Cdk5 and its activator protein p35 and p25 with the GR LBD could also mediate phosphorylation of hGR S203 and hGR S211, remarkably resulting in a decreased GR transcriptional activation of the MMTV and serum- and GC-inducible kinase (SGK) promoter via attenuated GR-cofactor interactions (328).
The p38 MAPK could possibly also contribute to the phosphorylation of hGR S211 from 20 h of GC exposure onwards, in lymphoid cells. However, in these cells pretreatment with p38 MAPK inhibitor SB203580 only slightly diminished hGR S211 phosphorylation. Furthermore, in these cells, overexpression of the hGR S211A mutant severely impaired the GR transactivation potential (329). Conversely, p38 MAPK-mediated phosphorylation of hGR at an undefined residue was also associated with a decreased GR ligand-binding affinity and a slightly reduced GC-dependent repression of GM-CSF production (330). Furthermore, activation of p38 MAPK via IL-1α administration or overexpression resulted in a diminished GR transactivation function and GR-GRE binding (331,332,333). Possibly, the p38 MAPK-mediated inhibition of GR activity is not mediated via a direct GR phosphorylation but is instigated via phosphorylation of a GR LBD-interacting factor (332).
Additionally, the direct interaction of GR via the JNK interaction motif with JNK MAPK accommodates JNK-mediated phosphorylation of rGR S246, corresponding to hGR S226. This phosphorylation inhibits GR transcriptional activation (334,335), but currently, the possible effect of this phosphorylation on GR transrepression mechanisms has not been researched. In correspondence with the above findings, overexpression of the JNK upstream activator MKK7 inhibits GR transactivation toward an MMTV reporter gene construct (332). Mechanistically, activated JNK-instigated export of GR to the cytoplasm via a leptomycin B-sensitive, CRM1-dependent mechanism could contribute to the inhibition of GR activity. In support, UV-induced JNK MAPK or overexpression of JNK expedited GR nuclear export and was associated with an inhibition of GR-mediated transcription, whereas expression of a GR S226A mutant shows no UV-induced export or associated diminished GR activity (334).
Furthermore, a GSK3-mediated phosphorylation of rGR at T171 was shown in vitro. Overexpression of GSK3 inhibited GR transcriptional activation but does not affect GR-mediated repression of an AP-1-driven reporter gene construct. However, this rGR T171 corresponds to a hGR A150, and GSK3 overexpression in human cells thus does not affect GR activity and shows a species-specific difference in GR phosphorylation. Nevertheless, when hGR A150 is mutated to T, the GSK3-mediated inhibition of GR transactivation can be restored (318). In contrast, hGR S404 was recently identified as a target of GSK3β. The nuclear phosphorylation of this residue would lead to nuclear export of GR, an enhanced down-regulation of GR and attenuated transactivation of GRE-containing promoters, and a hampered transrepression of NF-κB-regulated gene promoter activities (321).
Lastly, a ligand-independent association of GR with PKA has been reported, and overexpression of PKA can enhance basal and GC-induced MMTV reporter gene activity and GR-GRE binding (336,337,338). Although phosphorylation of GR by PKAc was suggested in vitro, this was never confirmed in vivo (339).
Although GRs display a low basal phosphorylation, these receptors get hyperphosphorylated upon the addition of agonist (316). In quiescent cells, hGR S211 phosphorylation count is lower than that of hGR S203. The GC-induced alterations in GR phosphorylation seem to depend on the preexisting phosphorylation status of GR because mutation of hGR S203 to A mildly impedes hGR S211 phosphorylation, while slightly enhancing hGR S226 phosphorylation, suggesting a possibly ordered, sequential phosphorylation of GR and an intersite dependency (320,340).
Although GCs strongly enhance both S203 and S211 phosphorylations, hGR S211 phosphorylation is proposed as a hallmark for the transactivation potential of GR because the antagonist RU486 still allows for GR S203 but not S211 phosphorylation (320). It should be noted, however, that RU486 can also behave as an agonist in a context-dependent manner (341). GR transactivation function is found to be at its peak when the relative phosphorylation of hGR S211 surpasses that of S226 (327). Although overexpression of hGR S211A only diminishes GR transcriptional activity (329), mutation analysis of mGR showed that S to A mutations for the S212 and S220 residues strongly decreased GR transactivation of a minimal GRE-regulated reporter gene construct (325). Not all phosphorylations lead to an increase in transactivation. The recently identified interaction of ERK8 with GRα via the LIM domain-containing Hic5 intermediate was suggested to function as a dampener of GR transactivation, the mechanism of which remains unknown (342).
The localization of phosphorylated GR can also differ. Ligand-induced S211 phosphorylated GR appears to be mainly nuclear, whereas S203 phosphorylated GR preferentially resides in the cytoplasm (320,326). In contrast, in the absence of ligand, basally phosphorylated GRs at S203 or S211 were both found in the cytoplasm (320,326). Conversely, a nuclear phosphorylation of hGR S404 by GSK3β and rGR S246 by JNK seems to expedite nucleocytoplasmic transport of these GRs (321,334). These findings demonstrate the impact of GR phosphorylation status on its subcellular localization. Interestingly, pharmacological inhibition of tyrosine phosphorylation—using genistein or tyrphostin AG126—is reported to stimulate nuclear export of GR (343). Currently, it is not clear whether this relocalization effect should be attributed to the direct modulation of an unknown GR Y residue phosphorylation. However, GR phosphorylation is not critical to the receptors’ nuclear import function because mGR, with all phosphorylatable sites mutated to A, still undergoes ligand-dependent nuclear translocation (325). In addition, RU486 can also elicit GR translocation (262).
A recent study has provided a link between cell compartment-specific phosphorylation of the GR, induced by acute or chronic stress, and GR-dependent transcriptional activity in rat central nervous system tissue. Only acute isolation stress resulted in an increase in serum corticosterone levels. Under the condition of chronic stress, despite unaltered levels of nuclear GR, a significant transcriptional activity was still observed. In fact, it turned out that GR-dependent gene regulation patterns in the central nervous system, for GR, corticotropin-releasing factor (CRF), and brain-derived neurotrophic factor (BDNF) were similar compared with the acute stress model. These results may suggest that the transcriptional activity of GR is not solely regulated by the levels of hormone. Rather, the transcriptional activity of GR under chronic isolation was proposed to result from an increased Cdk5 activation and phosphorylation of the nuclear GR at S232 and a decreased JNK activity, which was reflected in a decreased phosphorylation of nuclear GR at S246 (344).
GR phosphorylation can impact its half-life because hGR phosphorylated at S203 displays a more rapid decay than GR phosphorylated at S211, and various mutations ablating the phospho-acceptor sites of mGR lowered ligand-dependent down-regulation of mGR (320,325). In addition, association of GR with the tumor suppressor gene TSG101 (tumor susceptibility gene 101), which can bind ubiquitin groups and negatively affect ubiquitin-dependent proteasomal degradation, occurs preferentially with a nonphosphorylated GR. This interaction of TSG101 and hypophosphorylated GR then leads to unliganded GR protein stabilization (345,346).
The GRE-regulated tyrosine aminotransferase (TAT) and GILZ gene promoters appear to preferentially bind GR phosphorylated at S211 or S226 (326). Mechanistically, hGR phosphorylation of S211 would alter its conformation and thus accommodate interactions of GR with vitamin D receptor-interacting protein 150/mediator complex subunit 14 (DRIP150/MED14) (346). Indeed, in GR S211A-expressing cells, the expression of MED14-independent genes was not impaired, whereas the transcription of MED14-dependent genes was attenuated, suggesting a link between GR phosphorylation and MED14 involvement in a gene promoter-specific manner (327). Promoter selectivity was also previously shown by mutation analyses of mGR showing differential effects on various GRE-containing reporter gene constructs (325) and mutation analysis of hGR showing differential effects on various GC-activated and GC-repressed genes (321). In that respect, it is widely accepted that the different modification statuses of GR could lead to a variable cofactor interaction profile.
Thus, ligand-dependent phosphomodulation of GR could affect GR ligand binding, gene promoter-selective GR transactivation, GR DNA binding, cofactor recruitment, subcellular localization, and half-life.
B. GR dephosphorylation
Because phosphorylation is a reversible mechanism, GR function is also regulated by various phosphatases. In support, administration of a pharmacological inhibitor of PP1, PP2A, and PP5 function augments GR phosphorylation and blocks nuclear import of ligand-activated GR. These agents, however, allow export of GR to the cytoplasm but prevent its subsequent return to the nucleus, thus abrogating recycling of GR (340,347,348).
The tetratricopeptide repeat domain-containing PP5, via binding to Hsp90 in the GR chaperone complex, forms an indirect binding partner for GR (349,350,351). Knockdown of PP5 resulted in increased GR binding to DNA and GR transcriptional activity but did not affect the ability of the synthetic GC dexamethasone to bind to GR (352,353). In contrast, a similar experiment was recently reported to reduce GC-induced transcription of three endogenous genes (IRF8, IGF binding protein 1, Ladinin), while leaving GILZ expression unaffected. Concomitantly, this PP5 knockdown raised phosphorylation of hGR at S203, S211, and even more pronounced at the GR activity-inhibiting site S226 (340). This gene-specific control of PP5 over GR activity may be regulated by promoting the ligand-binding affinity of GR (354). The question arises whether these findings can be reconciled with one another on the basis of the existence of different contexts and/or a mix of primary and secondary effects. Alternatively, PP5 could mediate GC-instigated nuclear translocation of GR via the interaction between PP5 and the motor protein dynein (355). In this respect, PP5 is believed to dephosphorylate recycled GR proteins returning from the nucleus, thus resetting GR in a ligand-inducible state (340,356).
Very recently, estrogen has been described to inhibit GC induction of DUSP1 and GSK genes in breast cancer cells, providing a plausible explanation for why GC trials in breast cancer are not overtly successful (357). The mechanism involved a reduced ligand-induced GR phosphorylation at S211, which is associated with the active form of GR. Estrogen increased the expression of PP5, which mediates the dephosphorylation of GR at S211. After PP5 knockdown, the estrogen-promoted cell proliferation was significantly suppressed by GCs, providing proof for a crosstalk between estrogen-induced PP5 and GR action (357).
In short, GR phosphomodulation is thus a flexible mechanism, integrating cellular stimuli and modulating multiple receptor functionalities. Currently, no GR-targeting phosphatases have been linked to specific GR residues. Moreover, considering the plethora of kinases involved in GR phosphorylation, the small number of GR-targeting phosphatases, characterized until now, seems too constrained. Future research in this direction could thus possibly unveil additional information on these processes.
C. Other posttranslational modifications of GR
In this paragraph, we will discuss how other posttranslational modifications of the GR can affect its phosphorylation status. SUMOylation involves an E1-activating enzyme, an E2 conjugation enzyme, and an E3 ligase. The covalent attachment of a small ubiquitin-related modifier (SUMO) motif can affect protein stability, subcellular localization, and transcriptional activity (358). The following SUMO-target sites have been identified in GR: K703 in the C-terminal LBD and K277 and K293 in the N-terminal synergy control motif (which is defined to inhibit synergistic transcription conferred by GREs containing multiple GR binding sites). These latter SUMOylations can thus act as inhibitory elements controlling GR activity toward multiple, but not single, GREs (Fig. 4B) (359,360,361). Interestingly, the JNK-mediated phosphorylation of hGR S226 appears to facilitate subsequent GR SUMOylation at the N-terminal SUMOylation sites, and thus GR SUMOylation could be considered a phosphorylation-directed posttranslational GR modification (359). Overexpression of SUMO-1 can destabilize GR protein (362), whereas a mutation of the potential SUMOylation sites of GR or inhibition of the SUMOylation enhances GR transactivation in a gene-selective manner, with a preference for multiple GRE-containing gene promoters (359,360,361). Moreover, overexpression of the GR targeting SUMO-2 protein decreased GR-mediated expression of endogenous GRE-regulated genes (359). Although SUMOylation of GR can occur in a ligand-independent manner and does not require an intact GR LBD and DBD dimerization motif, the integrity of the latter domains and especially binding of GR to DNA appears necessary to implement SUMO-dependent transcriptional inhibition (360,362). Although binding of death domain-associated protein (DAXX) to SUMOylated GR has been proposed to mediate the described, inhibitory effects (363), a recent report finds no effect of death DAXX overexpression or knockdown on SUMOylated GR activity (360). Currently, it is unclear how GR SUMOylation controls its transcriptional activity, but SUMO-interacting proteins are most likely to be considered.
Additionally, it is interesting to note that the phosphorylatable hGR S404, which is phosphorylated by GSK3β, mGR S412, and rGR S424 are comprised in a PEST degradation motif (316,321). Ligand-activated mGR could be ubiquitinated at K426, leading to degradation of the GR protein (232,364). The residue K419 is the counterpart in hGR (Fig. 4B). Blocking mGR downmodulation via proteasomal inhibitors or via mutation of K426 enhances GR-mediated transactivation and retards GR mobility in the nucleus (232,364). Moreover, because GR becomes hyperphosphorylated upon ligand binding, it appears that this phosphoregulation is key to the onset of the ubiquitination-mediated proteasomal degradation of GR, since a mutant GR with all possible phospho-sites mutated to A does not undergo ligand-dependent downmodulation (325).
Because SUMOylation and ubiquitination appear to be affected by a differential phosphorylation of GR (325,359), it would prove interesting to research the possibility of other phospho-directed posttranslational modifications of GR. In this perspective, interesting research could focus on the acetylation of hGR at K494 and K495, which is a prerequisite for GR association with NF-κB p65 and assists the GC-instigated repression of GM-CSF gene expression (365).
III. Kinases Targeted by Glucocorticoid Receptor-Mediated Signaling
Because kinases such as MAPKs, MSKs, and Cdks are very important in controlling the expression of inflammatory cytokines, recent research has moved its focus to the modulation of these kinases as alternative antiinflammatory tools beside GCs. However, various aspects in kinase signaling and expression are already positively or negatively regulated by GCs. Therefore, this section will focus on the effects of activated GR on kinase signaling in the framework of inflammation.
A. Mitogen-activated protein kinases (MAPKs)
A dysregulated activation of MAPKs was traced in numerous inflammatory diseases such as rheumatoid arthritis, psoriasis, systemic lupus erythematosus, asthma, and inflammatory bowel disease (3,366). Interestingly, activated GR forms a complex regulatory loop with the activated MAPK signaling pathway. In short, whereas GR can directly and indirectly inactivate MAPKs, these MAPKs can affect GR via phosphorylating the receptor (see Section II.A). Depending on the cell type used, GCs have been shown to suppress p38, JNK, and/or ERK MAPK phosphorylation. The GC-induced dephosphorylations of p38, JNK, and ERK MAPK have all been suggested to occur via actions of the GC-induced DUSP1 phosphatase (see Section IV.A).
1. JNK MAPK
The GR crosstalk with the JNK signaling pathway, leading to repression of its downstream targets c-Jun, ATF2, and Elk-1, appears to have multiple mechanistic layers (367,368). Besides the GC-induced DUSP1-mediated dephosphorylation of JNK MAPK (369,370,371), GR can also directly interact with JNK and thus interfere with JNK activity without the necessity of de novo gene expression (367,372). Furthermore, the JNK-binding sequence of GR seemed to be required for GC-induced JNK inhibition and nuclear import of JNK (372). Notably, the interaction of GR with JNK can indirectly decrease AP-1 activity via binding of inactive JNK, together with GR, to the AP-1-bound elements of the c-jun gene promoter (372,373,374). However, binding of GR with the antagonist RU486 does not support a GR JNK interaction or GR and JNK recruitment to the c-jun gene promoter. Also, JNK signaling can be blocked by activated GR via interrupting the association between the activating MKK7 and JNK (372). Additionally, activation of the upstream MEKK1 can be hampered by GCs via its negative interference with the MEKK1 Hsp90 association, culminating in a disruption of the JNK activation (375). Recently, Kim et al. (376) demonstrated that the GC-inducible SGK1 kinase inhibits the activation of SAPK (stress-activated protein kinase)/ERK kinase 1 (SEK1) (the upstream JNK kinase, also called MKK4), thereby negatively regulating the JNK signaling pathway. SGK1 was further found to physically associate with SEK1. Because SGK1 has been implicated in the promotion of cell survival and the protection against cellular stress, the described mechanism explains how SGK1 negatively regulates stress-activated signaling, namely through the inhibition of SEK1 functionality (376). Finally, a GC-mediated inhibition of JNK MAPK activity has been shown to block the translation of TNF in murine monocytes (377).
2. ERK MAPK
Inhibition of ERK MAPK activation can occur via alternative GC-dependent mechanisms. The interaction of Raf-1, a MAP3K in the ERK signaling pathway, with its chaperone Hsp90 is inhibited by GCs. As a result, Raf-1 can no longer associate with Ras, the activation of which remained unaffected by GCs. Consequently, the impaired Raf-1 activation leads to a decrease in ERK activation (375,378).
GCs can elevate not only DUSP1 expression but also the expression of GRE-regulated GILZ in various cell lines (270,280,379,380,381,382,383,384,385,386,387,388,389). Interestingly, binding of GILZ to Raf-1 inhibits its phosphorylation and thus represses the phosphorylation of Raf-1’s downstream targets MKK1/2 and ultimately ERK1/2 MAPK (390,391). Of note, Raf-1 can also associate with liganded GR, together with 14-3-3 (392). Additionally, GILZ can directly bind to NF-κB and AP-1, and as such represses NF-κB- and AP-1-directed expression of proinflammatory genes (270,384,386,393,394). Other GC-induced genes, namely downstream of tyrosine kinase 1 (Dok-1), SLAP and Dexras1, have also been associated with a GC-mediated inhibition of ERK MAPK activation and inflammatory signaling (395,396,397,398).
3. p38 MAPK
Although in many cell lines GC treatment leads to a decrease in p38 MAPK phosphorylation and activation via a DUSP1-dependent regulation (275,399,400), prolonged GC exposure of lymphoid cells is actually known to increase p38 MAPK phosphorylation levels (329). Cell-specific effects should thus be considered.
B. MAPK-activated protein kinases (MKs)
Although p38 and ERK MAPKs have their own specific transcription factor and other protein targets, these MAPKs can continue the MAPK cascade by activating yet another level of kinases, the MKs (Fig. 5). Based on sequence homology, these MKs comprise MSKs, 90-kDa RSKs, MAPK-interacting kinases (MNKs), MK2, MK3, and as a final group MK5 (25). Only ERK MAPKs can activate RSKs and MNK2, and only p38 MAPKs can activate MK2, MK3, and MK5, whereas both ERK and p38 MAPKs can activate MSK1, MSK2, and MNK1 (25).
1. MSK1
The two MSK proteins, MSK1 and MSK2, consist of two kinase domains bound together by a linker region. The N-terminal kinase domain of MSK accommodates substrate and autophosphorylation, whereas the MSK (CTD) comprises a bipartite nuclear localization sequence (401), a MAPK-docking domain, and an autoinhibitory sequence (402,403) and can only mediate autophosphorylation of MSK (25). At the cellular level, MSK1 and MSK2 are predominantly nuclear kinases, most likely due to their bipartite NLS (401,404). MSKs are activated via p38α and/or ERK1/2 MAPK phosphorylation in response to various stimuli (84,401,405,406,407,408). Activation of these upstream kinases leads to the phosphorylation of MSK1 T581 or MSK2 T568, which is considered primordial to the activation of the MSK1 C-terminal kinase domain and the subsequent full activation of MSK1 (402,403,409,410). MSKs are involved in affecting chromatin structure, transcription factor accessibility, and ultimately gene transcription via the phosphorylation of CREB S133, ATF1 S63, and NF-κB S276, leading to their activation (73,83,84,405,407), and the phosphorylation of H3 S10, contributing to chromatin relaxation and thus facilitating transcription of select gene promoters (411,412,413,414). Interestingly, phosphorylation of H3 S10 and H3 S28 by MSKs creates confirmed binding sites for 14-3-3 regulatory proteins at c-jun, c-fos, and HDAC1 gene promoters (415,416,417). Furthermore, it was demonstrated that the concomitant acetylation of H3 K9 and K14 stabilizes the association between phospho-acetylated H3 and 14-3-3 (416,417,418). Lastly, the phosphorylation of H3 S10 and attraction of 14-3-3 correlates with dissociation of the transcription-repressive heterochromatin protein (HP) 1γ and recruitment of RNA pol II (417,419).
Recent investigation into the effect of GCs on MSK1 signaling revealed that, whereas GCs cannot profoundly affect MSK1 phosphorylation or activity, these steroids can effectively abolish MSK1 recruitment and thus H3 S10 phosphorylation at specific inflammatory gene promoters (420), which nuances an earlier report that GCs could not impact the overall H3 S10 phosphorylation in these cells (421). Moreover, the GC-induced lack of MSK1 occupancy at these TNF-activated inflammatory gene promoters contributes to a decline in NF-κB transactivation (420). Remarkably, GCs not only impede recruitment of MSK1 at inflammatory gene promoters, but actually drive certain MSK1 proteins to the cytoplasm in a GR- and CRM1-dependent manner and furthermore support a physical interaction between GC-activated GR and activated MSK1 (420). However, in-depth mechanistic studies of how GCs affect MSK1 localization are still warranted. Because the GR modulator compound A (CpdA), which can transrepress NF-κB-driven gene expression without displaying any transactivating properties (293,422), can export a fraction of the nuclear MSK1 to the cytoplasm similar to classical GCs, the active GR-instigated redistribution of MSK1 to the cytoplasm fits in the framework of a general cellular mechanism of GR-mediated transrepression of NF-κB-mediated transcription (420).
MSK1 has also been implicated in the cytoplasmic phosphorylation of the eukaryotic translation initiation factor 4E-binding protein 1 (4E-BP1) at S64 and possibly also T36 (423). In unstimulated cells, 4E-BP1 can bind to the eukaryotic translation initiation factor-4E (eIF-4E) complex, thus inhibiting functioning of the eIF-4E holocomplex in translation initiation. Phosphorylation of 4E-BP1 relieves the translational block by dissociating 4E-BP1 from the eIF-4F member eIF-4E at the mRNA cap (423). Interestingly, the administration of GCs blocked phosphorylation of the translational repressor 4E-BP1, thus allowing reassociation of 4E-BP1 and eIF-4E (424,425,426,427,428) (see Section V.A). The latter interaction prevents mRNA cap-dependent initiation of translation. Currently, crosstalk of GR with MSK1 in this mechanism has not been fully researched. As such, the possible involvement of GC-translocated cytoplasmic MSK1 in these events and the overall implications of these inhibitory effects for the antiinflammatory potential of GCs are not fully understood.
2. MK2
The GC-mediated MAPK inhibition results in an inhibition of proinflammatory gene transcription via repressing the phosphorylation of multiple factors, but it also affects proinflammatory protein production because MAPKs and the MK2 are involved in mRNA stability. However, only the select mRNAs that contain AREs at the 3′-untranslated end, which are often cytokine and chemokine transcripts, are affected by MAPK regulation (429). Mechanistically, TTP can bind and destabilize these ARE-containing transcripts and trigger their degradation via binding of exonucleases (430,431). However, TTP function can be inhibited via phosphorylation by the p38 MAPK-activated kinase MK2, thus stabilizing the ARE-containing transcripts such as TNF mRNA (432,433,434,435,436,437,438). Notably, TTP knockout (KO) mice display phenotypically inflammatory arthritis due to an increased stability of TNF mRNA and an enhanced production of TNF protein (439). Because GCs enhance the expression of TTP in various cell lines (243,269,274,440) and concomitantly inhibit p38 MAPK and thus most likely MK2 activity, these steroids promote the destabilization and degradation of, among others, TNF, COX-2, GM-CSF, vascular endothelial growth factor (VEGF), MMP1, MMP3, IL-6, and IL-8 mRNAs (243,268,279,283,441,442,443). In support, GCs repress TNF mRNA in lung epithelial cells and decrease luciferase expression of a TNF 3′ untranslated region reporter plasmid in an orientation-dependent manner. Small interfering RNAs to TTP significantly prevent this effect, and a cell line stably expressing a short-hairpin RNA to TTP showed that TTP is critical for GC-induced inhibition of TNF mRNA (269,274). Comparing knockdown approaches with TTP−/− murine embryonic fibroblasts, differential effects of the lack of TTP on the mRNA turnover of different target genes were observed. Despite this, a certain degree of loss of GC-induced repression of CCL2, CCL7, CXCL5, and IL-6, which demonstrated TTP binding, was found to be reproducible. The observed differences may be due to different levels of TTP repression and to the diverse phenotype of cells with a chronic vs. an acute factor depletion (269,274). Taken together, these studies reflect a novel inductive antiinflammatory signaling pathway for GCs that acts via posttranscriptional mechanisms (269,274). Not only p38 MAPK, but also JNK and ERK MAPK signaling has been linked to the posttranscriptional stabilization of TNF, IL-2, and IL-3 mRNA (444,445,446). Combined, the GC-induced mRNA destabilization together with the inhibited gene transcription causes a rapid clearance of existing inflammatory signaling molecules. Of note, the GC-stimulated destabilization of GR mRNA is mediated through this same mechanism (447), providing an adequate feedback response.
Currently, no link has been demonstrated between GR signaling and the RSKs, MNKs, MK3, or MK5.
C. Cyclin-dependent kinases (Cdks)
The proinflammatory stimulus TNF promotes the recruitment of RNA pol II at the IL-8 and ICAM1 promoter and the subsequent phosphorylation of the CTD of RNA pol II at S2 and S5. Conversely, GCs can interfere with the S2 phosphorylation of the RNA pol II CTD, while leaving its recruitment to the promoter unaffected (254). This S2 phosphorylation was mediated by Cdk9 of the Cdk9/ cyclinT1 P-TEFb complex and is absolutely necessary for (NF-κB-mediated) transcription (448,449). In support, knockdown or pharmacological blockage of P-TEFb severely impaired TNF stimulation of IL-8 gene transcription. Interestingly, at the IL-8 promoter, GR competes with P-TEFb for binding to NF-κB p65, thus inhibiting IL-8 gene transcription and the phosphorylation of the recruited RNA pol II S2 (253,254). Recently, it was also discovered that the association of P-TEFb with NF-κB required NF-κB p65 phosphorylation at S276 (81), thus integrating the GC-instigated blockage to recruit MSK1 (420) and P-TEFb (253,254) at the inflammatory IL-8 gene promoter.
Additionally, GCs can cell-dependently repress the gene expression of Cdk4 and Cdk 6 and their associating cyclin D3 (450) and induce expression of the Cdk inhibitor p21Cip1 (451,452). Furthermore, GCs can inhibit Cdk2 and Cdk4 activity, an event that is associated with the antiproliferative effects of GCs and decreased GRE- mediated transcription (453,454,455). Experiments with seliciclib, a Cdk inhibitor targeting Cdk2, Cdk7, and Cdk9, in a mouse model for the chronic inflammatory condition systemic lupus erythematosus revealed that this Cdk inhibitor can lower kidney inflammation and prolong survival. Upon combining seliciclib with GCs, however, the therapy elicited a greater beneficial effect than either therapy could alone (456). Additional research into the role of Cdks in the GC-mediated antiinflammatory mechanisms is still required.
D. IκB kinase α (IKKα)
The multifactorial kinase complex IKK is essential in relaying proinflammatory stimuli to NF-κB activation. When activated, IKKα and IKKβ can phosphorylate IκB, thus targeting this inhibitory molecule for degradation via the ubiquitin-proteasome degradation pathway and programming the subsequent release of NF-κB to translocate to the nucleus (10). Furthermore, IKKα can promote NF-κB DNA binding on specific gene promoters (457).
GCs can, in particular cell lines, up-regulate the expression of IκBα. The replenishment of the depleted IκBα pool thus redirects the activated NF-κB to the cytoplasm and ultimately counteracts the former actions of the NF-κB-activating IKK complex. A critical discussion of the role of GC-induced IκBα in the GR-mediated antiinflammatory mechanism is presented in Refs. 211 and 243.
In addition to MSK1 (411,414), IKKα has also been described to mediate H3 S10 phosphorylation (458,459). Constitutive shuttling of IKKα between cytoplasm and nucleus has been reported (460), but upon TNF induction, IKKα translocates into the nucleus (458,459). Ablation of IKKα halts H3 S10 phosphorylation and decreases proinflammatory gene expression, without affecting IκBα degradation or NF-κB DNA binding (458,459,461,462). Chromatin immunoprecipitation analysis of the NF-κB-regulated surfactant protein-A (SP-A) gene promoter in human fetal lung type II cells recently revealed that GCs can diminish H3 S10 phosphorylation in the SP-A promoter and can block the recruitment of the H3 S10 kinase IKKα (294). In this respect, it may also be interesting to investigate the recruitment dynamics of the H3 S10-phosphorylating kinase MSK1 at the SP-A gene promoter.
E. TANK-binding kinase 1 (TBK1)
In a recent report, TRAF family member-associated NF-κB activator (TANK)-binding kinase 1 (TBK1) was identified as a new target for GCs (463). This kinase is activated downstream of both TLR3 and TLR4 signaling and functions together with the inducible IκB kinase IKKε as an intermediate in the activation of the transcription factor IRF3 (464,465). Upon activation, IRF3 subsequently dimerizes, associates with CBP, and can then translocate into the nucleus where it binds onto specific IFN-stimulated response elements (ISREs), e.g., in the RANTES gene promoter (466,467). Currently, no effects of GCs on IKKε have been described. However, GCs can diminish LPS (TLR4)- and poly I-C (TLR3)-stimulated S127 phosphorylation of TBK1 and thus attenuate TBK1 activity. Possible involvement of DUSP10 and the GC-inducible DUSP4 (275), but not DUSP1, was suggested via analysis of the effects of their overexpression on ISRE-containing reporter gene constructs. However, in this respect, (double) knockdown studies are still lacking. Moreover, possible effects of GCs on the SH2-containing tyrosine phosphatase SHP-2, which is known to target the kinase domain of TBK1 (468), have not yet been researched. The described GC-mediated inhibition of TBK1 would thus contribute to the steroid-initiated inhibition of IRF3-regulated gene promoter activity of a reporter gene construct in U373 cells (463). Correspondingly, the LPS or poly(I-C)-induced expression of RANTES, of which the promoter contains an ISRE, can also be inhibited by addition of GCs. Moreover, the poly(I-C)/TLR3-induced expression of other IRF3-dependent genes such as IFN-β, IFN-inducible protein of 10 kDa (IP-10), ISG15, and ISG56 and ISRE-luc reporter gene constructs is also subject to a GC-mediated inhibition, which would be instigated via the disruption of IRF3 and GRIP1/nuclear coactivator-2) complexes in murine macrophages (469).
However, these events appear to be in contrast with findings of Ogawa et al. (299), who showed in macrophages that TLR4- or TLR9-mediated promoter activation of the chemokines IFN-inducible protein of 10 kDa (IP-10) or IFN-induced with tetratricopeptide repeats 1 (Ifit1) or of an IRF3-dependent reporter gene construct can be inhibited by GCs, whereas the TLR3-mediated activation of the same gene promoters is refractory to GCs (299). Mechanistically, Ifit1 depends on activation via the transcription factor IRF3, irrespective of the stimulus. However, the interaction of DNA-bound IRF3 with NF-κB p65 only occurs upon TLR4/TLR9-mediated Ifit1 gene promoter activation. Under these circumstances where p65 acts as a cofactor, the specific sensitivity of this promoter to GC-mediated repression in a TLR4- or TLR9-stimulated environment is being guaranteed. The basis of this repression was attributed to a binding competition between IRF3 and GR for the same site in NF-κB p65, in which GR has a greater affinity for NF-κB p65 than IRF3 (299). Interestingly, TBK1 is also a TNF-activated kinase for NF-κB p65 S536, which plays a role in the full transcriptional activation of this transcription factor (470). Further research is needed to resolve this apparent paradox between the results of McCoy et al. (463), Reily et al. (469), and Ogawa et al. (299).
F. Other kinases
The STAT transcription factors are phosphoproteins, activated by receptor-associated Janus kinases (JAKs), after cytokine receptor stimulation. Administration of GCs can inhibit the IL-2-induced phosphorylation, nuclear translocation, and DNA binding of STAT5 via a decreased expression of JAK3 and IL-2 receptor γ. As a consequence, IL-2, IL-4, IL-7, and IL-15 signaling and ultimately T cell proliferation was impeded (471). Furthermore, GCs can inhibit the IL-12-induced phosphorylation of STAT4 on position Y693 in T-lymphocytes, however, without affecting the IL-12-instigated JAK phosphorylation (472,473). The inhibition of IL-2-induced STAT5 phosphorylation by GCs in T cells was associated with a GC-mediated down-regulation of JAK3, but not JAK1, protein levels (471).
PKC is a family of S/T kinases, which are ubiquitously expressed under the control of diverse stimuli and have been implicated in the pathogenesis of asthma and COPD (474). As such, PKCs can function as an intermediate in NF-κB-dependent gene transcription (475,476). An overall GC-instigated decline in PKC activity was recently reported for mesenteric arteries from rats (477). In particular, PKCδ, which has been associated with the NF-κB-mediated activation of IL-8 gene promoter activity (476), is an interacting partner for the GC-induced Dexras1. This association results in a decline in PKCδ phosphorylation and activity (478).
Protein kinase B (PKB), also known as Akt, is a phosphatidylinositol-3-kinase (PI3K)-stimulated kinase, involved in cell cycle regulation. The GC-induced PKB activation can also result in the phosphorylation of its downstream target kinase, GSK3β, causing inhibition of its activity (479,480).
Upon GC administration, PKB can be rapidly activated via a PI3K-dependent pathway, leading to the release of vasorelaxing nitric oxide (479,481,482,483,484). In support, GR can interact with the p85α subunit of PI3K (484). These rapid nongenomic effects typically need high doses of GCs to occur.
Depending on the cell type, however, GCs may differentially target particular kinases. An important mechanism underlying long-term GC-induced bone loss is the impairment of osteoblast function and bone formation. The Wnt signaling pathway has been hypothesized to play a critical role in the osteoblast differentiation-related cell cycle. A key negative regulator in the Wnt signaling pathway is the serine/threonine kinase GSK3β. Consequently, GC-mediated activation of GSK3β in osteoblasts resulted in an inhibition of the Wnt signaling pathway. In osteoblasts, GCs do not stimulate but further inhibit phosphorylation of PKB/Akt on Ser473 and thus disrupt input from the PI3K/Akt pathway into the Wnt signaling pathway at the level of GSK3β (485). GSK3β, a known phosphorylator of NF-κB S468 (68), has recently also been shown to modulate GR (321), adding on complexity (see also Section II.A). In mammary epithelial tumor cells, GCs induce the phosphorylation of GSK3β, ultimately leading to its degradation via the ubiquitin 26S proteasome (480). This mechanism is believed to be involved in the control of tight junction formation in mammary epithelial tumor cells. Alternatively, GSK3β can also be activated via serum- and GC-inducible kinase 1 (SGK1). A high level of expression of SGK in breast cancer cells suggests that this kinase may function to protect tumor cells from apoptosis and thus can act as an oncogene. As its name suggests, the expression of SGK1 can be induced by GCs (486,487,488) and remarkably, this kinase can up-regulate also the transcriptional activity of NF-κB (489). In that respect, SGK1 was recently identified as the mediating kinase for PI3K-initiated phosphorylation of IKKα T23, which in turn facilitates the phosphorylation of IKKα S180 and the subsequent activation of NF-κB. Concomitantly, SGK1 was found to phosphorylate p300 S1834 and enhance NF-κB-mediated transcription (490). However, it is currently unknown how GCs could affect these processes and what their role could be in an inflammatory response. It may seem paradoxical that GCs are able to transcriptionally stimulate a kinase that subsequently serves to activate NF-κB, but it is important to realize that the functionality and role of SGK1 may be regulated not only transcriptionally but also posttranslationally by its upstream activating kinases, including PI3K and MAPK (490).
Additionally, GCs can affect various members of the Src family of tyrosine kinases. The c-Src kinase can—upon activation—contribute to IκBα phosphorylation and subsequent NF-κB translocation (491), whereas p56lck [lymphocyte kinase (Lck)] and p59fyn (Fyn) kinases and src-like spleen tyrosine kinase (Syk) have been reported to play a role in T cell receptor-mediated signal transduction (492). In contrast, GCs can rapidly inhibit the activity of these Lck and Fyn kinases via a membrane-associated GR, thus contributing to the GC-mediated inhibition of T-cell receptor signaling (493,494,495). Moreover, the GC-inducible increase in SLAP expression would inhibit Ag-induced phosphorylation of the Syk in mast cells (396). In contrast, GCs can augment the phosphorylation of Lck and the downstream activation of p59Fyn, Zap70, Rac1, and Vav in resting but not in activated T cells (496).
Furthermore, GCs can stimulate the protein and activity levels of the Rho-dependent protein kinase (ROCK) 2 but down-regulate the specific activity of ROCK1 in rat epithelial cells (497). The activation of ROCK2 is especially implicated in lung inflammation and the associated myosin light chain phosphorylation (498). In support, GCs can impede myosin light chain phosphorylation in airway hyperresponsive rats (499).
The molecular mechanisms that underlie nongenomic GC-induced immunosuppression remain to be precisely defined.
IV. Phosphatases Targeted by Glucocorticoid Receptor-Mediated Signaling
Phosphatases can be classified according to their substrate specificity. PPs targeting Ser/Thr and/or Tyr can be regarded as three groups: the tyrosine-specific phosphatases [protein tyrosine phosphatases (PTPs)], the Ser/Thr specific phosphatases, and the DUSPs, targeting phospho-Tyr/Ser/Thr (500,501). Currently, the most intensively researched GC-regulated phosphatase in the context of inflammation is DUSP1 (400,502). However, this phosphatase is not the only phosphatase that is subjected to GC-mediated modulation.
A. Dual specificity phosphatases (DUSPs)
The DUSP family comprises a subfamily of 10 enzymatically active DUSPs that target MAPKs, dephosphorylating T and Y residues, and therefore these phosphatases are also referred to as MAPK phosphatases. The MAPK family of protein kinases includes extracellular signal-related kinases ERK1 and ERK2; p38 MAPKs α, β, γ and δ; and the c-Jun N-terminal kinases JNK1, JNK2, and JNK3 (Fig. 5) (25,503). Structurally, these DUSPs are marked by a C-terminal catalytic domain, and DUSP1, DUSP2, DUSP4, and DUSP5 are considered inducible nuclear members of this DUSP subfamily (504).
The promoter of DUSP1 can be driven by ligand-activated GR, and overexpression of the dimerization-defective GR mutant (193) is incapable of mediating activation of the murine DUSP1 promoter by GCs (193,505). However, GCs enhance DUSP1 mRNA expression in murine macrophages expressing only this GRdim mutant (369). These findings per se are not necessarily in conflict because the group of Pearce (506) showed that GRdim can function even better than wild-type GR at promoters with multiple GR binding sites. Also, in the case of steroid receptors, the DBD dimer interface plays an important role because specific receptor mutations or noncanonical spacing of half-sites that disfavor this dimer interface lead to enhanced synergy (507). However, GRE sequences were never identified in DUSP1 promoters, and GCs failed to enhance human DUSP1 expression in cells solely expressing a rGR LS7 (P493R/A494S) mutant, which is incapable of transactivating classical GRE-regulated promoters (192,508,509). Alternatively, GC-mediated DUSP1 promoter activation was attributed to an ERK- and JNK-dependent mechanism involving AP-1 and CREB binding onto the DUSP1 promoter (510). Additionally, GR binding onto the DUSP promoter, 1.5 kb upstream of the transcription start site, was suggested to occur via a tethering mechanism onto C/EBPβ, and mutation of this C/EBP binding site attenuated GC-inducible DUSP1 reporter gene expression (509). In this context, it is interesting to note that GCs can enhance the function of C/EBPβ by inducing its phosphorylation (511). DUSP expression in response to GC signaling seems cell type-dependent. Besides DUSP1, also DUSP2, DUSP9, the PEST domain- enriched tyrosine phosphatase, and especially DUSP4 are up-regulated in response to GCs in bone marrow-derived mast cells (275), and GCs can also enhance the expression of DUSP4 in adipocytes (512), whereas the expression of DUSP2, DUSP4, and LC-PTP seems to be inhibited by GCs in various pre-B leukemia cell lines (513). Although, the above-mentioned DUSPs can also modulate ERK, p38, and JNK MAPK phosphorylation, thus impacting the inflammatory process (504), their role in the GR-mediated antiinflammatory mechanism is currently unaddressed. To date, no reports have been published on GC modulation of the related family members DUSP5, DUSP7, DUSP8, or DUSP16.
Besides GCs, mitogens and stressors can also elevate the expression of the inducible DUSPs, such as DUSP1, functioning as a negative feedback loop in MAPK regulation (504,514). Mechanistically, evidence from KO cells showed an involvement of the p38 and ERK MAPK- activated MSK1 and ATF1, and of the JNK1 MAPK in the LPS-mediated stimulation of DUSP1 gene transcription (514,515). However, stimulation of GR could further enhance the already LPS-induced DUSP1 promoter in a synergistic manner in macrophages (516). Very recently, a DUSP1/GRIP1-dependent combinatorial mechanism has been identified for repression by GR of LPS-induced COX-2, CXCL-5, and IL-6. Activated GR could inhibit TLR4-dependent COX-2 gene induction in macrophages via a mechanism that involves both DUSP1-mediated AP-1 inhibition and GR-GRIP1 recruitment to the p65 NF-κB DNA complex (517).
Interestingly, SOCS1, which appears to be up-regulated by GCs in hematopoietic cells and immune cell cancers (267,388,513,518,519), can hamper the transcription of DUSP1, probably via the association of GR and SOCS1 (520). In the inflammatory process, this SOCS is also involved in NF-κB p65 proteolysis (98) and can negatively interfere with TLR2- and TLR4-mediated signaling (521).
Cell-specific GC-elevated levels of DUSP1 can then target the activational phosphorylations of the MAPKs ERK, JNK, and p38, thus inactivating them (400). JNK and p38 MAPK are especially subject to DUSP1-mediated dephosphorylation because DUSP1 KO mice-derived immune cells display prolonged p38 and JNK activation, whereas the GC-induced ERK dephosphorylation was not altered (275,369). However, because the GC-induced ERK dephosphorylation depends on de novo protein production in RBL-2H3 mast cells, cell-specific effects may be at play here or possibly other GC-inducible DUSP family members, such as DUSP2, DUSP4, or DUSP9, may intervene (275,505).
Subsequent to the inactivation of the MAPKs, the ongoing proinflammatory signaling pathway is halted, and gene expression and mRNA translation of proinflammatory cytokines is attenuated (reviewed in Refs. 243,400 and 522) (see Sections III.A and III.B.2). Therefore, DUSP1 KO mice show an impaired GC-mediated repression of various proinflammatory cytokines. Nevertheless, GCs can still inhibit the expression of certain cytokines. The difference in GC-induced repression between DUSP1+/+ and DUSP1−/− cells depends on the selected cytokine but also cell type. Furthermore, inhibition of de novo protein synthesis in bone marrow-derived mast cells of DUSP1−/− mice can still partly reverse the antiinflammatory potential of GCs, suggesting the involvement of other GC-induced antiinflammatory proteins and of direct GC-dependent antiinflammatory mechanisms (275,369). Although DUSP1 KO mice are viable, they appear more susceptible to inflammation than their wild-type counterparts (275,369,523,524,525,526). However, the inflamed cells of these DUSP1 KO mice remain sensitive to the antiinflammatory mechanisms of GCs (275). In conclusion, GC-instigated up-regulation of DUSP1 is considered an additional layer in the overall antiinflammatory mechanism of GR.
In addition to GR and the MAPKs, DUSP1 itself is subject to posttranslational modifications. Although an extreme C-terminal phosphorylation of DUSP1 (S359, S364) by ERK MAPK instigates DUSP stabilization (527), prolonged ERK MAPK activity results in other C-terminal phosphorylations of DUSP1 (S296, S323). However, the latter phosphorylations promote DUSP degradation (528,529). Interestingly, GCs can inhibit the proteasomal degradation of DUSP1 (505). Moreover, LPS-mediated DUSP1 acetylation (K57) via p300 seems to stimulate the binding affinity of DUSP1 for p38 MAPK (530,531). It is currently unknown whether and how these processes are affected by GCs and connected to inflammation.
B. Other protein Y phosphatases
In the PTP family, PTP receptor type C (PTPRC, also known as LCA or CD45) is also regulated by GCs. As an essential regulator of T and B cell antigen receptor signaling, this membrane-associated tyrosine kinase is a marker for inflammation and is not surprisingly down-regulated by GCs in synovial membranes of arthritic rats and leukocytes of rheumatoid arthritis patients (532,533).
Alternatively, GCs have been reported to induce the expression of the SH2 domain-containing protein Y phosphatase PTP1C (also known as SHP1), but not PTP1D and PTP1 in pancreatic rat cells (534). This PTP1C has been implicated in the regulation of T cell antigen receptor signaling (535), and a deficiency in PTP1C function was recently linked to an increase in TLR-initiated inflammation (536).
C. Other phosphatases
The protein S/T phosphatases PP1 and PP2A family (PPP) are most abundantly present in the cell. The target-specific activity of PP1 and PP2A is encoded by five catalytical subunits and a vast abundance of interacting regulatory subunits (537). Although effects of PP1, PP2A, and PP5 on GR have been reported (see Section II.B), currently only a few effects of GCs on these PPs have been reported. PP1 and/or PP2A may be involved in the GC-mediated inhibition of translation via a GR-dependent dephosphorylation of p70 S6K (70-kDa ribosomal protein S6 kinase) (427,428) (see Section V.A). However, this involvement is yet to be confirmed via knockdown studies.
The S/T PP2A, which is composed of a catalytical subunit PP2Ac, a regulatory subunit PP2A-A (PR65), and a variable regulatory subunit PP2A-B (500), was suggested to target ERK, p38, JNK MAPK phosphorylations, and NF-κB phosphorylation in various cells (106,538,539,540,541,542,543,544). Furthermore, PP2A can impose a genome-wide dephosphorylation of H3 S10 in Drosophila (545). In light of the fact that GCs can also target H3 S10 phosphorylation (294,420), it could be interesting to investigate possible additional effects of GCs on PP2A.
PP2B (also known as calcineurin or PP3) is a Ca2+-dependent phosphatase comprising a catalytic A-subunit and a regulatory calmodulin-binding B-subunit (500), which has been implicated in the negative regulation of MyD88 and Trif and thus TLR-mediated signaling (546). Functionally, PP2B inhibitors are often used together with GCs as an immunosuppressant and antiinflammatory therapeutic (547,548). However, both pathways do not signal independently as GCs can also modulate PP2B activity (549,550). Because GCs can trigger the release of Ca2+, GCs could also rapidly and transiently enhance the activity of PP2B in T cells (549). Alternatively, GCs can stimulate PP2B activity in a Ca2+-independent mechanism, involving a GC-induced association of Hsp90 and PP2B in pancreatic β-cells (550,551). However, GCs do not seem to affect the protein levels of PP2B in various cells (551,552).
V. Kinase/Phosphatase Regulation in Glucocorticoid-Mediated Side Effects
Although GCs remain the mainstay in the treatment of acute and chronic inflammatory afflictions, long-term GC therapy of chronic inflammatory disorders could lead to a detrimental side-effect profile and possibly also GC resistance (see Section VI). Overall, prolonged GC treatment and high doses of potent GCs are the main risk factors in the onset of GC therapy-associated side effects. It is likely that both “desired effects” and “adverse effects” reflect GR actions, not only on transcription/translation directly but also on phosphorylation.
The most prevalent GC therapy-associated side effect is weight gain, followed by skin bruising/thinning and insomnia. From the patients’ view point, these skin problems together with a disturbed fat distribution are psychologically distressful. Clinically, however, hyperglycemia leading to diabetes mellitus, the enhanced susceptibility to infections, and osteoporosis are most worthy of attention and pose the greatest challenge and management issues in these patients. For instance, osteoporotic fractures occur in up to 50% of patients on long-term GC therapy, and the GC-induced patients’ sensitivity to infections could double their hospitalizations (553). Note that we call these secondary effects “side effects,” but it should be kept in mind that all of these effects are actually exaggerations of normal physiological GC actions.
Mechanistically, the onset and maintenance of GC- associated side effects is complex. Some GC therapy-associated side effects are mainly attributed to GR transactivation mechanisms (e.g., diabetes, glaucoma, myopathy, hypertension), whereas others originate from GR transrepression mechanisms [e.g., suppression of hypothalamic-pituitary-adrenocortical (HPA) axis]. However, the mechanisms of some side effects are not completely known or could be attributed to both GR transactivation and transrepression mechanisms (e.g., osteoporosis) (554). Further clinical and molecular research in this field is required to deepen our knowledge about the prevalence and mechanistic basis of GC therapy-associated side effects.
The GC-enhanced susceptibility to infections stems from the plethora of antiinflammatory and immunosuppressive actions of the activated GR, as discussed in the sections above. Below, we will highlight the involvement of phosphatase and kinase signaling in the mechanisms of GC-instigated side effects, with particular attention to the clinically most important ones.
A. Skeleton and muscle effects
Long-term GC therapy-induced osteoporosis originates from a dual mechanism: a decrease of osteoblast proliferation and activity and an increase in osteoclast activity (555,556).
Key to the activation status of osteoclastogenesis is the receptor activator of NF-κB ligand (RANKL)/osteoprotegerin (OPG) ratio. GCs can enhance the expression of RANKL while decreasing the expression of a RANKL-signaling inhibitor, namely osteoprotegerin (555,556). In the GC-mediated stimulatory mechanism of bone catabolism, abrogated expression of the IL-1R-associated pseudokinase IRAK-M is coupled to an enhanced differentiation of hematopoietic precursor cells into osteoclasts and an increase in osteoclast activity (557). In that respect, GCs can impede the expression of this IRAK-M and thus stimulate bone resorption (558).
In osteoblasts, ERK MAPK is essential to accommodate osteoblast proliferation. However, the GC-induced DUSP1 expression in these cells can dephosphorylate ERK MAPKs, an event that was thus linked to a tyrosine phosphatase-mediated inhibition of osteoblast proliferation. GCs directly affect osteoblasts by decreasing proliferation, and therefore this mechanism may contribute to the phenomenon of steroid-induced osteoporosis (559,560,561,562). Additionally, the GC-induced activation of GSK3β in osteoblasts was recently shown to contribute to their apoptosis, whereas GC-activated p38 MAPKs rather inhibited the apoptosis of these osteoblasts (563). Furthermore, the cell-detachment-induced apoptosis of osteocytes, which are bone-embedded cells that can regulate osteoblast activity via gap junctions, is regulated via the rapid activation of protein tyrosine kinase 2β (PTK2beta) (also known as RAFTK or Pyk2) and thus its downstream target JNK MAPK (564,565,566). Lastly, a GC-mediated inhibition of osteocalcin transcription, via a competitive nGRE (290,567) contributes to the inhibition of bone formation.
Long-term GC therapy in juvenile diseased patients also causes serious growth retardation (553). The GC-induced apoptosis of chondrocytes, linked to this process, has been attributed to GC-mediated caspase activation as well as GC-mediated inhibition of PKB phosphorylation (568).
When a high-dose GC treatment is sustained, patients may experience muscle atrophy, leading to myopathy. This GC-induced loss of muscle tissue is mainly attributed to enhanced protein degradation (catabolic effects) and diminished protein synthesis (antianabolic effects) (554,569).
One of the factors in the antianabolic effects of GCs, hampering the cellular protein synthesis mechanism, is the GC-mediated inhibition of 4E-BP1 phosphorylation, which facilitates the translation repressing association of 4E-BP1 with eIF-4E (424,425,426,427,428). However, okadaic acid could perturb the GC-mediated inhibition of p70 S6K and 4E-BP1 phosphorylation, thus pointing to a possible role for PP1 and/or PP2A as the PPs used (427,428). Alternatively, the GC-mediated decrease in phosphorylation of 4E-BP1 in myoblasts was suggested to occur via negative interference with upstream mammalian target of rapamycin (mTOR) signaling (570).
Additionally, the PI3K/PKB/GSKβ signaling pathway can play an important role in the regulation of muscle atrophy. Not surprisingly, GCs can inhibit PKB activity in myoblasts, thus allowing GSK3β activation, which is in turn associated with suppressed protein synthesis (569,571).
B. Hyperglycemia and diabetes
Patients who are subject to long-term GC therapy have a tendency to develop hyperglycemia, with the risk of diabetes because GCs not only decrease the stimulated insulin production but also lower their response to circulating insulin (554). The former decrease in insulin levels was attributed to a nGRE element in the insulin promoter as well as GC-mediated PP2B-dependent apoptosis of insulin-secreting cells (550,551,572). The latter insulin resistance could in part occur via a GC-induced rapid and transient inhibition of insulin receptor kinase activity and several downstream intermediates such as p70 S6K, PKB, 3-phosphoinositide-dependent protein kinase (PDK), Fyn, and GSK3 (494,512). Additionally, GCs could actually enhance JNK MAPK phosphorylation in adipocytes, which was associated with a perturbed insulin receptor-dependent signaling resulting in insulin resistance (494). In these same cells, an increase in DUSP1 expression was suggested to associate with a decrease in cellular glucose uptake (512). However, additional evidence for a role of DUSP1 in GC-induced hyperglycemia is currently lacking.
Lastly, GCs can stimulate the expression of various gluconeogenetic enzymes in the liver, among which TAT, pyruvate dehydrogenase kinase 4, glucose-6-phosphatase (G6Pase), and pyruvate carboxykinase (PEPCK), culminating in elevated glucose levels (573,574,575,576,577,578,579).
C. Other side effects
GC-induced hypertension is in part caused by a dysregulation of Na+ homeostasis. As an important factor in this event, GCs can enhance the GRE-regulated transcription of the epithelial Na+ channel (αENaC) gene and cell-specifically augment ENaC and Na(+)/H(+) exchanger 3 (NHE3) activity in a SGK1-dependent manner (554,580,581,582,583,584,585,586,587,588). The GC-activated rise in NHE3 activity would involve a direct SGK1-mediated NHE3 phosphorylation (588).
In the ENaC modulating mechanism, a SGK1-mediated inhibiting phosphorylation of ALL1-fused gene from chromosome 9 (Af9) of the histone-methylating Dot1a (disruptor of telomeric silencing alternative splice variant a)-Af9 complex can derepress the ENaC promoter, thus facilitating transcription (589). Furthermore, an aldosterone-induced SGK1 can phosphorylate the E3 ubiquitin ligase Nedd4-2, which is stabilized by 14-3-3, and can thus diminish the degradation rate of ENaC by reducing the interaction affinity between Nedd4-2 and ENaC (590,591,592,593). Alternatively, a specific and direct phosphorylation of ENaC by overexpressed SGK1 can directly enhance ENaC activity (594). Moreover, a steroid-mediated rise in GILZ expression and the concomitant inhibition of ERK MAPK signaling was recently linked to an increase of ENaC expression and activity (379,380,390). In addition, Na+ retention could possibly be enhanced by GCs via a SGK1-dependent increase in Na+/glucose cotransporter SGLT1 activity via a similar Nedd4-2- based mechanism (595). However, evidence to the specific GR-dependence of the above mechanisms is currently still lacking.
Furthermore, a GC-induced increase in gastric acid secretion, thus contributing to an enhanced risk of gastrointestinal bleeding and peptic ulcer development, occurs in a PI3K/SGK1-dependent manner of which the precise mechanism remains currently unclear (261,596).
Lastly, long-term GC therapy can in some cases lead to eye problems, such as cataract. A contributing mechanism in the GC-induced development of cataract is an increased rate of gluconeogenesis, thus implicating PEPCK and G6Pase regulation (554). Recently, it was discovered that GCs can enhance GILZ and DUSP1 expression in lens epithelial cells, coinciding with a decrease of Raf, ERK, p38 MAPK, and PKB phosphorylation (597). However, currently a role for this mechanism in cataractogenesis has not been firmly established.
VI. Kinase/Phosphatase Regulation in Glucocorticoid Resistance
Patients suffering from GC resistance are refractory to an antiinflammatory GC treatment. Although GC resistance can be innate, it can also be acquired due to a prolonged GC treatment (201). Here we will shortly discuss different mechanisms that possibly lie at the basis of GC resistance in inflammation, with special attention for GR phosphorylation in this phenomenon.
Foremost, the cause of innate GC resistance lies in a mutation of the GR itself, leading to abnormal GR concentrations, ligand-binding affinity, GR stability, GC- induced nuclear translocation, or GR-cofactor interactions (201). Currently, the research field concerning the several known GR haplotypes with regard to its specific effects on GR phosphorylation and GR effects on kinase and phosphatase regulation remains largely unexplored.
Furthermore, innate and acquired GC resistance was associated with a decrease in GRα protein levels via homologous down-regulation, an increase in the protein levels of the dominant-negatively acting GRβ, a decreased GR ligand-binding affinity, or GR DNA binding (598,599,600,601). Moreover, the sensitivity to GCs could be aligned with the degree of GC-induced GR nuclear translocation (602). Alternatively, GC resistance has been linked to an elevated expression of FKBP51, an element of the GR chaperone complex (603). At the level of cofactor regulation, GC resistance was associated with a reduced brahma-related gene (Brg1) 1 expression and a PI3K-regulated decrease in HDAC2 activity and expression (365,604,605,606). However, in some cases GC resistance was attributed to a failure of GCs to acetylate histone H4 K5 and thus transactivate gene expression, rather than to a disturbed GR transrepression mechanism (602). However, the existence of a multidrug resistance membrane transporter, extruding GCs out of the cell and thus limiting their activity, has also been reported in GC resistance and can function as a time-restricting control mechanism of activated GR (607).
In addition, GC-induced alterations in the GR phosphorylation status have been associated with acquired GC resistance. As such, GC resistance has also been linked to the inflammatory status of the diseased tissue via an enhanced kinase activity of JNK, ERK, and p38 MAPKs; an increased synthesis and/or activity of the transcription factors NF-κB and AP-1; and increased cytokine production (330,331,608,609,610,611,612,613,614,615). In this respect, p38 MAPK-, JNK MAPK-, and GSK3-mediated phosphorylations of GR have been linked to a decrease in GR transcriptional activity (321,330,331,332,333,334,335) (see Section II.A). Interestingly, a p38 MAPK- and GSK3-mediated phosphorylation of GR coincided with an attenuated repression of NF-κB-mediated gene expression by GCs (321,330). Conversely, GC resistance was associated with the inability of GCs to deactivate JNK MAPK, which was reflected in elevated phosphorylated c-Jun and c-fos gene expression in GC resistance and coincided with an observed decrease of GR-AP-1 interaction intensity in steroid-resistant asthma patients, when compared with samples from steroid-responsive patients (600,609,616,617,618). Moreover, the GC response of steroid-resistant patient samples could be restored via the addition of MAPK inhibitors (330,612,613,619), and thus the MAPK-mediated inhibition of GR function appears to be a central player in GC resistance. Of interest, GC-resistant T cells, in which the steroid responsiveness was restored via the addition of IFN, featured elevated DUSP1 expression levels and concomitantly an inhibition of p38 MAPK phosphorylation (619). In patients with severe asthma, which showed decreased responsiveness to GCs, an increased p38 MAPK phosphorylation corresponds to reduced induction of DUSP1 expression (614). Taken together, GC unresponsiveness features a central role for MAPK dysregulation and probably also impaired DUSP1 induction.
VII. Future Perspectives in the Combat of Inflammation
In this section, we will discuss new and upcoming GR-based therapeutics and therapeutic strategies, with special attention to the effect of these ligands on GR phosphorylation and GR-based kinase and phosphatase modulations.
A. New glucocorticoid receptor ligands
A long-standing hypothesis driving steroid development pharmacology strategies for the past decade was that the side effects of GCs are mainly attributed to GR transcriptional activation, whereas the antiinflammatory effects of GCs are predominantly mediated via GR transrepression mechanisms. This viewpoint was supported by the finding that GRdim, a GR variant with a mutation hampering GR homodimerization, thereby compromising DNA-binding at some GRE-driven genes, and thus preventing the stimulation of a classical GRE, still allows for GC-mediated repression of AP-1- and NF-κB-mediated proinflammatory gene expression (193,285,620). More recently, it was demonstrated that this model is too rigid and that GRdim still leads to transactivation of a number of genes, exemplified by phenyl-N-methyl-transferase. Thus, on a subset of GR-responsive promoters, GRs can form concerted multimers in a manner that is independent of the DBD-dimer interface (506). Indeed, GRdim mice still suffer from some side effects in response to GCs (276), and clearly not all GR-mediated side effects are solely controlled by classical GR transactivation mechanisms (621). Of course, a number of side effects are also attributed to transrepression mechanisms, e.g., HPA axis suppression.
Although it is recognized that a few antiinflammatory genes are under stimulatory control by GCs, the transcription of these genes sometimes depends on an atypical GR transactivation mechanism, and their role in the GC- induced antiinflammatory mechanism is not completely clear (243). Although the model in which GR transrepression mechanisms are associated with antiinflammatory effects and GR transactivation mechanisms are associated with undesired effects clearly has to be put into a more nuanced perspective, it is still believed that a selective modulation of GR, resulting in distinct GR mechanisms, could contribute to yielding a more beneficial side-effect profile in antiinflammatory therapies (268,285,622). It is paramount, however, to obtain further insights into the “context-dependency” phenomenon of GR-mediated regulation. Compounds that could activate select GR mechanisms and thus alter GR-mediated gene expression profiles are being designated as dissociated compounds, selective GR agonists (SEGRAs) or modulators (SEGRMs) (621,622,623,624). The terminology “SEGRMs” can have the most broad interpretation and can thus also include molecules that are GR antagonists, or even molecules that could activate particular functionalities of GR without actually being a ligand. Various compounds—steroidal and nonsteroidal—capable of activating GR transrepression without inducing GR transactivation mechanism are called dissociated compounds (625). However, when the dissociation is not absolute, it is semantically more correct to refer to them as SEGRAs or SEGRMs, depending on a reported binding of the compound in the ligand-binding pocket (623).
Multiple SEGRAs and SEGRMs have been reported: RU24858, RU24782, and RU40066 (286,626,627); A276575 (628); AL-438 (629,630); compound 25 (631); CpdA (293,422,632); ZK216348 (633,634); LGD-5552 (635,636); and various other compounds (622,637,638,639,640,641,642,643). When one of the above-mentioned compounds stimulates GR, cellular effects can mechanistically differ from classical GCs in affinity for the GR LBD, GR GRE DNA binding, dimerization, cofactor promoter occupancy, histone modifications at targeted promoters, but also GR phosphorylation—all resulting in a divergent gene expression profile (293,422,622,623,629,636).
Currently, GR phosphorylation-related research has only been conducted with CpdA. As a dissociative GR modulator, CpdA [i.e., 2-(4-acetoxyphenyl)-2-chloro-N-methylethylammonium chloride] can efficiently repress the transcription of inflammatory genes via diminishing NF-κB p65 DNA binding and the overall NF-κB p65 transactivation potential and promoting nuclear export of the transcription-facilitating kinase MSK1 (293,420). Conversely, CpdA cannot stimulate GRE-mediated gene promoter activation (293,422). Interestingly, unlike classical GCs, CpdA does not induce DUSP1 human or mouse reporter genes, nor can it elevate DUSP1 protein levels in various cell lines (I. M. E. Beck, unpublished results). Of special interest, CpdA instigates a differential phosphorylation profile of GR in comparison to classical GCs because it did not induce a hyperphosphorylation of hGR S211 (293). Although the N-terminal domain of GR is not necessary for CpdA- or classical GC-mediated transrepression of NF-κB-regulated gene expression, the lack of hGR S211 phosphorylation in CpdA-stimulated cells could possibly explain the deficiency in GR recruitment to and transactivation of GRE-regulated promoters (293). Taking into account that this phosphorylation site is considered crucial in the GR transactivation process (320,327), this leads to a question about whether all dissociated GR modulators share this feature. GR phosphorylation has also been implicated in ligand-mediated GR down-regulation (320,325,345,346). In this respect, it is interesting to note that CpdA does not evoke homologous GR down-regulation in fibroblast-like synovial cells, which is reflected in a diminished antiinflammatory therapy resistance in long-term treatment protocols (632). Currently, the mechanism lying at the basis of this CpdA-mediated GR preservation and thus the possible involvement of differential GR phosphorylation is unknown. Taken together, additional research into the phosphorylation profiles of GRs liganded with SEGRMS, could open up new and interesting perspectives in SEGRM research.
With respect to side effects, we can note that SEGRMs that cannot induce the GRE-regulated PEPCK and G6Pase, such as CpdA, typically do not induce hyperglycemia and hyperinsulinemia (293,422). Also, AL-438 and ZK216348 cannot instigate hyperglycemia (629,633). However, further investigations comparing the side-effect profile of classical GCs vs. the SEGRMs in a more elaborate manner and research into the in vivo antiinflammatory potential of these SEGRMs remain a future challenge in which special attention for SEGRM-induced effects on kinases and phosphatases could shed a new light on the mechanisms of these GR modulators.
B. Combination therapies
Considering the detrimental side-effect profile evoked by a long-term GC therapy, research efforts additionally focused on possible strategies to lower therapeutic GC concentrations while maintaining a similar antiinflammatory potential. Mechanistically, the idea of combining two or more different antiinflammatory compounds would allow lower dosages of each, while maintaining the antiinflammatory profile and attenuating possible side effects of these therapeutics (644).
The highly efficient antiinflammatory combination of long-acting β2 agonists (LABAs) with GCs is already clinically used for the treatment of asthma and COPD (645,646). Interestingly, LABAs can stimulate phosphorylation of hGR at, e.g., S211 and stimulate its nuclear translocation (647,648,649). Moreover, combining LABAs with GCs leads to a more pronounced inhibition of ERK MAPK, JNK MAPK, and IκBα phosphorylations (650) and increased expression levels of the antiinflammatory DUSP1, whereas the GC-induced expression of GILZ or TTP was not increased by the addition of LABAs (648,651).
Additionally, the antiinflammatory effects of combining Cdk, MAPK, or MSK inhibitors with GCs has been assessed, all of which resulted in an enhanced antiinflammatory treatment profile (330,331,456,652). Although these results appear promising, additional immunological research remains necessary to investigate the immunological and clinical aspects of these combination therapies.
Although it may seem temptingly simple to combine GCs with inhibitors targeted at inflammatory process-involved kinases to maximize the inflammatory effect, this review shows that because of the widespread crosstalk of kinases and phosphatases with all levels of GR signaling, caution is in order. Therefore, research into topical application or intermittent therapy strategies for GCs and kinase inhibitors, surpassing general systemic side effects; and more specific ligands for GR, such as the SEGRMs; and more specific kinase inhibitors and phosphatase modulators could broaden the array of available therapeutic agents. From this review, it is apparent that further research into the effects of kinases and phosphatases onto or by GC-mediated signaling could conduce the development of various new drug targets in the combat against inflammation and the control of GC-mediated side effects and GC resistance. In the treatment of chronic diseases, a more constrained therapeutic window is required than in acute afflictions, allowing for a short-term treatment. Preference should go to the targeting of kinases or phosphatases downstream or at the end of a signaling cascade, instead of targeting kinases at the top of the cascade. However, because of their actions in multiple pathways and because of a known difficulty in specific targeting, the decision to target kinases/phosphatases has to be approached with extreme caution, and the risk to off-target effects should be investigated and carefully weighed. As a prerequisite, the in-depth knowledge of the stereodynamic structure of the active enzymatic pocket of the kinase and phosphatase and pharmaceutical design will have to be broadened in the future.
C. MicroRNA-specific modulation of GR
The GC response varies among individuals, as well as within tissues from the same individual, depending on prereceptor stage, ligand metabolism, GR polymorphisms, selective expression of GR subtypes (197,199,653,654,655,656,657,658,659), kinase/phosphatase-dependent posttranslational modifications of the GR (as discussed in the current manuscript), the cofactor environment (531,660,661), and cell type-specific hormone receptor crosstalk with aryl hydrocarbon receptor, MR, and peroxisome proliferator-activated receptor (PPAR) (662,663,664).
MicroRNAs (miRNAs) are single-stranded small and noncoding RNA molecules that can regulate gene expression. miRNAs turn off target gene expression within cells by binding complementary regions in mRNA transcripts, consequently affecting mRNA translation. The possibility exists that various kinases/phosphatases may impact on miRNA-dependent regulation of GR function, involved in the control of developmental, tissue-specific, and individual-specific GC responses in human health and disease. Interestingly, miR-18 and miR-124a reduced GR-mediated transactivation in addition to decreasing GR protein levels and GC responsiveness in a cell type-specific fashion (665). Another target of miR-124a includes the mRNA of a small CTD phosphatase, an antineuronal factor expressed in nonneuronal tissues, playing a role during embryonic development (666).
Because GR ligands themselves can also change miRNA transcription profiles, miRNA control of GR and inflammatory stress-sensitive kinases and phosphatases will become a hot area of further research in GR stress biology and immune functionality (152,667,668,669,670).
D. Epigenetic approaches
Environmental exposure to low concentrations of hormones, air pollution, or toxicants can have heritable epigenetic DNA methylation effects in animals and humans and persistently change inflammatory stress responses later in life, or even in next generations (671,672,673,674,675,676,677). Interestingly, life events occurring during the prenatal, neonatal, and perinatal period have programming effects on the HPA axis, brain neurotransmitter systems, and cognitive abilities of the offspring and long-term effects on the behavioral and neuroendocrine response to stressors (678,679,680). Of special note, maternal stress during gestation, including maternal GCs, may predispose to immune-related pathologies later in life or in offspring (681,682). In male and female rats exposed to prenatal restraint stress, these effects include a long-lasting hyperactivation of the HPA response associated with an altered circadian rhythm of corticosterone secretion (682). More particularly, the methylation status of the hGR (NR3C1) gene promoter in newborns is sensitive to prenatal maternal mood and may stimulate a potential epigenetic process that links antenatal maternal mood and altered HPA stress reactivity during infancy (683). DNA methylation of CpG dinucleotides is generally associated with epigenetic silencing of transcription and is heritable through cell division. Multiple CpG sequences are rare in mammalian genomes, but frequently occur near the transcriptional start site of active genes, with clusters of CpGs (CpG islands) being hypomethylated. The GR CpG island contains seven alternative first exons, and their promoters show highly variable DNA methylation patterns between individuals. As such, methylation may orchestrate alternative first exon usage and silencing and may control tissue-specific expression of GR. The observed heterogeneity may reflect epigenetic mechanisms for fine-tuning GR activity, programmed by early life environment and events (684). Of particular interest, various epigenetic (co)factors, including HDACs, HATs, polycomb proteins, methyl CpG binding proteins [methyl CpG binding protein 2 (MeCP2), methyl CpG binding domain protein (MBD)], and even DNA methyltransferases themselves are regulated by kinase/phosphatase complexes during inflammatory stress (661,685,686,687,688,689,690,691,692,693,694,695,696). Reciprocally, lack of MeCP2 in a mouse model of Rett syndrome increases various GC-regulated genes, including the SGK1 (697). It remains to be further investigated whether kinase/phosphatase-dependent changes in DNA methylation at the GR gene promoter or GR target genes are cyclical or persistent (698,699). Consequently, combination therapy of GCs, kinase/phosphatase inhibitors, and epigenetic drugs may hold promises to modulate GR activity in a time-dependent, cell-specific, or gene-specific way.
VIII. Conclusions
GR activity is regulated by kinases and phosphatases at multiple levels. Alternatively, both kinases and phosphatases are subjected to regulation by the GR. The phosphomodulation of GR can affect its ligand affinity, DNA binding, cofactor recruitment, cellular localization, half-life and recycling, overall posttranslational modification profile, and ultimately transactivational and transrepressional properties. Because these receptors exist in a dynamic regulation, culminating in different receptor functions at different times, GRs cannot be considered as a uniform population, and researchers should be beware of this complication. In turn, GC-activated GRs can alter the expression level, activity, half-life, localization, and specific interactions of various kinases and the expression and activity of various phosphatases. In conclusion, the regulation and implications of GR phosphorylation and the effects of GR on various phosphorylation and dephosphorylation events in the framework of inflammatory models forms an intricate web. Because the current use of kinase inhibitors in both research and therapy has markedly increased in the field of inflammatory disease control, this web of regulations should be taken into account when interpreting results of kinase inhibitors with regard to the antiinflammatory mechanism of GCs.
Current scientific knowledge may lead to the identification of new kinase or phosphatase drug targets and the design of novel combination therapies that target GR as well as its associated kinases/phosphatases to promote the antiinflammatory efficacy, reduce side effects, tackle GC resistance, and ultimately culminate in a better therapeutic profile. Recently, kinases/phosphatases have also entered the field of epigenetic and miRNA-dependent regulation of GR function and are, as such, also involved in the control of developmental, tissue-specific, and individual-specific diversity in the GC pathway in human health and disease. However, further research, both at the basic and clinical levels, is still required to understand and deepen knowledge about the specificity and intertwinement of GR-, kinase-, and phosphatase-mediated events.
In conclusion, as the plethora of GR-regulated effects on and by kinase and phosphatase activities unfolds, the in-depth understanding of the antiinflammatory mechanism of GCs opens up new perspectives in manipulating this process therapeutically in a focused manner.
Footnotes
This work was financially supported by grants from Interuniversity Attraction Poles 6/18 (to G.H.), a Concerted Research Actions GOA from Ghent University (to G.H.), and U.S. National Institutes of Health Grant CA020535 (to K.R.Y.). I.M.E.B., W.V.B., L.V., and K.D.B. are postdoctoral fellows of the Research Foundation-Flanders (FWO–Vlaanderen).
Disclosure Summary: I.M.E.B., W.V.B., L.V., G.H., and K.D.B. have nothing to declare. K.R.Y. is a consultant for Merck and Co. and Sangamo Biosciences.
First Published Online November 4, 2009
Abbreviations: AF, Activation function; AP-1, activator protein-1; ARE, adenylate uridylate-rich element; ATF, activating transcription factor; C, carboxy; CBP, CREB-binding protein; cdc37, cell division cycle 37 protein; Cdk, cyclin-dependent kinase; C/EBP, CCAAT enhancer-binding protein; COPD, chronic obstructive pulmonary disease; COX-2, cyclooxygenase-2; CpdA, compound A; CREB, cAMP-responsive element-binding protein; CRM1, chromosome region maintenance (synonym, exportin1); CTD, C-terminal domain; DBD, DNA-binding domain; Dexras1, dexamethasone-induced Ras1; DUSP, dual specificity phosphatase; 4E-BP1, eukaryotic translation initiation factor 4E-binding protein 1; eIF-4E/F, eukaryotic translation initiation factor 4E/F; ELKS, protein rich in amino acids E, L, K and S; ENaC, epithelial Na+ channel; FKBP, FK506-binding protein; GC, glucocorticoid; GILZ, GC-induced leucine zipper; GM-CSF, granulocyte monocyte-colony stimulating factor; G6Pase, glucose-6-phosphatase; GR, GC receptor; GRE, GC response element; GRIP1, GR-interacting protein 1; GSK, glycogen synthase kinase; H3, histone H3; HAT, histone acetyl transferase; HDAC, histone deacetylase; hGR, human GR; HPA, hypothalamic-pituitary-adrenocortical; Hsp, heat shock protein; ICAM, intercellular adhesion molecule; IE, immediate-early; Ifit1, IFN-induced with tetratricopeptide repeats 1; IFN, interferon; IκB, inhibitor of NF-κB; IKK, IκB kinase; IL1RI, IL-1 receptor I; IRAK, IL-1 receptor-associated kinase; IRF, interferon regulatory factor; ISRE, IFN-stimulated response element; JAK, Janus kinase; JDP, Jun dimerization protein; JNK, c-Jun N-terminal kinase; KO, knockout; LABA, long-acting β2 agonist; LBD, ligand-binding domain; Lck, lymphocyte kinase; LPS, lipopolysaccharide; Mal, MyD88-adapter like protein; MED14, mediator complex subunit 14 (synonym, DRIP150); MEKK, MKK kinase (synonyms, MKKK, MAPKKK, MAP3K); mGR, mouse/murine GR; miRNA, microRNA; MK, MAPK-activated kinase; MKK, MAPK kinase (synonyms, MEK, MAPKK, MAP2K); MMP, matrix metalloproteinase; MMTV, mouse mammary tumor virus; MNK, MAPK-interacting kinase; MR, mineralocorticoid receptor; MSK, mitogen- and stress-activated protein kinase; MyD88, myeloid differentiation primary response gene 88; NCoR, nuclear corepressor; NF-κB, nuclear factor-κB; nGRE, negative GRE; NHE, Na+/H+ exchanger; NIK, NF-κB-inducing kinase; NLS, nuclear localization signal; p/CAF, p300/CBP-associated factor; PEPCK, phosphoenylpyruvate carboxykinase; PI3K, phosphatidylinositol-3-kinase; PKA, protein kinase A; PKB, protein kinase B (synonym, Akt); PKC, protein kinase C; PP, protein phosphatase; P-TEFb, positive transcription elongation factor b; PTP, protein tyrosine phosphatase; RANKL, receptor activator of NF-κB ligand; RANTES, regulated upon activation, normal T-cell expressed and secreted; rGR, rat GR; RIP, receptor-interacting protein; RNA pol II, RNA polymerase II; ROCK, Rho-dependent protein kinase; RSK, ribosomal S6 kinase; SEGRA, selective GR agonist; SEGRM, selective GR modulator; SEK1, SAPK/ERK kinase 1; SGK, serum and GC-inducible kinase; S6K, 70-kDa ribosomal protein S6 kinase; SLAP, Src-like adaptor protein; SOCS, suppressor of cytokine signaling; Sp1, specificity protein 1; SP-A, surfactant protein-A; SRC, steroid receptor coactivator; STAT, signal transducer and activator of transcription; SUMO, small ubiquitin-related modifier; TAD, transactivation domain; TAK1, TGF-activated kinase 1; TANK, TRAF family member-associated NF-κB activator; TBK1, TANK-binding kinase 1; TLR, Toll-like receptor; TNFR, TNF-α receptor; TPA, 12-O-tetradecanoylphorbol-13-acetate; TRADD, TNFR-associated death domain; TRAF, TNFR-associated factor; Tram, translocating chain-associating membrane protein; TRE, TPA-response element; Trif, TIR domain-containing adapter-inducing IFNβ; Trip6, thyroid receptor-interacting protein 6; TTP, tristetraprolin.
References
- Hench PS, Kendall EC, Slocumb CH 1949 The effect of a hormone of the adrenal cortex (17-hydroxy-11-dehydrocorticosterone; compound E) and of pituitary adrenocorticotropic hormone on rheumatoid arthritis. Mayo Clin Proc 24:181–197 [PubMed] [Google Scholar]
- Hollenberg SM, Weinberger C, Ong ES, Cerelli G, Oro A, Lebo R, Thompson EB, Rosenfeld MG, Evans RM 1985 Primary structure and expression of a functional human glucocorticoid receptor cDNA. Nature 318:635–641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweeney SE, Firestein GS 2006 Mitogen activated protein kinase inhibitors: where are we now and where are we going? Ann Rheum Dis 65(Suppl 3):iii83–iii88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascual G, Glass CK 2006 Nuclear receptors versus inflammation: mechanisms of transrepression. Trends Endocrinol Metab 17:321–327 [DOI] [PubMed] [Google Scholar]
- Polito AJ, Proud D 1998 Epithelia cells as regulators of airway inflammation. J Allergy Clin Immunol 102:714–718 [DOI] [PubMed] [Google Scholar]
- Ritchlin C 2000 Fibroblast biology. Effector signals released by the synovial fibroblast in arthritis. Arthritis Res 2:356–360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Standiford TJ, Kunkel SL, Basha MA, Chensue SW, Lynch 3rd JP, Toews GB, Westwick J, Strieter RM 1990 Interleukin-8 gene expression by a pulmonary epithelial cell line. A model for cytokine networks in the lung. J Clin Invest 86:1945–1953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes PJ, Chung KF, Page CP 1998 Inflammatory mediators of asthma: an update. Pharmacol Rev 50:515–596 [PubMed] [Google Scholar]
- Ghosh S, Karin M 2002 Missing pieces in the NF-κB puzzle. Cell 109 Suppl:S81–S96 [DOI] [PubMed] [Google Scholar]
- Hayden MS, Ghosh S 2008 Shared principles in NF-κB signaling. Cell 132:344–362 [DOI] [PubMed] [Google Scholar]
- O'Neill LA 2008 The interleukin-1 receptor/Toll-like receptor superfamily: 10 years of progress. Immunol Rev 226:10–18 [DOI] [PubMed] [Google Scholar]
- Verstrepen L, Bekaert T, Chau TL, Tavernier J, Chariot A, Beyaert R 2008 TLR-4, IL-1R and TNF-R signaling to NF-κB: variations on a common theme. Cell Mol Life Sci 65:2964–2978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandevoorde V, Haegeman G, Fiers W 1997 Induced expression of trimerized intracellular domains of the human tumor necrosis factor (TNF) p55 receptor elicits TNF effects. J Cell Biol 137:1627–1638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu H, Xiong J, Goeddel DV 1995 The TNF receptor 1-associated protein TRADD signals cell death and NF-κB activation. Cell 81:495–504 [DOI] [PubMed] [Google Scholar]
- Gaur U, Aggarwal BB 2003 Regulation of proliferation, survival and apoptosis by members of the TNF superfamily. Biochem Pharmacol 66:1403–1408 [DOI] [PubMed] [Google Scholar]
- Chen G, Goeddel DV 2002 TNF-R1 signaling: a beautiful pathway. Science 296:1634–1635 [DOI] [PubMed] [Google Scholar]
- Hsu H, Huang J, Shu HB, Baichwal V, Goeddel DV 1996 TNF-dependent recruitment of the protein kinase RIP to the TNF receptor-1 signaling complex. Immunity 4:387–396 [DOI] [PubMed] [Google Scholar]
- Hsu H, Shu HB, Pan MG, Goeddel DV 1996 TRADD-TRAF2 and TRADD-FADD interactions define two distinct TNF receptor 1 signal transduction pathways. Cell 84:299–308 [DOI] [PubMed] [Google Scholar]
- Ting AT, Pimentel-Muiños FX, Seed B 1996 RIP mediates tumor necrosis factor receptor 1 activation of NF-κB but not Fas/APO-1-initiated apoptosis. EMBO J 15:6189–6196 [PMC free article] [PubMed] [Google Scholar]
- Devin A, Lin Y, Yamaoka S, Li Z, Karin M, Liu Zg 2001 The α and β subunits of IκB kinase (IKK) mediate TRAF2-dependent IKK recruitment to tumor necrosis factor (TNF) receptor 1 in response to TNF. Mol Cell Biol 21:3986–3994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malinin NL, Boldin MP, Kovalenko AV, Wallach D 1997 MAP3K-related kinase involved in NF-κB induction by TNF, CD95 and IL-1. Nature 385:540–544 [DOI] [PubMed] [Google Scholar]
- Yang J, Lin Y, Guo Z, Cheng J, Huang J, Deng L, Liao W, Chen Z, Liu Z, Su B 2001 The essential role of MEKK3 in TNF-induced NF-κB activation. Nat Immunol 2:620–624 [DOI] [PubMed] [Google Scholar]
- Beyaert R, Van Loo G, Heyninck K, Vandenabeele P 2002 Signaling to gene activation and cell death by tumor necrosis factor receptors and Fas. Int Rev Cytol 214:225–272 [DOI] [PubMed] [Google Scholar]
- Boone E, Vandevoorde V, De Wilde G, Haegeman G 1998 Activation of p42/p44 mitogen-activated protein kinases (MAPK) and p38 MAPK by tumor necrosis factor (TNF) is mediated through the death domain of the 55-kDa TNF receptor. FEBS Lett 441:275–280 [DOI] [PubMed] [Google Scholar]
- Roux PP, Blenis J 2004 ERK and p38 MAPK-activated protein kinases: a family of protein kinases with diverse biological functions. Microbiol Mol Biol Rev 68:320–344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanden Berghe W, Plaisance S, Boone E, De Bosscher K, Schmitz ML, Fiers W, Haegeman G 1998 p38 and extracellular signal-regulated kinase mitogen-activated protein kinase pathways are required for nuclear factor-κB p65 transactivation mediated by tumor necrosis factor. J Biol Chem 273:3285–3290 [DOI] [PubMed] [Google Scholar]
- Baud V, Karin M 2001 Signal transduction by tumor necrosis factor and its relatives. Trends Cell Biol 11:372–377 [DOI] [PubMed] [Google Scholar]
- Kawai T, Akira S 2007 Signaling to NF-κB by Toll-like receptors. Trends Mol Med 13:460–469 [DOI] [PubMed] [Google Scholar]
- Kawai T, Akira S 2008 Toll-like receptor and RIG-I-like receptor signaling. Ann NY Acad Sci 1143:1–20 [DOI] [PubMed] [Google Scholar]
- Foster SL, Medzhitov R 2009 Gene-specific control of the TLR-induced inflammatory response. Clin Immunol 130:7–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes PJ 2006 Transcription factors in airway diseases. Lab Invest 86:867–872 [DOI] [PubMed] [Google Scholar]
- Bonizzi G, Karin M 2004 The two NF-κB activation pathways and their role in innate and adaptive immunity. Trends Immunol 25:280–288 [DOI] [PubMed] [Google Scholar]
- D'Acquisto F, Ianaro A 2006 From willow bark to peptides: the ever widening spectrum of NF-κB inhibitors. Curr Opin Pharmacol 6:387–392 [DOI] [PubMed] [Google Scholar]
- Kumar A, Takada Y, Boriek AM, Aggarwal BB 2004 Nuclear factor-κB: its role in health and disease. J Mol Med 82:434–448 [DOI] [PubMed] [Google Scholar]
- Liou HC 2002 Regulation of the immune system by NF-κB and IκB. J Biochem Mol Biol 35:537–546 [DOI] [PubMed] [Google Scholar]
- Smahi A, Courtois G, Rabia SH, Döffinger R, Bodemer C, Munnich A, Casanova JL, Israël A 2002 The NF-κB signalling pathway in human diseases: from incontinentia pigmenti to ectodermal dysplasias and immune-deficiency syndromes. Hum Mol Genet 11:2371–2375 [DOI] [PubMed] [Google Scholar]
- Bours V, Franzoso G, Azarenko V, Park S, Kanno T, Brown K, Siebenlist U 1993 The oncoprotein Bcl-3 directly transactivates through κB motifs via association with DNA-binding p50B homodimers. Cell 72:729–739 [DOI] [PubMed] [Google Scholar]
- Leeman JR, Gilmore TD 2008 Alternative splicing in the NF-κB signaling pathway. Gene 423:97–107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weih F, Carrasco D, Durham SK, Barton DS, Rizzo CA, Ryseck RP, Lira SA, Bravo R 1995 Multiorgan inflammation and hematopoietic abnormalities in mice with a targeted disruption of RelB, a member of the NF-κB/Rel family. Cell 80:331–340 [DOI] [PubMed] [Google Scholar]
- Hoffmann A, Leung TH, Baltimore D 2003 Genetic analysis of NF-κB/Rel transcription factors defines functional specificities. EMBO J 22:5530–5539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung TH, Hoffmann A, Baltimore D 2004 One nucleotide in a κB site can determine cofactor specificity for NF-κB dimers. Cell 118:453–464 [DOI] [PubMed] [Google Scholar]
- Chen-Park FE, Huang DB, Noro B, Thanos D, Ghosh G 2002 The κB DNA sequence from the HIV long terminal repeat functions as an allosteric regulator of HIV transcription. J Biol Chem 277:24701–24708 [DOI] [PubMed] [Google Scholar]
- Natoli G, Saccani S, Bosisio D, Marazzi I 2005 Interactions of NF-κB with chromatin: the art of being at the right place at the right time. Nat Immunol 6:439–445 [DOI] [PubMed] [Google Scholar]
- Phelps CB, Sengchanthalangsy LL, Malek S, Ghosh G 2000 Mechanism of κB DNA binding by Rel/NF-κB dimers. J Biol Chem 275:24392–24399 [DOI] [PubMed] [Google Scholar]
- Hayden MS, Ghosh S 2004 Signaling to NF-κB. Genes Dev 18:2195–2224 [DOI] [PubMed] [Google Scholar]
- Ben-Neriah Y 2002 Regulatory functions of ubiquitination in the immune system. Nat Immunol 3:20–26 [DOI] [PubMed] [Google Scholar]
- Brown K, Gerstberger S, Carlson L, Franzoso G, Siebenlist U 1995 Control of IκB-α proteolysis by site-specific, signal-induced phosphorylation. Science 267:1485–1488 [DOI] [PubMed] [Google Scholar]
- Häcker H, Karin M 2006 Regulation and function of IKK and IKK-related kinases. Sci STKE 2006:re13 [DOI] [PubMed] [Google Scholar]
- Chen G, Cao P, Goeddel DV 2002 TNF-induced recruitment and activation of the IKK complex require Cdc37 and Hsp90. Mol Cell 9:401–410 [DOI] [PubMed] [Google Scholar]
- Hinz M, Broemer M, Arslan SC, Otto A, Mueller EC, Dettmer R, Scheidereit C 2007 Signal responsiveness of IκB kinases is determined by Cdc37-assisted transient interaction with Hsp90. J Biol Chem 282:32311–32319 [DOI] [PubMed] [Google Scholar]
- Ducut Sigala JL, Bottero V, Young DB, Shevchenko A, Mercurio F, Verma IM 2004 Activation of transcription factor NF-κB requires ELKS, an IκB kinase regulatory subunit. Science 304:1963–1967 [DOI] [PubMed] [Google Scholar]
- Vanden Berghe T, Kalai M, van Loo G, Declercq W, Vandenabeele P 2003 Disruption of HSP90 function reverts tumor necrosis factor-induced necrosis to apoptosis. J Biol Chem 278:5622–5629 [DOI] [PubMed] [Google Scholar]
- Hu Y, Baud V, Delhase M, Zhang P, Deerinck T, Ellisman M, Johnson R, Karin M 1999 Abnormal morphogenesis but intact IKK activation in mice lacking the IKKα subunit of IκB kinase. Science 284:316–320 [DOI] [PubMed] [Google Scholar]
- Xiao G, Rabson AB, Young W, Qing G, Qu Z 2006 Alternative pathways of NF-κB activation: a double-edged sword in health and disease. Cytokine Growth Factor Rev 17:281–293 [DOI] [PubMed] [Google Scholar]
- Derudder E, Dejardin E, Pritchard LL, Green DR, Korner M, Baud V 2003 RelB/p50 dimers are differentially regulated by tumor necrosis factor-α and lymphotoxin-β receptor activation: critical roles for p100. J Biol Chem 278:23278–23284 [DOI] [PubMed] [Google Scholar]
- Solan NJ, Miyoshi H, Carmona EM, Bren GD, Paya CV 2002 RelB cellular regulation and transcriptional activity are regulated by p100. J Biol Chem 277:1405–1418 [DOI] [PubMed] [Google Scholar]
- Amir RE, Haecker H, Karin M, Ciechanover A 2004 Mechanism of processing of the NF-κB2 p100 precursor: identification of the specific polyubiquitin chain-anchoring lysine residue and analysis of the role of NEDD8-modification on the SCF(β-TrCP) ubiquitin ligase. Oncogene 23:2540–2547 [DOI] [PubMed] [Google Scholar]
- Fong A, Zhang M, Neely J, Sun SC 2002 S9, a 19 S proteasome subunit interacting with ubiquitinated NF-κB2/p100. J Biol Chem 277:40697–40702 [DOI] [PubMed] [Google Scholar]
- Liang C, Zhang M, Sun SC 2006 β-TrCP binding and processing of NF-κB2/p100 involve its phosphorylation at serines 866 and 870. Cell Signal 18:1309–1317 [DOI] [PubMed] [Google Scholar]
- Senftleben U, Cao Y, Xiao G, Greten FR, Krähn G, Bonizzi G, Chen Y, Hu Y, Fong A, Sun SC, Karin M 2001 Activation by IKKα of a second, evolutionary conserved, NF-κB signaling pathway. Science 293:1495–1499 [DOI] [PubMed] [Google Scholar]
- Xiao G, Fong A, Sun SC 2004 Induction of p100 processing by NF-κB-inducing kinase involves docking IκB kinase α (IKKα) to p100 and IKKα-mediated phosphorylation. J Biol Chem 279:30099–30105 [DOI] [PubMed] [Google Scholar]
- Chen LF, Williams SA, Mu Y, Nakano H, Duerr JM, Buckbinder L, Greene WC 2005 NF-κB RelA phosphorylation regulates RelA acetylation. Mol Cell Biol 25:7966–7975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perkins ND 2006 Post-translational modifications regulating the activity and function of the nuclear factor κB pathway. Oncogene 25:6717–6730 [DOI] [PubMed] [Google Scholar]
- Chen LF, Mu Y, Greene WC 2002 Acetylation of RelA at discrete sites regulates distinct nuclear functions of NF-κB. EMBO J 21:6539–6548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiernan R, Brès V, Ng RW, Coudart MP, El Messaoudi S, Sardet C, Jin DY, Emiliani S, Benkirane M 2003 Post-activation turn-off of NF-κB-dependent transcription is regulated by acetylation of p65. J Biol Chem 278:2758–2766 [DOI] [PubMed] [Google Scholar]
- Mabb AM, Miyamoto S 2007 SUMO and NF-κB ties. Cell Mol Life Sci 64:1979–1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann M, Naumann M 2007 Beyond IκBs: alternative regulation of NF-κB activity. FASEB J 21:2642–2654 [DOI] [PubMed] [Google Scholar]
- Buss H, Dörrie A, Schmitz ML, Frank R, Livingstone M, Resch K, Kracht M 2004 Phosphorylation of serine 468 by GSK-3β negatively regulates basal p65 NF-κB activity. J Biol Chem 279:49571–49574 [DOI] [PubMed] [Google Scholar]
- Chen Lf, Fischle W, Verdin E, Greene WC 2001 Duration of nuclear NF-κB action regulated by reversible acetylation. Science 293:1653–1657 [DOI] [PubMed] [Google Scholar]
- Mattioli I, Sebald A, Bucher C, Charles RP, Nakano H, Doi T, Kracht M, Schmitz ML 2004 Transient and selective NF-κB p65 serine 536 phosphorylation induced by T cell costimulation is mediated by IκB kinase β and controls the kinetics of p65 nuclear import. J Immunol 172:6336–6344 [DOI] [PubMed] [Google Scholar]
- Bohuslav J, Chen LF, Kwon H, Mu Y, Greene WC 2004 p53 induces NF-κB activation by an IκB kinase-independent mechanism involving phosphorylation of p65 by ribosomal S6 kinase 1. J Biol Chem 279:26115–26125 [DOI] [PubMed] [Google Scholar]
- Chen ZJ 2005 Ubiquitin signalling in the NF-κB pathway. Nat Cell Biol 7:758–765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vermeulen L, De Wilde G, Van Damme P, Vanden Berghe W, Haegeman G 2003 Transcriptional activation of the NF-κB p65 subunit by mitogen- and stress-activated protein kinase-1 (MSK1). EMBO J 22:1313–1324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong H, Voll RE, Ghosh S 1998 Phosphorylation of NF-κB p65 by PKA stimulates transcriptional activity by promoting a novel bivalent interaction with the coactivator CBP/p300. Mol Cell 1:661–671 [DOI] [PubMed] [Google Scholar]
- Duran A, Diaz-Meco MT, Moscat J 2003 Essential role of RelA Ser311 phosphorylation by ζPKC in NF-κB transcriptional activation. EMBO J 22:3910–3918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jamaluddin M, Wang S, Boldogh I, Tian B, Brasier AR 2007 TNF-α-induced NF-κB/RelA Ser(276) phosphorylation and enhanceosome formation is mediated by an ROS-dependent PKAc pathway. Cell Signal 19:1419–1433 [DOI] [PubMed] [Google Scholar]
- Leitges M, Sanz L, Martin P, Duran A, Braun U, García JF, Camacho F, Diaz-Meco MT, Rennert PD, Moscat J 2001 Targeted disruption of the ζPKC gene results in the impairment of the NF-κB pathway. Mol Cell 8:771–780 [DOI] [PubMed] [Google Scholar]
- Wang D, Baldwin Jr AS 1998 Activation of nuclear factor-κB-dependent transcription by tumor necrosis factor-α is mediated through phosphorylation of RelA/p65 on serine 529. J Biol Chem 273:29411–29416 [DOI] [PubMed] [Google Scholar]
- Schwabe RF, Sakurai H 2005 IKKβ phosphorylates p65 at S468 in transactivaton domain 2. FASEB J 19:1758–1760 [DOI] [PubMed] [Google Scholar]
- Okazaki T, Sakon S, Sasazuki T, Sakurai H, Doi T, Yagita H, Okumura K, Nakano H 2003 Phosphorylation of serine 276 is essential for p65 NF-κB subunit-dependent cellular responses. Biochem Biophys Res Commun 300:807–812 [DOI] [PubMed] [Google Scholar]
- Nowak DE, Tian B, Jamaluddin M, Boldogh I, Vergara LA, Choudhary S, Brasier AR 2008 RelA Ser276 phosphorylation is required for activation of a subset of NF-κB-dependent genes by recruiting cyclin-dependent kinase 9/cyclin T1 complexes. Mol Cell Biol 28:3623–3638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong H, SuYang H, Erdjument-Bromage H, Tempst P, Ghosh S 1997 The transcriptional activity of NF-κB is regulated by the IκB-associated PKAc subunit through a cyclic AMP-independent mechanism. Cell 89:413–424 [DOI] [PubMed] [Google Scholar]
- Reber L, Vermeulen L, Haegeman G, Frossard N 2009 Ser276 phosphorylation of NF-kB p65 by MSK1 controls SCF expression in inflammation. PLoS ONE 4:e4393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vermeulen L, Berghe WV, Beck IM, De Bosscher K, Haegeman G 2009 The versatile role of MSKs in transcriptional regulation. Trends Biochem Sci 34:311–318 [DOI] [PubMed] [Google Scholar]
- Zhong H, May MJ, Jimi E, Ghosh S 2002 The phosphorylation status of nuclear NF-κB determines its association with CBP/p300 or HDAC-1. Mol Cell 9:625–636 [DOI] [PubMed] [Google Scholar]
- Dong J, Jimi E, Zhong H, Hayden MS, Ghosh S 2008 Repression of gene expression by unphosphorylated NF-κB p65 through epigenetic mechanisms. Genes Dev 22:1159–1173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anrather J, Racchumi G, Iadecola C 2005 Cis-acting, element-specific transcriptional activity of differentially phosphorylated nuclear factor-κB. J Biol Chem 280:244–252 [DOI] [PubMed] [Google Scholar]
- Foster SL, Hargreaves DC, Medzhitov R 2007 Gene-specific control of inflammation by TLR-induced chromatin modifications. Nature 447:972–978 [DOI] [PubMed] [Google Scholar]
- Guan H, Hou S, Ricciardi RP 2005 DNA binding of repressor nuclear factor-κB p50/p50 depends on phosphorylation of Ser337 by the protein kinase A catalytic subunit. J Biol Chem 280:9957–9962 [DOI] [PubMed] [Google Scholar]
- Sakurai H, Miyoshi H, Toriumi W, Sugita T 1999 Functional interactions of transforming growth factor β-activated kinase 1 with IκB kinases to stimulate NF-κB activation. J Biol Chem 274:10641–10648 [DOI] [PubMed] [Google Scholar]
- Sakurai H, Chiba H, Miyoshi H, Sugita T, Toriumi W 1999 IκB kinases phosphorylate NF-κB p65 subunit on serine 536 in the transactivation domain. J Biol Chem 274:30353–30356 [DOI] [PubMed] [Google Scholar]
- Fujita F, Taniguchi Y, Kato T, Narita Y, Furuya A, Ogawa T, Sakurai H, Joh T, Itoh M, Delhase M, Karin M, Nakanishi M 2003 Identification of NAP1, a regulatory subunit of IκB kinase-related kinases that potentiates NF-κB signaling. Mol Cell Biol 23:7780–7793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki CY, Barberi TJ, Ghosh P, Longo DL 2005 Phosphorylation of RelA/p65 on serine 536 defines an IκBα-independent NF-κB pathway. J Biol Chem 280:34538–34547 [DOI] [PubMed] [Google Scholar]
- Jiang X, Takahashi N, Matsui N, Tetsuka T, Okamoto T 2003 The NF-κB activation in lymphotoxin β receptor signaling depends on the phosphorylation of p65 at serine 536. J Biol Chem 278:919–926 [DOI] [PubMed] [Google Scholar]
- Buss H, Dörrie A, Schmitz ML, Hoffmann E, Resch K, Kracht M 2004 Constitutive and interleukin-1-inducible phosphorylation of p65 NF-κB at serine 536 is mediated by multiple protein kinases including IκB kinase (IKK)-α, IKKβ, IKKε, TRAF family member-associated (TANK)-binding kinase 1 (TBK1), and an unknown kinase and couples p65 to TATA-binding protein-associated factor II31-mediated interleukin-8 transcription. J Biol Chem 279:55633–55643 [DOI] [PubMed] [Google Scholar]
- Adli M, Baldwin AS 2006 IKK-i/IKKε controls constitutive, cancer cell-associated NF-κB activity via regulation of Ser-536 p65/RelA phosphorylation. J Biol Chem 281:26976–26984 [DOI] [PubMed] [Google Scholar]
- Ryo A, Suizu F, Yoshida Y, Perrem K, Liou YC, Wulf G, Rottapel R, Yamaoka S, Lu KP 2003 Regulation of NF-κB signaling by Pin1-dependent prolyl isomerization and ubiquitin-mediated proteolysis of p65/RelA. Mol Cell 12:1413–1426 [DOI] [PubMed] [Google Scholar]
- Maine GN, Mao X, Komarck CM, Burstein E 2007 COMMD1 promotes the ubiquitination of NF-κB subunits through a cullin-containing ubiquitin ligase. EMBO J 26:436–447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saccani S, Marazzi I, Beg AA, Natoli G 2004 Degradation of promoter-bound p65/RelA is essential for the prompt termination of the nuclear factor κB response. J Exp Med 200:107–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bird TA, Schooley K, Dower SK, Hagen H, Virca GD 1997 Activation of nuclear transcription factor NF-κB by interleukin-1 is accompanied by casein kinase II-mediated phosphorylation of the p65 subunit. J Biol Chem 272:32606–32612 [DOI] [PubMed] [Google Scholar]
- Wang D, Westerheide SD, Hanson JL, Baldwin Jr AS 2000 Tumor necrosis factor α-induced phosphorylation of RelA/p65 on Ser529 is controlled by casein kinase II. J Biol Chem 275:32592–32597 [DOI] [PubMed] [Google Scholar]
- Yeh PY, Yeh KH, Chuang SE, Song YC, Cheng AL 2004 Suppression of MEK/ERK signaling pathway enhances cisplatin-induced NF-κB activation by protein phosphatase 4-mediated NF-κB p65 Thr dephosphorylation. J Biol Chem 279:26143–26148 [DOI] [PubMed] [Google Scholar]
- Rocha S, Garrett MD, Campbell KJ, Schumm K, Perkins ND 2005 Regulation of NF-κB and p53 through activation of ATR and Chk1 by the ARF tumour suppressor. EMBO J 24:1157–1169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell KJ, Witty JM, Rocha S, Perkins ND 2006 Cisplatin mimics ARF tumor suppressor regulation of RelA (p65) nuclear factor-κB transactivation. Cancer Res 66:929–935 [DOI] [PubMed] [Google Scholar]
- Mattioli I, Geng H, Sebald A, Hodel M, Bucher C, Kracht M, Schmitz ML 2006 Inducible phosphorylation of NF-κB p65 at serine 468 by T cell costimulation is mediated by IKKε. J Biol Chem 281:6175–6183 [DOI] [PubMed] [Google Scholar]
- Yang J, Fan GH, Wadzinski BE, Sakurai H, Richmond A 2001 Protein phosphatase 2A interacts with and directly dephosphorylates RelA. J Biol Chem 276:47828–47833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou G, Mihindukulasuriya KA, MacCorkle-Chosnek RA, Van Hooser A, Hu MC, Brinkley BR, Tan TH 2002 Protein phosphatase 4 is involved in tumor necrosis factor-α-induced activation of c-Jun N-terminal kinase. J Biol Chem 277:6391–6398 [DOI] [PubMed] [Google Scholar]
- Hu MC, Tang-Oxley Q, Qiu WR, Wang YP, Mihindukulasuriya KA, Afshar R, Tan TH 1998 Protein phosphatase X interacts with c-Rel and stimulates c-Rel/nuclear factor κB activity. J Biol Chem 273:33561–33565 [DOI] [PubMed] [Google Scholar]
- Nelson DE, Ihekwaba AE, Elliott M, Johnson JR, Gibney CA, Foreman BE, Nelson G, See V, Horton CA, Spiller DG, Edwards SW, McDowell HP, Unitt JF, Sullivan E, Grimley R, Benson N, Broomhead D, Kell DB, White MR 2004 Oscillations in NF-κB signaling control the dynamics of gene expression. Science 306:704–708 [DOI] [PubMed] [Google Scholar]
- Hoffmann A, Natoli G, Ghosh G 2006 Transcriptional regulation via the NF-κB signaling module. Oncogene 25:6706–6716 [DOI] [PubMed] [Google Scholar]
- Vogel CF, Matsumura F 2009 A new cross-talk between the aryl hydrocarbon receptor and RelB, a member of the NF-κB family. Biochem Pharmacol 77:734–745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian Y 2009 Ah receptor and NF-κB interplay on the stage of epigenome. Biochem Pharmacol 77:670–680 [DOI] [PubMed] [Google Scholar]
- Basak S, Hoffmann A 2008 Crosstalk via the NF-κB signaling system. Cytokine Growth Factor Rev 19:187–197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujioka S, Niu J, Schmidt C, Sclabas GM, Peng B, Uwagawa T, Li Z, Evans DB, Abbruzzese JL, Chiao PJ 2004 NF-κB and AP-1 connection: mechanism of NF-κB-dependent regulation of AP-1 activity. Mol Cell Biol 24:7806–7819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaisho T, Tanaka T 2008 Turning NF-κB and IRFs on and off in DC. Trends Immunol 29:329–336 [DOI] [PubMed] [Google Scholar]
- Kao YS, Fong JC 2008 Endothelin-1 induces glut1 transcription through enhanced interaction between Sp1 and NF-κB transcription factors. Cell Signal 20:771–778 [DOI] [PubMed] [Google Scholar]
- Park JM, Greten FR, Wong A, Westrick RJ, Arthur JS, Otsu K, Hoffmann A, Montminy M, Karin M 2005 Signaling pathways and genes that inhibit pathogen-induced macrophage apoptosis—CREB and NF-κB as key regulators. Immunity 23:319–329 [DOI] [PubMed] [Google Scholar]
- Yang J, Liao X, Agarwal MK, Barnes L, Auron PE, Stark GR 2007 Unphosphorylated STAT3 accumulates in response to IL-6 and activates transcription by binding to NFκB. Genes Dev 21:1396–1408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosisio D, Marazzi I, Agresti A, Shimizu N, Bianchi ME, Natoli G 2006 A hyper-dynamic equilibrium between promoter-bound and nucleoplasmic dimers controls NF-κB-dependent gene activity. EMBO J 25:798–810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Bosscher K, Haegeman G 2009 Minireview: latest perspectives on antiinflammatory actions of glucocorticoids. Mol Endocrinol 23:281–291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thanos D, Maniatis T 1995 Virus induction of human IFN β gene expression requires the assembly of an enhanceosome. Cell 83:1091–1100 [DOI] [PubMed] [Google Scholar]
- Edelstein LC, Pan A, Collins T 2005 Chromatin modification and the endothelial-specific activation of the E-selectin gene. J Biol Chem 280:11192–11202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerritsen ME, Williams AJ, Neish AS, Moore S, Shi Y, Collins T 1997 CREB-binding protein/p300 are transcriptional coactivators of p65. Proc Natl Acad Sci USA 94:2927–2932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perkins ND, Felzien LK, Betts JC, Leung K, Beach DH, Nabel GJ 1997 Regulation of NF-κB by cyclin-dependent kinases associated with the p300 coactivator. Science 275:523–527 [DOI] [PubMed] [Google Scholar]
- Na SY, Lee SK, Han SJ, Choi HS, Im SY, Lee JW 1998 Steroid receptor coactivator-1 interacts with the p50 subunit and coactivates nuclear factor κB-mediated transactivations. J Biol Chem 273:10831–10834 [DOI] [PubMed] [Google Scholar]
- Sheppard KA, Rose DW, Haque ZK, Kurokawa R, McInerney E, Westin S, Thanos D, Rosenfeld MG, Glass CK, Collins T 1999 Transcriptional activation by NF-κB requires multiple coactivators. Mol Cell Biol 19:6367–6378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito K, Charron CE, Adcock IM 2007 Impact of protein acetylation in inflammatory lung diseases. Pharmacol Ther 116:249–265 [DOI] [PubMed] [Google Scholar]
- Huang WC, Ju TK, Hung MC, Chen CC 2007 Phosphorylation of CBP by IKKα promotes cell growth by switching the binding preference of CBP from p53 to NF-κB. Mol Cell 26:75–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashburner BP, Westerheide SD, Baldwin Jr AS 2001 The p65 (RelA) subunit of NF-κB interacts with the histone deacetylase (HDAC) corepressors HDAC1 and HDAC2 to negatively regulate gene expression. Mol Cell Biol 21:7065–7077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito K, Barnes PJ, Adcock IM 2000 Glucocorticoid receptor recruitment of histone deacetylase 2 inhibits interleukin-1β-induced histone H4 acetylation on lysines 8 and 12. Mol Cell Biol 20:6891–6903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandl A, Heinzel T, Krämer OH 2009 Histone deacetylases: salesmen and customers in the post-translational modification market. Biol Cell 101:193–205 [DOI] [PubMed] [Google Scholar]
- Grabiec AM, Tak PP, Reedquist KA 2008 Targeting histone deacetylase activity in rheumatoid arthritis and asthma as prototypes of inflammatory disease: should we keep our HATs on? Arthritis Res Ther 10:226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calao M, Burny A, Quivy V, Dekoninck A, Van Lint C 2008 A pervasive role of histone acetyltransferases and deacetylases in an NF-κB-signaling code. Trends Biochem Sci 33:339–349 [DOI] [PubMed] [Google Scholar]
- Lee SK, Kim JH, Lee YC, Cheong J, Lee JW 2000 Silencing mediator of retinoic acid and thyroid hormone receptors, as a novel transcriptional corepressor molecule of activating protein-1, nuclear factor-κB, and serum response factor. J Biol Chem 275:12470–12474 [DOI] [PubMed] [Google Scholar]
- Espinosa L, Inglés-Esteve J, Robert-Moreno A, Bigas A 2003 IκBα and p65 regulate the cytoplasmic shuttling of nuclear corepressors: cross-talk between Notch and NFκB pathways. Mol Biol Cell 14:491–502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh S, May MJ, Kopp EB 1998 NF-κ B and Rel proteins: evolutionarily conserved mediators of immune responses. Annu Rev Immunol 16:225–260 [DOI] [PubMed] [Google Scholar]
- Saccani S, Pantano S, Natoli G 2001 Two waves of nuclear factor κB recruitment to target promoters. J Exp Med 193:1351–1359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian B, Nowak DE, Brasier AR 2005 A TNF-induced gene expression program under oscillatory NF-κB control. BMC Genomics 6:137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian B, Nowak DE, Jamaluddin M, Wang S, Brasier AR 2005 Identification of direct genomic targets downstream of the nuclear factor-κB transcription factor mediating tumor necrosis factor signaling. J Biol Chem 280:17435–17448 [DOI] [PubMed] [Google Scholar]
- Tam WF, Wang W, Sen R 2001 Cell-specific association and shuttling of IκBα provides a mechanism for nuclear NF-κB in B lymphocytes. Mol Cell Biol 21:4837–4846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh S, Hayden MS 2008 New regulators of NF-κB in inflammation. Nat Rev Immunol 8:837–848 [DOI] [PubMed] [Google Scholar]
- Jäättelä M, Mouritzen H, Elling F, Bastholm L 1996 A20 zinc finger protein inhibits TNF and IL-1 signaling. J Immunol 156:1166–1173 [PubMed] [Google Scholar]
- Wullaert A, Heyninck K, Janssens S, Beyaert R 2006 Ubiquitin: tool and target for intracellular NF-κB inhibitors. Trends Immunol 27:533–540 [DOI] [PubMed] [Google Scholar]
- Mauro C, Pacifico F, Lavorgna A, Mellone S, Iannetti A, Acquaviva R, Formisano S, Vito P, Leonardi A 2006 ABIN-1 binds to NEMO/IKKγ and co-operates with A20 in inhibiting NF-κB. J Biol Chem 281:18482–18488 [DOI] [PubMed] [Google Scholar]
- Gallagher J, Howlin J, McCarthy C, Murphy EP, Bresnihan B, FitzGerald O, Godson C, Brady HR, Martin F 2003 Identification of Naf1/ABIN-1 among TNF-α-induced expressed genes in human synoviocytes using oligonucleotide microarrays. FEBS Lett 551:8–12 [DOI] [PubMed] [Google Scholar]
- Heyninck K, Kreike MM, Beyaert R 2003 Structure-function analysis of the A20-binding inhibitor of NF-κB activation, ABIN-1. FEBS Lett 536:135–140 [DOI] [PubMed] [Google Scholar]
- Lantz M, Malik S, Slevin ML, Olsson I 1990 Infusion of tumor necrosis factor (TNF) causes an increase in circulating TNF-binding protein in humans. Cytokine 2:402–406 [DOI] [PubMed] [Google Scholar]
- Reddy P, Slack JL, Davis R, Cerretti DP, Kozlosky CJ, Blanton RA, Shows D, Peschon JJ, Black RA 2000 Functional analysis of the domain structure of tumor necrosis factor-α converting enzyme. J Biol Chem 275:14608–14614 [DOI] [PubMed] [Google Scholar]
- Kovalenko A, Chable-Bessia C, Cantarella G, Israël A, Wallach D, Courtois G 2003 The tumour suppressor CYLD negatively regulates NF-κB signalling by deubiquitination. Nature 424:801–805 [DOI] [PubMed] [Google Scholar]
- Fukuda M, Fukuda F, Horiuchi Y, Oku Y, Suzuki S, Kusama K, Sakashita H 2006 Expression of CYLD, NF-κB and NF-κB-related factors in salivary gland tumors. In Vivo 20:467–472 [PubMed] [Google Scholar]
- Platzer C, Meisel C, Vogt K, Platzer M, Volk HD 1995 Up-regulation of monocytic IL-10 by tumor necrosis factor-α and cAMP elevating drugs. Int Immunol 7:517–523 [DOI] [PubMed] [Google Scholar]
- Taganov KD, Boldin MP, Chang KJ, Baltimore D 2006 NF-κB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc Natl Acad Sci USA 103:12481–12486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werner SL, Kearns JD, Zadorozhnaya V, Lynch C, O'Dea E, Boldin MP, Ma A, Baltimore D, Hoffmann A 2008 Encoding NF-κB temporal control in response to TNF: distinct roles for the negative regulators IκBα and A20. Genes Dev 22:2093–2101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zenz R, Eferl R, Scheinecker C, Redlich K, Smolen J, Schonthaler HB, Kenner L, Tschachler E, Wagner EF 2008 Activator protein 1 (Fos/Jun) functions in inflammatory bone and skin disease. Arthritis Res Ther 10:201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess J, Angel P, Schorpp-Kistner M 2004 AP-1 subunits: quarrel and harmony among siblings. J Cell Sci 117:5965–5973 [DOI] [PubMed] [Google Scholar]
- Eychène A, Rocques N, Pouponnot C 2008 A new MAFia in cancer. Nat Rev Cancer 8:683–693 [DOI] [PubMed] [Google Scholar]
- van Dam H, Castellazzi M 2001 Distinct roles of Jun:Fos and Jun:ATF dimers in oncogenesis. Oncogene 20:2453–2464 [DOI] [PubMed] [Google Scholar]
- Eferl R, Wagner EF 2003 AP-1: a double-edged sword in tumorigenesis. Nat Rev Cancer 3:859–868 [DOI] [PubMed] [Google Scholar]
- Hai T, Hartman MG 2001 The molecular biology and nomenclature of the activating transcription factor/cAMP responsive element binding family of transcription factors: activating transcription factor proteins and homeostasis. Gene 273:1–11 [DOI] [PubMed] [Google Scholar]
- Xiao W, Hodge DR, Wang L, Yang X, Zhang X, Farrar WL 2004 NF-κB activates IL-6 expression through cooperation with c-Jun and IL6-AP1 site, but is independent of its IL6-NFκB regulatory site in autocrine human multiple myeloma cells. Cancer Biol Ther 3:1007–1017 [DOI] [PubMed] [Google Scholar]
- Hazzalin CA, Mahadevan LC 2002 MAPK-regulated transcription: a continuously variable gene switch? Nat Rev Mol Cell Biol 3:30–40 [DOI] [PubMed] [Google Scholar]
- Wagner EF, Eferl R 2005 Fos/AP-1 proteins in bone and the immune system. Immunol Rev 208:126–140 [DOI] [PubMed] [Google Scholar]
- Bannister AJ, Kouzarides T 1995 CBP-induced stimulation of c-Fos activity is abrogated by E1A. EMBO J 14:4758–4762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janknecht R, Nordheim A 1996 MAP kinase-dependent transcriptional coactivation by Elk-1 and its cofactor CBP. Biochem Biophys Res Commun 228:831–837 [DOI] [PubMed] [Google Scholar]
- Cahill MA, Janknecht R, Nordheim A 1996 Signalling pathways: jack of all cascades. Curr Biol 6:16–19 [DOI] [PubMed] [Google Scholar]
- Janknecht R, Cahill MA, Nordheim A 1995 Signal integration at the c-fos promoter. Carcinogenesis 16:443–450 [DOI] [PubMed] [Google Scholar]
- Tanos T, Marinissen MJ, Leskow FC, Hochbaum D, Martinetto H, Gutkind JS, Coso OA 2005 Phosphorylation of c-Fos by members of the p38 MAPK family. Role in the AP-1 response to UV light. J Biol Chem 280:18842–18852 [DOI] [PubMed] [Google Scholar]
- Espino PS, Li L, He S, Yu J, Davie JR 2006 Chromatin modification of the trefoil factor 1 gene in human breast cancer cells by the Ras/mitogen-activated protein kinase pathway. Cancer Res 66:4610–4616 [DOI] [PubMed] [Google Scholar]
- Kallunki T, Deng T, Hibi M, Karin M 1996 c-Jun can recruit JNK to phosphorylate dimerization partners via specific docking interactions. Cell 87:929–939 [DOI] [PubMed] [Google Scholar]
- Chalmers CJ, Gilley R, March HN, Balmanno K, Cook SJ 2007 The duration of ERK1/2 activity determines the activation of c-Fos and Fra-1 and the composition and quantitative transcriptional output of AP-1. Cell Signal 19:695–704 [DOI] [PubMed] [Google Scholar]
- Sutherland JA, Cook A, Bannister AJ, Kouzarides T 1992 Conserved motifs in Fos and Jun define a new class of activation domain. Genes Dev 6:1810–1819 [DOI] [PubMed] [Google Scholar]
- Rogatsky I, Zarember KA, Yamamoto KR 2001 Factor recruitment and TIF2/GRIP1 corepressor activity at a collagenase-3 response element that mediates regulation by phorbol esters and hormones. EMBO J 20:6071–6083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozanne BW, Spence HJ, McGarry LC, Hennigan RF 2006 Invasion is a genetic program regulated by transcription factors. Curr Opin Genet Dev 16:65–70 [DOI] [PubMed] [Google Scholar]
- Ozanne BW, Spence HJ, McGarry LC, Hennigan RF 2007 Transcription factors control invasion: AP-1 the first among equals. Oncogene 26:1–10 [DOI] [PubMed] [Google Scholar]
- Chrousos GP, Kino T 2007 Glucocorticoid action networks and complex psychiatric and/or somatic disorders. Stress 10:213–219 [DOI] [PubMed] [Google Scholar]
- Germain P, Staels B, Dacquet C, Spedding M, Laudet V 2006 Overview of nomenclature of nuclear receptors. Pharmacol Rev 58:685–704 [DOI] [PubMed] [Google Scholar]
- Datson NA, van der Perk J, de Kloet ER, Vreugdenhil E 2001 Identification of corticosteroid-responsive genes in rat hippocampus using serial analysis of gene expression. Eur J Neurosci 14:675–689 [DOI] [PubMed] [Google Scholar]
- Kumar R, Thompson EB 1999 The structure of the nuclear hormone receptors. Steroids 64:310–319 [DOI] [PubMed] [Google Scholar]
- Luisi BF, Xu WX, Otwinowski Z, Freedman LP, Yamamoto KR, Sigler PB 1991 Crystallographic analysis of the interaction of the glucocorticoid receptor with DNA. Nature 352:497–505 [DOI] [PubMed] [Google Scholar]
- Dahlman-Wright K, Wright A, Gustafsson JA, Carlstedt-Duke J 1991 Interaction of the glucocorticoid receptor DNA-binding domain with DNA as a dimer is mediated by a short segment of five amino acids. J Biol Chem 266:3107–3112 [PubMed] [Google Scholar]
- Härd T, Kellenbach E, Boelens R, Maler BA, Dahlman K, Freedman LP, Carlstedt-Duke J, Yamamoto KR, Gustafsson JA, Kaptein R 1990 Solution structure of the glucocorticoid receptor DNA-binding domain. Science 249:157–160 [DOI] [PubMed] [Google Scholar]
- van Tilborg MA, Bonvin AM, Hård K, Davis AL, Maler B, Boelens R, Yamamoto KR, Kaptein R 1995 Structure refinement of the glucocorticoid receptor-DNA binding domain from NMR data by relaxation matrix calculations. J Mol Biol 247:689–700 [DOI] [PubMed] [Google Scholar]
- van Tilborg MA, Lefstin JA, Kruiskamp M, Teuben J, Boelens R, Yamamoto KR, Kaptein R 2000 Mutations in the glucocorticoid receptor DNA-binding domain mimic an allosteric effect of DNA. J Mol Biol 301:947–958 [DOI] [PubMed] [Google Scholar]
- Robinson-Rechavi M, Escriva Garcia H, Laudet V 2003 The nuclear receptor superfamily. J Cell Sci 116:585–586 [DOI] [PubMed] [Google Scholar]
- Bledsoe RK, Stewart EL, Pearce KH 2004 Structure and function of the glucocorticoid receptor ligand binding domain. Vitam Horm 68:49–91 [DOI] [PubMed] [Google Scholar]
- Pratt WB, Galigniana MD, Morishima Y, Murphy PJ 2004 Role of molecular chaperones in steroid receptor action. Essays Biochem 40:41–58 [DOI] [PubMed] [Google Scholar]
- Darimont BD, Wagner RL, Apriletti JW, Stallcup MR, Kushner PJ, Baxter JD, Fletterick RJ, Yamamoto KR 1998 Structure and specificity of nuclear receptor-coactivator interactions. Genes Dev 12:3343–3356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong H, Darimont BD, Ma H, Yang L, Yamamoto KR, Stallcup MR 1999 An additional region of coactivator GRIP1 required for interaction with the hormone-binding domains of a subset of nuclear receptors. J Biol Chem 274:3496–3502 [DOI] [PubMed] [Google Scholar]
- Ricketson D, Hostick U, Fang L, Yamamoto KR, Darimont BD 2007 A conformational switch in the ligand-binding domain regulates the dependence of the glucocorticoid receptor on Hsp90. J Mol Biol 368:729–741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meijsing SH, Pufall MA, So AY, Bates DL, Chen L, Yamamoto KR 2009 DNA binding site sequence directs glucocorticoid receptor structure and activity. Science 324:407–410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bladh LG, Lidén J, Dahlman-Wright K, Reimers M, Nilsson S, Okret S 2005 Identification of endogenous glucocorticoid repressed genes differentially regulated by a glucocorticoid receptor mutant able to separate between nuclear factor-κB and activator protein-1 repression. Mol Pharmacol 67:815–826 [DOI] [PubMed] [Google Scholar]
- Liden J, Delaunay F, Rafter I, Gustafsson J, Okret S 1997 A new function for the C-terminal zinc finger of the glucocorticoid receptor. Repression of RelA transactivation. J Biol Chem 272:21467–21472 [DOI] [PubMed] [Google Scholar]
- Heck S, Kullmann M, Gast A, Ponta H, Rahmsdorf HJ, Herrlich P, Cato AC 1994 A distinct modulating domain in glucocorticoid receptor monomers in the repression of activity of the transcription factor AP-1. EMBO J 13:4087–4095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picard D, Yamamoto KR 1987 Two signals mediate hormone-dependent nuclear localization of the glucocorticoid receptor. EMBO J 6:3333–3340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savory JG, Hsu B, Laquian IR, Giffin W, Reich T, Haché RJ, Lefebvre YA 1999 Discrimination between NL1- and NL2-mediated nuclear localization of the glucocorticoid receptor. Mol Cell Biol 19:1025–1037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duma D, Jewell CM, Cidlowski JA 2006 Multiple glucocorticoid receptor isoforms and mechanisms of post-translational modification. J Steroid Biochem Mol Biol 102:11–21 [DOI] [PubMed] [Google Scholar]
- Lu NZ, Cidlowski JA 2006 Glucocorticoid receptor isoforms generate transcription specificity. Trends Cell Biol 16:301–307 [DOI] [PubMed] [Google Scholar]
- Lu NZ, Collins JB, Grissom SF, Cidlowski JA 2007 Selective regulation of bone cell apoptosis by translational isoforms of the glucocorticoid receptor. Mol Cell Biol 27:7143–7160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross KL, Cidlowski JA 2008 Tissue-specific glucocorticoid action: a family affair. Trends Endocrinol Metab 19:331–339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross KL, Lu NZ, Cidlowski JA 2009 Molecular mechanisms regulating glucocorticoid sensitivity and resistance. Mol Cell Endocrinol 300:7–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charmandari E, Kino T, Ichijo T, Chrousos GP 2008 Generalized glucocorticoid resistance: clinical aspects, molecular mechanisms, and implications of a rare genetic disorder. J Clin Endocrinol Metab 93:1563–1572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gametchu B, Chen F, Sackey F, Powell C, Watson CS 1999 Plasma membrane-resident glucocorticoid receptors in rodent lymphoma and human leukemia models. Steroids 64:107–119 [DOI] [PubMed] [Google Scholar]
- Guo Z, Chen YZ, Xu RB, Fu H 1995 Binding characteristics of glucocorticoid receptor in synaptic plasma membrane from rat brain. Funct Neurol 10:183–194 [PubMed] [Google Scholar]
- Sionov RV, Cohen O, Kfir S, Zilberman Y, Yefenof E 2006 Role of mitochondrial glucocorticoid receptor in glucocorticoid-induced apoptosis. J Exp Med 203:189–201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans SJ, Murray TF, Moore FL 2000 Partial purification and biochemical characterization of a membrane glucocorticoid receptor from an amphibian brain. J Steroid Biochem Mol Biol 72:209–221 [DOI] [PubMed] [Google Scholar]
- Gametchu B, Watson CS, Wu S 1993 Use of receptor antibodies to demonstrate membrane glucocorticoid receptor in cells from human leukemic patients. FASEB J 7:1283–1292 [DOI] [PubMed] [Google Scholar]
- Tasker JG, Di S, Malcher-Lopes R 2006 Minireview: rapid glucocorticoid signaling via membrane-associated receptors. Endocrinology 147:5549–5556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grad I, Picard D 2007 The glucocorticoid responses are shaped by molecular chaperones. Mol Cell Endocrinol 275:2–12 [DOI] [PubMed] [Google Scholar]
- Haller J, Mikics E, Makara GB 2008 The effects of non-genomic glucocorticoid mechanisms on bodily functions and the central neural system. A critical evaluation of findings. Front Neuroendocrinol 29:273–291 [DOI] [PubMed] [Google Scholar]
- Bledsoe RK, Montana VG, Stanley TB, Delves CJ, Apolito CJ, McKee DD, Consler TG, Parks DJ, Stewart EL, Willson TM, Lambert MH, Moore JT, Pearce KH, Xu HE 2002 Crystal structure of the glucocorticoid receptor ligand binding domain reveals a novel mode of receptor dimerization and coactivator recognition. Cell 110:93–105 [DOI] [PubMed] [Google Scholar]
- De Bosscher K, Vanden Berghe W, Haegeman G 2003 The interplay between the glucocorticoid receptor and nuclear factor-κB or activator protein-1: molecular mechanisms for gene repression. Endocr Rev 24:488–522 [DOI] [PubMed] [Google Scholar]
- De Bosscher K, Vanden Berghe W, Haegeman G 2006 Cross-talk between nuclear receptors and nuclear factor κB. Oncogene 25:6868–6886 [DOI] [PubMed] [Google Scholar]
- Song IH, Buttgereit F 2006 Non-genomic glucocorticoid effects to provide the basis for new drug developments. Mol Cell Endocrinol 246:142–146 [DOI] [PubMed] [Google Scholar]
- Löwenberg M, Verhaar AP, van den Brink GR, Hommes DW 2007 Glucocorticoid signaling: a non-genomic mechanism for T-cell immunosuppression. Trends Mol Med 13:158–163 [DOI] [PubMed] [Google Scholar]
- Falkenstein E, Wehling M 2000 Nongenomically initiated steroid actions. Eur J Clin Invest 30(Suppl 3):51–54 [DOI] [PubMed] [Google Scholar]
- Haché RJ, Tse R, Reich T, Savory JG, Lefebvre YA 1999 Nucleocytoplasmic trafficking of steroid-free glucocorticoid receptor. J Biol Chem 274:1432–1439 [DOI] [PubMed] [Google Scholar]
- Freedman ND, Yamamoto KR 2004 Importin 7 and importin α/importin β are nuclear import receptors for the glucocorticoid receptor. Mol Biol Cell 15:2276–2286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao T, Lan J, Lukacs GL, Haché RJ, Kaplan F 2006 Importin 13 regulates nuclear import of the glucocorticoid receptor in airway epithelial cells. Am J Respir Cell Mol Biol 35:668–680 [DOI] [PubMed] [Google Scholar]
- Kumar S, Chaturvedi NK, Nishi M, Kawata M, Tyagi RK 2004 Shuttling components of nuclear import machinery involved in nuclear translocation of steroid receptors exit nucleus via exportin-1/CRM-1 independent pathway. Biochim Biophys Acta 1691:73–77 [DOI] [PubMed] [Google Scholar]
- Holaska JM, Black BE, Rastinejad F, Paschal BM 2002 Ca2+-dependent nuclear export mediated by calreticulin. Mol Cell Biol 22:6286–6297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walther RF, Lamprecht C, Ridsdale A, Groulx I, Lee S, Lefebvre YA, Haché RJ 2003 Nuclear export of the glucocorticoid receptor is accelerated by cell fusion-dependent release of calreticulin. J Biol Chem 278:37858–37864 [DOI] [PubMed] [Google Scholar]
- Carrigan A, Walther RF, Salem HA, Wu D, Atlas E, Lefebvre YA, Haché RJ 2007 An active nuclear retention signal in the glucocorticoid receptor functions as a strong inducer of transcriptional activation. J Biol Chem 282:10963–10971 [DOI] [PubMed] [Google Scholar]
- McNally JG, Müller WG, Walker D, Wolford R, Hager GL 2000 The glucocorticoid receptor: rapid exchange with regulatory sites in living cells. Science 287:1262–1265 [DOI] [PubMed] [Google Scholar]
- Sackey FN, Haché RJ, Reich T, Kwast-Welfeld J, Lefebvre YA 1996 Determinants of subcellular distribution of the glucocorticoid receptor. Mol Endocrinol 10:1191–1205 [DOI] [PubMed] [Google Scholar]
- Schaaf MJ, Lewis-Tuffin LJ, Cidlowski JA 2005 Ligand-selective targeting of the glucocorticoid receptor to nuclear subdomains is associated with decreased receptor mobility. Mol Endocrinol 19:1501–1515 [DOI] [PubMed] [Google Scholar]
- Burnstein KL, Jewell CM, Sar M, Cidlowski JA 1994 Intragenic sequences of the human glucocorticoid receptor complementary DNA mediate hormone-inducible receptor messenger RNA down-regulation through multiple mechanisms. Mol Endocrinol 8:1764–1773 [DOI] [PubMed] [Google Scholar]
- Okret S, Poellinger L, Dong Y, Gustafsson JA 1986 Down-regulation of glucocorticoid receptor mRNA by glucocorticoid hormones and recognition by the receptor of a specific binding sequence within a receptor cDNA clone. Proc Natl Acad Sci USA 83:5899–5903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimojo M, Hiroi N, Yakushiji F, Ueshiba H, Yamaguchi N, Miyachi Y 1995 Differences in down-regulation of glucocorticoid receptor mRNA by cortisol, prednisolone and dexamethasone in HeLa cells. Endocr J 42:629–636 [DOI] [PubMed] [Google Scholar]
- Vedeckis WV, Ali M, Allen HR 1989 Regulation of glucocorticoid receptor protein and mRNA levels. Cancer Res 49:2295s–2302s [PubMed] [Google Scholar]
- Alarid ET 2006 Lives and times of nuclear receptors. Mol Endocrinol 20:1972–1981 [DOI] [PubMed] [Google Scholar]
- Hoeck W, Rusconi S, Groner B 1989 Down-regulation and phosphorylation of glucocorticoid receptors in cultured cells. Investigations with a monospecific antiserum against a bacterially expressed receptor fragment. J Biol Chem 264:14396–14402 [PubMed] [Google Scholar]
- Wallace AD, Cidlowski JA 2001 Proteasome-mediated glucocorticoid receptor degradation restricts transcriptional signaling by glucocorticoids. J Biol Chem 276:42714–42721 [DOI] [PubMed] [Google Scholar]
- Liu J, DeFranco DB 2000 Protracted nuclear export of glucocorticoid receptor limits its turnover and does not require the exportin 1/CRM1-directed nuclear export pathway. Mol Endocrinol 14:40–51 [DOI] [PubMed] [Google Scholar]
- La Baer J, Yamamoto KR 1994 Analysis of the DNA-binding affinity, sequence specificity and context dependence of the glucocorticoid receptor zinc finger region. J Mol Biol 239:664–688 [DOI] [PubMed] [Google Scholar]
- Schena M, Freedman LP, Yamamoto KR 1989 Mutations in the glucocorticoid receptor zinc finger region that distinguish interdigitated DNA binding and transcriptional enhancement activities. Genes Dev 3:1590–1601 [DOI] [PubMed] [Google Scholar]
- Rhen T, Cidlowski JA 2005 Antiinflammatory action of glucocorticoids—new mechanisms for old drugs. N Engl J Med 353:1711–1723 [DOI] [PubMed] [Google Scholar]
- Lefstin JA, Thomas JR, Yamamoto KR 1994 Influence of a steroid receptor DNA-binding domain on transcriptional regulatory functions. Genes Dev 8:2842–2856 [DOI] [PubMed] [Google Scholar]
- Lefstin JA, Yamamoto KR 1998 Allosteric effects of DNA on transcriptional regulators. Nature 392:885–888 [DOI] [PubMed] [Google Scholar]
- Ringold GM, Yamamoto KR, Tomkins GM, Bishop M, Varmus HE 1975 Dexamethasone-mediated induction of mouse mammary tumor virus RNA: a system for studying glucocorticoid action. Cell 6:299–305 [DOI] [PubMed] [Google Scholar]
- Fryer CJ, Archer TK 1998 Chromatin remodelling by the glucocorticoid receptor requires the BRG1 complex. Nature 393:88–91 [DOI] [PubMed] [Google Scholar]
- Diamond MI, Miner JN, Yoshinaga SK, Yamamoto KR 1990 Transcription factor interactions: selectors of positive or negative regulation from a single DNA element. Science 249:1266–1272 [DOI] [PubMed] [Google Scholar]
- Miner JN, Yamamoto KR 1991 Regulatory crosstalk at composite response elements. Trends Biochem Sci 16:423–426 [DOI] [PubMed] [Google Scholar]
- Clark AR 2007 Anti-inflammatory functions of glucocorticoid-induced genes. Mol Cell Endocrinol 275:79–97 [DOI] [PubMed] [Google Scholar]
- Rogatsky I, Wang JC, Derynck MK, Nonaka DF, Khodabakhsh DB, Haqq CM, Darimont BD, Garabedian MJ, Yamamoto KR 2003 Target-specific utilization of transcriptional regulatory surfaces by the glucocorticoid receptor. Proc Natl Acad Sci USA 100:13845–13850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- So AY, Chaivorapol C, Bolton EC, Li H, Yamamoto KR 2007 Determinants of cell- and gene-specific transcriptional regulation by the glucocorticoid receptor. PLoS Genet 3:e94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- So AY, Cooper SB, Feldman BJ, Manuchehri M, Yamamoto KR 2008 Conservation analysis predicts in vivo occupancy of glucocorticoid receptor-binding sequences at glucocorticoid-induced genes. Proc Natl Acad Sci USA 105:5745–5749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Martino MU, Bhattachryya N, Alesci S, Ichijo T, Chrousos GP, Kino T 2004 The glucocorticoid receptor and the orphan nuclear receptor chicken ovalbumin upstream promoter-transcription factor II interact with and mutually affect each other’s transcriptional activities: implications for intermediary metabolism. Mol Endocrinol 18:820–833 [DOI] [PubMed] [Google Scholar]
- Doppler W, Windegger M, Soratroi C, Tomasi J, Lechner J, Rusconi S, Cato AC, Almlöf T, Liden J, Okret S, Gustafsson JA, Richard-Foy H, Starr DB, Klocker H, Edwards D, Geymayer S 2001 Expression level-dependent contribution of glucocorticoid receptor domains for functional interaction with STAT5. Mol Cell Biol 21:3266–3279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stöcklin E, Wissler M, Gouilleux F, Groner B 1996 Functional interactions between Stat5 and the glucocorticoid receptor. Nature 383:726–728 [DOI] [PubMed] [Google Scholar]
- Pearce D, Matsui W, Miner JN, Yamamoto KR 1998 Glucocorticoid receptor transcriptional activity determined by spacing of receptor and nonreceptor DNA sites. J Biol Chem 273:30081–30085 [DOI] [PubMed] [Google Scholar]
- Kassel O, Herrlich P 2007 Crosstalk between the glucocorticoid receptor and other transcription factors: molecular aspects. Mol Cell Endocrinol 275:13–29 [DOI] [PubMed] [Google Scholar]
- Rogatsky I, Ivashkiv LB 2006 Glucocorticoid modulation of cytokine signaling. Tissue Antigens 68:1–12 [DOI] [PubMed] [Google Scholar]
- Luecke HF, Yamamoto KR 2005 The glucocorticoid receptor blocks P-TEFb recruitment by NFκB to effect promoter-specific transcriptional repression. Genes Dev 19:1116–1127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nissen RM, Yamamoto KR 2000 The glucocorticoid receptor inhibits NFκB by interfering with serine-2 phosphorylation of the RNA polymerase II carboxy-terminal domain. Genes Dev 14:2314–2329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caldenhoven E, Liden J, Wissink S, Van de Stolpe A, Raaijmakers J, Koenderman L, Okret S, Gustafsson JA, Van der Saag PT 1995 Negative cross-talk between RelA and the glucocorticoid receptor: a possible mechanism for the antiinflammatory action of glucocorticoids. Mol Endocrinol 9:401–412 [DOI] [PubMed] [Google Scholar]
- McKay LI, Cidlowski JA 1998 Cross-talk between nuclear factor-κB and the steroid hormone receptors: mechanisms of mutual antagonism. Mol Endocrinol 12:45–56 [DOI] [PubMed] [Google Scholar]
- Li X, Wong J, Tsai SY, Tsai MJ, O'Malley BW 2003 Progesterone and glucocorticoid receptors recruit distinct coactivator complexes and promote distinct patterns of local chromatin modification. Mol Cell Biol 23:3763–3773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins BD, Pullen CB, Darimont BD 2001 Novel glucocorticoid receptor coactivator effector mechanisms. Trends Endocrinol Metab 12:122–126 [DOI] [PubMed] [Google Scholar]
- Perissi V, Rosenfeld MG 2005 Controlling nuclear receptors: the circular logic of cofactor cycles. Nat Rev Mol Cell Biol 6:542–554 [DOI] [PubMed] [Google Scholar]
- McKenna NJ, O'Malley BW 2002 Minireview: nuclear receptor coactivators—an update. Endocrinology 143:2461–2465 [DOI] [PubMed] [Google Scholar]
- Sandu C, Artunc F, Grahammer F, Rotte A, Boini KM, Friedrich B, Sandulache D, Metzger M, Just L, Mack A, Skutella T, Rexhepaj R, Risler T, Wulff P, Kuhl D, Lang F 2007 Role of the serum and glucocorticoid inducible kinase SGK1 in glucocorticoid stimulation of gastric acid secretion. Pflugers Arch 455:493–503 [DOI] [PubMed] [Google Scholar]
- Garside H, Stevens A, Farrow S, Normand C, Houle B, Berry A, Maschera B, Ray D 2004 Glucocorticoid ligands specify different interactions with NF-κB by allosteric effects on the glucocorticoid receptor DNA binding domain. J Biol Chem 279:50050–50059 [DOI] [PubMed] [Google Scholar]
- Chen W, Rogatsky I, Garabedian MJ 2006 MED14 and MED1 differentially regulate target-specific gene activation by the glucocorticoid receptor. Mol Endocrinol 20:560–572 [DOI] [PubMed] [Google Scholar]
- Freeman BC, Yamamoto KR 2002 Disassembly of transcriptional regulatory complexes by molecular chaperones. Science 296:2232–2235 [DOI] [PubMed] [Google Scholar]
- Stavreva DA, Müller WG, Hager GL, Smith CL, McNally JG 2004 Rapid glucocorticoid receptor exchange at a promoter is coupled to transcription and regulated by chaperones and proteasomes. Mol Cell Biol 24:2682–2697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meijsing SH, Elbi C, Luecke HF, Hager GL, Yamamoto KR 2007 The ligand binding domain controls glucocorticoid receptor dynamics independent of ligand release. Mol Cell Biol 27:2442–2451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinenov Y, Rogatsky I 2007 Glucocorticoids and the innate immune system: crosstalk with the toll-like receptor signaling network. Mol Cell Endocrinol 275:30–42 [DOI] [PubMed] [Google Scholar]
- Newton R, Holden NS 2007 Separating transrepression and transactivation: a distressing divorce for the glucocorticoid receptor? Mol Pharmacol 72:799–809 [DOI] [PubMed] [Google Scholar]
- Ishmael FT, Fang X, Galdiero MR, Atasoy U, Rigby WF, Gorospe M, Cheadle C, Stellato C 2008 Role of the RNA-binding protein tristetraprolin in glucocorticoid-mediated gene regulation. J Immunol 180:8342–8353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eddleston J, Herschbach J, Wagelie-Steffen AL, Christiansen SC, Zuraw BL 2007 The anti-inflammatory effect of glucocorticoids is mediated by glucocorticoid-induced leucine zipper in epithelial cells. J Allergy Clin Immunol 119:115–122 [DOI] [PubMed] [Google Scholar]
- Deroo BJ, Archer TK 2001 Glucocorticoid receptor activation of the IκBα promoter within chromatin. Mol Biol Cell 12:3365–3374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang N, Zhang W, Shi XM 2008 Glucocorticoid-induced leucine zipper (GILZ) mediates glucocorticoid action and inhibits inflammatory cytokine-induced COX-2 expression. J Cell Biochem 103:1760–1771 [DOI] [PubMed] [Google Scholar]
- D'Acquisto F, Perretti M, Flower RJ 2008 Annexin-A1: a pivotal regulator of the innate and adaptive immune systems. Br J Pharmacol 155:152–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smoak K, Cidlowski JA 2006 Glucocorticoids regulate tristetraprolin synthesis and posttranscriptionally regulate tumor necrosis factor α inflammatory signaling. Mol Cell Biol 26:9126–9135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maier JV, Brema S, Tuckermann J, Herzer U, Klein M, Stassen M, Moorthy A, Cato AC 2007 Dual specificity phosphatase 1 knockout mice show enhanced susceptibility to anaphylaxis but are sensitive to glucocorticoids. Mol Endocrinol 21:2663–2671 [DOI] [PubMed] [Google Scholar]
- Kleiman A, Tuckermann JP 2007 Glucocorticoid receptor action in beneficial and side effects of steroid therapy: lessons from conditional knockout mice. Mol Cell Endocrinol 275:98–108 [DOI] [PubMed] [Google Scholar]
- De Bosscher K, Schmitz ML, Vanden Berghe W, Plaisance S, Fiers W, Haegeman G 1997 Glucocorticoid-mediated repression of nuclear factor-κB-dependent transcription involves direct interference with transactivation. Proc Natl Acad Sci USA 94:13504–13509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wissink S, van Heerde EC, vand der Burg B, van der Saag PT 1998 A dual mechanism mediates repression of NF-κB activity by glucocorticoids. Mol Endocrinol 12:355–363 [DOI] [PubMed] [Google Scholar]
- Chaudhary LR, Avioli LV 1996 Regulation of interleukin-8 gene expression by interleukin-1β, osteotropic hormones, and protein kinase inhibitors in normal human bone marrow stromal cells. J Biol Chem 271:16591–16596 [DOI] [PubMed] [Google Scholar]
- Chivers JE, Gong W, King EM, Seybold J, Mak JC, Donnelly LE, Holden NS, Newton R 2006 Analysis of the dissociated steroid RU24858 does not exclude a role for inducible genes in the anti-inflammatory actions of glucocorticoids. Mol Pharmacol 70:2084–2095 [DOI] [PubMed] [Google Scholar]
- Henderson BR, Kefford RF 1993 Dexamethasone decreases urokinase plasminogen activator mRNA stability in MAT 13762 rat mammary carcinoma cells. Br J Cancer 67:99–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newton R, Seybold J, Kuitert LM, Bergmann M, Barnes PJ 1998 Repression of cyclooxygenase-2 and prostaglandin E2 release by dexamethasone occurs by transcriptional and post-transcriptional mechanisms involving loss of polyadenylated mRNA. J Biol Chem 273:32312–32321 [DOI] [PubMed] [Google Scholar]
- Tobler A, Meier R, Seitz M, Dewald B, Baggiolini M, Fey MF 1992 Glucocorticoids downregulate gene expression of GM-CSF, NAP-1/IL-8, and IL-6, but not of M-CSF in human fibroblasts. Blood 79:45–51 [PubMed] [Google Scholar]
- Chang MM, Juarez M, Hyde DM, Wu R 2001 Mechanism of dexamethasone-mediated interleukin-8 gene suppression in cultured airway epithelial cells. Am J Physiol Lung Cell Mol Physiol 280:L107–L115 [DOI] [PubMed] [Google Scholar]
- Reichardt HM, Tuckermann JP, Göttlicher M, Vujic M, Weih F, Angel P, Herrlich P, Schütz G 2001 Repression of inflammatory responses in the absence of DNA binding by the glucocorticoid receptor. EMBO J 20:7168–7173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belvisi MG, Wicks SL, Battram CH, Bottoms SE, Redford JE, Woodman P, Brown TJ, Webber SE, Foster ML 2001 Therapeutic benefit of a dissociated glucocorticoid and the relevance of in vitro separation of transrepression from transactivation activity. J Immunol 166:1975–1982 [DOI] [PubMed] [Google Scholar]
- Dostert A, Heinzel T 2004 Negative glucocorticoid receptor response elements and their role in glucocorticoid action. Curr Pharm Des 10:2807–2816 [DOI] [PubMed] [Google Scholar]
- Ki SH, Cho IJ, Choi DW, Kim SG 2005 Glucocorticoid receptor (GR)-associated SMRT binding to C/EBPβ TAD and Nrf2 Neh4/5: role of SMRT recruited to GR in GSTA2 gene repression. Mol Cell Biol 25:4150–4165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malkoski SP, Dorin RI 1999 Composite glucocorticoid regulation at a functionally defined negative glucocorticoid response element of the human corticotropin-releasing hormone gene. Mol Endocrinol 13:1629–1644 [DOI] [PubMed] [Google Scholar]
- Strömstedt PE, Poellinger L, Gustafsson JA, Carlstedt-Duke J 1991 The glucocorticoid receptor binds to a sequence overlapping the TATA box of the human osteocalcin promoter: a potential mechanism for negative regulation. Mol Cell Biol 11:3379–3383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramaniam N, Cairns W, Okret S 1998 Glucocorticoids repress transcription from a negative glucocorticoid response element recognized by two homeodomain-containing proteins, Pbx and Oct-1. J Biol Chem 273:23567–23574 [DOI] [PubMed] [Google Scholar]
- Liberman AC, Druker J, Refojo D, Holsboer F, Arzt E 2009 Glucocorticoids inhibit GATA-3 phosphorylation and activity in T cells. FASEB J 23:1558–1571 [DOI] [PubMed] [Google Scholar]
- De Bosscher K, Vanden Berghe W, Beck IM, Van Molle W, Hennuyer N, Hapgood J, Libert C, Staels B, Louw A, Haegeman G 2005 A fully dissociated compound of plant origin for inflammatory gene repression. Proc Natl Acad Sci USA 102:15827–15832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Islam KN, Mendelson CR 2008 Glucocorticoid/glucocorticoid receptor inhibition of surfactant protein-A (SP-A) gene expression in lung type II cells is mediated by repressive changes in histone modification at the SP-A promoter. Mol Endocrinol 22:585–596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kassel O, Schneider S, Heilbock C, Litfin M, Göttlicher M, Herrlich P 2004 A nuclear isoform of the focal adhesion LIM-domain protein Trip6 integrates activating and repressing signals at AP-1- and NF-κB-regulated promoters. Genes Dev 18:2518–2528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schüle R, Rangarajan P, Kliewer S, Ransone LJ, Bolado J, Yang N, Verma IM, Evans RM 1990 Functional antagonism between oncoprotein c-Jun and the glucocorticoid receptor. Cell 62:1217–1226 [DOI] [PubMed] [Google Scholar]
- Rogatsky I, Luecke HF, Leitman DC, Yamamoto KR 2002 Alternate surfaces of transcriptional coregulator GRIP1 function in different glucocorticoid receptor activation and repression contexts. Proc Natl Acad Sci USA 99:16701–16706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollenberg SM, Evans RM 1988 Multiple and cooperative trans-activation domains of the human glucocorticoid receptor. Cell 55:899–906 [DOI] [PubMed] [Google Scholar]
- Ogawa S, Lozach J, Benner C, Pascual G, Tangirala RK, Westin S, Hoffmann A, Subramaniam S, David M, Rosenfeld MG, Glass CK 2005 Molecular determinants of crosstalk between nuclear receptors and toll-like receptors. Cell 122:707–721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diefenbacher M, Sekula S, Heilbock C, Maier JV, Litfin M, van Dam H, Castellazzi M, Herrlich P, Kassel O 2008 Restriction to Fos family members of Trip6-dependent coactivation and glucocorticoid receptor-dependent trans-repression of activator protein-1. Mol Endocrinol 22:1767–1780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito K, Lim S, Caramori G, Chung KF, Barnes PJ, Adcock IM 2001 Cigarette smoking reduces histone deacetylase 2 expression, enhances cytokine expression, and inhibits glucocorticoid actions in alveolar macrophages. FASEB J 15:1110–1112 [PubMed] [Google Scholar]
- Tsaprouni LG, Ito K, Adcock IM, Punchard N 2007 Suppression of lipopolysaccharide- and tumour necrosis factor-α-induced interleukin (IL)-8 expression by glucocorticoids involves changes in IL-8 promoter acetylation. Clin Exp Immunol 150:151–157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouzarides T 2002 Histone methylation in transcriptional control. Curr Opin Genet Dev 12:198–209 [DOI] [PubMed] [Google Scholar]
- Sheppard KA, Phelps KM, Williams AJ, Thanos D, Glass CK, Rosenfeld MG, Gerritsen ME, Collins T 1998 Nuclear integration of glucocorticoid receptor and nuclear factor-κB signaling by CREB-binding protein and steroid receptor coactivator-1. J Biol Chem 273:29291–29294 [DOI] [PubMed] [Google Scholar]
- Burkhart BA, Hebbar PB, Trotter KW, Archer TK 2005 Chromatin-dependent E1A activity modulates NF-κB RelA-mediated repression of glucocorticoid receptor-dependent transcription. J Biol Chem 280:6349–6358 [DOI] [PubMed] [Google Scholar]
- De Bosscher K, Vanden Berghe W, Haegeman G 2001 Glucocorticoid repression of AP-1 is not mediated by competition for nuclear coactivators. Mol Endocrinol 15:219–227 [DOI] [PubMed] [Google Scholar]
- De Bosscher K, Vanden Berghe W, Vermeulen L, Plaisance S, Boone E, Haegeman G 2000 Glucocorticoids repress NF-κB-driven genes by disturbing the interaction of p65 with the basal transcription machinery, irrespective of coactivator levels in the cell. Proc Natl Acad Sci USA 97:3919–3924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKay LI, Cidlowski JA 2000 CBP (CREB binding protein) integrates NF-κB (nuclear factor-κB) and glucocorticoid receptor physical interactions and antagonism. Mol Endocrinol 14:1222–1234 [DOI] [PubMed] [Google Scholar]
- Wu J, Li Y, Dietz J, Lala DS 2004 Repression of p65 transcriptional activation by the glucocorticoid receptor in the absence of receptor-coactivator interactions. Mol Endocrinol 18:53–62 [DOI] [PubMed] [Google Scholar]
- Sun Y, Tao YG, Kagan BL, He Y, Jr SS 2008 Modulation of transcription parameters in glucocorticoid receptor-mediated repression. Mol Cell Endocrinol 295:59–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonas BA, Varlakhanova N, Hayakawa F, Goodson M, Privalsky ML 2007 Response of SMRT (silencing mediator of retinoic acid and thyroid hormone receptor) and N-CoR (nuclear receptor corepressor) corepressors to mitogen-activated protein kinase kinase kinase cascades is determined by alternative mRNA splicing. Mol Endocrinol 21:1924–1939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu RC, Feng Q, Lonard DM, O'Malley BW 2007 SRC-3 coactivator functional lifetime is regulated by a phospho-dependent ubiquitin time clock. Cell 129:1125–1140 [DOI] [PubMed] [Google Scholar]
- Wu RC, Smith CL, O'Malley BW 2005 Transcriptional regulation by steroid receptor coactivator phosphorylation. Endocr Rev 26:393–399 [DOI] [PubMed] [Google Scholar]
- Faus H, Haendler B 2006 Post-translational modifications of steroid receptors. Biomed Pharmacother 60:520–528 [DOI] [PubMed] [Google Scholar]
- Rochette-Egly C 2003 Nuclear receptors: integration of multiple signalling pathways through phosphorylation. Cell Signal 15:355–366 [DOI] [PubMed] [Google Scholar]
- Bodwell JE, Hu JM, Orti E, Munck A 1995 Hormone-induced hyperphosphorylation of specific phosphorylated sites in the mouse glucocorticoid receptor. J Steroid Biochem Mol Biol 52:135–140 [DOI] [PubMed] [Google Scholar]
- Bodwell JE, Ortí E, Coull JM, Pappin DJ, Smith LI, Swift F 1991 Identification of phosphorylated sites in the mouse glucocorticoid receptor. J Biol Chem 266:7549–7555 [PubMed] [Google Scholar]
- Rogatsky I, Waase CL, Garabedian MJ 1998 Phosphorylation and inhibition of rat glucocorticoid receptor transcriptional activation by glycogen synthase kinase-3 (GSK-3). Species-specific differences between human and rat glucocorticoid receptor signaling as revealed through GSK-3 phosphorylation. J Biol Chem 273:14315–14321 [DOI] [PubMed] [Google Scholar]
- Ismaili N, Garabedian MJ 2004 Modulation of glucocorticoid receptor function via phosphorylation. Ann NY Acad Sci 1024:86–101 [DOI] [PubMed] [Google Scholar]
- Wang Z, Frederick J, Garabedian MJ 2002 Deciphering the phosphorylation “code” of the glucocorticoid receptor in vivo. J Biol Chem 277:26573–26580 [DOI] [PubMed] [Google Scholar]
- Galliher-Beckley AJ, Williams JG, Collins JB, Cidlowski JA 2008 Glycogen synthase kinase 3β-mediated serine phosphorylation of the human glucocorticoid receptor redirects gene expression profiles. Mol Cell Biol 28:7309–7322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dephoure N, Zhou C, Villén J, Beausoleil SA, Bakalarski CE, Elledge SJ, Gygi SP 2008 A quantitative atlas of mitotic phosphorylation. Proc Natl Acad Sci USA 105:10762–10767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krstic MD, Rogatsky I, Yamamoto KR, Garabedian MJ 1997 Mitogen-activated and cyclin-dependent protein kinases selectively and differentially modulate transcriptional enhancement by the glucocorticoid receptor. Mol Cell Biol 17:3947–3954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Garabedian MJ 2003 Modulation of glucocorticoid receptor transcriptional activation, phosphorylation, and growth inhibition by p27Kip1. J Biol Chem 278:50897–50901 [DOI] [PubMed] [Google Scholar]
- Webster JC, Jewell CM, Bodwell JE, Munck A, Sar M, Cidlowski JA 1997 Mouse glucocorticoid receptor phosphorylation status influences multiple functions of the receptor protein. J Biol Chem 272:9287–9293 [DOI] [PubMed] [Google Scholar]
- Blind RD, Garabedian MJ 2008 Differential recruitment of glucocorticoid receptor phospho-isoforms to glucocorticoid-induced genes. J Steroid Biochem Mol Biol 109:150–157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W, Dang T, Blind RD, Wang Z, Cavasotto CN, Hittelman AB, Rogatsky I, Logan SK, Garabedian MJ 2008 Glucocorticoid receptor phosphorylation differentially affects target gene expression. Mol Endocrinol 22:1754–1766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kino T, Ichijo T, Amin ND, Kesavapany S, Wang Y, Kim N, Rao S, Player A, Zheng YL, Garabedian MJ, Kawasaki E, Pant HC, Chrousos GP 2007 Cyclin-dependent kinase 5 differentially regulates the transcriptional activity of the glucocorticoid receptor through phosphorylation: clinical implications for the nervous system response to glucocorticoids and stress. Mol Endocrinol 21:1552–1568 [DOI] [PubMed] [Google Scholar]
- Miller AL, Webb MS, Copik AJ, Wang Y, Johnson BH, Kumar R, Thompson EB 2005 p38 Mitogen-activated protein kinase (MAPK) is a key mediator in glucocorticoid-induced apoptosis of lymphoid cells: correlation between p38 MAPK activation and site-specific phosphorylation of the human glucocorticoid receptor at serine 211. Mol Endocrinol 19:1569–1583 [DOI] [PubMed] [Google Scholar]
- Irusen E, Matthews JG, Takahashi A, Barnes PJ, Chung KF, Adcock IM 2002 p38 Mitogen-activated protein kinase-induced glucocorticoid receptor phosphorylation reduces its activity: role in steroid-insensitive asthma. J Allergy Clin Immunol 109:649–657 [DOI] [PubMed] [Google Scholar]
- Bantel H, Schmitz ML, Raible A, Gregor M, Schulze-Osthoff K 2002 Critical role of NF-κB and stress-activated protein kinases in steroid unresponsiveness. FASEB J 16:1832–1834 [DOI] [PubMed] [Google Scholar]
- Szatmáry Z, Garabedian MJ, Vilcek J 2004 Inhibition of glucocorticoid receptor-mediated transcriptional activation by p38 mitogen-activated protein (MAP) kinase. J Biol Chem 279:43708–43715 [DOI] [PubMed] [Google Scholar]
- Wang X, Wu H, Miller AH 2004 Interleukin 1α (IL-1α) induced activation of p38 mitogen-activated protein kinase inhibits glucocorticoid receptor function. Mol Psychiatry 9:65–75 [DOI] [PubMed] [Google Scholar]
- Itoh M, Adachi M, Yasui H, Takekawa M, Tanaka H, Imai K 2002 Nuclear export of glucocorticoid receptor is enhanced by c-Jun N-terminal kinase-mediated phosphorylation. Mol Endocrinol 16:2382–2392 [DOI] [PubMed] [Google Scholar]
- Rogatsky I, Logan SK, Garabedian MJ 1998 Antagonism of glucocorticoid receptor transcriptional activation by the c-Jun N-terminal kinase. Proc Natl Acad Sci USA 95:2050–2055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espinás ML, Roux J, Pictet R, Grange T 1995 Glucocorticoids and protein kinase A coordinately modulate transcription factor recruitment at a glucocorticoid-responsive unit. Mol Cell Biol 15:5346–5354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doucas V, Shi Y, Miyamoto S, West A, Verma I, Evans RM 2000 Cytoplasmic catalytic subunit of protein kinase A mediates cross-repression by NF-κB and the glucocorticoid receptor. Proc Natl Acad Sci USA 97:11893–11898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rangarajan PN, Umesono K, Evans RM 1992 Modulation of glucocorticoid receptor function by protein kinase A. Mol Endocrinol 6:1451–1457 [DOI] [PubMed] [Google Scholar]
- Haske T, Nakao M, Moudgil VK 1994 Phosphorylation of immunopurified rat liver glucocorticoid receptor by the catalytic subunit of cAMP-dependent protein kinase. Mol Cell Biochem 132:163–171 [DOI] [PubMed] [Google Scholar]
- Wang Z, Chen W, Kono E, Dang T, Garabedian MJ 2007 Modulation of glucocorticoid receptor phosphorylation and transcriptional activity by a C-terminal-associated protein phosphatase. Mol Endocrinol 21:625–634 [DOI] [PubMed] [Google Scholar]
- Zhang S, Jonklaas J, Danielsen M 2007 The glucocorticoid agonist activities of mifepristone (RU486) and progesterone are dependent on glucocorticoid receptor levels but not on EC50 values. Steroids 72:600–608 [DOI] [PubMed] [Google Scholar]
- Saelzler MP, Spackman CC, Liu Y, Martinez LC, Harris JP, Abe MK 2006 ERK8 down-regulates transactivation of the glucocorticoid receptor through Hic-5. J Biol Chem 281:16821–16832 [DOI] [PubMed] [Google Scholar]
- Yang J, Liu J, DeFranco DB 1997 Subnuclear trafficking of glucocorticoid receptors in vitro: chromatin recycling and nuclear export. J Cell Biol 137:523–538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adzic M, Djordjevic J, Djordjevic A, Niciforovic A, Demonacos C, Radojcic M, Krstic-Demonacos M 2009 Acute or chronic stress induce cell compartment-specific phosphorylation of glucocorticoid receptor and alter its transcriptional activity in Wistar rat brain. J Endocrinol 202:87–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ismaili N, Blind R, Garabedian MJ 2005 Stabilization of the unliganded glucocorticoid receptor by TSG101. J Biol Chem 280:11120–11126 [DOI] [PubMed] [Google Scholar]
- Hittelman AB, Burakov D, Iñiguez-Lluhí JA, Freedman LP, Garabedian MJ 1999 Differential regulation of glucocorticoid receptor transcriptional activation via AF-1-associated proteins. EMBO J 18:5380–5388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeFranco DB, Qi M, Borror KC, Garabedian MJ, Brautigan DL 1991 Protein phosphatase types 1 and/or 2A regulate nucleocytoplasmic shuttling of glucocorticoid receptors. Mol Endocrinol 5:1215–1228 [DOI] [PubMed] [Google Scholar]
- Galigniana MD, Housley PR, DeFranco DB, Pratt WB 1999 Inhibition of glucocorticoid receptor nucleocytoplasmic shuttling by okadaic acid requires intact cytoskeleton. J Biol Chem 274:16222–16227 [DOI] [PubMed] [Google Scholar]
- Banerjee A, Periyasamy S, Wolf IM, Hinds Jr TD, Yong W, Shou W, Sanchez ER 2008 Control of glucocorticoid and progesterone receptor subcellular localization by the ligand-binding domain is mediated by distinct interactions with tetratricopeptide repeat proteins. Biochemistry 47:10471–10480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silverstein AM, Galigniana MD, Chen MS, Owens-Grillo JK, Chinkers M, Pratt WB 1997 Protein phosphatase 5 is a major component of glucocorticoid receptor.hsp90 complexes with properties of an FK506-binding immunophilin. J Biol Chem 272:16224–16230 [DOI] [PubMed] [Google Scholar]
- Chen MS, Silverstein AM, Pratt WB, Chinkers M 1996 The tetratricopeptide repeat domain of protein phosphatase 5 mediates binding to glucocorticoid receptor heterocomplexes and acts as a dominant negative mutant. J Biol Chem 271:32315–32320 [DOI] [PubMed] [Google Scholar]
- Somers JP, DeFranco DB 1992 Effects of okadaic acid, a protein phosphatase inhibitor, on glucocorticoid receptor-mediated enhancement. Mol Endocrinol 6:26–34 [DOI] [PubMed] [Google Scholar]
- Zuo Z, Urban G, Scammell JG, Dean NM, McLean TK, Aragon I, Honkanen RE 1999 Ser/Thr protein phosphatase type 5 (PP5) is a negative regulator of glucocorticoid receptor-mediated growth arrest. Biochemistry 38:8849–8857 [DOI] [PubMed] [Google Scholar]
- Davies TH, Ning YM, Sánchez ER 2005 Differential control of glucocorticoid receptor hormone-binding function by tetratricopeptide repeat (TPR) proteins and the immunosuppressive ligand FK506. Biochemistry 44:2030–2038 [DOI] [PubMed] [Google Scholar]
- Galigniana MD, Radanyi C, Renoir JM, Housley PR, Pratt WB 2001 Evidence that the peptidylprolyl isomerase domain of the hsp90-binding immunophilin FKBP52 is involved in both dynein interaction and glucocorticoid receptor movement to the nucleus. J Biol Chem 276:14884–14889 [DOI] [PubMed] [Google Scholar]
- Hinds Jr TD, Sánchez ER 2008 Protein phosphatase 5. Int J Biochem Cell Biol 40:2358–2362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Leung DY, Nordeen SK, Goleva E 2009 Estrogen inhibits glucocorticoid action via protein phosphatase 5 (PP5) mediated glucocorticoid receptor dephosphorylation. J Biol Chem 284:24542–24552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hay RT 2006 Role of ubiquitin-like proteins in transcriptional regulation. Ernst Schering Res Found Workshop 57:173–192 [DOI] [PubMed] [Google Scholar]
- Davies L, Karthikeyan N, Lynch JT, Sial EA, Gkourtsa A, Demonacos C, Krstic-Demonacos M 2008 Cross talk of signaling pathways in the regulation of the glucocorticoid receptor function. Mol Endocrinol 22:1331–1344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmstrom SR, Chupreta S, So AY, Iñiguez-Lluhí JA 2008 SUMO-mediated inhibition of glucocorticoid receptor synergistic activity depends on stable assembly at the promoter but not on DAXX. Mol Endocrinol 22:2061–2075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian S, Poukka H, Palvimo JJ, Jänne OA 2002 Small ubiquitin-related modifier-1 (SUMO-1) modification of the glucocorticoid receptor. Biochem J 367:907–911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Drean Y, Mincheneau N, Le Goff P, Michel D 2002 Potentiation of glucocorticoid receptor transcriptional activity by sumoylation. Endocrinology 143:3482–3489 [DOI] [PubMed] [Google Scholar]
- Lin DY, Huang YS, Jeng JC, Kuo HY, Chang CC, Chao TT, Ho CC, Chen YC, Lin TP, Fang HI, Hung CC, Suen CS, Hwang MJ, Chang KS, Maul GG, Shih HM 2006 Role of SUMO-interacting motif in Daxx SUMO modification, subnuclear localization, and repression of sumoylated transcription factors. Mol Cell 24:341–354 [DOI] [PubMed] [Google Scholar]
- Deroo BJ, Rentsch C, Sampath S, Young J, DeFranco DB, Archer TK 2002 Proteasomal inhibition enhances glucocorticoid receptor transactivation and alters its subnuclear trafficking. Mol Cell Biol 22:4113–4123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito K, Yamamura S, Essilfie-Quaye S, Cosio B, Ito M, Barnes PJ, Adcock IM 2006 Histone deacetylase 2-mediated deacetylation of the glucocorticoid receptor enables NF-κB suppression. J Exp Med 203:7–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thalhamer T, McGrath MA, Harnett MM 2008 MAPKs and their relevance to arthritis and inflammation. Rheumatology (Oxford) 47:409–414 [DOI] [PubMed] [Google Scholar]
- Caelles C, González-Sancho JM, Muñoz A 1997 Nuclear hormone receptor antagonism with AP-1 by inhibition of the JNK pathway. Genes Dev 11:3351–3364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ventura JJ, Roncero C, Fabregat I, Benito M 1999 Glucocorticoid receptor down-regulates c-Jun amino terminal kinases induced by tumor necrosis factor α in fetal rat hepatocyte primary cultures. Hepatology 29:849–857 [DOI] [PubMed] [Google Scholar]
- Abraham SM, Lawrence T, Kleiman A, Warden P, Medghalchi M, Tuckermann J, Saklatvala J, Clark AR 2006 Antiinflammatory effects of dexamethasone are partly dependent on induction of dual specificity phosphatase 1. J Exp Med 203:1883–1889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen P, Li J, Barnes J, Kokkonen GC, Lee JC, Liu Y 2002 Restraint of proinflammatory cytokine biosynthesis by mitogen-activated protein kinase phosphatase-1 in lipopolysaccharide-stimulated macrophages. J Immunol 169:6408–6416 [DOI] [PubMed] [Google Scholar]
- Issa R, Xie S, Khorasani N, Sukkar M, Adcock IM, Lee KY, Chung KF 2007 Corticosteroid inhibition of growth-related oncogene protein-α via mitogen-activated kinase phosphatase-1 in airway smooth muscle cells. J Immunol 178:7366–7375 [DOI] [PubMed] [Google Scholar]
- Bruna A, Nicolàs M, Muñoz A, Kyriakis JM, Caelles C 2003 Glucocorticoid receptor-JNK interaction mediates inhibition of the JNK pathway by glucocorticoids. EMBO J 22:6035–6044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- González MV, Jiménez B, Berciano MT, González-Sancho JM, Caelles C, Lafarga M, Muñoz A 2000 Glucocorticoids antagonize AP-1 by inhibiting the activation/phosphorylation of JNK without affecting its subcellular distribution. J Cell Biol 150:1199–1208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirasawa N, Izumi S, Linwong W, Ohuchi K 2003 Inhibition by dexamethasone of interleukin 13 production via glucocorticoid receptor-mediated inhibition of c-Jun phosphorylation. FEBS Lett 554:489–493 [DOI] [PubMed] [Google Scholar]
- Cissel DS, Beaven MA 2000 Disruption of Raf-1/heat shock protein 90 complex and Raf signaling by dexamethasone in mast cells. J Biol Chem 275:7066–7070 [DOI] [PubMed] [Google Scholar]
- Kim MJ, Chae JS, Kim KJ, Hwang SG, Yoon KW, Kim EK, Yun HJ, Cho JH, Kim J, Kim BW, Kim HC, Kang SS, Lang F, Cho SG, Choi EJ 2007 Negative regulation of SEK1 signaling by serum- and glucocorticoid-inducible protein kinase 1. EMBO J 26:3075–3085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swantek JL, Cobb MH, Geppert TD 1997 Jun N-terminal kinase/stress-activated protein kinase (JNK/SAPK) is required for lipopolysaccharide stimulation of tumor necrosis factor α (TNF-α) translation: glucocorticoids inhibit TNF-α translation by blocking JNK/SAPK. Mol Cell Biol 17:6274–6282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rider LG, Hirasawa N, Santini F, Beaven MA 1996 Activation of the mitogen-activated protein kinase cascade is suppressed by low concentrations of dexamethasone in mast cells. J Immunol 157:2374–2380 [PubMed] [Google Scholar]
- Bhalla V, Soundararajan R, Pao AC, Li H, Pearce D 2006 Disinhibitory pathways for control of sodium transport: regulation of ENaC by SGK1 and GILZ. Am J Physiol Renal Physiol 291:F714–F721 [DOI] [PubMed] [Google Scholar]
- Soundararajan R, Zhang TT, Wang J, Vandewalle A, Pearce D 2005 A novel role for glucocorticoid-induced leucine zipper protein in epithelial sodium channel-mediated sodium transport. J Biol Chem 280:39970–39981 [DOI] [PubMed] [Google Scholar]
- Wang JC, Derynck MK, Nonaka DF, Khodabakhsh DB, Haqq C, Yamamoto KR 2004 Chromatin immunoprecipitation (ChIP) scanning identifies primary glucocorticoid receptor target genes. Proc Natl Acad Sci USA 101:15603–15608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen N, Mouly E, Hamdi H, Maillot MC, Pallardy M, Godot V, Capel F, Balian A, Naveau S, Galanaud P, Lemoine FM, Emilie D 2006 GILZ expression in human dendritic cells redirects their maturation and prevents antigen-specific T lymphocyte response. Blood 107:2037–2044 [DOI] [PubMed] [Google Scholar]
- Ehrchen J, Steinmüller L, Barczyk K, Tenbrock K, Nacken W, Eisenacher M, Nordhues U, Sorg C, Sunderkötter C, Roth J 2007 Glucocorticoids induce differentiation of a specifically activated, anti-inflammatory subtype of human monocytes. Blood 109:1265–1274 [DOI] [PubMed] [Google Scholar]
- Berrebi D, Bruscoli S, Cohen N, Foussat A, Migliorati G, Bouchet-Delbos L, Maillot MC, Portier A, Couderc J, Galanaud P, Peuchmaur M, Riccardi C, Emilie D 2003 Synthesis of glucocorticoid-induced leucine zipper (GILZ) by macrophages: an anti-inflammatory and immunosuppressive mechanism shared by glucocorticoids and IL-10. Blood 101:729–738 [DOI] [PubMed] [Google Scholar]
- D'Adamio F, Zollo O, Moraca R, Ayroldi E, Bruscoli S, Bartoli A, Cannarile L, Migliorati G, Riccardi C 1997 A new dexamethasone-induced gene of the leucine zipper family protects T lymphocytes from TCR/CD3-activated cell death. Immunity 7:803–812 [DOI] [PubMed] [Google Scholar]
- Mittelstadt PR, Ashwell JD 2001 Inhibition of AP-1 by the glucocorticoid-inducible protein GILZ. J Biol Chem 276:29603–29610 [DOI] [PubMed] [Google Scholar]
- Galon J, Franchimont D, Hiroi N, Frey G, Boettner A, Ehrhart-Bornstein M, O'Shea JJ, Chrousos GP, Bornstein SR 2002 Gene profiling reveals unknown enhancing and suppressive actions of glucocorticoids on immune cells. FASEB J 16:61–71 [DOI] [PubMed] [Google Scholar]
- Tonko M, Ausserlechner MJ, Bernhard D, Helmberg A, Kofler R 2001 Gene expression profiles of proliferating vs. G1/G0 arrested human leukemia cells suggest a mechanism for glucocorticoid-induced apoptosis. FASEB J 15:693–699 [DOI] [PubMed] [Google Scholar]
- Godot V, Garcia G, Capel F, Arock M, Durand-Gasselin I, Asselin-Labat ML, Emilie D, Humbert M 2006 Dexamethasone and IL-10 stimulate glucocorticoid-induced leucine zipper synthesis by human mast cells. Allergy 61:886–890 [DOI] [PubMed] [Google Scholar]
- Ayroldi E, Zollo O, Macchiarulo A, Di Marco B, Marchetti C, Riccardi C 2002 Glucocorticoid-induced leucine zipper inhibits the Raf-extracellular signal-regulated kinase pathway by binding to Raf-1. Mol Cell Biol 22:7929–7941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayroldi E, Zollo O, Bastianelli A, Marchetti C, Agostini M, Di Virgilio R, Riccardi C 2007 GILZ mediates the antiproliferative activity of glucocorticoids by negative regulation of Ras signaling. J Clin Invest 117:1605–1615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Widén C, Zilliacus J, Gustafsson JA, Wikström AC 2000 Glucocorticoid receptor interaction with 14-3-3 and Raf-1, a proposed mechanism for cross-talk of two signal transduction pathways. J Biol Chem 275:39296–39301 [DOI] [PubMed] [Google Scholar]
- Di Marco B, Massetti M, Bruscoli S, Macchiarulo A, Di Virgilio R, Velardi E, Donato V, Migliorati G, Riccardi C 2007 Glucocorticoid-induced leucine zipper (GILZ)/NF-κB interaction: role of GILZ homo-dimerization and C-terminal domain. Nucleic Acids Res 35:517–528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayroldi E, Migliorati G, Bruscoli S, Marchetti C, Zollo O, Cannarile L, D'Adamio F, Riccardi C 2001 Modulation of T-cell activation by the glucocorticoid-induced leucine zipper factor via inhibition of nuclear factor κB. Blood 98:743–753 [DOI] [PubMed] [Google Scholar]
- Hiragun T, Peng Z, Beaven MA 2005 Dexamethasone up-regulates the inhibitory adaptor protein Dok-1 and suppresses downstream activation of the mitogen-activated protein kinase pathway in antigen-stimulated RBL-2H3 mast cells. Mol Pharmacol 67:598–603 [DOI] [PubMed] [Google Scholar]
- Hiragun T, Peng Z, Beaven MA 2006 Cutting edge: dexamethasone negatively regulates Syk in mast cells by up-regulating SRC-like adaptor protein. J Immunol 177:2047–2050 [DOI] [PubMed] [Google Scholar]
- Graham TE, Prossnitz ER, Dorin RI 2002 Dexras1/AGS-1 inhibits signal transduction from the Gi-coupled formyl peptide receptor to Erk-1/2 MAP kinases. J Biol Chem 277:10876–10882 [DOI] [PubMed] [Google Scholar]
- Kemppainen RJ, Behrend EN 1998 Dexamethasone rapidly induces a novel ras superfamily member-related gene in AtT-20 cells. J Biol Chem 273:3129–3131 [DOI] [PubMed] [Google Scholar]
- Bhattacharyya S, Brown DE, Brewer JA, Vogt SK, Muglia LJ 2007 Macrophage glucocorticoid receptors regulate Toll-like receptor 4-mediated inflammatory responses by selective inhibition of p38 MAP kinase. Blood 109:4313–4319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark AR, Martins JR, Tchen CR 2008 Role of dual specificity phosphatases in biological responses to glucocorticoids. J Biol Chem 283:25765–25769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deak M, Clifton AD, Lucocq LM, Alessi DR 1998 Mitogen- and stress-activated protein kinase-1 (MSK1) is directly activated by MAPK and SAPK2/p38, and may mediate activation of CREB. EMBO J 17:4426–4441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy CE, Macdonald A, Morrice NA, Campbell DG, Deak M, Toth R, McIlrath J, Arthur JS 2007 Identification of novel phosphorylation sites in MSK1 by precursor ion scanning MS. Biochem J 402:491–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomás-Zuber M, Mary JL, Lamour F, Bur D, Lesslauer W 2001 C-terminal elements control location, activation threshold, and p38 docking of ribosomal S6 kinase B (RSKB). J Biol Chem 276:5892–5899 [DOI] [PubMed] [Google Scholar]
- Dunn KL, Espino PS, Drobic B, He S, Davie JR 2005 The Ras-MAPK signal transduction pathway, cancer and chromatin remodeling. Biochem Cell Biol 83:1–14 [DOI] [PubMed] [Google Scholar]
- Arthur JS 2008 MSK activation and physiological roles. Front Biosci 13:5866–5879 [DOI] [PubMed] [Google Scholar]
- Darragh J, Soloaga A, Beardmore VA, Wingate AD, Wiggin GR, Peggie M, Arthur JS 2005 MSKs are required for the transcription of the nuclear orphan receptors Nur77, Nurr1 and Nor1 downstream of MAPK signalling. Biochem J 390:749–759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiggin GR, Soloaga A, Foster JM, Murray-Tait V, Cohen P, Arthur JS 2002 MSK1 and MSK2 are required for the mitogen- and stress-induced phosphorylation of CREB and ATF1 in fibroblasts. Mol Cell Biol 22:2871–2881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beardmore VA, Hinton HJ, Eftychi C, Apostolaki M, Armaka M, Darragh J, McIlrath J, Carr JM, Armit LJ, Clacher C, Malone L, Kollias G, Arthur JS 2005 Generation and characterization of p38β (MAPK11) gene-targeted mice. Mol Cell Biol 25:10454–10464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy CE, Campbell DG, Deak M, Bloomberg GB, Arthur JS 2005 MSK1 activity is controlled by multiple phosphorylation sites. Biochem J 387:507–517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomás-Zuber M, Mary JL, Lesslauer W 2000 Control sites of ribosomal S6 kinase B and persistent activation through tumor necrosis factor. J Biol Chem 275:23549–23558 [DOI] [PubMed] [Google Scholar]
- Davie JR 2003 MSK1 and MSK2 mediate mitogen- and stress-induced phosphorylation of histone H3: a controversy resolved. Sci STKE 2003:PE33 [DOI] [PubMed] [Google Scholar]
- Dunn KL, Davie JR 2005 Stimulation of the Ras-MAPK pathway leads to independent phosphorylation of histone H3 on serine 10 and 28. Oncogene 24:3492–3502 [DOI] [PubMed] [Google Scholar]
- Dyson MH, Thomson S, Inagaki M, Goto H, Arthur SJ, Nightingale K, Iborra FJ, Mahadevan LC 2005 MAP kinase-mediated phosphorylation of distinct pools of histone H3 at S10 or S28 via mitogen- and stress-activated kinase 1/2. J Cell Sci 118:2247–2259 [DOI] [PubMed] [Google Scholar]
- Soloaga A, Thomson S, Wiggin GR, Rampersaud N, Dyson MH, Hazzalin CA, Mahadevan LC, Arthur JS 2003 MSK2 and MSK1 mediate the mitogen- and stress-induced phosphorylation of histone H3 and HMG-14. EMBO J 22:2788–2797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macdonald N, Welburn JP, Noble ME, Nguyen A, Yaffe MB, Clynes D, Moggs JG, Orphanides G, Thomson S, Edmunds JW, Clayton AL, Endicott JA, Mahadevan LC 2005 Molecular basis for the recognition of phosphorylated and phosphoacetylated histone h3 by 14-3-3. Mol Cell 20:199–211 [DOI] [PubMed] [Google Scholar]
- Winter S, Fischle W, Seiser C 2008 Modulation of 14-3-3 interaction with phosphorylated histone H3 by combinatorial modification patterns. Cell Cycle 7:1336–1342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter S, Simboeck E, Fischle W, Zupkovitz G, Dohnal I, Mechtler K, Ammerer G, Seiser C 2008 14-3-3 proteins recognize a histone code at histone H3 and are required for transcriptional activation. EMBO J 27:88–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter W, Clynes D, Tang Y, Marmorstein R, Mellor J, Berger SL 2008 14-3-3 interaction with histone H3 involves a dual modification pattern of phosphoacetylation. Mol Cell Biol 28:2840–2849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vicent GP, Ballaré C, Nacht AS, Clausell J, Subtil-Rodríguez A, Quiles I, Jordan A, Beato M 2006 Induction of progesterone target genes requires activation of Erk and Msk kinases and phosphorylation of histone H3. Mol Cell 24:367–381 [DOI] [PubMed] [Google Scholar]
- Beck IM, Vanden Berghe W, Vermeulen L, Bougarne N, Vander Cruyssen B, Haegeman G, De Bosscher K 2008 Altered subcellular distribution of MSK1 induced by glucocorticoids contributes to NF-κB inhibition. EMBO J 27:1682–1693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasegawa Y, Tomita K, Watanabe M, Yamasaki A, Sano H, Hitsuda Y, Shimizu E 2005 Dexamethasone inhibits phosphorylation of histone H3 at serine 10. Biochem Biophys Res Commun 336:1049–1055 [DOI] [PubMed] [Google Scholar]
- Dewint P, Gossye V, De Bosscher K, Vanden Berghe W, Van Beneden K, Deforce D, Van Calenbergh S, Müller-Ladner U, Vander Cruyssen B, Verbruggen G, Haegeman G, Elewaut D 2008 A plant-derived ligand favoring monomeric glucocorticoid receptor conformation with impaired transactivation potential attenuates collagen-induced arthritis. J Immunol 180:2608–2615 [DOI] [PubMed] [Google Scholar]
- Liu G, Zhang Y, Bode AM, Ma WY, Dong Z 2002 Phosphorylation of 4E-BP1 is mediated by the p38/MSK1 pathway in response to UVB irradiation. J Biol Chem 277:8810–8816 [DOI] [PubMed] [Google Scholar]
- Otulakowski G, Duan W, Gandhi S, O'brodovich H 2007 Steroid and oxygen effects on eIF4F complex, mTOR, and ENaC translation in fetal lung epithelia. Am J Respir Cell Mol Biol 37:457–466 [DOI] [PubMed] [Google Scholar]
- Liu Z, Jahn LA, Long W, Fryburg DA, Wei L, Barrett EJ 2001 Branched chain amino acids activate messenger ribonucleic acid translation regulatory proteins in human skeletal muscle, and glucocorticoids blunt this action. J Clin Endocrinol Metab 86:2136–2143 [DOI] [PubMed] [Google Scholar]
- Liu Z, Li G, Kimball SR, Jahn LA, Barrett EJ 2004 Glucocorticoids modulate amino acid-induced translation initiation in human skeletal muscle. Am J Physiol Endocrinol Metab 287:E275–E281 [DOI] [PubMed] [Google Scholar]
- Shah OJ, Iniguez-Lluhi JA, Romanelli A, Kimball SR, Jefferson LS 2002 The activated glucocorticoid receptor modulates presumptive autoregulation of ribosomal protein S6 protein kinase, p70 S6K. J Biol Chem 277:2525–2533 [DOI] [PubMed] [Google Scholar]
- Shah OJ, Kimball SR, Jefferson LS 2000 Glucocorticoids abate p70(S6k) and eIF4E function in L6 skeletal myoblasts. Am J Physiol Endocrinol Metab 279:E74–E82 [DOI] [PubMed] [Google Scholar]
- Dong C, Davis RJ, Flavell RA 2002 MAP kinases in the immune response. Annu Rev Immunol 20:55–72 [DOI] [PubMed] [Google Scholar]
- Carballo E, Lai WS, Blackshear PJ 1998 Feedback inhibition of macrophage tumor necrosis factor-α production by tristetraprolin. Science 281:1001–1005 [DOI] [PubMed] [Google Scholar]
- Carrick DM, Lai WS, Blackshear PJ 2004 The tandem CCCH zinc finger protein tristetraprolin and its relevance to cytokine mRNA turnover and arthritis. Arthritis Res Ther 6:248–264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carballo E, Cao H, Lai WS, Kennington EA, Campbell D, Blackshear PJ 2001 Decreased sensitivity of tristetraprolin-deficient cells to p38 inhibitors suggests the involvement of tristetraprolin in the p38 signaling pathway. J Biol Chem 276:42580–42587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chrestensen CA, Schroeder MJ, Shabanowitz J, Hunt DF, Pelo JW, Worthington MT, Sturgill TW 2004 MAPKAP kinase 2 phosphorylates tristetraprolin on in vivo sites including Ser178, a site required for 14-3-3 binding. J Biol Chem 279:10176–10184 [DOI] [PubMed] [Google Scholar]
- Hitti E, Iakovleva T, Brook M, Deppenmeier S, Gruber AD, Radzioch D, Clark AR, Blackshear PJ, Kotlyarov A, Gaestel M 2006 Mitogen-activated protein kinase-activated protein kinase 2 regulates tumor necrosis factor mRNA stability and translation mainly by altering tristetraprolin expression, stability, and binding to adenine/uridine-rich element. Mol Cell Biol 26:2399–2407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brook M, Tchen CR, Santalucia T, McIlrath J, Arthur JS, Saklatvala J, Clark AR 2006 Posttranslational regulation of tristetraprolin subcellular localization and protein stability by p38 mitogen-activated protein kinase and extracellular signal-regulated kinase pathways. Mol Cell Biol 26:2408–2418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neininger A, Kontoyiannis D, Kotlyarov A, Winzen R, Eckert R, Volk HD, Holtmann H, Kollias G, Gaestel M 2002 MK2 targets AU-rich elements and regulates biosynthesis of tumor necrosis factor and interleukin-6 independently at different post-transcriptional levels. J Biol Chem 277:3065–3068 [DOI] [PubMed] [Google Scholar]
- Stoecklin G, Stubbs T, Kedersha N, Wax S, Rigby WF, Blackwell TK, Anderson P 2004 MK2-induced tristetraprolin:14-3-3 complexes prevent stress granule association and ARE-mRNA decay. EMBO J 23:1313–1324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dean JL, Sarsfield SJ, Tsounakou E, Saklatvala J 2003 p38 Mitogen-activated protein kinase stabilizes mRNAs that contain cyclooxygenase-2 and tumor necrosis factor AU-rich elements by inhibiting deadenylation. J Biol Chem 278:39470–39476 [DOI] [PubMed] [Google Scholar]
- Taylor GA, Carballo E, Lee DM, Lai WS, Thompson MJ, Patel DD, Schenkman DI, Gilkeson GS, Broxmeyer HE, Haynes BF, Blackshear PJ 1996 A pathogenetic role for TNF α in the syndrome of cachexia, arthritis, and autoimmunity resulting from tristetraprolin (TTP) deficiency. Immunity 4:445–454 [DOI] [PubMed] [Google Scholar]
- Stojadinovic O, Lee B, Vouthounis C, Vukelic S, Pastar I, Blumenberg M, Brem H, Tomic-Canic M 2007 Novel genomic effects of glucocorticoids in epidermal keratinocytes: inhibition of apoptosis, interferon-γ pathway, and wound healing along with promotion of terminal differentiation. J Biol Chem 282:4021–4034 [DOI] [PubMed] [Google Scholar]
- Lasa M, Abraham SM, Boucheron C, Saklatvala J, Clark AR 2002 Dexamethasone causes sustained expression of mitogen-activated protein kinase (MAPK) phosphatase 1 and phosphatase-mediated inhibition of MAPK p38. Mol Cell Biol 22:7802–7811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasa M, Brook M, Saklatvala J, Clark AR 2001 Dexamethasone destabilizes cyclooxygenase 2 mRNA by inhibiting mitogen-activated protein kinase p38. Mol Cell Biol 21:771–780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stellato C 2004 Post-transcriptional and non-genomic effects of glucocorticoids. Proc Am Thorac Soc 1:255–263 [DOI] [PubMed] [Google Scholar]
- Chen CY, Del Gatto-Konczak F, Wu Z, Karin M 1998 Stabilization of interleukin-2 mRNA by the c-Jun NH2-terminal kinase pathway. Science 280:1945–1949 [DOI] [PubMed] [Google Scholar]
- Dumitru CD, Ceci JD, Tsatsanis C, Kontoyiannis D, Stamatakis K, Lin JH, Patriotis C, Jenkins NA, Copeland NG, Kollias G, Tsichlis PN 2000 TNF-α induction by LPS is regulated posttranscriptionally via a Tpl2/ERK-dependent pathway. Cell 103:1071–1083 [DOI] [PubMed] [Google Scholar]
- Ming XF, Kaiser M, Moroni C 1998 c-jun N-terminal kinase is involved in AUUUA-mediated interleukin-3 mRNA turnover in mast cells. EMBO J 17:6039–6048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaaf MJ, Cidlowski JA 2003 Molecular determinants of glucocorticoid receptor mobility in living cells: the importance of ligand affinity. Mol Cell Biol 23:1922–1934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price DH 2000 P-TEFb, a cyclin-dependent kinase controlling elongation by RNA polymerase II. Mol Cell Biol 20:2629–2634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barboric M, Nissen RM, Kanazawa S, Jabrane-Ferrat N, Peterlin BM 2001 NF-κB binds P-TEFb to stimulate transcriptional elongation by RNA polymerase II. Mol Cell 8:327–337 [DOI] [PubMed] [Google Scholar]
- Rogatsky I, Trowbridge JM, Garabedian MJ 1997 Glucocorticoid receptor-mediated cell cycle arrest is achieved through distinct cell-specific transcriptional regulatory mechanisms. Mol Cell Biol 17:3181–3193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cha HH, Cram EJ, Wang EC, Huang AJ, Kasler HG, Firestone GL 1998 Glucocorticoids stimulate p21 gene expression by targeting multiple transcriptional elements within a steroid responsive region of the p21waf1/cip1 promoter in rat hepatoma cells. J Biol Chem 273:1998–2007 [DOI] [PubMed] [Google Scholar]
- Cram EJ, Ramos RA, Wang EC, Cha HH, Nishio Y, Firestone GL 1998 Role of the CCAAT/enhancer binding protein-α transcription factor in the glucocorticoid stimulation of p21waf1/cip1 gene promoter activity in growth-arrested rat hepatoma cells. J Biol Chem 273:2008–2014 [DOI] [PubMed] [Google Scholar]
- Greenberg AK, Hu J, Basu S, Hay J, Reibman J, Yie TA, Tchou-Wong KM, Rom WN, Lee TC 2002 Glucocorticoids inhibit lung cancer cell growth through both the extracellular signal-related kinase pathway and cell cycle regulators. Am J Respir Cell Mol Biol 27:320–328 [DOI] [PubMed] [Google Scholar]
- Bhattacharjee RN, Banks GC, Trotter KW, Lee HL, Archer TK 2001 Histone H1 phosphorylation by Cdk2 selectively modulates mouse mammary tumor virus transcription through chromatin remodeling. Mol Cell Biol 21:5417–5425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banks GC, Deterding LJ, Tomer KB, Archer TK 2001 Hormone-mediated dephosphorylation of specific histone H1 isoforms. J Biol Chem 276:36467–36473 [DOI] [PubMed] [Google Scholar]
- Zoja C, Casiraghi F, Conti S, Corna D, Rottoli D, Cavinato RA, Remuzzi G, Benigni A 2007 Cyclin-dependent kinase inhibition limits glomerulonephritis and extends lifespan of mice with systemic lupus. Arthritis Rheum 56:1629–1637 [DOI] [PubMed] [Google Scholar]
- Gloire G, Horion J, El Mjiyad N, Bex F, Chariot A, Dejardin E, Piette J 2007 Promoter-dependent effect of IKKα on NF-κB/p65 DNA binding. J Biol Chem 282:21308–21318 [DOI] [PubMed] [Google Scholar]
- Anest V, Hanson JL, Cogswell PC, Steinbrecher KA, Strahl BD, Baldwin AS 2003 A nucleosomal function for IκB kinase-α in NF-κB-dependent gene expression. Nature 423:659–663 [DOI] [PubMed] [Google Scholar]
- Yamamoto Y, Verma UN, Prajapati S, Kwak YT, Gaynor RB 2003 Histone H3 phosphorylation by IKK-α is critical for cytokine-induced gene expression. Nature 423:655–659 [DOI] [PubMed] [Google Scholar]
- Park GY, Wang X, Hu N, Pedchenko TV, Blackwell TS, Christman JW 2006 NIK is involved in nucleosomal regulation by enhancing histone H3 phosphorylation by IKKα. J Biol Chem 281:18684–18690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Lu Q, Hwang JY, Büscher D, Lee KF, Izpisua-Belmonte JC, Verma IM 1999 IKK1-deficient mice exhibit abnormal development of skin and skeleton. Genes Dev 13:1322–1328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sizemore N, Lerner N, Dombrowski N, Sakurai H, Stark GR 2002 Distinct roles of the Iκ B kinase α and β subunits in liberating nuclear factor κ B (NF-κ B) from Iκ B and in phosphorylating the p65 subunit of NF-κB. J Biol Chem 277:3863–3869 [DOI] [PubMed] [Google Scholar]
- McCoy CE, Carpenter S, Pålsson-McDermott EM, Gearing LJ, O'Neill LA 2008 Glucocorticoids inhibit IRF3 phosphorylation in response to Toll-like receptor-3 and -4 by targeting TBK1 activation. J Biol Chem 283:14277–14285 [DOI] [PubMed] [Google Scholar]
- McWhirter SM, Fitzgerald KA, Rosains J, Rowe DC, Golenbock DT, Maniatis T 2004 IFN-regulatory factor 3-dependent gene expression is defective in Tbk1-deficient mouse embryonic fibroblasts. Proc Natl Acad Sci USA 101:233–238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemmi H, Takeuchi O, Sato S, Yamamoto M, Kaisho T, Sanjo H, Kawai T, Hoshino K, Takeda K, Akira S 2004 The roles of two IκB kinase-related kinases in lipopolysaccharide and double stranded RNA signaling and viral infection. J Exp Med 199:1641–1650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honda K, Taniguchi T 2006 IRFs: master regulators of signalling by Toll-like receptors and cytosolic pattern-recognition receptors. Nat Rev Immunol 6:644–658 [DOI] [PubMed] [Google Scholar]
- Servant MJ, Grandvaux N, tenOever BR, Duguay D, Lin R, Hiscott J 2003 Identification of the minimal phosphoacceptor site required for in vivo activation of interferon regulatory factor 3 in response to virus and double-stranded RNA. J Biol Chem 278:9441–9447 [DOI] [PubMed] [Google Scholar]
- An H, Zhao W, Hou J, Zhang Y, Xie Y, Zheng Y, Xu H, Qian C, Zhou J, Yu Y, Liu S, Feng G, Cao X 2006 SHP-2 phosphatase negatively regulates the TRIF adaptor protein-dependent type I interferon and proinflammatory cytokine production. Immunity 25:919–928 [DOI] [PubMed] [Google Scholar]
- Reily MM, Pantoja C, Hu X, Chinenov Y, Rogatsky I 2006 The GRIP1:IRF3 interaction as a target for glucocorticoid receptor-mediated immunosuppression. EMBO J 25:108–117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buss H, Dörrie A, Schmitz ML, Hoffmann E, Resch K, Kracht M 2004 Constitutive and interleukin-1-inducible phosphorylation of p65 NF-κB at serine 536 is mediated by multiple protein kinases including IκB kinase (IKK)-α, IKKβ, IKKε, TRAF family member-associated (TANK)-binding kinase 1 (TBK1), and an unknown kinase and couples p65 to TATA-binding protein-associated factor II31-mediated interleukin-8 transcription. J Biol Chem 279:55633–55643 [DOI] [PubMed] [Google Scholar]
- Bianchi M, Meng C, Ivashkiv LB 2000 Inhibition of IL-2-induced Jak-STAT signaling by glucocorticoids. Proc Natl Acad Sci USA 97:9573–9578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franchimont D, Galon J, Gadina M, Visconti R, Zhou Y, Aringer M, Frucht DM, Chrousos GP, O'Shea JJ 2000 Inhibition of Th1 immune response by glucocorticoids: dexamethasone selectively inhibits IL-12-induced Stat4 phosphorylation in T lymphocytes. J Immunol 164:1768–1774 [DOI] [PubMed] [Google Scholar]
- Fahey AJ, Robins RA, Kindle KB, Heery DM, Constantinescu CS 2006 Effects of glucocorticoids on STAT4 activation in human T cells are stimulus-dependent. J Leukoc Biol 80:133–144 [DOI] [PubMed] [Google Scholar]
- Dempsey EC, Cool CD, Littler CM 2007 Lung disease and PKCs. Pharmacol Res 55:545–559 [DOI] [PubMed] [Google Scholar]
- Catley MC, Cambridge LM, Nasuhara Y, Ito K, Chivers JE, Beaton A, Holden NS, Bergmann MW, Barnes PJ, Newton R 2004 Inhibitors of protein kinase C (PKC) prevent activated transcription: role of events downstream of NF-κB DNA binding. J Biol Chem 279:18457–18466 [DOI] [PubMed] [Google Scholar]
- Page K, Li J, Zhou L, Iasvovskaia S, Corbit KC, Soh JW, Weinstein IB, Brasier AR, Lin A, Hershenson MB, Iasvoyskaia S 2003 Regulation of airway epithelial cell NF-κB-dependent gene expression by protein kinase C δ. J Immunol 170:5681–5689 [DOI] [PubMed] [Google Scholar]
- Aras-López R, Xavier FE, Ferrer M, Balfagón G 2009 Dexamethasone decreases neuronal nitric oxide release in mesenteric arteries from hypertensive rats through decreased protein kinase C activation. Clin Sci (Lond) 117:305–312 [DOI] [PubMed] [Google Scholar]
- Nguyen CH, Watts VJ 2006 Dexamethasone-induced Ras protein 1 negatively regulates protein kinase Cδ: implications for adenylyl cyclase 2 signaling. Mol Pharmacol 69:1763–1771 [DOI] [PubMed] [Google Scholar]
- Matthews L, Berry A, Ohanian V, Ohanian J, Garside H, Ray D 2008 Caveolin mediates rapid glucocorticoid effects and couples glucocorticoid action to the antiproliferative program. Mol Endocrinol 22:1320–1330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Failor KL, Desyatnikov Y, Finger LA, Firestone GL 2007 Glucocorticoid-induced degradation of glycogen synthase kinase-3 protein is triggered by serum- and glucocorticoid-induced protein kinase and Akt signaling and controls β-catenin dynamics and tight junction formation in mammary epithelial tumor cells. Mol Endocrinol 21:2403–2415 [DOI] [PubMed] [Google Scholar]
- Hafezi-Moghadam A, Simoncini T, Yang Z, Limbourg FP, Plumier JC, Rebsamen MC, Hsieh CM, Chui DS, Thomas KL, Prorock AJ, Laubach VE, Moskowitz MA, French BA, Ley K, Liao JK 2002 Acute cardiovascular protective effects of corticosteroids are mediated by non-transcriptional activation of endothelial nitric oxide synthase. Nat Med 8:473–479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Limbourg FP, Huang Z, Plumier JC, Simoncini T, Fujioka M, Tuckermann J, Schütz G, Moskowitz MA, Liao JK 2002 Rapid nontranscriptional activation of endothelial nitric oxide synthase mediates increased cerebral blood flow and stroke protection by corticosteroids. J Clin Invest 110:1729–1738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langdown ML, Holness MJ, Sugden MC 2001 Early growth retardation induced by excessive exposure to glucocorticoids in utero selectively increases cardiac GLUT1 protein expression and Akt/protein kinase B activity in adulthood. J Endocrinol 169:11–22 [DOI] [PubMed] [Google Scholar]
- Limbourg FP, Liao JK 2003 Nontranscriptional actions of the glucocorticoid receptor. J Mol Med 81:168–174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith E, Frenkel B 2005 Glucocorticoids inhibit the transcriptional activity of LEF/TCF in differentiating osteoblasts in a glycogen synthase kinase-3β-dependent and -independent manner. J Biol Chem 280:2388–2394 [DOI] [PubMed] [Google Scholar]
- Wu W, Zou M, Brickley DR, Pew T, Conzen SD 2006 Glucocorticoid receptor activation signals through forkhead transcription factor 3a in breast cancer cells. Mol Endocrinol 20:2304–2314 [DOI] [PubMed] [Google Scholar]
- Itani OA, Liu KZ, Cornish KL, Campbell JR, Thomas CP 2002 Glucocorticoids stimulate human sgk1 gene expression by activation of a GRE in its 5′-flanking region. Am J Physiol Endocrinol Metab 283:E971–E979 [DOI] [PubMed] [Google Scholar]
- Mikosz CA, Brickley DR, Sharkey MS, Moran TW, Conzen SD 2001 Glucocorticoid receptor-mediated protection from apoptosis is associated with induction of the serine/threonine survival kinase gene, sgk-1. J Biol Chem 276:16649–16654 [DOI] [PubMed] [Google Scholar]
- Zhang L, Cui R, Cheng X, Du J 2005 Antiapoptotic effect of serum and glucocorticoid-inducible protein kinase is mediated by novel mechanism activating IκB kinase. Cancer Res 65:457–464 [PubMed] [Google Scholar]
- Tai DJ, Su CC, Ma YL, Lee EH 2009 SGK1 phosphorylation of IκB kinase α and p300 up-regulates NF-κB activity and increases N-methyl-D-aspartate receptor NR2A and NR2B expression. J Biol Chem 284:4073–4089 [DOI] [PubMed] [Google Scholar]
- Abu-Amer Y, Ross FP, McHugh KP, Livolsi A, Peyron JF, Teitelbaum SL 1998 Tumor necrosis factor-α activation of nuclear transcription factor-κB in marrow macrophages is mediated by c-Src tyrosine phosphorylation of Iκ Bα. J Biol Chem 273:29417–29423 [DOI] [PubMed] [Google Scholar]
- Wong WS 2005 Inhibitors of the tyrosine kinase signaling cascade for asthma. Curr Opin Pharmacol 5:264–271 [DOI] [PubMed] [Google Scholar]
- Löwenberg M, Tuynman J, Bilderbeek J, Gaber T, Buttgereit F, van Deventer S, Peppelenbosch M, Hommes D 2005 Rapid immunosuppressive effects of glucocorticoids mediated through Lck and Fyn. Blood 106:1703–1710 [DOI] [PubMed] [Google Scholar]
- Löwenberg M, Tuynman J, Scheffer M, Verhaar A, Vermeulen L, van Deventer S, Hommes D, Peppelenbosch M 2006 Kinome analysis reveals non-genomic glucocorticoid receptor-dependent inhibition of insulin signaling. Endocrinology 147:3555–3562 [DOI] [PubMed] [Google Scholar]
- Löwenberg M, Verhaar AP, Bilderbeek J, Marle J, Buttgereit F, Peppelenbosch MP, van Deventer SJ, Hommes DW 2006 Glucocorticoids cause rapid dissociation of a T-cell-receptor-associated protein complex containing LCK and FYN. EMBO Rep 7:1023–1029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh MC, Baatar D, Collins G, Carter A, Indig F, Biragyn A, Taub DD 2009 Dexamethasone augments CXCR4-mediated signaling in resting human T cells via the activation of the Src kinase Lck. Blood 113:575–584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubenstein NM, Callahan JA, Lo DH, Firestone GL 2007 Selective glucocorticoid control of Rho kinase isoforms regulate cell-cell interactions. Biochem Biophys Res Commun 354:603–607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mong PY, Wang Q 2009 Activation of Rho kinase isoforms in lung endothelial cells during inflammation. J Immunol 182:2385–2394 [DOI] [PubMed] [Google Scholar]
- Goto K, Chiba Y, Sakai H, Misawa M 2008 Glucocorticoids inhibited airway hyperresponsiveness through downregulation of CPI-17 in bronchial smooth muscle. Eur J Pharmacol 591:231–236 [DOI] [PubMed] [Google Scholar]
- Barford D 1996 Molecular mechanisms of the protein serine/threonine phosphatases. Trends Biochem Sci 21:407–412 [DOI] [PubMed] [Google Scholar]
- Camps M, Nichols A, Arkinstall S 2000 Dual specificity phosphatases: a gene family for control of MAP kinase function. FASEB J 14:6–16 [PubMed] [Google Scholar]
- Abraham SM, Clark AR 2006 Dual-specificity phosphatase 1: a critical regulator of innate immune responses. Biochem Soc Trans 34:1018–1023 [DOI] [PubMed] [Google Scholar]
- Turjanski AG, Vaqué JP, Gutkind JS 2007 MAP kinases and the control of nuclear events. Oncogene 26:3240-3253 [DOI] [PubMed] [Google Scholar]
- Dickinson RJ, Keyse SM 2006 Diverse physiological functions for dual-specificity MAP kinase phosphatases. J Cell Sci 119:4607–4615 [DOI] [PubMed] [Google Scholar]
- Kassel O, Sancono A, Krätzschmar J, Kreft B, Stassen M, Cato AC 2001 Glucocorticoids inhibit MAP kinase via increased expression and decreased degradation of MKP-1. EMBO J 20:7108–7116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams M, Meijer OC, Wang J, Bhargava A, Pearce D 2003 Homodimerization of the glucocorticoid receptor is not essential for response element binding: activation of the phenylethanolamine N-methyltransferase gene by dimerization-defective mutants. Mol Endocrinol 17:2583–2592 [DOI] [PubMed] [Google Scholar]
- Iñiguez-Lluhí JA, Pearce D 2000 A common motif within the negative regulatory regions of multiple factors inhibits their transcriptional synergy. Mol Cell Biol 20:6040-6050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helmberg A, Auphan N, Caelles C, Karin M 1995 Glucocorticoid-induced apoptosis of human leukemic cells is caused by the repressive function of the glucocorticoid receptor. EMBO J 14:452–460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansson-Haque K, Palanichamy E, Okret S 2008 Stimulation of MAPK-phosphatase 1 gene expression by glucocorticoids occurs through a tethering mechanism involving C/EBP. J Mol Endocrinol 41:239–249 [DOI] [PubMed] [Google Scholar]
- Fürst R, Zahler S, Vollmar AM 2008 Dexamethasone-induced expression of endothelial mitogen-activated protein kinase phosphatase-1 involves activation of the transcription factors activator protein-1 and 3′,5′-cyclic adenosine 5′-monophosphate response element-binding protein and the generation of reactive oxygen species. Endocrinology 149:3635–3642 [DOI] [PubMed] [Google Scholar]
- Berg T, Didon L, Barton J, Andersson O, Nord M 2005 Glucocorticoids increase C/EBPβ activity in the lung epithelium via phosphorylation. Biochem Biophys Res Commun 334:638–645 [DOI] [PubMed] [Google Scholar]
- Bazuine M, Carlotti F, Tafrechi RS, Hoeben RC, Maassen JA 2004 Mitogen-activated protein kinase (MAPK) phosphatase-1 and -4 attenuate p38 MAPK during dexamethasone-induced insulin resistance in 3T3–L1 adipocytes. Mol Endocrinol 18:1697–1707 [DOI] [PubMed] [Google Scholar]
- Yoshida NL, Miyashita T, U M, Yamada M, Reed JC, Sugita Y, Oshida T 2002 Analysis of gene expression patterns during glucocorticoid-induced apoptosis using oligonucleotide arrays. Biochem Biophys Res Commun 293:1254–1261 [DOI] [PubMed] [Google Scholar]
- Sánchez-Tilló E, Comalada M, Xaus J, Farrera C, Valledor AF, Caelles C, Lloberas J, Celada A 2007 JNK1 is required for the induction of Mkp1 expression in macrophages during proliferation and lipopolysaccharide-dependent activation. J Biol Chem 282:12566–12573 [DOI] [PubMed] [Google Scholar]
- Ananieva O, Darragh J, Johansen C, Carr JM, McIlrath J, Park JM, Wingate A, Monk CE, Toth R, Santos SG, Iversen L, Arthur JS 2008 The kinases MSK1 and MSK2 act as negative regulators of Toll-like receptor signaling. Nat Immunol 9:1028–1036 [DOI] [PubMed] [Google Scholar]
- Hammer M, Mages J, Dietrich H, Schmitz F, Striebel F, Murray PJ, Wagner H, Lang R 2005 Control of dual-specificity phosphatase-1 expression in activated macrophages by IL-10. Eur J Immunol 35:2991–3001 [DOI] [PubMed] [Google Scholar]
- Cho IJ, Kim SG 2009 A novel mitogen-activated protein kinase phosphatase-1 and glucocorticoid receptor (GR) interacting protein-1-dependent combinatorial mechanism of gene transrepression by GR. Mol Endocrinol 23:86–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chauhan S, Leach CH, Kunz S, Bloom JW, Miesfeld RL 2003 Glucocorticoid regulation of human eosinophil gene expression. J Steroid Biochem Mol Biol 84:441–452 [DOI] [PubMed] [Google Scholar]
- Schmidt S, Rainer J, Riml S, Ploner C, Jesacher S, Achmüller C, Presul E, Skvortsov S, Crazzolara R, Fiegl M, Raivio T, Jänne OA, Geley S, Meister B, Kofler R 2006 Identification of glucocorticoid-response genes in children with acute lymphoblastic leukemia. Blood 107:2061–2069 [DOI] [PubMed] [Google Scholar]
- Haffner MC, Jurgeit A, Berlato C, Geley S, Parajuli N, Yoshimura A, Doppler W 2008 Interaction and functional interference of glucocorticoid receptor and SOCS1. J Biol Chem 283:22089–22096 [DOI] [PubMed] [Google Scholar]
- Mansell A, Smith R, Doyle SL, Gray P, Fenner JE, Crack PJ, Nicholson SE, Hilton DJ, O'Neill LA, Hertzog PJ 2006 Suppressor of cytokine signaling 1 negatively regulates Toll-like receptor signaling by mediating Mal degradation. Nat Immunol 7:148–155 [DOI] [PubMed] [Google Scholar]
- Wang X, Liu Y 2007 Regulation of innate immune response by MAP kinase phosphatase-1. Cell Signal 19:1372–1382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi H, Barry SP, Roth RJ, Wu JJ, Jones EA, Bennett AM, Flavell RA 2006 Dynamic regulation of pro- and anti-inflammatory cytokines by MAPK phosphatase 1 (MKP-1) in innate immune responses. Proc Natl Acad Sci USA 103:2274–2279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammer M, Mages J, Dietrich H, Servatius A, Howells N, Cato AC, Lang R 2006 Dual specificity phosphatase 1 (DUSP1) regulates a subset of LPS-induced genes and protects mice from lethal endotoxin shock. J Exp Med 203:15–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salojin KV, Owusu IB, Millerchip KA, Potter M, Platt KA, Oravecz T 2006 Essential role of MAPK phosphatase-1 in the negative control of innate immune responses. J Immunol 176:1899–1907 [DOI] [PubMed] [Google Scholar]
- Zhao Q, Wang X, Nelin LD, Yao Y, Matta R, Manson ME, Baliga RS, Meng X, Smith CV, Bauer JA, Chang CH, Liu Y 2006 MAP kinase phosphatase 1 controls innate immune responses and suppresses endotoxic shock. J Exp Med 203:131–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brondello JM, Pouysségur J, McKenzie FR 1999 Reduced MAP kinase phosphatase-1 degradation after p42/p44MAPK-dependent phosphorylation. Science 286:2514–2517 [DOI] [PubMed] [Google Scholar]
- Lin YW, Chuang SM, Yang JL 2003 ERK1/2 achieves sustained activation by stimulating MAPK phosphatase-1 degradation via the ubiquitin-proteasome pathway. J Biol Chem 278:21534–21541 [DOI] [PubMed] [Google Scholar]
- Lin YW, Yang JL 2006 Cooperation of ERK and SCFSkp2 for MKP-1 destruction provides a positive feedback regulation of proliferating signaling. J Biol Chem 281:915–926 [DOI] [PubMed] [Google Scholar]
- Cao W, Bao C, Padalko E, Lowenstein CJ 2008 Acetylation of mitogen-activated protein kinase phosphatase-1 inhibits Toll-like receptor signaling. J Exp Med 205:1491–1503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi H, Flavell RA 2008 Acetylation of MKP-1 and the control of inflammation. Sci Signal 1:pe44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelegrí C, Franch A, Castellote C, Castell M 1995 Immunohistochemical changes in synovial tissue during the course of adjuvant arthritis. J Rheumatol 22:124–132 [PubMed] [Google Scholar]
- Nielsen H, Petersen AA, Skjødt H, Hørslev-Petersen K, Bendtzen K 1999 Blood levels of CD11b+ memory T lymphocytes are selectively upregulated in patients with active rheumatoid arthritis. APMIS 107:1124–1130 [DOI] [PubMed] [Google Scholar]
- Cambillau C, Rauly I, Sarfati P, Saint-Laurent N, Esteve JP, Fanjul M, Svoboda M, Prats H, Hollande E, Vaysse N 1995 Regulation of the src homology 2 domain-containing protein tyrosine phosphatase PTP1C by glucocorticoids in rat pancreatic AR42J cells. Endocrinology 136:5476–5484 [DOI] [PubMed] [Google Scholar]
- Pani G, Fischer KD, Mlinaric-Rascan I, Siminovitch KA 1996 Signaling capacity of the T cell antigen receptor is negatively regulated by the PTP1C tyrosine phosphatase. J Exp Med 184:839–852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croker BA, Lawson BR, Rutschmann S, Berger M, Eidenschenk C, Blasius AL, Moresco EM, Sovath S, Cengia L, Shultz LD, Theofilopoulos AN, Pettersson S, Beutler BA 2008 Inflammation and autoimmunity caused by a SHP1 mutation depend on IL-1, MyD88, and a microbial trigger. Proc Natl Acad Sci USA 105:15028–15033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Virshup DM, Shenolikar S 2009 From promiscuity to precision: protein phosphatases get a makeover. Mol Cell 33:537–545 [DOI] [PubMed] [Google Scholar]
- Boudreau RT, Hoskin DW, Lin TJ 2004 Phosphatase inhibition potentiates IL-6 production by mast cells in response to FcεRI-mediated activation: involvement of p38 MAPK. J Leukoc Biol 76:1075–1081 [DOI] [PubMed] [Google Scholar]
- Kins S, Kurosinski P, Nitsch RM, Götz J 2003 Activation of the ERK and JNK signaling pathways caused by neuron-specific inhibition of PP2A in transgenic mice. Am J Pathol 163:833–843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millward TA, Zolnierowicz S, Hemmings BA 1999 Regulation of protein kinase cascades by protein phosphatase 2A. Trends Biochem Sci 24:186–191 [DOI] [PubMed] [Google Scholar]
- Silverstein AM, Barrow CA, Davis AJ, Mumby MC 2002 Actions of PP2A on the MAP kinase pathway and apoptosis are mediated by distinct regulatory subunits. Proc Natl Acad Sci USA 99:4221–4226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonoda Y, Kasahara T, Yamaguchi Y, Kuno K, Matsushima K, Mukaida N 1997 Stimulation of interleukin-8 production by okadaic acid and vanadate in a human promyelocyte cell line, an HL-60 subline. Possible role of mitogen-activated protein kinase on the okadaic acid-induced NF-κB activation. J Biol Chem 272:15366–15372 [DOI] [PubMed] [Google Scholar]
- Shanley TP, Vasi N, Denenberg A, Wong HR 2001 The serine/threonine phosphatase, PP2A: endogenous regulator of inflammatory cell signaling. J Immunol 166:966–972 [DOI] [PubMed] [Google Scholar]
- Amaral MC, Casillas AM, Nel AE 1993 Contrasting effects of two tumour promoters, phorbol myristate acetate and okadaic acid, on T-cell responses and activation of p42 MAP-kinase/ERK-2. Immunology 79:24–31 [PMC free article] [PubMed] [Google Scholar]
- Nowak SJ, Pai CY, Corces VG 2003 Protein phosphatase 2A activity affects histone H3 phosphorylation and transcription in Drosophila melanogaster. Mol Cell Biol 23:6129–6138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang YJ, Kusler B, Otsuka M, Hughes M, Suzuki N, Suzuki S, Yeh WC, Akira S, Han J, Jones PP 2007 Calcineurin negatively regulates TLR-mediated activation pathways. J Immunol 179:4598–4607 [DOI] [PubMed] [Google Scholar]
- Braun WE 2003 Renal transplantation: basic concepts and evolution of therapy. J Clin Apher 18:141–152 [DOI] [PubMed] [Google Scholar]
- Kitahara K, Kawai S 2007 Cyclosporine and tacrolimus for the treatment of rheumatoid arthritis. Curr Opin Rheumatol 19:238–245 [DOI] [PubMed] [Google Scholar]
- Tong DC, Buck SM, Roberts BR, Klein JD, Tumlin JA 2004 Calcineurin phosphatase activity: activation by glucocorticoids and role of intracellular calcium. Transplantation 77:259–267 [DOI] [PubMed] [Google Scholar]
- Ranta F, Avram D, Berchtold S, Düfer M, Drews G, Lang F, Ullrich S 2006 Dexamethasone induces cell death in insulin-secreting cells, an effect reversed by exendin-4. Diabetes 55:1380–1390 [DOI] [PubMed] [Google Scholar]
- Ranta F, Düfer M, Stork B, Wesselborg S, Drews G, Häring HU, Lang F, Ullrich S 2008 Regulation of calcineurin activity in insulin-secreting cells: stimulation by Hsp90 during glucocorticoid-induced apoptosis. Cell Signal 20:1780–1786 [DOI] [PubMed] [Google Scholar]
- Lumbers ER, Boyce AC, Joulianos G, Kumarasamy V, Barner E, Segar JL, Burrell JH 2005 Effects of cortisol on cardiac myocytes and on expression of cardiac genes in fetal sheep. Am J Physiol Regul Integr Comp Physiol 288:R567–R574 [DOI] [PubMed] [Google Scholar]
- McDonough AK, Curtis JR, Saag KG 2008 The epidemiology of glucocorticoid-associated adverse events. Curr Opin Rheumatol 20:131–137 [DOI] [PubMed] [Google Scholar]
- Schäcke H, Döcke WD, Asadullah K 2002 Mechanisms involved in the side effects of glucocorticoids. Pharmacol Ther 96:23–43 [DOI] [PubMed] [Google Scholar]
- Vega D, Maalouf NM, Sakhaee K 2007 Clinical Review: the role of receptor activator of nuclear factor-κB (RANK)/RANK ligand/osteoprotegerin: clinical implications. J Clin Endocrinol Metab 92:4514–4521 [DOI] [PubMed] [Google Scholar]
- Cooper MS 2004 Sensitivity of bone to glucocorticoids. Clin Sci (Lond) 107:111–123 [DOI] [PubMed] [Google Scholar]
- Li H, Cuartas E, Cui W, Choi Y, Crawford TD, Ke HZ, Kobayashi KS, Flavell RA, Vignery A 2005 IL-1 receptor-associated kinase M is a central regulator of osteoclast differentiation and activation. J Exp Med 201:1169–1177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soares-Schanoski A, Gómez-Piña V, del Fresno C, Rodríguez-Rojas A, García F, Glaría A, Sánchez M, Vallejo-Cremades MT, Baos R, Fuentes-Prior P, Arnalich F, López-Collazo E 2007 6-Methylprednisolone down-regulates IRAK-M in human and murine osteoclasts and boosts bone-resorbing activity: a putative mechanism for corticoid-induced osteoporosis. J Leukoc Biol 82:700–709 [DOI] [PubMed] [Google Scholar]
- Horsch K, de Wet H, Schuurmans MM, Allie-Reid F, Cato AC, Cunningham J, Burrin JM, Hough FS, Hulley PA 2007 Mitogen-activated protein kinase phosphatase 1/dual specificity phosphatase 1 mediates glucocorticoid inhibition of osteoblast proliferation. Mol Endocrinol 21:2929–2940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelbrecht Y, de Wet H, Horsch K, Langeveldt CR, Hough FS, Hulley PA 2003 Glucocorticoids induce rapid up-regulation of mitogen-activated protein kinase phosphatase-1 and dephosphorylation of extracellular signal-regulated kinase and impair proliferation in human and mouse osteoblast cell lines. Endocrinology 144:412–422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conradie MM, de Wet H, Kotze DD, Burrin JM, Hough FS, Hulley PA 2007 Vanadate prevents glucocorticoid-induced apoptosis of osteoblasts in vitro and osteocytes in vivo. J Endocrinol 195:229–240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulley PA, Conradie MM, Langeveldt CR, Hough FS 2002 Glucocorticoid-induced osteoporosis in the rat is prevented by the tyrosine phosphatase inhibitor, sodium orthovanadate. Bone 31:220–229 [DOI] [PubMed] [Google Scholar]
- Yun SI, Yoon HY, Jeong SY, Chung YS 2009 Glucocorticoid induces apoptosis of osteoblast cells through the activation of glycogen synthase kinase 3β. J Bone Miner Metab 27:140–148 [DOI] [PubMed] [Google Scholar]
- Plotkin LI, Manolagas SC, Bellido T 2007 Glucocorticoids induce osteocyte apoptosis by blocking focal adhesion kinase-mediated survival. Evidence for inside-out signaling leading to anoikis. J Biol Chem 282:24120–24130 [DOI] [PubMed] [Google Scholar]
- Chauhan D, Hideshima T, Pandey P, Treon S, Teoh G, Raje N, Rosen S, Krett N, Husson H, Avraham S, Kharbanda S, Anderson KC 1999 RAFTK/PYK2-dependent and -independent apoptosis in multiple myeloma cells. Oncogene 18:6733–6740 [DOI] [PubMed] [Google Scholar]
- Tokiwa G, Dikic I, Lev S, Schlessinger J 1996 Activation of Pyk2 by stress signals and coupling with JNK signaling pathway. Science 273:792–794 [DOI] [PubMed] [Google Scholar]
- Meyer T, Carlstedt-Duke J, Starr DB 1997 A weak TATA box is a prerequisite for glucocorticoid-dependent repression of the osteocalcin gene. J Biol Chem 272:30709–30714 [DOI] [PubMed] [Google Scholar]
- Chrysis D, Zaman F, Chagin AS, Takigawa M, Sävendahl L 2005 Dexamethasone induces apoptosis in proliferative chondrocytes through activation of caspases and suppression of the Akt-phosphatidylinositol 3′-kinase signaling pathway. Endocrinology 146:1391–1397 [DOI] [PubMed] [Google Scholar]
- Schakman O, Gilson H, Thissen JP 2008 Mechanisms of glucocorticoid-induced myopathy. J Endocrinol 197:1–10 [DOI] [PubMed] [Google Scholar]
- Wang H, Kubica N, Ellisen LW, Jefferson LS, Kimball SR 2006 Dexamethasone represses signaling through the mammalian target of rapamycin in muscle cells by enhancing expression of REDD1. J Biol Chem 281:39128–39134 [DOI] [PubMed] [Google Scholar]
- Schakman O, Kalista S, Bertrand L, Lause P, Verniers J, Ketelslegers JM, Thissen JP 2008 Role of Akt/GSK-3β/β-catenin transduction pathway in the muscle anti-atrophy action of insulin-like growth factor-I in glucocorticoid-treated rats. Endocrinology 149:3900–3908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman PA, Medina-Martinez O, Fernandez-Mejia C 1996 Identification of the human insulin negative regulatory element as a negative glucocorticoid response element. Mol Cell Endocrinol 120:139–146 [DOI] [PubMed] [Google Scholar]
- Yoshiuchi I, Shingu R, Nakajima H, Hamaguchi T, Horikawa Y, Yamasaki T, Oue T, Ono A, Miyagawa JI, Namba M, Hanafusa T, Matsuzawa Y 1998 Mutation/polymorphism scanning of glucose-6-phosphatase gene promoter in noninsulin-dependent diabetes mellitus patients. J Clin Endocrinol Metab 83:1016–1019 [DOI] [PubMed] [Google Scholar]
- Kwon HS, Huang B, Unterman TG, Harris RA 2004 Protein kinase B-α inhibits human pyruvate dehydrogenase kinase-4 gene induction by dexamethasone through inactivation of FOXO transcription factors. Diabetes 53:899–910 [DOI] [PubMed] [Google Scholar]
- Nakae J, Kitamura T, Silver DL, Accili D 2001 The forkhead transcription factor Foxo1 (Fkhr) confers insulin sensitivity onto glucose-6-phosphatase expression. J Clin Invest 108:1359–1367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rigaud G, Roux J, Pictet R, Grange T 1991 In vivo footprinting of rat TAT gene: dynamic interplay between the glucocorticoid receptor and a liver-specific factor. Cell 67:977–986 [DOI] [PubMed] [Google Scholar]
- Arizmendi C, Liu S, Croniger C, Poli V, Friedman JE 1999 The transcription factor CCAAT/enhancer-binding protein β regulates gluconeogenesis and phosphoenolpyruvate carboxykinase (GTP) gene transcription during diabetes. J Biol Chem 274:13033–13040 [DOI] [PubMed] [Google Scholar]
- Crosson SM, Roesler WJ 2000 Hormonal regulation of the phosphoenolpyruvate carboxykinase gene. Role of specific CCAAT/enhancer-binding protein isoforms. J Biol Chem 275:5804–5809 [DOI] [PubMed] [Google Scholar]
- Yamada K, Duong DT, Scott DK, Wang JC, Granner DK 1999 CCAAT/enhancer-binding protein β is an accessory factor for the glucocorticoid response from the cAMP response element in the rat phosphoenolpyruvate carboxykinase gene promoter. J Biol Chem 274:5880–5887 [DOI] [PubMed] [Google Scholar]
- Itani OA, Auerbach SD, Husted RF, Volk KA, Ageloff S, Knepper MA, Stokes JB, Thomas CP 2002 Glucocorticoid-stimulated lung epithelial Na(+) transport is associated with regulated ENaC and sgk1 expression. Am J Physiol Lung Cell Mol Physiol 282:L631–L641 [DOI] [PubMed] [Google Scholar]
- Sayegh R, Auerbach SD, Li X, Loftus RW, Husted RF, Stokes JB, Thomas CP 1999 Glucocorticoid induction of epithelial sodium channel expression in lung and renal epithelia occurs via trans-activation of a hormone response element in the 5′-flanking region of the human epithelial sodium channel α subunit gene. J Biol Chem 274:12431–12437 [DOI] [PubMed] [Google Scholar]
- Brennan FE, Fuller PJ 2000 Rapid upregulation of serum and glucocorticoid-regulated kinase (sgk) gene expression by corticosteroids in vivo. Mol Cell Endocrinol 166:129–136 [DOI] [PubMed] [Google Scholar]
- Chen SY, Bhargava A, Mastroberardino L, Meijer OC, Wang J, Buse P, Firestone GL, Verrey F, Pearce D 1999 Epithelial sodium channel regulated by aldosterone-induced protein sgk. Proc Natl Acad Sci USA 96:2514–2519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyd C, Náray-Fejes-Tóth A 2007 Steroid-mediated regulation of the epithelial sodium channel subunits in mammary epithelial cells. Endocrinology 148:3958–3967 [DOI] [PubMed] [Google Scholar]
- Vallon V, Lang F 2005 New insights into the role of serum- and glucocorticoid-inducible kinase SGK1 in the regulation of renal function and blood pressure. Curr Opin Nephrol Hypertens 14:59–66 [DOI] [PubMed] [Google Scholar]
- Choi JY, Kim SY, Son EJ, Kim JL, Shin JH, Song MH, Moon UY, Yoon JH 2006 Dexamethasone increases fluid absorption via Na+/H+ exchanger (NHE) 3 activation in normal human middle ear epithelial cells. Eur J Pharmacol 536:12–18 [DOI] [PubMed] [Google Scholar]
- Wang D, Zhang H, Lang F, Yun CC 2007 Acute activation of NHE3 by dexamethasone correlates with activation of SGK1 and requires a functional glucocorticoid receptor. Am J Physiol Cell Physiol 292:C396–C404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D, Sun H, Lang F, Yun CC 2005 Activation of NHE3 by dexamethasone requires phosphorylation of NHE3 at Ser663 by SGK1. Am J Physiol Cell Physiol 289:C802–C810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Xia X, Reisenauer MR, Rieg T, Lang F, Kuhl D, Vallon V, Kone BC 2007 Aldosterone-induced Sgk1 relieves Dot1a-Af9-mediated transcriptional repression of epithelial Na+ channel α. J Clin Invest 117:773–783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debonneville C, Flores SY, Kamynina E, Plant PJ, Tauxe C, Thomas MA, Münster C, Chraïbi A, Pratt JH, Horisberger JD, Pearce D, Loffing J, Staub O 2001 Phosphorylation of Nedd4-2 by Sgk1 regulates epithelial Na(+) channel cell surface expression. EMBO J 20:7052–7059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder PM, Olson DR, Thomas BC 2002 Serum and glucocorticoid-regulated kinase modulates Nedd4-2-mediated inhibition of the epithelial Na+ channel. J Biol Chem 277:5–8 [DOI] [PubMed] [Google Scholar]
- Ichimura T, Yamamura H, Sasamoto K, Tominaga Y, Taoka M, Kakiuchi K, Shinkawa T, Takahashi N, Shimada S, Isobe T 2005 14-3-3 proteins modulate the expression of epithelial Na+ channels by phosphorylation-dependent interaction with Nedd4-2 ubiquitin ligase. J Biol Chem 280:13187–13194 [DOI] [PubMed] [Google Scholar]
- Nagaki K, Yamamura H, Shimada S, Saito T, Hisanaga S, Taoka M, Isobe T, Ichimura T 2006 14-3-3 Mediates phosphorylation-dependent inhibition of the interaction between the ubiquitin E3 ligase Nedd4-2 and epithelial Na+ channels. Biochemistry 45:6733–6740 [DOI] [PubMed] [Google Scholar]
- Diakov A, Korbmacher C 2004 A novel pathway of epithelial sodium channel activation involves a serum- and glucocorticoid-inducible kinase consensus motif in the C terminus of the channel's α-subunit. J Biol Chem 279:38134–38142 [DOI] [PubMed] [Google Scholar]
- Dieter M, Palmada M, Rajamanickam J, Aydin A, Busjahn A, Boehmer C, Luft FC, Lang F 2004 Regulation of glucose transporter SGLT1 by ubiquitin ligase Nedd4-2 and kinases SGK1, SGK3, and PKB. Obes Res 12:862–870 [DOI] [PubMed] [Google Scholar]
- Lang PA, Schniepp R, Kirchhoff P, Socrates T, Sidani SM, Geibel JP 2007 PI3 kinase dependent stimulation of gastric acid secretion by dexamethasone. Cell Physiol Biochem 20:527–534 [DOI] [PubMed] [Google Scholar]
- Gupta V, Awasthi N, Wagner BJ 2007 Specific activation of the glucocorticoid receptor and modulation of signal transduction pathways in human lens epithelial cells. Invest Ophthalmol Vis Sci 48:1724–1734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webster JC, Oakley RH, Jewell CM, Cidlowski JA 2001 Proinflammatory cytokines regulate human glucocorticoid receptor gene expression and lead to the accumulation of the dominant negative β isoform: a mechanism for the generation of glucocorticoid resistance. Proc Natl Acad Sci USA 98:6865–6870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sousa AR, Lane SJ, Cidlowski JA, Staynov DZ, Lee TH 2000 Glucocorticoid resistance in asthma is associated with elevated in vivo expression of the glucocorticoid receptor β-isoform. J Allergy Clin Immunol 105:943–950 [DOI] [PubMed] [Google Scholar]
- Adcock IM, Lane SJ, Brown CR, Lee TH, Barnes PJ 1995 Abnormal glucocorticoid receptor-activator protein 1 interaction in steroid-resistant asthma. J Exp Med 182:1951–1958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adcock IM, Lane SJ, Brown CR, Peters MJ, Lee TH, Barnes PJ 1995 Differences in binding of glucocorticoid receptor to DNA in steroid-resistant asthma. J Immunol 154:3500–3505 [PubMed] [Google Scholar]
- Matthews JG, Ito K, Barnes PJ, Adcock IM 2004 Defective glucocorticoid receptor nuclear translocation and altered histone acetylation patterns in glucocorticoid-resistant patients. J Allergy Clin Immunol 113:1100–1108 [DOI] [PubMed] [Google Scholar]
- Woodruff PG, Boushey HA, Dolganov GM, Barker CS, Yang YH, Donnelly S, Ellwanger A, Sidhu SS, Dao-Pick TP, Pantoja C, Erle DJ, Yamamoto KR, Fahy JV 2007 Genome-wide profiling identifies epithelial cell genes associated with asthma and with treatment response to corticosteroids. Proc Natl Acad Sci USA 104:15858–15863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilodeau S, Vallette-Kasic S, Gauthier Y, Figarella-Branger D, Brue T, Berthelet F, Lacroix A, Batista D, Stratakis C, Hanson J, Meij B, Drouin J 2006 Role of Brg1 and HDAC2 in GR trans-repression of the pituitary POMC gene and misexpression in Cushing disease. Genes Dev 20:2871–2886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito K, Herbert C, Siegle JS, Vuppusetty C, Hansbro N, Thomas PS, Foster PS, Barnes PJ, Kumar RK 2008 Steroid-resistant neutrophilic inflammation in a mouse model of an acute exacerbation of asthma. Am J Respir Cell Mol Biol 39:543–550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marwick JA, Caramori G, Stevenson CS, Casolari P, Jazrawi E, Barnes PJ, Ito K, Adcock IM, Kirkham PA, Papi A 2009 Inhibition of PI3Kδ restores glucocorticoid function in smoking-induced airway inflammation in mice. Am J Respir Crit Care Med 179:542–548 [DOI] [PubMed] [Google Scholar]
- Pariante CM 2008 The role of multi-drug resistance p-glycoprotein in glucocorticoid function: studies in animals and relevance in humans. Eur J Pharmacol 583:263–271 [DOI] [PubMed] [Google Scholar]
- Pace TW, Hu F, Miller AH 2007 Cytokine-effects on glucocorticoid receptor function: relevance to glucocorticoid resistance and the pathophysiology and treatment of major depression. Brain Behav Immun 21:9–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sousa AR, Lane SJ, Soh C, Lee TH 1999 In vivo resistance to corticosteroids in bronchial asthma is associated with enhanced phosphorylation of JUN N-terminal kinase and failure of prednisolone to inhibit JUN N-terminal kinase phosphorylation. J Allergy Clin Immunol 104:565–574 [DOI] [PubMed] [Google Scholar]
- Adcock IM, Lane SJ 2003 Corticosteroid-insensitive asthma: molecular mechanisms. J Endocrinol 178:347–355 [DOI] [PubMed] [Google Scholar]
- Leung DY, Bloom JW 2003 Update on glucocorticoid action and resistance. J Allergy Clin Immunol 111:3–22; quiz 23 [DOI] [PubMed] [Google Scholar]
- Tsitoura DC, Rothman PB 2004 Enhancement of MEK/ERK signaling promotes glucocorticoid resistance in CD4+ T cells. J Clin Invest 113:619–627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li LB, Goleva E, Hall CF, Ou LS, Leung DY 2004 Superantigen-induced corticosteroid resistance of human T cells occurs through activation of the mitogen-activated protein kinase kinase/extracellular signal-regulated kinase (MEK-ERK) pathway. J Allergy Clin Immunol 114:1059–1069 [DOI] [PubMed] [Google Scholar]
- Bhavsar P, Hew M, Khorasani N, Torrego A, Barnes PJ, Adcock I, Chung KF 2008 Relative corticosteroid insensitivity of alveolar macrophages in severe asthma compared with non-severe asthma. Thorax 63:784–790 [DOI] [PubMed] [Google Scholar]
- Goleva E, Hauk PJ, Hall CF, Liu AH, Riches DW, Martin RJ, Leung DY 2008 Corticosteroid-resistant asthma is associated with classical antimicrobial activation of airway macrophages. J Allergy Clin Immunol 122:550–559.e3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loke TK, Mallett KH, Ratoff J, O'Connor BJ, Ying S, Meng Q, Soh C, Lee TH, Corrigan CJ 2006 Systemic glucocorticoid reduces bronchial mucosal activation of activator protein 1 components in glucocorticoid-sensitive but not glucocorticoid-resistant asthmatic patients. J Allergy Clin Immunol 118:368–375 [DOI] [PubMed] [Google Scholar]
- Lane SJ, Adcock IM, Richards D, Hawrylowicz C, Barnes PJ, Lee TH 1998 Corticosteroid-resistant bronchial asthma is associated with increased c-fos expression in monocytes and T lymphocytes. J Clin Invest 102:2156–2164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi E, Onda K, Hirano T, Oka K, Maruoka N, Tsuyuguchi M, Matsumura Y, Niitsuma T, Hayashi T 2002 Expression of c-fos, rather than c-jun or glucocorticoid-receptor mRNA, correlates with decreased glucocorticoid response of peripheral blood mononuclear cells in asthma. Int Immunopharmacol 2:1419–1427 [DOI] [PubMed] [Google Scholar]
- Goleva E, Li LB, Leung DY 2009 IFN-γ reverses IL-2- and IL-4-mediated T-cell steroid resistance. Am J Respir Cell Mol Biol 40:223–230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichardt HM, Kaestner KH, Tuckermann J, Kretz O, Wessely O, Bock R, Gass P, Schmid W, Herrlich P, Angel P, Schütz G 1998 DNA binding of the glucocorticoid receptor is not essential for survival. Cell 93:531–541 [DOI] [PubMed] [Google Scholar]
- Schäcke H, Hennekes H, Schottelius A, Jaroch S, Lehmann M, Schmees N, Rehwinkel H, Asadullah K 2002 SEGRAs: a novel class of anti-inflammatory compounds. Ernst Schering Res Found Workshop 40:357–371 [DOI] [PubMed] [Google Scholar]
- Schäcke H, Berger M, Rehwinkel H, Asadullah K 2007 Selective glucocorticoid receptor agonists (SEGRAs): novel ligands with an improved therapeutic index. Mol Cell Endocrinol 275:109–117 [DOI] [PubMed] [Google Scholar]
- Rosen J, Miner JN 2005 The search for safer glucocorticoid receptor ligands. Endocr Rev 26:452–464 [DOI] [PubMed] [Google Scholar]
- Song IH, Gold R, Straub RH, Burmester GR, Buttgereit F 2005 New glucocorticoids on the horizon: repress, don’t activate! J Rheumatol 32:1199–1207 [PubMed] [Google Scholar]
- Hudson AR, Roach SL, Higuchi RI 2008 Recent developments in the discovery of selective glucocorticoid receptor modulators (SGRMs). Curr Top Med Chem 8:750–765 [DOI] [PubMed] [Google Scholar]
- Vanden Berghe W, Francesconi E, De Bosscher K, Resche-Rigon M, Haegeman G 1999 Dissociated glucocorticoids with anti-inflammatory potential repress interleukin-6 gene expression by a nuclear factor-κB-dependent mechanism. Mol Pharmacol 56:797–806 [PubMed] [Google Scholar]
- Vayssière BM, Dupont S, Choquart A, Petit F, Garcia T, Marchandeau C, Gronemeyer H, Resche-Rigon M 1997 Synthetic glucocorticoids that dissociate transactivation and AP-1 transrepression exhibit antiinflammatory activity in vivo. Mol Endocrinol 11:1245–1255 [DOI] [PubMed] [Google Scholar]
- Lin CW, Nakane M, Stashko M, Falls D, Kuk J, Miller L, Huang R, Tyree C, Miner JN, Rosen J, Kym PR, Coghlan MJ, Carter G, Lane BC 2002 Trans-activation and repression properties of the novel nonsteroid glucocorticoid receptor ligand 2,5-dihydro-9-hydroxy-10-methoxy-2,2,4-trimethyl-5-(1-methylcyclohexen-3-y 1)-1H-[1]benzopyrano[3,4-f]quinoline (A276575) and its four stereoisomers. Mol Pharmacol 62:297–303 [DOI] [PubMed] [Google Scholar]
- Coghlan MJ, Jacobson PB, Lane B, Nakane M, Lin CW, Elmore SW, Kym PR, Luly JR, Carter GW, Turner R, Tyree CM, Hu J, Elgort M, Rosen J, Miner JN 2003 A novel antiinflammatory maintains glucocorticoid efficacy with reduced side effects. Mol Endocrinol 17:860–869 [DOI] [PubMed] [Google Scholar]
- Owen HC, Miner JN, Ahmed SF, Farquharson C 2007 The growth plate sparing effects of the selective glucocorticoid receptor modulator, AL-438. Mol Cell Endocrinol 264:164–170 [DOI] [PubMed] [Google Scholar]
- Ali A, Thompson CF, Balkovec JM, Graham DW, Hammond ML, Quraishi N, Tata JR, Einstein M, Ge L, Harris G, Kelly TM, Mazur P, Pandit S, Santoro J, Sitlani A, Wang C, Williamson J, Miller DK, Thompson CM, Zaller DM, Forrest MJ, Carballo-Jane E, Luell S 2004 Novel N-arylpyrazolo[3,2-c]-based ligands for the glucocorticoid receptor: receptor binding and in vivo activity. J Med Chem 47:2441–2452 [DOI] [PubMed] [Google Scholar]
- Gossye V, Elewaut D, Van Beneden K, Dewint P, Haegeman G, De Bosscher K 9 February 2009 A plant-derived glucocorticoid receptor modulator attenuates inflammation without provoking ligand-induced resistance. Ann Rheum Dis doi:10.1136/ard.2008.102871 [DOI] [PubMed] [Google Scholar]
- Schäcke H, Schottelius A, Döcke WD, Strehlke P, Jaroch S, Schmees N, Rehwinkel H, Hennekes H, Asadullah K 2004 Dissociation of transactivation from transrepression by a selective glucocorticoid receptor agonist leads to separation of therapeutic effects from side effects. Proc Natl Acad Sci USA 101:227–232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphrey EL, Williams JH, Davie MW, Marshall MJ 2006 Effects of dissociated glucocorticoids on OPG and RANKL in osteoblastic cells. Bone 38:652–661 [DOI] [PubMed] [Google Scholar]
- López FJ, Ardecky RJ, Bebo B, Benbatoul K, De Grandpre L, Liu S, Leibowitz MD, Marschke K, Rosen J, Rungta D, Viveros HO, Yen WC, Zhi L, Negro-Vilar A, Miner JN 2008 LGD-5552, an antiinflammatory glucocorticoid receptor ligand with reduced side effects, in vivo. Endocrinology 149:2080–2089 [DOI] [PubMed] [Google Scholar]
- Miner JN, Ardecky B, Benbatoul K, Griffiths K, Larson CJ, Mais DE, Marschke K, Rosen J, Vajda E, Zhi L, Negro-Vilar A 2007 Antiinflammatory glucocorticoid receptor ligand with reduced side effects exhibits an altered protein-protein interaction profile. Proc Natl Acad Sci USA 104:19244–19249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker M, Clackers M, Copley R, Demaine DA, Humphreys D, Inglis GG, Johnston MJ, Jones HT, Haase MV, House D, Loiseau R, Nisbet L, Pacquet F, Skone PA, Shanahan SE, Tape D, Vinader VM, Washington M, Uings I, Upton R, McLay IM, Macdonald SJ 2006 Dissociated nonsteroidal glucocorticoid receptor modulators; discovery of the agonist trigger in a tetrahydronaphthalene-benzoxazine series. J Med Chem 49:4216–4231 [DOI] [PubMed] [Google Scholar]
- Shah N, Scanlan TS 2004 Design and evaluation of novel nonsteroidal dissociating glucocorticoid receptor ligands. Bioorg Med Chem Lett 14:5199–5203 [DOI] [PubMed] [Google Scholar]
- Thompson CF, Quraishi N, Ali A, Mosley RT, Tata JR, Hammond ML, Balkovec JM, Einstein M, Ge L, Harris G, Kelly TM, Mazur P, Pandit S, Santoro J, Sitlani A, Wang C, Williamson J, Miller DK, Yamin TT, Thompson CM, O'Neill EA, Zaller D, Forrest MJ, Carballo-Jane E, Luell S 2007 Novel glucocorticoids containing a 6,5-bicyclic core fused to a pyrazole ring: synthesis, in vitro profile, molecular modeling studies, and in vivo experiments. Bioorg Med Chem Lett 17:3354–3361 [DOI] [PubMed] [Google Scholar]
- Thompson CF, Quraishi N, Ali A, Tata JR, Hammond ML, Balkovec JM, Einstein M, Ge L, Harris G, Kelly TM, Mazur P, Pandit S, Santoro J, Sitlani A, Wang C, Williamson J, Miller DK, Yamin TT, Thompson CM, O'Neill EA, Zaller D, Forrest MJ, Carballo-Jane E, Luell S 2005 Novel heterocyclic glucocorticoids: in vitro profile and in vivo efficacy. Bioorg Med Chem Lett 15:2163–2167 [DOI] [PubMed] [Google Scholar]
- Wang JC, Shah N, Pantoja C, Meijsing SH, Ho JD, Scanlan TS, Yamamoto KR 2006 Novel arylpyrazole compounds selectively modulate glucocorticoid receptor regulatory activity. Genes Dev 20:689–699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biggadike K, Boudjelal M, Clackers M, Coe DM, Demaine DA, Hardy GW, Humphreys D, Inglis GG, Johnston MJ, Jones HT, House D, Loiseau R, Needham D, Skone PA, Uings I, Veitch G, Weingarten GG, McLay IM, Macdonald SJ 2007 Nonsteroidal glucocorticoid agonists: tetrahydronaphthalenes with alternative steroidal A-ring mimetics possessing dissociated (transrepression/transactivation) efficacy selectivity. J Med Chem 50:6519–6534 [DOI] [PubMed] [Google Scholar]
- Robinson RP, Buckbinder L, Haugeto AI, McNiff PA, Millham ML, Reese MR, Schaefer JF, Abramov YA, Bordner J, Chantigny YA, Kleinman EF, Laird ER, Morgan BP, Murray JC, Salter ED, Wessel MD, Yocum SA 2009 Octahydrophenanthrene-2,7-diol analogues as dissociated glucocorticoid receptor agonists: discovery and lead exploration. J Med Chem 52:1731–1743 [DOI] [PubMed] [Google Scholar]
- Chou TC 2006 Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol Rev 58:621–681 [DOI] [PubMed] [Google Scholar]
- Barnes PJ 2006 Corticosteroids: the drugs to beat. Eur J Pharmacol 533:2–14 [DOI] [PubMed] [Google Scholar]
- Calverley PM, Anderson JA, Celli B, Ferguson GT, Jenkins C, Jones PW, Yates JC, Vestbo J 2007 Salmeterol and fluticasone propionate and survival in chronic obstructive pulmonary disease. N Engl J Med 356:775–789 [DOI] [PubMed] [Google Scholar]
- Eickelberg O, Roth M, Lörx R, Bruce V, Rüdiger J, Johnson M, Block LH 1999 Ligand-independent activation of the glucocorticoid receptor by β2-adrenergic receptor agonists in primary human lung fibroblasts and vascular smooth muscle cells. J Biol Chem 274:1005–1010 [DOI] [PubMed] [Google Scholar]
- Usmani OS, Ito K, Maneechotesuwan K, Ito M, Johnson M, Barnes PJ, Adcock IM 2005 Glucocorticoid receptor nuclear translocation in airway cells after inhaled combination therapy. Am J Respir Crit Care Med 172:704–712 [DOI] [PubMed] [Google Scholar]
- Adcock IM, Maneechotesuwan K, Usmani O 2002 Molecular interactions between glucocorticoids and long-acting β2-agonists. J Allergy Clin Immunol 110:S261–268 [DOI] [PubMed] [Google Scholar]
- Fragaki K, Kileztky C, Trentesaux C, Zahm JM, Bajolet O, Johnson M, Puchelle E 2006 Downregulation by a long-acting β2-adrenergic receptor agonist and corticosteroid of Staphylococcus aureus-induced airway epithelial inflammatory mediator production. Am J Physiol Lung Cell Mol Physiol 291:L11–L18 [DOI] [PubMed] [Google Scholar]
- Kaur M, Chivers JE, Giembycz MA, Newton R 2008 Long-acting β2-adrenoceptor agonists synergistically enhance glucocorticoid-dependent transcription in human airway epithelial and smooth muscle cells. Mol Pharmacol 73:203–214 [DOI] [PubMed] [Google Scholar]
- Beck IM, Berghe WV, Gerlo S, Bougarne N, Vermeulen L, De Bosscher K, Haegeman G 2009 Glucocorticoids and mitogen- and stress-activated protein kinase 1 inhibitors: possible partners in the combat against inflammation. Biochem Pharmacol 77:1194–1205 [DOI] [PubMed] [Google Scholar]
- Lim-Tio SS, Fuller PJ 1998 Intracellular signaling pathways confer specificity of transactivation by mineralocorticoid and glucocorticoid receptors. Endocrinology 139:1653–1661 [DOI] [PubMed] [Google Scholar]
- Derijk RH, de Kloet ER 2008 Corticosteroid receptor polymorphisms: determinants of vulnerability and resilience. Eur J Pharmacol 583:303–311 [DOI] [PubMed] [Google Scholar]
- Aubry EM, Odermatt A 2009 Retinoic acid reduces glucocorticoid sensitivity in C2C12 myotubes by decreasing 11β-hydroxysteroid dehydrogenase type 1 and glucocorticoid receptor activities. Endocrinology 150:2700–2708 [DOI] [PubMed] [Google Scholar]
- Odermatt A, Gumy C 2008 Glucocorticoid and mineralocorticoid action: why should we consider influences by environmental chemicals? Biochem Pharmacol 76:1184–1193 [DOI] [PubMed] [Google Scholar]
- Atanasov AG, Odermatt A 2007 Readjusting the glucocorticoid balance: an opportunity for modulators of 11β-hydroxysteroid dehydrogenase type 1 activity? Endocr Metab Immune Disord Drug Targets 7:125–140 [DOI] [PubMed] [Google Scholar]
- Kolber BJ, Wieczorek L, Muglia LJ 2008 Hypothalamic-pituitary-adrenal axis dysregulation and behavioral analysis of mouse mutants with altered glucocorticoid or mineralocorticoid receptor function. Stress 11:321–338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruber G, Carlet M, Türtscher E, Meister B, Irving JA, Ploner C, Kofler R 2009 Levels of glucocorticoid receptor and its ligand determine sensitivity and kinetics of glucocorticoid-induced leukemia apoptosis. Leukemia 23:820–823 [DOI] [PubMed] [Google Scholar]
- Rosenfeld MG, Lunyak VV, Glass CK 2006 Sensors and signals: a coactivator/corepressor/epigenetic code for integrating signal-dependent programs of transcriptional response. Genes Dev 20:1405–1428 [DOI] [PubMed] [Google Scholar]
- Brush MH, Guardiola A, Connor JH, Yao TP, Shenolikar S 2004 Deactylase inhibitors disrupt cellular complexes containing protein phosphatases and deacetylases. J Biol Chem 279:7685–7691 [DOI] [PubMed] [Google Scholar]
- Pascussi JM, Gerbal-Chaloin S, Duret C, Daujat-Chavanieu M, Vilarem MJ, Maurel P 2008 The tangle of nuclear receptors that controls xenobiotic metabolism and transport: crosstalk and consequences. Annu Rev Pharmacol Toxicol 48:1–32 [DOI] [PubMed] [Google Scholar]
- Bougarne N, Paumelle R, Caron S, Hennuyer N, Mansouri R, Gervois P, Staels B, Haegeman G, De Bosscher K 2009 PPARα blocks glucocorticoid receptor α-mediated transactivation but cooperates with the activated glucocorticoid receptor α for transrepression on NF-κB. Proc Natl Acad Sci USA 106:7397–7402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishi M, Kawata M 2007 Dynamics of glucocorticoid receptor and mineralocorticoid receptor: implications from live cell imaging studies. Neuroendocrinology 85:186–192 [DOI] [PubMed] [Google Scholar]
- Vreugdenhil E, Verissimo CS, Mariman R, Kamphorst JT, Barbosa JS, Zweers T, Champagne DL, Schouten T, Meijer OC, de Kloet ER, Fitzsimons CP 2009 MicroRNA 18 and 124a down-regulate the glucocorticoid receptor: implications for glucocorticoid responsiveness in the brain. Endocrinology 150:2220–2228 [DOI] [PubMed] [Google Scholar]
- Visvanathan J, Lee S, Lee B, Lee JW, Lee SK 2007 The microRNA miR-124 antagonizes the anti-neural REST/SCP1 pathway during embryonic CNS development. Genes Dev 21:744–749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rainer J, Ploner C, Jesacher S, Ploner A, Eduardoff M, Mansha M, Wasim M, Panzer-Grümayer R, Trajanoski Z, Niederegger H, Kofler R 2009 Glucocorticoid-regulated microRNAs and mirtrons in acute lymphoblastic leukemia. Leukemia 23:746–752 [DOI] [PubMed] [Google Scholar]
- Bethke A, Fielenbach N, Wang Z, Mangelsdorf DJ, Antebi A 2009 Nuclear hormone receptor regulation of microRNAs controls developmental progression. Science 324:95–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai R, Phillips RA, Zhang Y, Khan D, Crasta O, Ahmed SA 2008 Suppression of LPS-induced interferon-γ and nitric oxide in splenic lymphocytes by select estrogen-regulated microRNAs: a novel mechanism of immune modulation. Blood 112:4591–4597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asirvatham AJ, Gregorie CJ, Hu Z, Magner WJ, Tomasi TB 2008 MicroRNA targets in immune genes and the Dicer/Argonaute and ARE machinery components. Mol Immunol 45:1995–2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burkhart BA, Ivey ML, Archer TK 2009 Long-term low level glucocorticoid exposure induces persistent repression in chromatin. Mol Cell Endocrinol 298:66–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adcock IM, Tsaprouni L, Bhavsar P, Ito K 2007 Epigenetic regulation of airway inflammation. Curr Opin Immunol 19:694–700 [DOI] [PubMed] [Google Scholar]
- Nawrot TS, Adcock I 2009 The detrimental health effects of traffic-related air pollution: a role for DNA methylation? Am J Respir Crit Care Med 179:523–524 [DOI] [PubMed] [Google Scholar]
- Kagoshima M, Wilcke T, Ito K, Tsaprouni L, Barnes PJ, Punchard N, Adcock IM 2001 Glucocorticoid-mediated transrepression is regulated by histone acetylation and DNA methylation. Eur J Pharmacol 429:327–334 [DOI] [PubMed] [Google Scholar]
- Baccarelli A, Wright RO, Bollati V, Tarantini L, Litonjua AA, Suh HH, Zanobetti A, Sparrow D, Vokonas PS, Schwartz J 2009 Rapid DNA methylation changes after exposure to traffic particles. Am J Respir Crit Care Med 179:572–578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bäckdahl L, Bushell A, Beck S 2009 Inflammatory signalling as mediator of epigenetic modulation in tissue-specific chronic inflammation. Int J Biochem Cell Biol 41:176–184 [DOI] [PubMed] [Google Scholar]
- Vanden Berghe W, Ndlovu MN, Hoya-Arias R, Dijsselbloem N, Gerlo S, Haegeman G 2006 Keeping up NF-κB appearances: epigenetic control of immunity or inflammation-triggered epigenetics. Biochem Pharmacol 72:1114–1131 [DOI] [PubMed] [Google Scholar]
- van Bel F, Heijnen CJ 2009 Perinatal programming and reprogramming by glucocorticoid therapy and perinatal stress. Semin Fetal Neonatal Med 14:127–129 [DOI] [PubMed] [Google Scholar]
- Champagne DL, de Kloet ER, Joëls M 2009 Fundamental aspects of the impact of glucocorticoids on the (immature) brain. Semin Fetal Neonatal Med 14:136–142 [DOI] [PubMed] [Google Scholar]
- Weaver IC 2009 Epigenetic effects of glucocorticoids. Semin Fetal Neonatal Med 14:143–150 [DOI] [PubMed] [Google Scholar]
- Merlot E, Couret D, Otten W 2008 Prenatal stress, fetal imprinting and immunity. Brain Behav Immun 22:42–51 [DOI] [PubMed] [Google Scholar]
- Darnaudéry M, Maccari S 2008 Epigenetic programming of the stress response in male and female rats by prenatal restraint stress. Brain Res Rev 57:571–585 [DOI] [PubMed] [Google Scholar]
- Oberlander TF, Weinberg J, Papsdorf M, Grunau R, Misri S, Devlin AM 2008 Prenatal exposure to maternal depression, neonatal methylation of human glucocorticoid receptor gene (NR3C1) and infant cortisol stress responses. Epigenetics 3:97–106 [DOI] [PubMed] [Google Scholar]
- Turner JD, Pelascini LP, Macedo JA, Muller CP 2008 Highly individual methylation patterns of alternative glucocorticoid receptor promoters suggest individualized epigenetic regulatory mechanisms. Nucleic Acids Res 36:7207–7218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwermann J, Rathinam C, Schubert M, Schumacher S, Noyan F, Koseki H, Kotlyarov A, Klein C, Gaestel M 2009 MAPKAP kinase MK2 maintains self-renewal capacity of haematopoietic stem cells. EMBO J 28:1392–1406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voncken JW, Niessen H, Neufeld B, Rennefahrt U, Dahlmans V, Kubben N, Holzer B, Ludwig S, Rapp UR 2005 MAPKAP kinase 3pK phosphorylates and regulates chromatin association of the polycomb group protein Bmi1. J Biol Chem 280:5178–5187 [DOI] [PubMed] [Google Scholar]
- Fischle W 2008 Talk is cheap—cross-talk in establishment, maintenance, and readout of chromatin modifications. Genes Dev 22:3375–3382 [DOI] [PubMed] [Google Scholar]
- Adhvaryu KK, Selker EU 2008 Protein phosphatase PP1 is required for normal DNA methylation in Neurospora. Genes Dev 22:3391–3396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodge DR, Cho E, Copeland TD, Guszczynski T, Yang E, Seth AK, Farrar WL 2007 IL-6 enhances the nuclear translocation of DNA cytosine-5-methyltransferase 1 (DNMT1) via phosphorylation of the nuclear localization sequence by the AKT kinase. Cancer Genomics Proteomics 4:387–398 [PubMed] [Google Scholar]
- Gazin C, Wajapeyee N, Gobeil S, Virbasius CM, Green MR 2007 An elaborate pathway required for Ras-mediated epigenetic silencing. Nature 449:1073–1077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merienne K, Pannetier S, Harel-Bellan A, Sassone-Corsi P 2001 Mitogen-regulated RSK2-CBP interaction controls their kinase and acetylase activities. Mol Cell Biol 21:7089–7096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thambirajah AA, Eubanks JH, Ausió J 2009 MeCP2 post-translational regulation through PEST domains: two novel hypotheses: potential relevance and implications for Rett syndrome. Bioessays 31:561–569 [DOI] [PubMed] [Google Scholar]
- Chao HT, Zoghbi HY 2009 The yin and yang of MeCP2 phosphorylation. Proc Natl Acad Sci USA 106:4577–4578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao J, Hu K, Chang Q, Wu H, Sherman NE, Martinowich K, Klose RJ, Schanen C, Jaenisch R, Wang W, Sun YE 2009 Phosphorylation of MeCP2 at serine 80 regulates its chromatin association and neurological function. Proc Natl Acad Sci USA 106:4882–4887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kameshita I, Sekiguchi M, Hamasaki D, Sugiyama Y, Hatano N, Suetake I, Tajima S, Sueyoshi N 2008 Cyclin-dependent kinase-like 5 binds and phosphorylates DNA methyltransferase 1. Biochem Biophys Res Commun 377:1162–1167 [DOI] [PubMed] [Google Scholar]
- Miyake K, Nagai K 2007 Phosphorylation of methyl-CpG binding protein 2 (MeCP2) regulates the intracellular localization during neuronal cell differentiation. Neurochem Int 50:264–270 [DOI] [PubMed] [Google Scholar]
- Nuber UA, Kriaucionis S, Roloff TC, Guy J, Selfridge J, Steinhoff C, Schulz R, Lipkowitz B, Ropers HH, Holmes MC, Bird A 2005 Up-regulation of glucocorticoid-regulated genes in a mouse model of Rett syndrome. Hum Mol Genet 14:2247–2256 [DOI] [PubMed] [Google Scholar]
- Kangaspeska S, Stride B, Métivier R, Polycarpou-Schwarz M, Ibberson D, Carmouche RP, Benes V, Gannon F, Reid G 2008 Transient cyclical methylation of promoter DNA. Nature 452:112–115 [DOI] [PubMed] [Google Scholar]
- Métivier R, Gallais R, Tiffoche C, Le Péron C, Jurkowska RZ, Carmouche RP, Ibberson D, Barath P, Demay F, Reid G, Benes V, Jeltsch A, Gannon F, Salbert G 2008 Cyclical DNA methylation of a transcriptionally active promoter. Nature 452:45–50 [DOI] [PubMed] [Google Scholar]