

Abstract
There is at present no cure or effective therapy for spinal muscular atrophy (SMA), a neurodegenerative disease that is the leading genetic cause of infant mortality. SMA usually results from loss of the SMN1 (survival of motor neuron 1) gene, which leads to selective motor neuron degeneration. SMN2 is nearly identical to SMN1 but has a nucleotide replacement that causes exon 7 skipping, resulting in a truncated, unstable version of the SMA protein. SMN2 is present in all SMA patients, and correcting SMN2 splicing is a promising approach for SMA therapy. We identified a tetracycline-like compound, PTK-SMA1, which stimulates exon 7 splicing and increases SMN protein levels in vitro and in vivo in mice. Unlike previously identified molecules that stimulate SMN production via SMN2 promoter activation or undefined mechanisms, PTK-SMA1 is a unique therapeutic candidate in that it acts by directly stimulating splicing of exon 7. Synthetic small-molecule compounds such as PTK-SMA1 offer an alternative to antisense oligonucleotide therapies that are being developed as therapeutics for a number of disease-associated splicing defects.
INTRODUCTION
Spinal muscular atrophy (SMA) is an autosomal-recessive disease caused by homozygous point mutations or deletions in the SMN1 (survival of motor neuron 1) gene (1). The reduction in functional SMN protein resulting from SMN1 mutations leads to the selective degeneration of the lower motor neurons in the ventral horn of the spinal cord, causing progressive muscle weakness and atrophy. SMA affects 1 in ~6000 live births and is the leading genetic cause of infant mortality. Most children with the most severe form of the disease, which accounts for 50 to 60% of affected individuals, die before the age of 2 years in the absence of supportive respiratory care (2). There is no cure for SMA at present.
SMN1 encodes SMN protein, which is part of a molecular complex essential for the assembly of small nuclear ribonucleoprotein particles (snRNPs) (3, 4). snRNPs are integral components of the spliceosome, a large multiprotein-RNA complex that processes precursor messenger RNA (pre-mRNA) transcripts into mature mRNA by splicing out intronic sequences (5). Splicing is a required step in gene expression, and the loss of a key component of the pathway would be expected to be incompatible with life. However, the homozygous loss of SMN1 is partially compensated for by the presence of a second gene, SMN2, which also codes for SMN protein. SMN2 cannot fully compensate for the loss of SMN1 in SMA because the expression of full-length protein generated from SMN2 is substantially lower than that from SMN1. This difference is due to a single nucleotide change in SMN2 exon 7 relative to SMN1. This nucleotide difference, although resulting in a synonymous codon, causes exon 7 to be excluded from most of the mRNA expressed from the SMN2 gene (6, 7). This Δ7 mRNA codes for a truncated protein that is highly unstable. A small percentage of SMN2 spliced transcripts retains exon 7 and codes for full-length, functional SMN protein. Disease severity in SMA correlates inversely with SMN2 gene copy number (8) and SMN protein abundance (9, 10).
SMA is likely caused by insufficient SMN protein in affected tissues, although it is not clear why spinal cord motor neurons are especially vulnerable. Thus, increasing functional SMN protein production from SMN2 is expected to have therapeutic value for SMA. One promising approach to achieve this goal is to improve the efficiency of SMN2 exon 7 splicing.
The pre-mRNA splicing machinery, especially factors involved in alternative splicing, is a viable target for altering gene expression as a treatment for human diseases (11–13). Pre-mRNA splicing catalyzed by the spliceosome is a two-step reaction (5, 14). The spliceosome is a complex machine composed of ~200 protein components and five small nuclear RNAs (snRNAs) (15). The role of each individual factor in splicing is largely unknown, although it is clear that interactions of various factors with cis-acting pre-mRNA sequences contribute to alternative splice-site selection.
Methods for targeting cis-acting sequence elements to therapeutically alter splicing patterns are becoming more common, although targeting of trans-acting splicing factors is relatively undeveloped. Small molecules make up the majority of traditional therapeutics in part because they can be modified and designed to increase potency, efficacy, and bioavailability. In the case of SMA, an ideal drug would have the ability to cross the blood-brain barrier and target spinal motor neurons, the cells that are affected by the disease. Several compounds have been found to increase full-length SMN protein production. However, their activities are largely nonspecific, likely eliciting widespread changes in transcription, localization, or phosphorylation of splicing factors as well as unrelated components (16). In addition, a number of compounds, such as aclarubicin, are toxic to cells (17, 18). Drugs that target specific splicing factors may be less toxic and more potent than nonspecific compounds.
Tetracyclines, a widely used drug class of demonstrated safety, bind to specific RNAs and proteins and modify their structure and function (19, 20). Moreover, small-molecule drugs, including aclarubicin, have been shown to increase full-length SMN2 mRNA and protein in cell culture (17). The structural similarity between tetracyclines and aclarubicin suggested that the former might represent a nontoxic means of altering the splicing event. We present here the characterization of a tetracycline-like molecule, discovered as a result of screening a diverse collection of tetracycline derivatives. PTK-SMA1 represents a new class of compounds that may be useful for the treatment of SMA.
RESULTS
Identification of a small-molecule activator of SMN2 exon 7 splicing
To identify compounds that specifically target the splicing machinery to increase the extent of SMN2 exon 7 splicing, we used a cell-free splicing system and tested a number of small molecules that had been reported to increase SMN protein concentration in cells. A cell-free splicing assay has an advantage over cell-based assays in that it allows the identification of compounds that directly alter the splicing reaction in the absence of other steps in gene expression, such as transcription, 3′-end processing, mRNA export, and translation; in addition, any effects on the turnover of precursor or product RNAs can be simultaneously assessed. For the cell-free assay, SMN2 pre-mRNA transcribed in vitro from an SMN2 minigene comprising exon 6, exon 7, the 5′ portion of exon 8, and the intervening introns was used as a substrate to assess exon 7 splicing in HeLa cell nuclear extract, with an antisense peptide nucleic acid (PNA) as a positive control (21).
Indoprofen, valproic acid, and salbutamol, all compounds that increase full-length SMN protein and/or mRNA concentration in cells (16, 22–24), did not affect splicing activity in the cell-free splicing assays (Fig. 1). Kinetin, a compound that has been shown to increase exon inclusion in IKBKAP and a number of other gene transcripts in cells (25), also did not significantly improve exon 7 inclusion. The results with aclarubicin were particularly interesting because this compound has been reported to increase the proportion of transcripts including exon 7 relative to those lacking exon 7 in cells (17), yet it had no effect on exon 7 splicing in our cell-free assay. Because aclarubicin is structurally similar to tetracycline, we also screened a number of tetracycline derivatives from Paratek Pharmaceuticals' chemical library (26). One of these derivatives, PTK-SMA1, increased the amount of full-length SMN2 transcripts relative to the exon 7–skipped form (Fig. 1). The effect of PTK-SMA1 was dose dependent, with the highest concentration of exon 7 splicing observed at 10 μM.
Fig. 1.

Identification of compounds that increase SMN2 exon 7 splicing in vitro. (A) Schematic of the 3′ region of the SMN1 and SMN2 genes. SMN1 has a C at position 6 of exon 7 and SMN2 has a T. Hatched lines depict predominant splicing pathway for the respective gene transcripts. (B) Cell-free splicing analysis of SMN2 minigene transcript in HeLa cell nuclear extract in the presence of PTK-SMA1 (2.5, 5, and 10 μM), aclarubicin (Acla) (40 and 80 μM), and indoprofen (Ind) (20 and 40 μM). Lane 1, addition of vehicle only; lane 2, positive control with an antisense PNA peptide that promotes exon 7 inclusion (65). (C) Quantitation of SMN2 exon 7 splicing in vitro. Bars indicate the % exon 7 inclusion [included / (included + skipped) × 100]. Triangles indicate increasing concentrations of compounds: PTK-SMA1 (5 and 10 μM), aclarubicin (40 and 80 μM), valproic acid (VPA) (20 and 40 μM), kinetin (5 and 10 μM), and salbutamol (Sal) (5 and 10 μM). Error bars represent SEM (n = 3). **P < 0.005, t test.
PTK-SMA1 is a novel compound generated by derivatization of the tetracycline scaffold at the C7 position of the naphthacene ring (26) (Fig. 2A). The position 7 substitution is critical for PTK-SMA1 activity in splicing. Compounds that are structurally related to PTK-SMA1 but lacking the position 7 substitution, such as doxycycline and minocycline, had no effect on exon 7 splicing (Fig. 2). Doses of PTK-SMA1 higher than 40 μM consistently showed a general inhibition of SMN2 splicing (Fig. 2C). Overall, at its maximum active concentration, PTK-SMA1 elicited a fourfold increase in the percent of SMN2 transcripts that included exon 7. Moreover, PTK-SMA1 improved SMN2 exon 7 splicing 30% above that of the SMN1 control (Fig. 2D). Together, these results demonstrate that the tetracycline derivative PTK-SMA1 is a highly effective activator of SMN2 exon 7 splicing.
Fig. 2.

The tetracycline derivative PTK-SMA1 promotes SMN2 exon 7 inclusion. (A) Chemical structures of PTK-SMA1, minocycline, and doxycycline. (B) Quantitation of SMN2 exon 7 in vitro splicing in untreated reactions or reactions incubated with PTK-SMA1 (n = 4), minocycline (n = 3), or doxycycline (n = 4) at each of the indicated final concentrations. Results are plotted as the % exon 7 inclusion, as in Fig. 1C. Error bars represent SEM. **P < 0.005, ***P < 0.0005 relative to untreated, t test. (C) SMN exon 7 splicing in vitro. SMN2 was incubated in the absence (−) or presence of increasing amounts of PTK-SMA1 (lanes 5 to 13: 0.625, 1.25, 2.5, 5, 10, 20, 40, 80, and 160 μM, respectively). PNA indicates the addition of SMN-RS10 PNA peptide (65). SMN1 pre-mRNA was spliced in lanes 1 and 2 as a control. (D) Quantitation of in vitro splicing of SMN1 and SMN2 exon 7 splicing and the stimulation of SMN2 exon 7 by PTK-SMA1 at peak activity. Error bars represent SEM (n = 380). ***P < 0.0005 relative to untreated.
PTK-SMA1 specifically promotes exon 7 inclusion
To determine whether the activity of PTK-SMA1 is specific to SMN2 exon 7 splicing, we tested its effect on the splicing of additional substrates with the cell-free assay. SMN1 exon 7 splicing was stimulated to some degree by PTK-SMA1 (Fig. 3). A BRCA1 minigene transcript with a G to T point mutation in exon 18 (E1694X) that results in skipping of the exon (27) was also tested. This mutation is similar to the C to T change in SMN2 exon 7 relative to SMN1: both the BRCA1 E1694X mutation and the SMN2 change are at position 6 of the affected exon and both disrupt an SF2/ASF exonic splicing enhancer (ESE) motif (21, 28). However, when PTK-SMA1 was included in the BRCA1 pre-mRNA splicing reaction, only a subtle change in exon 18 splicing was observed (Fig. 3). PTK-SMA1 similarly did not substantially enhance constitutive splicing of β-globin or immunoglobulin M (IgM) M1–M2 splicing substrates (Fig. 3). Alternative splicing of a model substrate with two competing 5′ splice sites (5′Dup) also was not affected by PTK-SMA1. Thus, PTK-SMA1 does not appear to be a general activator of splicing. Instead, the molecule acts in a specific manner to stimulate SMN2 exon 7 splicing, and not other splicing events, at least for the substrates we have tested to date.
Fig. 3.
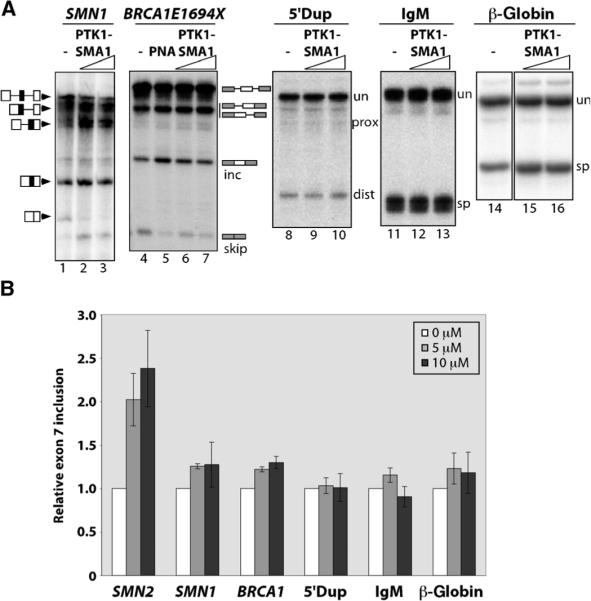
PTK-SMA1 acts specifically on SMN2 exon 7 splicing. (A) The effect of PTK-SMA1 on splicing in vitro. Splicing reactions with the indicated pre-mRNA substrates were incubated without PTK-SMA1 (−, lanes 1, 4, 8, 11, and 14) or with 5 μM PTK-SMA1 (lanes 2, 6, 9, 12, and 15) or 10 μM PTK-SMA1 (lanes 3, 7, 10, 13, and 16). PNA peptide was used as a positive control for splice-site switching (lane 5) and, in this case, refers to a PNA sequence targeted to the BRCA1 transcript, as previously described (65). Unspliced (un) and spliced (sp) products are indicated. inc and skip refer to spliced products that include or skip exon 18, and prox and dist refer to splicing to proximal or distal alternative 5′ splice sites. (B) Quantification of in vitro splicing. Splicing was calculated as either included / (included + skipped) × 100 (for SMN1 and BRCA1E1694X) or spliced / (unspliced + spliced) × 100 (for 5′Dup, IgM, and β-globin) and then normalized to the value for the corresponding untreated control. SMN2 values are included for comparison and are identical to Fig. 1. Error bars represent SEM (n = 3).
PTK-SMA1 may stimulate splicing of SMN2 exon 7 either by promoting intron 6 or intron 7 removal or by blocking splicing from exon 6 to exon 8. To distinguish among these possibilities, RNA transcripts were made from minigenes comprising exon 6, intron 6, and exon 7 (SMN67)or exon 7, intron7, and exon 8(SMN78)of the SMN2 gene, and splicing was tested in the cell-free assay in the presence or absence of PTK-SMA1. Treatment resulted in a modest stimulation of exon 6 to exon 7 splicing, peaking at 2.5 to 5 μM (Fig. 4, A and D). Exon 7 to exon 8 splicing was not stimulated by PTK-SMA1 and, in fact, a modest reduction in spliced product relative to unspliced transcript was observed (Fig. 4, B and D). This reduction reflects, at least in part, stabilization of unspliced pre-mRNA without further conversion of precursor into spliced products. These results suggest that PTK-SMA1 may act by shifting the balance of splicing paths through selective inhibition of specific splicing events that are normally competing. This idea is supported by results from splicing of an RNA transcript equivalent to SMN2 with exons 6, 7, and 8, except that the 5′ splice site following exon 7 was mutated, such that the only expected splicing event is from exon 6 to exon 8 (SMN2Δex7-5′ss). This splicing event was inhibited at a concentration of 5 μM PTK-SMA1 (Fig. 4, C and D). Together, these results suggest that PTK-SMA1 exerts its pronounced effect on exon 7 splicing by stimulating splicing from exon 6 to exon 7 while also inhibiting exon 7 skipping.
Fig. 4.

PTK-SMA1 stimulates exon 6 to exon 7 splicing and blocks exon 7 skipping. (A) In vitro splicing analysis of SMN2 intron 6. PTK-SMA1 was included in the reactions at final concentrations of 1.25, 2.5, 5, 10, 20, and 40 μM (lanes 2 to 7, respectively). Arrowhead indicates the released intron lariat. (B) Splicing of SMN2 intron 7. PTK-SMA1 was included in the reactions at increasing doses of 1.25, 2.5, 5, 10, and 20 μM (lanes 2 to 6, respectively). (C) SMN2 exon 7 skipping with the SMN2Δex7-5′ss transcript. PTK-SMA1 was included in the reaction at final concentrations of 2.5, 5, 10, 20, 40, and 80 μM (lanes 2 to 7, respectively). Unspliced pre-mRNA transcripts and spliced products are indicated to the right of each gel. (D) Quantitation of splicing from in vitro reactions like those in (A) to (C). Average values are graphed from SMN67 (n = 7;except 2.5 and 40 μM, n = 6), SMN78 (n = 3;except 2.5 and 40 μM, n = 2), and SMN2Δex7-5′ss (n = 2). Error bars represent SEM.
PTK-SMA1 increases SMN protein concentrations in SMA patient fibroblasts
To have therapeutic value, an increase in SMN2 exon 7 splicing must ultimately result in an increase in full-length SMN protein concentration. To test whether PTK-SMA1 can elevate SMN protein abundance, we cultured fibroblast cells derived from an SMA patient in the presence of the compound. Reverse transcription polymerase chain reaction (RT-PCR) analysis of SMN2 exon 7 splicing from the treated and untreated cells confirmed a dose-dependent increase in exon 7 splicing (Fig. 5A). Western blot analysis of total cellular proteins from treated and untreated cells also revealed a significant increase in SMN protein in cells treated with PTK-SMA1. A ~40% increase in the amount of SMN protein was observed in cells treated with 10 μM PTK-SMA1 relative to vehicle-treated control cells (Fig. 5B). SMN protein localizes to Cajal bodies or gems, which are observed as distinct foci in normal cells (29, 30). In SMA patient cells, however, a dramatic reduction in SMN-containing gems is typically observed (9, 10) (Fig. 5C). PTK-SMA1 treatment of cells for 48 hours led to an increase in SMN-containing gems. PTK-SMA1 was more potent than valproic acid (Fig. 5C) (31), a compound that is known to elevate SMN protein, and is now in clinical trials for SMA therapy (23, 31, 32). The maximum number of gems was observed upon treatment of cells with 2.5 μM PTK-SMA1.
Fig. 5.

PTK-SMA1 increases SMN protein concentration in SMA patient fibroblast cells. Cells were treated with the indicated concentrations of PTK-SMA1. Total protein or RNA was collected after 48 hours. (A) RNA was analyzed by RT-PCR and SMN2 spliced products were separated on a 2% agarose gel. A representative gel is shown. The graph represents the mean % of exon 7 splicing [(included / (included + skipped) × 100] normalized to untreated cells in a series of independent experiments. Error bars show SEM. **P < 0.005. (B) Western blotting with SMN and β-actin antibodies. The plotted data represent the mean change in SMN relative to β-actin protein concentrations in drug-treated cells relative to untreated cells. Error bars represent SEM (n = 5). *P < 0.05, **P < 0.001 relative to control, t test. (C) Gems or Cajal bodies were detected by immunofluorescence microscopy with an SMN antibody. A number of gems were quantitated in untreated 3814 control cells or in 3813 cells treated with the indicated concentrations of PTK-SMA1 or valproic acid. The graph plots the number of gems per 100 cells. Each circle represents an independent experiment and the bar indicates the average of all measurements.
PTK-SMA1 increases SMN2 exon 7 splicing and SMN protein concentration in mice
To determine whether PTK-SMA1 can increase SMN2 exon 7 splicing and SMN protein concentration in vivo, we tested PTK-SMA1 activity in a mouse model of SMA. These mice have a homozygous deletion of the single mouse Smn gene and have four copies of a human SMN2 transgene (33). The mice have short tails and small ears, typical of a mild type III–like SMA phenotype in mice (34). Mice were treated by daily intraperitoneal injection of PTK-SMA1 (25 or 50 mg/kg) over the course of 6 days. Because of the mild phenotype and normal life span of these SMA mice, phenotypic rescue by PTK-SMA1 could not be evaluated; however, we chose this strain so that mRNA splicing and protein expression from the transgene could be measured in adult mice. Two hours after the final PTK-SMA1 dosing, RNA was isolated from the liver tissue and semiquantitative radioactive RT-PCR analysis was performed to detect changes in SMN2 exon 7 splicing. A factor of 1.5 increase in exon 7 inclusion was observed in the PTK-SMA1–treated mice (Fig. 6A). Quantitative Western blot analysis of protein lysates from the liver of treated and untreated mice showed an increase by a factor of >5 in SMN protein concentration in PTK-SMA1–treated mice (Fig. 6B). SMN protein abundance in the liver of SMA mice was ~10% of that in normal mice. Thus, treatment with PTK-SMA1 increased the amount of SMN protein to ~50% of the concentration found in wild-type mice.
Fig. 6.

PTK-SMA1 increases SMN2 exon 7 splicing and SMN protein concentration in SMA mice. (A) RT-PCR analysis of RNA from liver of type III SMA adult mice (hSMN2+/+; Smn−/−) treated with PTK-SMA1 or vehicle control. Transcripts including and skipping human SMN2 exon 7 are labeled. The plotted data represent the mean % exon 7 inclusion in PTK-SMA1-treated and control mice. Error bars represent the SEM, with n displayed above the respective bars. (B) Western blot analysis of protein lysates from liver isolated from mice treated with PTK-SMA1. β-Actin was used as a loading control. Mean SMN to β-actin ratios are shown graphically. Error bars represent the SEM. (C) RT-PCR analysis of RNA from liver of transgenic SMA adult mice (hSMN2+/+; Smn+/+ or hSMN2+/+; Smn+/−) treated with PTK-SMA1 by intravenous (iv) or intraperitoneal (ip) injection. Representative samples are shown. The graph represents the mean % exon 7 inclusion in PTK-SMA1–treated and control mice. Error bars represent SEM, with n displayed above the respective bars. (D) Western blot analysis of liver protein lysates probed with a human-specific SMN antibody. β-Actin was used as a loading control. Mean SMN to β-actin ratios normalized to untreated samples are shown graphically. Error bars represent the SEM of values from samples shown in blots.
Similar results were obtained with a different mouse model of type I SMA that has two copies of a human SMN2 transgene (Fig. 6, C and D) (7). Adult transgenic mice with one or both copies of the wild-type mouse Smn gene were treated by daily intraperitoneal or intravenous injection of PTK-SMA1 over the course of 4 days. RNA and protein from the liver of treated animals were analyzed by RT-PCR and Western blotting with human-specific primers and antibody, respectively. A factor of 1.4 increase in exon 7 inclusion was seen in the intraperitoneally treated mice (Fig. 6C). This increase in exon 7 splicing corresponded with a nearly twofold increase in SMN protein concentration (Fig. 6D). This effect on SMN protein was lower than the effect we observed with the Smn−/− type III SMA mice. This difference likely reflects the longer duration of treatment and higher dosing in those mice. Type I SMA mice treated by intravenous injection of PTK-SMA showed smaller increases in full-length mRNA and protein expression compared to intraperitoneal administration (Fig. 6, C and D).
DISCUSSION
SMA is caused by loss of the SMN1 gene, which results in reduced cellular concentration of full-length SMN protein, expressed from the paralogous gene SMN2. One promising strategy for SMA therapy is to increase SMN protein abundance or function in SMA patients. A logical approach to achieve this goal is to improve the efficiency of splicing of exon 7 from SMN2 and thereby increase full-length SMN2 mRNA and protein concentration expressed from the gene. To this end, we performed a directed screening of small-molecule compounds for their ability to improve SMN2 exon 7 splicing in a cell-free splicing assay. We identified a novel small-molecule compound, PTK-SMA1, which increases SMN2 exon 7 splicing and SMN protein concentrations in vitro and in vivo. To our knowledge, this compound is the only molecule identified to date that has been demonstrated to alter splicing by directly targeting the splicing reaction to promote a specific splicing pathway.
The degree to which SMN2 mRNA and SMN protein concentrations must increase to achieve a therapeutically valuable effect is not clear. SMA carriers who only have one functional copy of SMN1 are asymptomatic. Individuals with homozygous deletions of SMN1 but multiple copies of SMN2 have complete protection from the disease or are less severely affected (8, 35–37). These studies suggest that increasing SMN2 expression by a factor of 2 could be clinically beneficial. In the cell-free assay, PTK-SMA1 improved splicing of exon 7 in SMN2 pre-mRNA to a concentration exceeding that seen with SMN1 pre-mRNA, suggesting that the molecule has the potential to completely rescue the splicing defect in SMN2. PTK-SMA1 administration to type III SMA mice resulted in a 50% increase in SMN2 exon 7 inclusion in the liver, which translated into a nearly fivefold increase in SMN protein concentrations compared to untreated animals. The fact that a modest increase in SMN2 exon 7 splicing results in a dramatic increase in SMN protein concentrations suggests additional levels of regulation in SMN gene expression and highlights the value of targeting the splicing reaction for SMA therapy. Although these results indicate that PTK-SMA1 is a promising SMA therapeutic candidate, the activity of this molecule in motor neurons must still be determined.
PTK-SMA1 is a tetracycline derivative that has been modified at the C7 position of the tetracycline backbone. Tetracyclines are anthracycline-type antibiotics that inhibit binding of bacterial transfer RNA to ribosomes (38). These molecules are safe, well-characterized, and commonly used antibacterial drugs in humans. PTK-SMA1 has reduced antibacterial activity and phototoxicity relative to other tetracycline-derived molecules, such as doxycycline and minocycline, and these characteristics may improve its tolerability in humans.
The fact that PTK-SMA1 promotes SMN2 exon 7 splicing in our cell-free splicing assay indicates that the compound is targeting the splicing reaction directly to improve exon 7 inclusion. Tetracyclines in general can bind RNA and affect RNA structure, synthesis, and stability (19, 20). Tetracyclines are also able to bind and activate proteins, such as the tetracycline repressor (39). The mechanism by which PTK-SMA1 stimulates SMN2 exon 7 splicing could be by direct binding to the SMN2 pre-mRNA or an RNA component of the splicing machinery (such as a U snRNA) or it could bind to a protein involved in splicing. It is possible that PTK-SMA1 could alter structure or activity in either of these scenarios. It is also possible that the tetracycline backbone binds to RNA nonspecifically and the PTK-SMA1–specific side chain contributes binding specificity or a protein interaction that alters splicing. Support for this idea comes from studies that have shown that tetracycline and other antibiotics can inhibit splicing at very high (>100 μM) concentrations (40). Indeed, PTK-SMA1 appears to function in such a way as to stimulate a particular splicing reaction in a specific manner at lower concentrations and to nonspecifically inhibit splicing at higher concentrations.
One speculative idea for the mechanism of action of PTK-SMA1 is that it is fortuitously acting as part of a riboswitch to control exon 7 splicing. Riboswitches are structured RNA sequences within a tran script that change structure after the binding of a regulatory molecule. This change in structure interferes with gene expression, typically by altering transcription, translation, or RNA processing. Natural riboswitches have not yet been identified in mammalian cells. However, they are a common mechanism of gene control in bacteria and have been identified in fungi and plants (41–43). It is intriguing that the thiamin pyrophosphate–responsive riboswitch that has been identified in eukaryotes controls splicing and alternative 3′-end processing of mRNAs (43). This finding indicates that natural riboswitches can regulate RNA splicing. Synthetic riboswitches that are recognized by tetracycline have been engineered in yeast to regulate pre-mRNA splicing (44), demonstrating the ability of the tetracycline compound class to alter splicing through recognition of a specific RNA structure. Indeed, secondary structure is an important splicing-regulatory feature of the SMN1 or SMN2 RNA transcript (45).
The structure of PTK-SMA1 is critical for splicing activity. Aclarubicin, the tetracycline derivative that has activity in cells and provided the impetus for investigating the tetracycline scaffold as a platform for screening SMN2 exon 7 activators, did not improve splicing of exon 7 in the cell-free splicing assay. Aclarubicin is most commonly used as a chemotherapeutic agent for cancer therapy and is extremely cytotoxic (46). It is possible that the effect of aclarubicin on SMN2 splicing and SMN protein concentrations in cells (17) is a secondary effect resulting from global changes in the cell. Nonetheless, when modified with particular side chains, such as that on PTK-SMA1, the tetracycline scaffold forms the basis of a potent activator of SMN2 exon 7 splicing.
SMN2 exon 7 splicing is controlled by a number of cis-acting sequence elements and trans-acting factors. The C residue at position 6 of SMN1 exon 7 is part of an ESE element that is recognized by the serine-arginine-rich (SR) protein SF2/ASF (21). This ESE is critical for recognition and splicing of exon 7. In SMN2 pre-mRNA, position 6 of exon 7 is a U rather than aC. The U changes the ESE such that it is not effectively recognized by SF2/ASF and exon 7 is skipped most of the time. In the absence of the ESE, inhibitory interactions between splicing silencers and heterogeneous nuclear RNP A1 predominate and result in exon skipping (21, 47). Although the single-nucleotide difference between SMN1 and SMN2 is sufficient to prevent efficient SMN2 exon 7 splicing, other cis-acting motifs are involved in splicing of exon 7, including an ESE that is recognized by the Tra2 family of SR-like proteins (6, 48–51). Additional cis-acting elements within the flanking introns and in exon 7 are also important for exon 7 inclusion. Some of these elements are recognized by trans-acting splicing factors and others appear to be regulatory secondary structures (52–54). Binding of splicing factors to the 3′ splice site is also impaired in SMN2 relative to SMN1 (55). Any of the splicing factors or sequence elements that control exon 7 splicing could be a potential target for PTK-SMA1.
A number of molecules have been identified that modulate alternative splicing in general (12), and some have also been identified that increase the concentration of full-length SMN2 mRNA in cells (18, 56). However, there is no evidence that these act directly on splicing as opposed to, for example, mRNA stability. We have tested a number of these compounds in the cell-free splicing assay and found that none of them altered splicing of SMN2 exon 7 (Fig. 1). These molecules may influence splicing by modulating transcription, nuclear transport, stability, signaling pathways, or some other cellular process that can affect splicing indirectly. Although kinetin improves exon inclusion of a number of transcripts (25), indicating that the compound may have a general effect on the splicing reaction, it did not significantly affect SMN2 exon 7 splicing in our assay (Fig. 1). Several small molecules that act specifically on components of the splicing machinery have been described. To date, these compounds have all been demonstrated to be inhibitors of splicing (57–61). PTK-SMA1 appears to be unique in its ability to influence splicing directly and promote a specific splicing event.
PTK-SMA1 improves exon 7 splicing in two ways: by stimulating splicing of the intron upstream of exon 7 and by inhibiting splicing from exon 6 to exon 8 (exon 7 skipping) (Fig. 4). It is possible that both of these activities are mediated by a single protein or sequence element. ESEs, for example, which typically function to promote splicing of nearby splice sites, also block skipping of the exon in which they are located (62).
Although the precise molecular target of PTK-SMA1 is not yet known, our screening assay has narrowed the target down to the splicing reaction itself. Discovering the mechanism of action of PTK-SMA1 in splicing is an important goal toward its development as a therapeutic and would also increase our understanding of how splicing can be targeted by small molecules. Because many diseases are caused by defects in RNA splicing (63), the therapeutic impact of this drug class could be significant. Tetracycline may be a chemical scaffold that can be manipulated to generate agents for context-specific repair of aberrant splicing. Of more immediate impact, PTK-SMA1 and its derivatives are promising therapeutic candidates for the treatment of SMA.
A major goal for SMA therapy is the development of a drug that can penetrate the blood-brain barrier and increase SMN protein concentration in the motor neurons to stop or reverse disease progression. PTK-SMA1 does not cross the blood-brain barrier, and hence, we examined its activity in the liver of transgenic mice as a proof of principle for the compound's ability to promote SMN2 exon 7 splicing in vivo. Ongoing structure-activity relation studies are focused on modifying PTK-SMA1 to produce a molecule that efficiently crosses the blood-brain barrier and improves SMN protein expression in the central nervous system.
MATERIALS AND METHODS
Cell-free splicing
Plasmids pCI-SMN1 and pCI-SMN2 linearized with Sal I (64) and BRCA1E1694X (28) were used as templates for in vitro transcription with T7 RNA polymerase (Promega) to make SMN1, SMN2, and BRCA1 transcripts. SMN67 and SMN78 were made by RT-PCR using pCI-SMN2 as a template. The forward primer T7SMNex6-5′-TAATACGACTCACTATAGGATAATTCCCCCACCACCTC-3′ or T7SMN2ex7-5′-TAATACGACTCACTATAGGTTTTAGACAAAATCAAAAAG-3′ includes a 5′ T7 RNA polymerase promoter sequence and the 5′ end of SMN2 exon 6 or exon 7, respectively. Reverse primers were specific to the 3′ end of exon 7 with a 5′ splice site at the 3′ end (SMNexon7L+5′-5′-GCAGACTTACTCCTTAATTTAAGGAATGTG-3′) or 75 nucleotides of exon 8 with a 5′ splice site (SMNex8-75R+5′R-5′-AAGTACTTACCTGTAACGCTTCACATTCCAGATCTGTC-3′). SMN2Δex7-5′ss was made by overlap extension PCR using pCI-SMN2 as a template and the primers (SMNex6-5′-ATTCCCCCACCACCTC-3′ and SMNCL-5′-GCTGGCAGACTTAGTCCTTAATTTAAGG-3′) in one reaction and the primers (SMNex8-75R+5′R and SMNCR-5′-CCTTAAATTAAGGACTAAGTCTGCCAGC) in a second reaction. The products from the two reactions were gel isolated and combined in a second RT-PCR reaction with the primers (T7SMNex6-5′ and SMNex8-75R+5′R) to produce the T7 SMN2ex7-5′ss. pSP65-μM1–M2 and pSP64-HβΔ6 were linearized with Xba I and Bam HI, respectively, and transcribed with SP6 RNA polymerase. Transcription reactions were carried out in the presence of [32P]uridine 5′-triphosphate and 7Me-GpppG cap analog (New England Biolabs) to make pre-mRNAs for in vitro splicing analysis. RNAs were purified by denaturing polyacrylamide gel electrophoresis (PAGE) and spliced in HeLa cell nuclear extract as previously described (21). Specifically, in a 10-μl reaction, RNA (10 fmol) was incubated with 3 μl of nuclear extract dialyzed into Buffer D [20 mM Hepes-KOH (pH 8), 100 mM KCl, 0.2 mM EDTA, and 20% (v/v) glycerol], 1.3% (w/v) polyvinyl alcohol, 0.5 mM ATP, 20 mM creatine phosphate, 1.6 mM MgCl2, and 30% (v/v) Buffer D at 30°C for 3 hours.
Cell culture and treatments
SMA type I homozygous and carrier fibroblasts (3813 and 3814, Coriell Cell Repositories) were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% (v/v) fetal bovine serum. For treatments with compounds, cells were split at a density of 1 × 105 to 2 × 105 cells per well in six-well plates the day before addition of compounds. Compounds or vehicle control [0.176 mM NaOH or 0.05% (v/v) dimethyl sulfoxide] was added directly to cells, which were then grown for an additional 48 hours. Cells were then washed with 1× phosphate-buffered saline (PBS) and collected by scraping into 1× PBS. After centrifugation, PBS was removed and cells were lysed in Laemmli buffer and heated at 99°C for 10 min.
Western blot analysis
Protein samples were separated by SDS-PAGE and transferred onto Immobilon-P membranes (Millipore). Blots were probed with mouse monoclonal antibodies specific for SMN (BD Biosciences) and mouse monoclonal antibodies specific for human SMN protein or β-actin (Sigma) followed by Alexa Fluor 594-conjugated secondary antibody against mouse IgG (Invitrogen) or horseradish peroxidase (HRP)–conjugated goat secondary antibody against mouse IgG. Detection and quantitation was performed with a Typhoon 9400 Variable Mode Imager (GE Healthcare) and ImageQuant T software for fluorescence-labeled blots or with Lumi-Light Western Blotting Substrate (Roche Diagnostics) for HRP-labeled blots.
Immunofluorescence
Fibroblast cells were plated on coverslips and grown in DMEM for 12 hours and then treated with compound or vehicle control. After 12 hours, the medium was removed and cells were washed with PBS and treated with fresh compound. Cells were treated every 12 hours, and 48 hours after the first treatment, they were washed with PBS and fixed with 4% (v/v) paraformaldehyde in PBS for 10 min. The fixed cells were washed with PBS, permeabilized in 0.2% (v/v) Triton X-100 and 0.5% (w/v) bovine serum albumin (BSA) in PBS for 5 min on ice, blocked for 30 min in blocking buffer [2% (v/v) normal goat serum and 2% (w/v) BSA], and incubated for 1 hour in blocking buffer containing mouse monoclonal antibody against SMN (Sigma). Cells were washed with PBS and incubated for 1 hour in blocking buffer containing Alexa Fluor 594–conjugated goat secondary antibody against mouse IgG (Invitrogen). Cells were washed with PBS and coverslips were mounted with Prolong Gold mounting solution containing 4′,6-diamidino-2-phenylindole (DAPI) for staining the nucleus (Invitrogen). Cells were analyzed with an Axioplan 2i fluorescence microscope (Carl Zeiss) equipped with Chroma filters (Chroma Technology). OpenLab software (Improvision) was used to collect images from a charge-coupled device camera (Hamamatsu). Cajal bodies or gems were counted by random field selection. Cell number was determined based on the number of DAPI-stained nuclei. At least three fields of view were counted.
Mice
Transgenic mice carrying four copies of human SMN2 were homo-zygous null at the mouse Smn locus (hSMN2+/+; Smn−/−)(33) or had two copies of the transgene and were heterozygote or wild type at the mouse Smn locus (7). Mice were maintained in accordance with the Cold Spring Harbor Laboratory Animal Care and Use regulations.
For experiments with hSMN2+/+; Smn−/− mice (33), PTK-SMA1 was dissolved in 10% (w/v) polyethylene glycol 400 in PBS and given to mice at 25 mg/kg by intraperitoneal injection once per day (25 mg/kg) or twice per day (50 mg/kg). Control animals received equal volumes of vehicle alone. Mice were injected for 6 days and killed 2 hours after the first injection on the final day. Treatment was well tolerated, with the exception of one mouse that died after two injections of PTK-SMA1 (50 mg/kg) and one mouse that died after four injections of PTK-SMA1 (25 mg/kg). These mice were excluded from the analysis. Mouse livers were perfused with ice-cold saline and then snap-frozen in liquid nitrogen and stored at −70°C.
For experiments with hSMN2+/+; Smn+/− or hSMN2+/+; Smn+/+ mice, PTK-SMA1 was dissolved in water and injected into the tail vein or peritoneum once a day for 4 days. Mice injected with PTK-SMA1 (50 mg/kg, intravenously) were injected with 25 mg/kg for the final treatment. One of these mice died after the third treatment. Mice were killed 24 hours after the final injection and tissues were collected and snap-frozen in liquid nitrogen.
RNA was extracted from livers using Trizol reagent (Invitrogen) according to the manufacturer's protocol. To prepare protein lysates, 100 mg of liver tissue was sonicated in 900 μl of radioimmunoprecipitation assay buffer [1× PBS, 0.25% (w/v) sodium deoxycholate, 0.1% (w/v) SDS, 1 mM EDTA, and complete mini protease inhibitor cocktail (Roche)].
RT-PCR
Human-specific primers for the amplification of human SMN2 transcripts in RNA samples from transgenic mouse liver were E4-33to55-F (5′-AAGTGAGAACTCCAGGTCTCCTG-3′) and E8-15to36-R (5′-GTGGTGTCATTTAGTGCTGCTC-3′). The RT-PCR reactions were performed as previously described (64). PCR products were labeled with [α-32P]2′-deoxycytidine 5′-triphosphate and analyzed by 6% native PAGE followed by phophorimage analysis.
For analysis of SMN2 exon 7 splicing in 3813 fibroblasts, total RNA was isolated from cells with RNeasy kit (Qiagen) according to the manufacturer's instructions. The following primers were used to amplify endogenous SMN2 mRNA: SMN2 FwdP (5′-ATAATTCCCCCACCACCTC-3′)and SMN2 RevP (5′-GCCTCACCACCGTGCTGG-3′). The PCR cycling conditions were set as follows: 95°C for 5 min, 23 cycles of 1 min at 95°C, 2 min at 55°C, 3 min at 72°C, and a final extension time of 10 min at 72°C. PCR products were resolved by electrophoresis through 2% agarose gels and visualized by ethidium bromide staining (Invitrogen). The band intensities were quantitated with ImageJ and AlphaImager 2200 version 5.5 software.
Statistical analysis
The data are presented as the mean and error bars represent the SEM. Data points were compared by the two-tailed t test, and the P values and number of independent experiments are indicated in the figures and/or legends.
Acknowledgments
We thank A. Burghes, E. Androphy, L. Simard, and K. Sahashi for invaluable advice; J. Jarecki and X. Roca for helpful discussions, advice, and comments on the manuscript; Y. Hua for the human-specific SMN antibody and critical comments on the manuscript; and Y. Dai for advice on statistical analysis.
Funding: Families of SMA research awards to M.L.H., A.R.K., and Paratek Pharmaceuticals and NIH grants 1U01NS061801 (P.H.) and GM42699 (A.R.K.).
Footnotes
Competing interests: J.B., P.A., C.D., K.L., J.E., T.B., M.D., and P.H. are employees of Paratek Pharmaceuticals Inc., the owner of PTK-SMA1 used in this study, and materially benefit either directly or indirectly through stock options. A.R.K. serves on the scientific advisory board of two nonprofit SMA foundations. Paratek Pharmaceuticals holds a patent on the PTK-SMA1 molecule used in this study.
REFERENCES AND NOTES
- 1.Lefebvre S, Bürglen L, Reboullet S, Clermont O, Burlet P, Viollet L, Benichou B, Cruaud C, Millasseau P, Zeviani M, Le Paslier D, Frézal J, Cohen D, Weissenbach J, Munnich A, Melki J. Identification and characterization of a spinal muscular atrophy-determining gene. Cell. 1995;80:155–165. doi: 10.1016/0092-8674(95)90460-3. [DOI] [PubMed] [Google Scholar]
- 2.Munsat TL, Davies KE. Neuromuscul. Disord; International SMA consortium meeting; Bonn, Germany. 26–28 June 1992; 1992. pp. 423–428. [DOI] [PubMed] [Google Scholar]
- 3.Neuenkirchen N, Chari A, Fischer U. Deciphering the assembly pathway of Sm-class U snRNPs. FEBS Lett. 2008;582:1997–2003. doi: 10.1016/j.febslet.2008.03.009. [DOI] [PubMed] [Google Scholar]
- 4.Pellizzoni L. Chaperoning ribonucleoprotein biogenesis in health and disease. EMBO Rep. 2007;8:340–345. doi: 10.1038/sj.embor.7400941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wahl MC, Will CL, Lührmann R. The spliceosome: Design principles of a dynamic RNP machine. Cell. 2009;136:701–718. doi: 10.1016/j.cell.2009.02.009. [DOI] [PubMed] [Google Scholar]
- 6.Lorson CL, Hahnen E, Androphy EJ, Wirth B. A single nucleotide in the SMN gene regulates splicing and is responsible for spinal muscular atrophy. Proc. Natl. Acad. Sci. U.S.A. 1999;96:6307–6311. doi: 10.1073/pnas.96.11.6307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Monani UR, Sendtner M, Coovert DD, Parsons DW, Andreassi C, Le TT, Jablonka S, Schrank B, Rossoll W, Prior TW, Morris GE, Burghes AH. The human centromeric survival motor neuron gene (SMN2) rescues embryonic lethality in Smn−/− mice and results in a mouse with spinal muscular atrophy. Hum. Mol. Genet. 2000;9:333–339. doi: 10.1093/hmg/9.3.333. [DOI] [PubMed] [Google Scholar]
- 8.Feldkötter M, Schwarzer V, Wirth R, Wienker TF, Wirth B. Quantitative analyses of SMN1 and SMN2 based on real-time LightCycler PCR: Fast and highly reliable carrier testing and prediction of severity of spinal muscular atrophy. Am. J. Hum. Genet. 2002;70:358–368. doi: 10.1086/338627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Coovert DD, Le TT, McAndrew PE, Strasswimmer J, Crawford TO, Mendell JR, Coulson SE, Androphy EJ, Prior TW, Burghes AH. The survival motor neuron protein in spinal muscular atrophy. Hum. Mol. Genet. 1997;6:1205–1214. doi: 10.1093/hmg/6.8.1205. [DOI] [PubMed] [Google Scholar]
- 10.Lefebvre S, Burlet P, Liu Q, Bertrandy S, Clermont O, Munnich A, Dreyfuss G, Melki J. Correlation between severity and SMN protein level in spinal muscular atrophy. Nat. Genet. 1997;16:265–269. doi: 10.1038/ng0797-265. [DOI] [PubMed] [Google Scholar]
- 11.Hagiwara M. Alternative splicing: A new drug target of the post-genome era. Biochim. Biophys. Acta. 2005;1754:324–331. doi: 10.1016/j.bbapap.2005.09.010. [DOI] [PubMed] [Google Scholar]
- 12.Sumanasekera C, Watt DS, Stamm S. Substances that can change alternative splice-site selection. Biochem. Soc. Trans. 2008;36:483–490. doi: 10.1042/BST0360483. [DOI] [PubMed] [Google Scholar]
- 13.Tazi J, Durand S, Jeanteur P. The spliceosome: A novel multi-faceted target for therapy. Trends Biochem. Sci. 2005;30:469–478. doi: 10.1016/j.tibs.2005.06.002. [DOI] [PubMed] [Google Scholar]
- 14.Hastings ML, Krainer AR. Pre-mRNA splicing in the new millennium. Curr. Opin. Cell Biol. 2001;13:302–309. doi: 10.1016/s0955-0674(00)00212-x. [DOI] [PubMed] [Google Scholar]
- 15.Jurica MS, Moore MJ. Pre-mRNA splicing: Awash in a sea of proteins. Mol. Cell. 2003;12:5–14. doi: 10.1016/s1097-2765(03)00270-3. [DOI] [PubMed] [Google Scholar]
- 16.Sumner CJ. Therapeutics development for spinal muscular atrophy. NeuroRx. 2006;3:235–245. doi: 10.1016/j.nurx.2006.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Andreassi C, Jarecki J, Zhou J, Coovert DD, Monani UR, Chen X, Whitney M, Pollok B, Zhang M, Androphy E, Burghes AH. Aclarubicin treatment restores SMN levels to cells derived from type I spinal muscular atrophy patients. Hum. Mol. Genet. 2001;10:2841–2849. doi: 10.1093/hmg/10.24.2841. [DOI] [PubMed] [Google Scholar]
- 18.Jarecki J, Chen X, Bernardino A, Coovert DD, Whitney M, Burghes A, Stack J, Pollok BA. Diverse small-molecule modulators of SMN expression found by high-throughput compound screening: Early leads towards a therapeutic for spinal muscular atrophy. Hum. Mol. Genet. 2005;14:2003–2018. doi: 10.1093/hmg/ddi205. [DOI] [PubMed] [Google Scholar]
- 19.Chopra I, Roberts M. Tetracycline antibiotics: Mode of action, applications, molecular biology, and epidemiology of bacterial resistance. Microbiol. Mol. Biol. Rev. 2001;65:232–260. doi: 10.1128/MMBR.65.2.232-260.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gallego J, Varani G. Targeting RNA with small-molecule drugs: Therapeutic promise and chemical challenges. Acc. Chem. Res. 2001;34:836–843. doi: 10.1021/ar000118k. [DOI] [PubMed] [Google Scholar]
- 21.Cartegni L, Hastings ML, Calarco JA, de Stanchina E, Krainer AR. Determinants of exon 7 splicing in the spinal muscular atrophy genes, SMN1 and SMN2. Am. J. Hum. Genet. 2006;78:63–77. doi: 10.1086/498853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Angelozzi C, Borgo F, Tiziano FD, Martella A, Neri G, Brahe C. Salbutamol increases SMN mRNA and protein levels in spinal muscular atrophy cells. J. Med. Genet. 2008;45:29–31. doi: 10.1136/jmg.2007.051177. [DOI] [PubMed] [Google Scholar]
- 23.Brichta L, Hofmann Y, Hahnen E, Siebzehnrubl FA, Raschke H, Blumcke I, Eyupoglu IY, Wirth B. Valproic acid increases the SMN2 protein level: A well-known drug as a potential therapy for spinal muscular atrophy. Hum. Mol. Genet. 2003;12:2481–2489. doi: 10.1093/hmg/ddg256. [DOI] [PubMed] [Google Scholar]
- 24.Lunn MR, Root DE, Martino AM, Flaherty SP, Kelley BP, Coovert DD, Burghes AH, Man NT, Morris GE, Zhou J, Androphy EJ, Sumner CJ, Stockwell BR. Indoprofen upregulates the survival motor neuron protein through a cyclooxygenase-independent mechanism. Chem. Biol. 2004;11:1489–1493. doi: 10.1016/j.chembiol.2004.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hims MM, Ibrahim EC, Leyne M, Mull J, Liu L, Lazaro C, Shetty RS, Gill S, Gusella JF, Reed R, Slaugenhaupt SA. Therapeutic potential and mechanism of kinetin as a treatment for the human splicing disease familial dysautonomia. J. Mol. Med. 2007;85:149–161. doi: 10.1007/s00109-006-0137-2. [DOI] [PubMed] [Google Scholar]
- 26.Nelson ML, Ismail MY, McIntyre L, Bhatia B, Viski P, Hawkins P, Rennie G, Andorsky D, Messersmith D, Stapleton K, Dumornay J, Sheahan P, Verma AK, Warchol T, Levy SB. Versatile and facile synthesis of diverse semisynthetic tetracycline derivatives via Pd-catalyzed reactions. J. Org. Chem. 2003;68:5838–5851. doi: 10.1021/jo030047d. [DOI] [PubMed] [Google Scholar]
- 27.Mazoyer S, Puget N, Perrin-Vidoz L, Lynch HT, Serova-Sinilnikova OM, Lenoir GM. A BRCA1 nonsense mutation causes exon skipping. Am. J. Hum. Genet. 1998;62:713–715. doi: 10.1086/301768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu HX, Cartegni L, Zhang MQ, Krainer AR. A mechanism for exon skipping caused by nonsense or missense mutations in BRCA1 and other genes. Nat. Genet. 2001;27:55–58. doi: 10.1038/83762. [DOI] [PubMed] [Google Scholar]
- 29.Carvalho T, Almeida F, Calapez A, Lafarga M, Berciano MT, Carmo-Fonseca M. The spinal muscular atrophy disease gene product, SMN: A link between snRNP biogenesis and the Cajal (coiled) body. J. Cell Biol. 1999;147:715–728. doi: 10.1083/jcb.147.4.715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu Q, Dreyfuss G. A novel nuclear structure containing the survival of motor neurons protein. EMBO J. 1996;15:3555–3565. [PMC free article] [PubMed] [Google Scholar]
- 31.Sumner CJ, Huynh TN, Markowitz JA, Perhac JS, Hill B, Coovert DD, Schussler K, Chen X, Jarecki J, Burghes AH, Taylor JP, Fischbeck KH. Valproic acid increases SMN levels in spinal muscular atrophy patient cells. Ann. Neurol. 2003;54:647–654. doi: 10.1002/ana.10743. [DOI] [PubMed] [Google Scholar]
- 32.Swoboda KJ, Scott CB, Teyna SP, Prior TW, LaSalle B, Sorenson SL, Wood J, Acsadi G, Crawford TO, Kissel JT, Krosschell KJ, D'Anjou G, Bromberg MB, Schroth MK, Chan GM, Elsheikh B, Simard LR. Phase II open label study of valproic acid in spinal muscular atrophy. PLoS One. 2009;4:e5268. doi: 10.1371/journal.pone.0005268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hsieh-Li HM, Chang JG, Jong YJ, Wu MH, Wang NM, Tsai CH, Li H. A mouse model for spinal muscular atrophy. Nat. Genet. 2000;24:66–70. doi: 10.1038/71709. [DOI] [PubMed] [Google Scholar]
- 34.Monani UR, Coovert DD, Burghes AH. Animal models of spinal muscular atrophy. Hum. Mol. Genet. 2000;9:2451–2457. doi: 10.1093/hmg/9.16.2451. [DOI] [PubMed] [Google Scholar]
- 35.Mailman MD, Heinz JW, Papp AC, Snyder PJ, Sedra MS, Wirth B, Burghes AH, Prior TW. Molecular analysis of spinal muscular atrophy and modification of the phenotype by SMN2. Genet. Med. 2002;4:20–26. doi: 10.1097/00125817-200201000-00004. [DOI] [PubMed] [Google Scholar]
- 36.Ogino S, Gao S, Leonard DG, Paessler M, Wilson RB. Inverse correlation between SMN1 and SMN2 copy numbers: Evidence for gene conversion from SMN2 to SMN1. Eur. J. Hum. Genet. 2003;11:275–277. doi: 10.1038/sj.ejhg.5200957. [DOI] [PubMed] [Google Scholar]
- 37.Wirth B, Brichta L, Schrank B, Lochmüller H, Blick S, Baasner A, Heller R. Mildly affected patients with spinal muscular atrophy are partially protected by an increased SMN2 copy number. Hum. Genet. 2006;119:422–428. doi: 10.1007/s00439-006-0156-7. [DOI] [PubMed] [Google Scholar]
- 38.Hierowski M. Inhibition of protein synthesis by chlortetracycline in the E. coli in vitro system. Proc. Natl. Acad. Sci. U.S.A. 1965;53:594–599. doi: 10.1073/pnas.53.3.594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Scholz O, Köstner M, Reich M, Gastiger S, Hillen W. Teaching TetR to recognize a new inducer. J. Mol. Biol. 2003;329:217–227. doi: 10.1016/s0022-2836(03)00427-3. [DOI] [PubMed] [Google Scholar]
- 40.Hertweck M, Hiller R, Mueller MW. Inhibition of nuclear pre-mRNA splicing by antibiotics in vitro. Eur. J. Biochem. 2002;269:175–183. doi: 10.1046/j.0014-2956.2001.02636.x. [DOI] [PubMed] [Google Scholar]
- 41.Cheah MT, Wachter A, Sudarsan N, Breaker RR. Control of alternative RNA splicing and gene expression by eukaryotic riboswitches. Nature. 2007;447:497–500. doi: 10.1038/nature05769. [DOI] [PubMed] [Google Scholar]
- 42.Henkin TM. Riboswitch RNAs: Using RNA to sense cellular metabolism. Genes Dev. 2008;22:3383–3390. doi: 10.1101/gad.1747308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wachter A, Tunc-Ozdemir M, Grove BC, Green PJ, Shintani DK, Breaker RR. Riboswitch control of gene expression in plants by splicing and alternative 3′ end processing of mRNAs. Plant Cell. 2007;19:3437–3450. doi: 10.1105/tpc.107.053645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Weigand JE, Suess B. Tetracycline aptamer-controlled regulation of pre-mRNA splicing in yeast. Nucleic Acids Res. 2007;35:4179–4185. doi: 10.1093/nar/gkm425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Singh NN, Singh RN, Androphy EJ. Modulating role of RNA structure in alternative splicing of a critical exon in the spinal muscular atrophy genes. Nucleic Acids Res. 2007;35:371–389. doi: 10.1093/nar/gkl1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Van Echo DA, Whitacre MY, Aisner J, Applefeld MM, Wiernik PH. Phase I trial of aclacinomycin A. Cancer Treat. Rep. 1982;66:1127–1132. [PubMed] [Google Scholar]
- 47.Kashima T, Manley JL. A negative element in SMN2 exon 7 inhibits splicing in spinal muscular atrophy. Nat. Genet. 2003;34:460–463. doi: 10.1038/ng1207. [DOI] [PubMed] [Google Scholar]
- 48.Frugier T, Nicole S, Cifuentes-Diaz C, Melki J. The molecular bases of spinal muscular atrophy. Curr. Opin. Genet. Dev. 2002;12:294–298. doi: 10.1016/s0959-437x(02)00301-5. [DOI] [PubMed] [Google Scholar]
- 49.Young PJ, DiDonato CJ, Hu D, Kothary R, Androphy EJ, Lorson CL. SRp30c-dependent stimulation of survival motor neuron (SMN) exon 7 inclusion is facilitated by a direct interaction with hTra2b1. Hum. Mol. Genet. 2002;11:577–587. doi: 10.1093/hmg/11.5.577. [DOI] [PubMed] [Google Scholar]
- 50.Hofmann Y, Lorson CL, Stamm S, Androphy EJ, Wirth B. Htra2-β1 stimulates an exonic splicing enhancer and can restore full-length SMN expression to survival motor neuron 2 (SMN2) Proc. Natl. Acad. Sci. U.S.A. 2000;97:9618–9623. doi: 10.1073/pnas.160181697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hofmann Y, Wirth B. hnRNP-G promotes exon 7 inclusion of survival motor neuron (SMN) via direct interaction with Htra2-β1. Hum. Mol. Genet. 2002;11:2037–2049. doi: 10.1093/hmg/11.17.2037. [DOI] [PubMed] [Google Scholar]
- 52.Chen HH, Chang JG, Lu RM, Peng TY, Tarn WY. The RNA binding protein hnRNP Q modulates the utilization of exon 7 in the survival motor neuron 2 (SMN2) gene. Mol. Cell. Biol. 2008;28:6929–6938. doi: 10.1128/MCB.01332-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hua Y, Vickers TA, Okunola HL, Bennett CF, Krainer AR. Antisense masking of an hnRNP A1/A2 intronic splicing silencer corrects SMN2 splicing in transgenic mice. Am. J. Hum. Genet. 2008;82:834–848. doi: 10.1016/j.ajhg.2008.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Singh NK, Singh NN, Androphy EJ, Singh RN. Splicing of a critical exon of human Survival Motor Neuron is regulated by a unique silencer element located in the last intron. Mol. Cell. Biol. 2006;26:1333–1346. doi: 10.1128/MCB.26.4.1333-1346.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Martins de Araújo M, Bonnal S, Hastings ML, Krainer AR, Valcárcel J. Differential 3′ splice site recognition of SMN1 and SMN2 transcripts by U2AF and U2 snRNP. RNA. 2009;15:515–523. doi: 10.1261/rna.1273209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Avila AM, Burnett BG, Taye AA, Gabanella F, Knight MA, Hartenstein P, Cizman Z, Di Prospero NA, Pellizzoni L, Fischbeck KH, Sumner CJ. Trichostatin A increases SMN expression and survival in a mouse model of spinal muscular atrophy. J. Clin. Invest. 2007;117:659–671. doi: 10.1172/JCI29562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kaida D, Motoyoshi H, Tashiro E, Nojima T, Hagiwara M, Ishigami K, Watanabe H, Kitahara T, Yoshida T, Nakajima H, Tani T, Horinouchi S, Yoshida M. Spliceostatin A targets SF3b and inhibits both splicing and nuclear retention of pre-mRNA. Nat. Chem. Biol. 2007;3:576–583. doi: 10.1038/nchembio.2007.18. [DOI] [PubMed] [Google Scholar]
- 58.Kotake Y, Sagane K, Owa T, Mimori-Kiyosue Y, Shimizu H, Uesugi M, Ishihama Y, Iwata M, Mizui Y. Splicing factor SF3b as a target of the antitumor natural product pladienolide. Nat. Chem. Biol. 2007;3:570–575. doi: 10.1038/nchembio.2007.16. [DOI] [PubMed] [Google Scholar]
- 59.Kuhn AN, van Santen MA, Schwienhorst A, Urlaub H, Lührmann R. Stalling of spliceosome assembly at distinct stages by small-molecule inhibitors of protein acetylation and deacetylation. RNA. 2009;15:153–175. doi: 10.1261/rna.1332609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pilch B, Allemand E, Facompré M, Bailly C, Riou JF, Soret J, Tazi J. Specific inhibition of serine- and arginine-rich splicing factors phosphorylation, spliceosome assembly, and splicing by the antitumor drug NB-506. Cancer Res. 2001;61:6876–6884. [PubMed] [Google Scholar]
- 61.Soret J, Bakkour N, Maire S, Durand S, Zekri L, Gabut M, Fic W, Divita G, Rivalle C, Dauzonne D, Nguyen CH, Jeanteur P, Tazi J. Selective modification of alternative splicing by indole derivatives that target serine-arginine-rich protein splicing factors. Proc. Natl. Acad. Sci. U.S.A. 2005;102:8764–8769. doi: 10.1073/pnas.0409829102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ibrahim EC, Schaal TD, Hertel KJ, Reed R, Maniatis T. Serine/arginine-rich protein-dependent suppression of exon skipping by exonic splicing enhancers. Proc. Natl. Acad. Sci. U.S.A. 2005;102:5002–5007. doi: 10.1073/pnas.0500543102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cooper TA, Wan L, Dreyfuss G. RNA and disease. Cell. 2009;136:777–793. doi: 10.1016/j.cell.2009.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hua Y, Vickers TA, Baker BF, Bennett CF, Krainer AR. Enhancement of SMN2 exon 7 inclusion by antisense oligonucleotides targeting the exon. PLoS Biol. 2007;5:e73. doi: 10.1371/journal.pbio.0050073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cartegni L, Krainer AR. Correction of disease-associated exon skipping by synthetic exon-specific activators. Nat. Struct. Biol. 2003;10:120–125. doi: 10.1038/nsb887. [DOI] [PubMed] [Google Scholar]