

Abstract
Metal complexation can have a major influence on the antiviral and co-receptor binding properties of cyclam and bicyclam macrocycles. We report the synthesis of the vanadyl cyclam complexes [V(IV) O(cyclam)SO4] (1) and [V(IV)O(cyclam)Cl]Cl - (2), and the analogous xylylbicyclam sulfato (3) and chlorido (4) complexes. The X-ray crystal structures of 1·1.33CH3OH and 2·CH3OH1.5H2O show short V=O bonds (1.6093(19) and 1.599(3) Å, respectively) with monodentate sulfate H-bonded to ring NH groups for 1, but a long V-Cl bond (2.650(12) Å) for 2. The solid-state structures of 3 and 4 were compared to those of 1 and 2 using vanadium K-edge EXAFS data. These suggested that complex 4 was oligomeric and contained bridging chlorido ligands. EPR studies suggested that the SO42- (from 1) and Cl- (from 2) ligands are readily substituted by water in solution, whereas these remain partially bound for the VIV xylylbicyclam complexes 3 and 4. The vanadyl xylylbicyclam complexes were highly active against HIV-1 (IIIB) and HIV-2 (ROD) strains with IC50 values in the range 1-5 μM for 3 and 0.1-0.3 μM for 4; in contrast the vanadyl cyclam complexes 1 and 2 were inactive. The factors which contribute to the activity of these complexes are discussed. Studies of vanadyl cyclam docked into a model of the human CXCR4 co-receptor revealed that the coordination of vanadium to the carboxylate of Asp171 may be accompanied by H-bonding to the macrocycle and an attractive V=O··H interaction involving the backbone Trp195 α-carbon proton of CXCR4. In addition, hydrophobic interactions with Trp195 are present. Both ring configuration and the xylyl linker may play roles in determining the higher activity of the bicyclam complexes.
Introduction
Macrocycles such as cyclam derivatives show promise as stem cell mobilizers and antivirals, including activity against HIV and related AIDS disease. Drug treatments used today against HIV infection are primarily associated with post-infection and classified as (i) reverse transcriptase inhibitors (RTIs) - antiretroviral drugs that inhibit the enzyme reverse transcriptase, essential for successful replication of HIV, (ii) protease inhibitors that inhibit the function of HIV-1 protease, and (iii) integrase inhibitors such as Raltegravir, FDA-approved in 2007 for the treatment of HIV-infections.1 A fourth class of antiviral agents known as entry inhibitors, target the pre-infection cycle of HIV. Maraviroc is an entry inhibitor that specifically targets the chemokine receptor, CCR5. Importantly, Pfizer's Celsentri® (maraviroc), is the first new oral class of HIV treatment and was licensed by the European Agency for the Evaluation of Medicinal Products (EMEA) in September 2007. Currently, all other available oral HIV medicines target HIV only after it has entered the immune cells.2
The biological targets of entry inhibitor drugs are specific protein receptors expressed on the surface of helper T-cells (a type of white blood cell or leukocyte) found in the immune system. CD4 is a glycoprotein receptor and the primary receptor used by HIV-1 to gain entry to host cells.3-5 Binding of the virus to CD4 occurs through attachment of the viral envelope glycoprotein gp120, and through this association the virus gains access to the coreceptor CXCR4, an important coreceptor for HIV-1.1 Interaction with CXCR4 enables the virus to fuse with the cell membrane, gaining entry to the cell where it effectively discharges the viral RNA, leading to replication and the onset of infection. CXCR4 is associated with HIV infection during the later stages of AIDS disease when the immune system deteriorates rapidly.
The small molecule CXCR4 chemokine antagonist xylylbicyclam (Chart 1; AMD3100) reached Phase II clinical trials as an entry inhibitor drug against HIV (IC50 < 0.1 μM). It has since been further pursued as a stem cell mobilizer.6 The product has since been relaunched commercially under the name of Mozobil having completed Phase III clinical trials as a stem cell mobilizer and was approved for clinical use in the US in December 2008. The same membrane protein CXCR4 that aids entry of HIV to cells also anchors stem cells in the bone marrow. Mobilization of stem cells is beneficial during, for example, transplant therapy.
Chart 1.
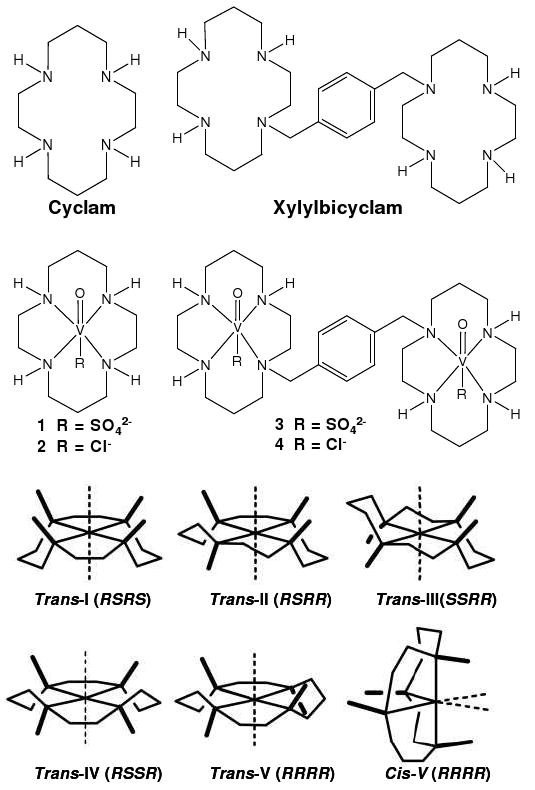
Configurations of metallocyclams and structures of cyclam, xylylbicyclam and complexes 1-4. AMD3100 is the octahydrochloride salt (xylylbicyclam.8HCl).
Cyclam macrocycles can bind strongly to d-block metal ions.7-10 The specific configurations adopted by metal cyclam complexes11 (Chart 1) may be important for receptor recognition and biological activity12 and it seems likely that the various configurations of metallo-cyclams are recognised differently by the co-receptor. Zinc(II) in particular is abundant in the blood plasma (ca. 19 μM) and the binding of xylylbicyclam to zinc(II) may play a role in its mechanism of action.13-17 Indeed zinc complexes with configurationally constrained bicyclam ligands9 can exhibit higher activity against HIV-1 than AMD3100. Recognition of metallocyclams such as Zn(II)-xylylbicyclam by the CXCR4 coreceptor is thought to involve a combination of direct coordination to side chains (Asp, Glu), hydrogen-bonding to cyclam NH groups and hydrophobic interactions (with the periphery of the cyclam rings and the xylyl linker).12, 18 Here we describe the role of axial interactions with the metal by introducing the vanadyl fragment (VO2+) into the cyclam ring. This new class of complexes introduces a different application for the bioinorganic chemistry of vanadium, and complements the wealth of vanadium research involving insulin-enhancing agents, cofactors in haloperoxidases, catalysts and nitrogenase cofactors.19-21 There appears to be little previously reported work on vanadyl cyclams. We have studied both cyclam and xylylbicyclam complexes with additional chloride and sulfate ligands using a variety of techniques including X-ray crystallography, XANES, EXAFS, EPR and molecular modelling. The anti-HIV activity of these vanadyl cyclam and xylylbicyclam complexes was also determined.
Experimental Section
Materials
Cyclam (1,4,8,11-tetraazacyclotetradecane), α,α′-dibromo-p-xylene, ethyl trifluoroacetate, triethylamine, barium chloride, vanadyl sulfate hydrate were purchased from Aldrich Chemical Co. and were reagent grade unless otherwise stated.
Synthesis of oxovanadium(IV)-cyclam sulfate, 1
Cyclam, (0.91 g, 4.56 mmol) was dissolved in HPLC grade methanol (35 mL) and refluxed under N2 for 10 min. Vanadyl sulfate hydrate (1.07 g, 4.56 mmol, 1 mol. equiv.) was added and the reaction refluxed for 24 h. On stirring, a color change was observed from blue to green after 1 min and to grey after 3 min. After 15 min the solution turned cloudy green and after 30 min was blue/grey in color. After 2.5 h this color deepened to a dark blue/grey. The solution settled into two layers. The top layer was a vivid green in color and a blue precipitate settled out as the bottom layer. The top layer was separated by centrifugation and then filtration, and the solvent removed under reduced pressure to give a green powder (yield: 1.50 g, 4.13 mmol, 90.6%). The product was further dried under vacuum.
Anal. Calc. for C10H24N4VSO5·MeOH·0.5H2O: %C, 32.70; %H, 6.23; %N, 13.86. Found %C, 32.43; %H, 6.49; %N, 14.46. ESI-MS: m/z = 363.90 [M+H]+; νmax (KBr)/cm-1 V=O, 974; SO4, ν3 1106, 1064, 1028; ν4 619, 598.
Crystals of 1·1.33CH3OH suitable for X-ray diffraction were obtained by slow diffusion of ether into a methanolic solution of the product at 277 K.
Synthesis of oxovanadium(IV)-cyclam dichloride, 2
Oxovanadium(IV)-cyclam, 1 (0.73 g, 0.21 mmol), was dissolved in distilled water (10 mL) and barium chloride (0.63 g, 0.30 mmol, 3 mol. equiv. excess) added as an aqueous solution (4 mL). A white precipitate formed immediately and the mixture was stirred at room temperature in air for 16 h. The mixture was separated using centrifugal force (4000 rpm, 1 h) and the green solution filtered. The solvent was removed under reduced pressure and further dried under vacuum to give a crystalline green powder. Anal. Calc. for C10H24N4VOCl2·H2O %C, 33.70; %H, 7.35; %N, 15.70. Found %C, 34.04; %H, 7.22; %N, 16.05. ESI-MS: m/z = 302.10 (Cl35), 304.00 (Cl37) [M]+; νmax (KBr)/cm-1 V=O, 945. Crystals of 2·CH3OH·1.5H2O suitable for X-ray diffraction were formed by slow diffusion of ether into a methanolic solution of the product at 277 K. Yield of crystals 0.03 g, 0.08 mmol, 36.6%.
Synthesis of oxovanadium(IV)-(xylylbicyclam) disulfate, 3
Xylylbicyclam (0.05 g, 0.10 mmol) was dissolved in 5 mL anhydrous methanol and refluxed gently under argon for 5 min. Vanadyl sulfate hydrate (0.05 g, 0.20 mmol, 2 mol. equiv.) was added and a color change was observed, both in the solution and in the solid added. After 3 min the initial clear solution began to turn green and the blue solid (vanadyl sulfate hydrate) turned grey. After 15 min, the reaction became more homogenous and the color darkened to brown. On standing, the flask contents settled into two layers and a pale green solution formed on top of the dark solid. The reaction was refluxed for 16 h and the solution on standing was green in color. The dark solid changed color to light blue. The green solution was separated from the blue solid under centrifugal force, 4000 rpm, 1 h and the solvent removed under reduced pressure and further dried under vacuum. The product dried to give a dark green powder (0.05 g, 0.06 mmol, 61.9%).
Anal. calc. for C28H54N8V2S2O10·MeOH·3H2O, %C, 38.07; %H, 7.05; %N, 12.25. Found %C, 37.53; %H, 7.29; %N, 11.67. ESI-MS: m/z = 829.1 [M+H]+; νmax (K/Br)cm-1 V=O 945; SO42- ν3 1120, 1034; ν4 615.
Synthesis of oxovanadium(IV)-(xylylbicyclam) tetrachloride, 4
Oxovanadium(IV)-(xylylbicyclam)-sulfate 3, (0.05 g, 0.06 mmol) was dissolved in water (3 mL) and heated until fully dissolved. Barium chloride (0.03 g, 0.12 mmol, 2 mol. equiv.) was added as an aqueous solution (2 mL) and a cloudy solution formed immediately. The mixture was stirred for 16 h at room temperature then separated under centrifugal force to give a green solution and a blue solid. The green solution was decanted, filtered and the solvent was removed by freeze-drying. On drying, a color change was noted from green to brown, and on redissolving in water a green solution was obtained (yield: 0.04 g, 4.50 mmol, 75%).
Anal. calc. for C28H54N8V2Cl2O2·5H2O·2NaCl, %C, 36.77; %H, 7.05; %N, 12.25. Found %C, 37.09; %H, 7.02; %N, 11.53.
ESI-MS: m/z = 705.4 [M-2Cl]2+ (100%), 741.3 [M-Cl]+; νmax (K/Br)cm-1 V=O 964.
X-ray crystallography
Diffraction data for compound 1 and 2 were collected at 150 K using a Bruker Smart Apex CCD diffractometer. Absorption corrections for all data sets were performed with the multi-scan procedure SADABS.22 Both structures were solved by direct methods (SHELXS or SIR92).23, 24 Refinement was against F2 using all data (CRYSTALS).25 Unless stated otherwise H-atoms were placed in geometrically calculated positions. In complex 1, one of the molecules of methanol of solvation was disordered over 3 sites about a three-fold axis, and no H-atoms were placed on this part of the structure. A second molecule of methanol of solvation was on a general position; the hydroxyl H-atom on this molecule was located in a difference map and refined. In complex 2, H atoms attached to oxygen were also located in a difference map. H-atom positions were initially refined subject to restraints and thereafter allowed to ride on their parent atoms. The programs Diamond 3.02026, Mercury 1.4.127 and ORTEP 3228 were used for analysis of data and production of graphics. X-ray crystallographic data have been deposited in the Cambridge Crystallographic Data Centre under the accession numbers CCDC 748340 and 748341.
IR spectroscopy
Infrared spectra were recorded as KBr pellets in the range 4000–400 cm-1 on a Perkin-Elmer Paragon 1000 Fourier-transform spectrometer.
X-ray absorption spectroscopy
X-ray absorption spectra were recorded at Stanford Synchrotron Radiation Laboratory (SSRL) on the 20-pole wiggler beam line 7-3, under ring operating conditions of 80-100 mA and 3 GeV. Data were collected using a Si (220) phi = 0° double crystal monochromator and the signal was detected using a 30 element Ge detector array. All samples were prepared by lightly grinding 5 mg of vanadium complex with boron nitride in a mortar and pestle until a fine uniform powder was formed. XAS data were collected in the energy range from 5235-6485 eV using XAS Collect software. Ten scans of each compound were obtained at the V K edge, to ensure reproducibility. Energy calibration was performed using the V K-edge spectrum of a vanadium metal foil, assuming the lowest energy peak to be 5465 eV. Data processing, including averaging scans, energy calibration, background subtraction and normalization was performed in SixPACK.29 EXAFS analysis was carried out using Artemis.30
EPR spectroscopy
EPR spectra were recorded on an X-band Bruker ER200D-SCR spectrometer connected to a Datalink 486DX PC with EPR Acquisition System, version 2.42 software. Measurements were recorded in fluid solution at 298 K and in frozen solution at 77 K. Data were simulated using Bruker WINEPR Simphonia version 1.26 software by the National EPR Centre, University of Manchester, UK and corrected as for the DPPH (2,2-diphenyl-1-picrylhydrazyl) standard.31
Antiviral testing
Experimental protocols for anti-HIV testing are detailed in Supporting Information.32
Molecular modeling
Our previously reported homology model of CXCR412 was used for manual docking of oxovanadium(IV) cyclam complex 1 under the SYBYL version 6.9 (Tripos Associates, St. Louis, MO) modelling environment. Complex 1 was first superimposed onto one ring of the previously docked [Zn2-xylylbicyclam]-CXCR4 interaction12 using all equivalent Cα atoms (crystal structures for both vanadium bicyclams complexes 3,4 were unavailable). The vanadium cyclam complex 1 was then “merged” with the CXCR4 protein model into a separate MOL2 file using SYBYLv6.9. Expected clashes were observed between Asp171 and the sulfate group that were subsequently removed and two models of possible interaction were produced. In Model 1 (Figure 7a), parameters were set with a view to investigate the possibility of metal-carboxylate interactions with Asp171 in the binding site. In this case, the vanadium center was fixed relative to the receptor and distance restraints of 2.14 Å (V-sulfate bond length) with an average force constant of 500 kcal mol-1 Å-2 were applied to the postulated V-O bond with Asp171, to represent metal-carboxylate binding. A second distance (1.609 Å) was set for V=O to maintain the bond length and prevent stretching during the minimization process. Bad contacts and geometries were then relieved by energy minimizing the region of the CXCR4 receptor surrounding the oxovanadium(IV) cyclam complex 1, (all amino acid residues with a sphere radius of 5 Å from the cyclam ligand). In Model 2 (Figure 7b), no distance restraint was applied between the vanadium center and carboxylate oxygens of Asp171, but docking was otherwise undertaken as in Model 1.
Figure 7.
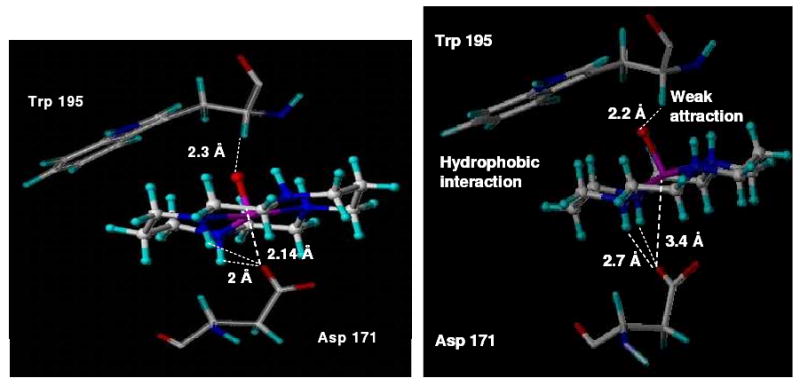
Molecular model of {VIVO(cyclam)} bound to the co-receptor CXCR4, based on the trans-III configuration of [VIVO(cyclam)SO4] 1 found in its X-ray structure. (a) Model 1 (left), vanadyl cyclam forms a coordinative bond (2.14 Å) and other H-bonds with Asp171. A weak V=O⋯H (2.3 Å) attraction is also seen between the α-carbon proton from Trp195 and the vanadyl oxygen atom. (b) Model 2 (right), lacks the coordination bond to the carboxylate of Asp171 (V⋯O 3.4 Å).
Results
Synthesis and structure
The synthesis of the sulfate complex 1 was achieved by addition of 1 mol equiv of VOSO4·xH2O to cyclam in methanol and refluxing for 24 h. The chloride complex 2 was synthesized by addition of 1 mol equiv BaCl2 to 1 in aqueous solution to precipitate the sulfate ligand as BaSO4. For complexes 3 and 4, xylylbicyclam was synthesized according to a previously published procedure33 and the sulfate and chloride complexes were obtained by procedures similar to those used for the corresponding cyclam complexes.
X-ray crystallographic data for complexes 1 and 2 were obtained at 150 K. Complex 1 (Figure 1a) crystallized as [V(IV)O(cyclam)SO4]·1.33CH3OH as the trans-III isomer (space group R3c). A definition of the six possible configurations cyclam can adopt on complexing with a metal ion (Chart 1) was proposed over 40 years ago11 and the trans-III configuration is generally considered to be the most thermodynamically stable isomer. Similarly complex 2, [V(IV)O(cyclam)Cl]Cl·CH3OH·1.5H2O (Figure 1b), also exhibited the trans-III configuration. This complex crystallized with two molecules in the asymmetric unit (space group Cc). Both cyclam complexes have a distorted octahedral geometry with sulfate in 1 and chloride in 2 as ligands trans- to the axial V=O group. Complex 1 is neutral and the sulfate ligand coordinates as a monodentate ligand. Two of the remaining sulfate oxygens form hydrogen bonds to two NH protons of the cyclam ring (Figure 1a). Crystal packing along the c axis of the unit cell shows hydrogen bonding between adjacent macrocycles via the sulfate ligand as NH⋯O⋯HN. In complex 2, crystal packing shows networks of hydrogen bonding between chlorine atoms, ring protons and solvent molecules, contributing to the stability of the trans-III configuration. The Cl-V=O angle (176.99(12)°) is significantly smaller than the corresponding O3SO-V=O angle in the sulfate complex, 1 (177.83(9)°). The N(1)-V(15)-N(8) angle (165.46(16)°) is also significantly smaller than the corresponding angle in complex 1, (166.88(7)°). Selected bond lengths and angles are given in Table 1 for comparison with EXAFS data. V-N bond lengths are comparable with similar examples for vanadium complexes found on searching the CCDC. No previous examples of double H-bonding by sulfate as an axial ligand in cyclam-type macrocycles appear to have been reported.
Figure 1.
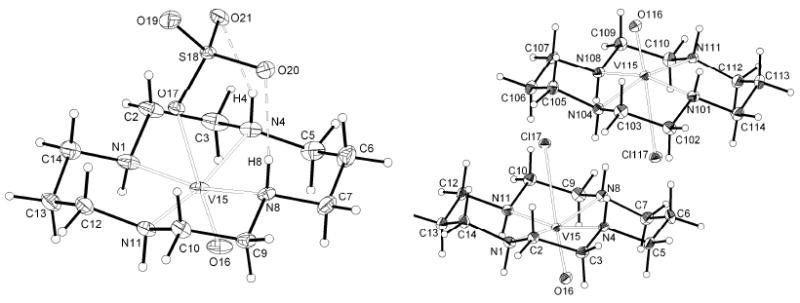
ORTEP (30% probability thermal ellipsoids) views of the sulfato complex 1 (left), and the chlorido complex 2 (right).
Table 1.
Selected Bond Distances (Å) and Angles (degrees) for Complexes 1 and 2
| Bond/Angle | Length (Å) / Angle (deg) | |
|---|---|---|
| [VO(cyclam)SO4].1.33CH3OH (1) |
[VO(cyclam)Cl]Cl.CH3OH.1.5H2O (2) |
|
| N(1)—V(15) | 2.107(2) | 2.097(4) |
| N(4)—V(15) | 2.114(2) | 2.097(4) |
| N(8)—V(15) | 2.0841(19) | 2.081(4) |
| N(11)—V(15) | 2.0976(19) | 2.082(4) |
| V(15)—O(16) | 1.6093(17) | 1.599(3) |
| V(15)—O(17) | 2.1359(16) | - |
| V(15)—Cl(17) | - | 2.6501(12) |
| O(16)—V(15)—O(17) | 177.83(9) | - |
| O(16)—V(15)—Cl(17) | - | 176.99(12) |
Infrared spectroscopy
IR spectroscopy for 1 showed a fairly high characteristic V(IV)=O vibrational stretch at 974 cm-1 suggesting that sulfate is weakly bound.20 The V(IV)=O stretch for the chloride species 2 is found at 988 cm-1. The V(IV)=O stretch for the sulfate bicyclam 3 was at 945 cm-1 and for chlorido bicyclam 4 at 964 cm-1. Both stretches are lower for the bicyclams, consistent with stronger binding of sulfate and chloride as suggested by the EPR data (vide infra).
X-ray absorption spectroscopy
Vanadium K-edge EXAFS data for the two bicyclam complexes 3 and 4 are compared with those of the cyclam analogues 1 and 2 in Figure 2 which shows the R-space EXAFS and calculated fits. Data were Fourier-transformed over k-ranges of at least 12.5 Ǻ-1; EXAFS-derived parameters were obtained from R-space fits and are provided in Table 2. For the sulfate complexes 1 and 3, two main first-coordination shells are clearly observed and are assigned as V=O and V-N/O single scattering peaks. The anticipated scattering peak for the axial V-OSO3 single scattering path is seemingly indistinguishable from that of the intense equatorial V-N scattering feature due to similarities in scattering path length and phase. For the chlorido complexes 2 and 4, three main first-coordination shells are observed in the data, assigned to the V=O, V-N and V-Cl scattering pathways. The presence of the axial V-Cl scattering peak reflects the marked differences between second-row and third-row scatterers and more pronounced differences in bond distances. Statistical parameters from the best R-space fits and all k-space data for the four complexes are given in the Supporting Information, Table S2, Figure S2. The V K-edge XANES spectra for complexes 1-4 (Figure 3) are dominated by intense pre-edge features due to formally electric dipole forbidden V3d←V1s transitions. The non-centrosymmetric environment at the V center, enforced primarily by the presence of the short V=O bond, allows for large 4p/3d mixing and thus intense pre-edge features for all complexes. The overall nature of the ligand field is very similar for all cases, with subtle differences in both transition intensities and energies (vide infra).
Figure 2.
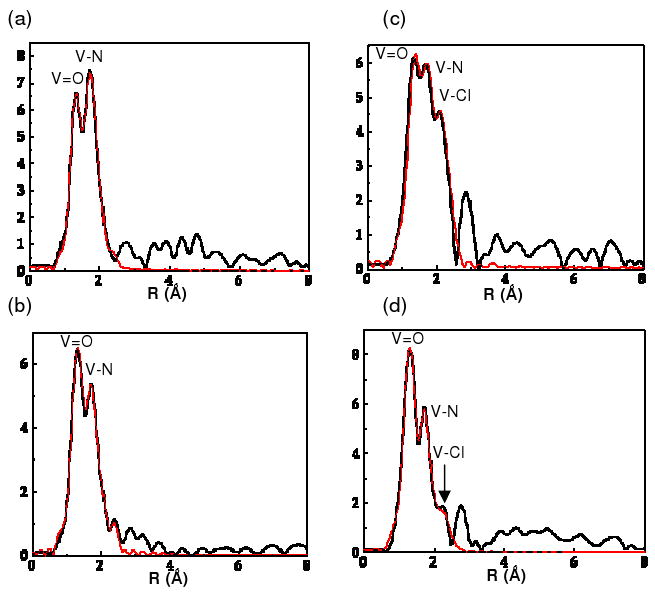
R-space (non-phase shift corrected) with calculated fits EXAFS data for (a) [VIVO(cyclam)SO4)] 1, (b) [(VIVO)2(xylylbicyclam)(SO4)2] 3, (c) [VIVO(cyclam)Cl]+ 2, and (d) [(VIVO)2(xylylbicyclam)(Cl2)]2+ 4. Key: black line, R-space; red line, R-space fit.
Table 2.
EXAFS Parameters for Oxovanadium(IV) Cyclam Complexes 1 (sulfate) and 2 (chloride) Compared with Bicyclam Complexes 3 (sulfate) and 4 (chloride)
| Complex | Scattering Pair | Na | R (Å)a | σ2a | SO2a | ΔEoa | CNc | Global SO2d |
|---|---|---|---|---|---|---|---|---|
| 1 | V-O | 1b | 1.63 | 0.00096 | 0.88 | 0.88 | 0.88 | 1.0 |
| V-N | 4b | 2.11 | 0.0016 | 0.96 | 0.19 | 3.84 | ||
| V-O (sulfate) | 1b | 2.46 | 0.0023 | 0.84 | 1.37 | 0.84 | ||
| 2 | V-O | 1b | 1.63 | 0.0034 | 1.23 | 6.94 | 1.23 | 1.0 |
| V-N | 4b | 2.11 | 0.0017 | 0.81 | 5.69 | 3.24 | ||
| V-Cl | 1b | 2.58 | 0.0044 | 1.43 | -3.38 | 1.43 | ||
| 3 | V-O | 1b | 1.62 | 0.00083 | 1.03 | -2.07 | 1.03 | 1.0 |
| V-N | 4b | 2.12 | 0.0020 | 0.80b | 1.99 | 3.20 | ||
| V-O (sulfate) | 1b | 2.59 | 0.0073 | 0.80b | 19.81* | 0.80 | ||
| 4 Fit 1 | V-O | 1b | 1.61 | 0.0011 | 1.31 | -1.40 | 1.05 | 0.8 |
| V-N | 4b | 2.13 | 0.0054 | 1.26 | 1.33 | 4.03 | ||
| V-Cl | 1b | 2.63 | 0.0052 | 0.59 | -6.45 | 0.47 | ||
| 4 Fit 2 | V-O | 1b | 1.61 | 0.0011 | 1.31 | -1.59 | 1.05 | 0.8 |
| V-N | 4b | 2.13 | 0.0059 | 1.36 | 1.68 | 4.35 | ||
| V-Cl | 1b | 2.63 | 0.011 | 1.20b | -8.37 | 0.96 |
N = path degeneracy, R = distance in Ǻ, σ2 = bond variance (related to Debye-Waller factor), SO2 = amplitude reduction parameter and ΔEo = internal reference energy.
These parameters were fixed during the fitting procedures.
Estimated coordination number for each path, where CN = N*SO2*(GSO2)
Global SO2 (GSO2) is defined as a global amplitude reduction factor (equivalent for all paths). GSO2 was adjusted in cases where CN for V-O deviated significantly from 1.
Note the ΔEo is large for this sample; however, this provided the best possible fit.
1 (k3, Δk = 2-13 Å-1, Δr = 0.7-2.5 Å), 2 (k3, Δk = 1.6-12.5 Å-1, Δr = 0.5-2.6 Å), 3 (k3, Δk = 1.7-12.5 Å-1, Δr = 0.7-2.5 Å), 4 (k3, Δk = 2-13 Å-1, Δr = 0.7-2.5 Å)
Figure 3.
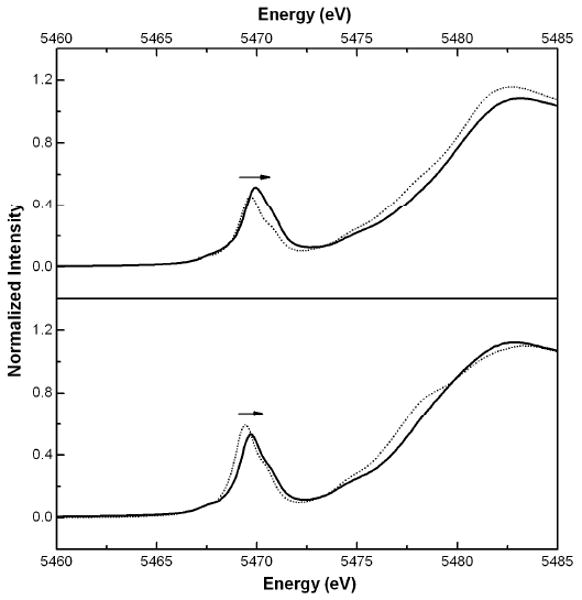
Normalized solid-state XANES spectra of the pre-edge region of the four vanadyl compounds. Top: [VIVO(cyclam)SO4)] 1 (⋯·), [(VIVO)2(xylylbicyclam(SO4)2] 3 (—). Bottom: [VIVO(cyclam)Cl]+ 2 (⋯·), [(VIVO)2(xylylbicyclam(Cl)] + 4 (—). The arrow indicates the slight increase in energy of the pre-edge feature when moving from vanadyl cyclam to bicyclam for both the Cl and SO4 compounds.
EPR spectroscopy
EPR spectroscopy revealed 8-line spectra associated with the 51V (I =7/2, 99.76% natural abundance) quadrupolar nucleus, as expected for these 3d1 complexes. Calculated A and g-values in spectra of room temperature and frozen solutions are shown in Tables 3 and 4 for cyclam and bicyclam complexes respectively, and spectra for the cyclam complexes are shown in Figure 4. Spectra for the bicyclam complexes are shown in Figure 5 (room temperature) and Figure 6 (frozen solutions). Simulated EPR spectra for all samples are shown in Figure S3 of the Supporting Information. Data were simulated using Bruker WINEPR Simphonia version 1.25 software and corrected as for the DPPH standard.31 Spectra were obtained in water and methanol and compared with a reference complex where the 6th axial ligand X is expected to be a solvent molecule. Synthesis procedures for solvent-exchanged complexes (solvent ligand denoted X) are detailed in the Supporting Information (S4). These complexes were synthesized for EPR study from the species isolated by removing the chloride ligands from complexes 2 and 4.
Table 3.
EPR parameters A(G) and g-values for Cyclam and Bicyclam Complexes in Water at 298 K and Aqua Complexes (axial ligand, X = solvent)
| Cyclam complexes | Bicyclam complexes | ||||||
|---|---|---|---|---|---|---|---|
| Axial ligand X trans- to V=O | Axial ligand X trans- to V=O | ||||||
| Parameter / Temp | Sulfate (1) | Chloride (2) | H2O | Parameter / Temp | Sulfate (3) | Chloride (4) | H2O |
| Aiso (G) | 88 | 88 | 88 | Aiso (G) | 84 | 87 | 88 |
| 298 K | 298 K | 95 | 115 | - | |||
| giso | 1.972 | 1.972 | 1.972 | giso | 1.974 | 1.972 | 1.974 |
| 298 K | 298 K | 1.972 | 1.959 | - | |||
Table 4.
EPR Parameters A(G) and g-values for Cyclam and Bicyclam Complexes in Methanol at 298 K and 77 K
| Cyclam complexes | Bicyclam complexes | |||||
|---|---|---|---|---|---|---|
| Axial ligand X trans- to V=O | Axial ligand X trans- to V=O | |||||
| Parameter / Temp | Sulfate (1) | Chloride (2) | MeOH | Parameter / Temp | Sulfate (3) | Chloride (4) |
| Aiso (G) | 88 | 88 | 88 | Aiso (G) | 90 | 90 |
| 298 K | 298 K | |||||
| giso | 1.972 | 1.972 | 1.972 | giso | 1.967 | 1.973 |
| 298 K | 298 K | |||||
| A‖ | 167 zz | 167 zz | 167 zz | A‖ | 167 zz A1 | 167 zz A1 |
| 77 K | 77 K | 174 zz A2 | ||||
| A⊥ | 53 xx | 53 xx | 53 xx | A⊥ | 53 xx A1 | 53 xx A1 |
| 77 K | 77 K | 53 xx A2 | ||||
| g | 1.968 zz | 1.968 zz | 1.968 zz | g | 1.957 zz A1 | 1.957 zz A1 |
| 77 K | 1.983 xx | 1.983 xx | 1.983 xx | 77 K | 1.972 xx A1 | 1.971 xx A1 |
| 1.950 zz A2 | ||||||
| 1.971 xx A2 | ||||||
A1 and A2 represent A(G) parameters for the overlapping sets of signals seen for oxovanadium(IV)-xylylbicyclam, 3 (1 set) and 4 (2 sets) in methanol at 77 K
Figure 4.

EPR spectra of sulfato oxovanadium(IV) cyclam 1. (a) In water at 298 K (the aqua complex, see Table 3). (b) In frozen methanol at 77 K (sulfato complex).
Figure 5.

EPR solution spectra at 298 K of oxovanadium(IV) bicyclam complexes [(VO)2(bicyclam)X2] where X = solvent, sulfate or chloride. (a) Aqua complex, (b) sulfato complex 3, and (c) chloride complex, 4. The spectra clearly show two different environments for vanadium for both the sulfato and chlorido complexes. The chlorido complex in particular shows significant overlapping signals and may be due to different ring configurations in solution, or replacement of one chlorido ligand by H2O.
Figure 6.

EPR spectra of oxovanadium(IV) bicyclam complexes [(VO)2(bicyclam)X2], where X = (a) sulfate (complex 3) and (b) chloride (complex 4), as frozen methanolic solutions at 77 K. Hyperfine couplings (A‖, z-axis) are seen for two sets of signals from complex 4 (A1 and A2) and one set of signals from complex 3 (A1). These signals cannot all be attributed to the solvent-exchanged complex [(VO)2(bicyclam)X2] where X = methanol, as only one set of signals would be seen and both complexes would give rise to signals with the same A and g values. It seems likely that only one V center of the bicyclam complexes undergoes exchange.
Antiviral testing
All four compounds were tested for antiviral activity and results are shown in Supporting Information Table S1 (cyclam complexes) and Table 5 (bicyclam complexes). The cyclam complexes 1 and 2 were inactive (IC50 values >340 μM and >360 μM respectively). The xylylbicyclam complexes 3 and 4 were highly active, although less active than AMD3100 and the Zn-complex, Zn2-xylylbicyclam. Cytotoxicity levels are low, but slightly higher than for AMD3100 and Zn2-xylylbicyclam.
Table 5.
Anti-HIV Activity and Cytotoxicity Evaluation in MT-4 Cell Cultures for Bicyclam Complexes 3 (sulfate) and 4 (chloride)
| Complex | Strain | IC50 μMa | CC50 μMb | SIc | av. IC50 μM | av. CC50 μM | SI |
|---|---|---|---|---|---|---|---|
| 3 | HIV-1 (IIIB) | 1.149 | > 151 | > 131 | |||
| 1.57 | > 151 | > 96 | 1.365 | > 151 | > 111 | ||
| HIV-2 (ROD) | 4.457 | > 151 | > 34 | ||||
| 2.138 | > 151 | > 71 | 3.297 | > 151 | > 46 | ||
| 4 | HIV-1 (IIIB) | 0.119 | > 161 | > 1351 | |||
| 0.117 | > 161 | >1379 | 0.116 | > 161 | >1365 | ||
| HIV-2 (ROD) | 0.236 | > 161 | > 683 | ||||
| 0.142 | > 161 | >1139 | 0.193 | > 161 | > 853 |
IC50: 50% inhibitory concentration of the compound required to protect 50% of the cells against the cytopathicity of HIV. For details of the method : see Ref (29)
CC50: 50% cytotoxic concentration of the compound required to reduce viability of uninfected MT-4 cells by 50%.
SI: Selectivity index or ratio of CC50 to IC50.
Co-receptor docking
Oxovanadium(IV) cyclam complex 1 was docked into an homology model of CXCR4 and two models of possible interaction were produced, in keeping with experimental observations from EPR data. Differences in bonding-interactions for the two models were noticed.
In Model 1 (Figure 7a), parameters were set with a view to investigate the possibility of metal-carboxylate interactions with Asp171 in the binding site. The vanadium center was fixed relative to the receptor and distance restraints of 2.14 Ǻ (V-sulfate bond length) with an average force constant of 500 kcal mol-1 Ǻ-2 were applied to the postulated V-O bond with Asp171. A second distance (1.609 Ǻ) was set for V=O to maintain the bond length and prevent stretching during the minimization process. Energy minimization was performed for the region of the CXCR4 receptor surrounding the oxovanadium(IV) cyclam complex 1, i.e. all amino acid residues with a sphere radius of 5 Ǻ from the cyclam ligand.
In the energy-minimized Model 1 (Figure 7a), hydrogen bonds are observed between carboxylate oxygen atoms of Asp171 and two NH protons of the cyclam ring (∼2.0 Å and 2.2 Å) and a weak attraction is observed between V=O⋯H (2.3 Å) of the α-carbon proton from Trp195 in the CXCR4 protein backbone. It is now recognised that CH⋯O hydrogen bonding can play an important role in stabilizing protein structures.34 Additionally, Trp195 is involved in a putative stacking (hydrophobic) interaction with the hydrocarbon backbone of the cyclam ring, as previously suggested for the Zn2-bicyclam adduct.18 An optimum distance between the vanadium center and the carboxylate oxygens of Asp171 gives rise to the possibility of a coordinative bond.
The EPR data suggest that complexes 1 and 2 (unlike complexes 3 and 4) may have the 6th (axial) ligand replaced by solvent. In light of these findings, Model 2 (Figure 7b) was produced, such that no distance restraint was applied between the vanadium center and carboxylate oxygens of Asp171 but docking was otherwise undertaken as in Model 1. In the energy-minimized Model 2, potential hydrogen bonds are again observed between the carboxylate oxygens and NH protons on the cyclam ring. Another weak attraction between the oxygen of V=O and the proton of the α-carbon of Trp195 is also seen, but the distance between the vanadium center and carboxylate oxygens of Asp171 (3.4Ǻ) is not compatible with formation of a coordination bond.
Discussion
Crystallographic Studies
There appear to be no previous reports of the structures of 6-coordinate oxovanadium(IV) complexes of cyclam or xylylbicyclam, although several vanadium structures do exist with ligands such as chloride trans- to the V=O group. A 5-coordinate square-pyramidal oxo-vanadium(IV) cyclam has been reported,35 but no crystal structure is available for comparison of V-N bond lengths. The V=O bonds in both 1 and 2 are short (1.6093(19) Å and 1.599(3) Å, respectively)36 and somewhat intermediate between reported bond lengths for VIV and VV. A search of the Cambridge Crystallographic Database (CCDC) revealed 21 six-coordinate complexes with V(IV)=O groups and an additional 10 containing the V(V)=O group. Bond lengths varied between 1.575 Å and 1.623 Å for VIV=O complexes, and between 1.614 Å and 1.640 Å for VV=O complexes. The V=O bond lengths of 1 and 2 are comparable with the V=O bond in the square-pyramidal oxovanadium(IV) complex [VIVO(acac)2] (1.58 Å).37 The V—O(sulfate) bond length of 2.1359(16) Å, is slightly longer than the V—O bond in [V(IV)O(acac)2] (1.97 Å).37 In complex 2, the V—Cl bond length (2.6501(12) Ǻ) is significantly longer those in examples of octahedral vanadium(IV) complexes with a chloride ligand trans- to V=O (2.465 Ǻ,38 2.468 Ǻ39) and suggests that this ligand is weakly bound. A CCDC search for similar vanadyl macrocyclic complexes with sulfate and chloride ligands in the 6th axial position did not reveal any reported examples. Three vanadyl non-manrocyclic complexes with a sulfate ligand were found and their bond lengths are compared in Table 6. In 1, the V=O bond length is comparable with the trans-V=O(SO4) complex a36 trans-[VIVO(SO4)(C23H34N4O2).5H2O, but significantly shorter than the trans-V=O(SO4) complex b38 trans-[VIVO(SO4)(bipy)2] and longer than the cis-V=O(SO4) complex c40 cis-[VIVO(SO4)(phen)2]. In 1, the V-O bond of the metal-sulfate interaction is shorter than that found for complex a where the bond length is thought to be due to a significant trans-influence of the vanadyl group. The V-O bond length in 1 is found to be significantly longer than for b and c. In 1, the corresponding O-S bond length is longer than that found in a, but shorter than in b and c.
Table 6.
Distribution of V=O Bond Lengths for CN = 6 Complexes with Sulfate as an Axial Ligand (data from the CCDC)
| Bond | [V=O cyclam] (Å) | Range (Å) | [V=O SO4]a (Å)36 | Range (Å) | [V=O SO4]b (Å)38 | [V=O SO4]c (Å)40 |
|---|---|---|---|---|---|---|
| V=O | 1.6093(17) | 1.6144-1.6042 | 1.602(5) | 1.617-1.587 | 1.638 | 1.585 |
| V-O | 2.1359(16) | 2.1407-2.1311 | 2.255(5) | 2.27-2.24 | 1.981 | 1.959 |
| O-S | 1.5050(15) | 1.5095-1.5005 | 1.483(5) | 1.498-1.468 | 1.830 | 1.523 |
trans-[VIVO(SO4)-(C23H34N4O2).5H2O
trans-[VIVO(SO4)(bipy)2]
cis-[VIVO(SO4)(phen)2]
Single crystals suitable for X-ray diffraction studies were not obtained for either the oxovanadium(IV) xylylbicyclam complexes 3 or 4, although the latter did afford a polycrystalline material.
Spectroscopic Studies
In the absence of crystallographic data, we have used V K-edge XAS to characterize structurally and compare the solid-state structures of the cyclam and bicyclam complexes. Most notably, a drastic change in the intensity of the V-Cl feature from the cyclam complex 2 to the bicyclam complex 4 is seen (see Figure 2c,d) with a concomitant increase in the V-Cl distance from 2.58 Ǻ to 2.63 Ǻ, respectively. The observed drop in intensity for the bicyclam complex suggests either the loss of chloride-ligand occupancy in the solid state structure of 4 or a large increase in disorder of the chloride ligand. To explore these two possibilities, different fits to the EXAFS data were attempted. The best fit to the first coordination sphere yielded a coordination environment consistent with VON4Cl0.5 corresponding to the loss of chloride from one of the vanadyl units in the bicyclam complex. Enforcing the anticipated VON4Cl coordination environment for each ring yielded a significantly poorer fit with a concomitant doubling of σ2 for the V-Cl pathway (Table 2). Comparisons of these fits and their associated goodness-of-fit parameters, suggests that partial loss of Cl ligation provides the most reasonable interpretation of the data. The VON4Cl0.5 formulation could indicate partial occupancy of the sixth coordination position or result from bridging chloride ligands to generate oligomeric species in the solid state. This interpretation is consistent with the observed lengthening of the V-Cl bonds by 0.05 Ǻ, and provides a rationale for problems encountered during the synthetic work-up and recrystallization process, where precipitation of insoluble products was observed. This is further supported by the near-edge XAS spectra (see Figure 3), which show only subtle changes in the 3d manifold. The most notable change is a small shift to higher energy (0.1-0.2 eV) in the pre-edge features of the bicyclams complexes as compared to their cyclam analogues. Partial loss of the axial chloride ligand in 4 as compared to 2 would be expected to result in a significant increase in the electric dipole-allowed component of the 1s→3dz2 transition associated with the short V=O bond.41, 42 Given that the overall intensity and relative energy distribution of the 1s→3d transitions remains essentially unchanged on going from the cyclam to the bicyclam, implies that the overall ligand field is not perturbed significantly.
EPR data for water and methanol solutions show that the g-values are close to 2 in all cases as expected, as the unpaired electron is in a non-degenerate orbital. Any orbital angular momentum is subsequently quenched resulting in a “spin-only” system. In addition, there should be little mixing in of an orbital component from excited states through spin-orbit coupling, leading to typical g-values close to, but slightly less than, 2. The unpaired electron is in the dxy orbital and oriented between the N atoms in the cyclam plane. As dxy is essentially non-bonding, any interaction with the nitrogen orbitals is very small and no resolved coupling to nitrogen is seen. The cyclam complexes 1, 2 show very similar spectra to the X = H2O aqua complex [VO(cyclam)(H2O)]2+ and may suggest that the 6th ligand has been replaced by solvent. As the pH values of the solutions were acidic (pH = 3), it seems likely that the axial ligand is water rather than hydroxide. This proposed ligand exchange is consistent with the X-ray crystal structure data (Table 1) where bond lengths of the axial ligand trans- to the V=O group suggest that the ligand is weakly bound. For the sulfate 3, and chloride 4 complexes, significant differences are observed. Neither set of peaks for complex 3 show values of g and A similar to that of the aqua complex, which suggests there is little substitution of the 6th ligand by solvent unlike that proposed for the cyclam complexes. The low Aiso value of 84 (G) is at the lower end of those reported for other vanadium(IV) species with O-donor ligands (85 to 110 G), although not the lowest reported (71 G).43-45 The values of giso (1.985, 1.983) fall within reported ranges.44, 45 The separate set of signals observed and confirmed by simulation may also suggest that each vanadium center is in a different environment, indicating that the macrocyclic rings may adopt different configurations. The possibility of greater delocalization onto the sulfate ligand may also result in the observed low Aiso value. Complex 4 clearly shows two sets of peaks in the room temperature spectrum. Some degree of partial substitution of chloride by water is indicated by the similar g-value and Aiso value (87 G) shown in Table 3. A larger Aiso value was found for the second set of peaks found for complex 4, (Aiso = 115 G) and suggests that a stronger interaction exists between the unpaired electron and the vanadium center. The A-value, the hyperfine coupling constant, is related to the unpaired electron density at the vanadium center. For this particular configuration, the unpaired electron may be more localized on the vanadium than for the other configuration where Aiso = 87 G. An interesting observation is that the average g and A values (energy units of cm-1) used to describe the frozen solution spectra should approximate to the isotropic g and A values (cm-1) used to describe the fluid solution spectra. There are some differences between these values in the complexes described which may be explained by different solvent interactions of the complexes with water and methanol, or may indicate that certain configurations are being ‘frozen out’ in the frozen solution spectra. Large deviations from square-pyramidal geometry and also the nature of the 6th ligand can influence the A-value and π-donation into the dxy orbital can have a large effect. In the light of these observations, it would appear that for the bicyclam complexes the retention of, or alternatively slow exchange of the 6th axial ligand, appears to affect the value of A. Previously, 2D NMR studies on zinc xylylbicyclam12 revealed an equilibrium mixture of trans-I, trans-III and cis-V configurations in solution, but such experiments were not possible with the paramagnetic vanadyl complexes 1 - 4.
Antiviral Testing
Transition-metal complexes show varying levels of anti-HIV activity which may arise partly from a relationship between the metal-cyclam geometry and the number of binding site interactions possible. Those favouring square planar geometries are found predominantly in the trans-I configuration while octahedral complexes favour trans-III. Analysis of all nickel(II) cyclam complexes in the Cambridge Structural Database (CSD) shows the most common configuration to be trans-III, so deemed the most stable configuration. Less than 23% of octahedral nickel(II) complexes with a 1,4,8,11-substituted cyclam backbone adopted the trans-I configuration compared with more than 75% preferring trans-III.46 This situation is reversed for square-planar, square-pyramidal and trigonal-bipyramidal complexes with less than 30% adopting trans-III and the majority showing a preference for the trans-I configuration. Repeated analyses for copper(II) complexes resulted in similar findings.47 A study of protein recognition of macrocycles has shown how the affinity of xylylbicyclam for CXCR4 is enhanced further by binding to Ni(II) or Zn(II).48 Anti-HIV activity is very much dependent upon the bound metal and a ZnII-xylylbicyclam complex showed slightly higher biological activity than the metal-free ligand.
Our oxovanadium(IV)-xylylbicyclam complexes 3 and 4 possess high anti-HIV activity against HIV-1 (IIIB) and HIV-2 (ROD) strains, with IC50 values in the range 1-5 μM for 3 and 0.1-0.3 μM for 4. The latter values are comparable with that of xylylbicyclam itself (AMD3100; 0.01 μM), but higher than that of Zn2-xylylbicyclam complex (0.003-0.009 μM). The reported activity of metal xylylbicyclams follows the order: Zn(II) > xylylbicyclam ∼ Ni(II) > Cu(II) > Co(III) ⋙ Pd(II), with the Pd(II) complex being virtually inactive.14, 17, 49 The oxovanadium(IV)-cyclam complexes 1 and 2 are also inactive. By exploring the structures of protein-cyclam adducts, insight into specific hydrophilic and hydrophobic interactions involved in the binding of bicyclam to CXCR4 can be gained. In addition to interactions with aspartate and glutamate residues, there is a possibility that the hydrocarbon backbone of the bicyclam rings is involved in hydrophobic interactions with tryptophan residues, Trp195 and Trp283.50 This type of interaction is comparable to that of the hydrophobic contact of the side-chain of proline with tryptophan found at some protein-protein interfaces, commonly described as a Trp-Pro-Trp sandwich.51 Recently, a configurationally-restricted bismacrocyclic CXCR4 receptor antagonist was reported to have very high activity against HIV.9 The zinc complex of an ethylene-bridged analogue of AMD3100 was found to have a lower EC50 value than AMD3100 and [Zn2-AMD3100]4+ and the highest reported activity against HIV (0.0025 μM vs 0.08 μM). It is expected that particular ring configurations may play a crucial part in determining the antiviral activity of macrocyclic complexes. Metal complexes of cyclam and bicyclam in solution give rise to several different configurational isomers, each dependent on the chirality of the 4 ring nitrogens.8 The trans-I, II, III and cis-V configurations may all have a significant influence on the bioactivity of metallo-macrocyclic antiviral compounds.9, 10, 12 The inactivity of the cyclam complexes 1, 2 suggests that the aromatic linker of the bicyclam complexes may play an important role in the co-receptor binding of complexes 3 and 4 and the anti-HIV potency. Antiviral activity of cyclam and its derivatives is influenced by metal complexation and subsequent binding to carboxylate oxygens of acidic (Glu and Asp) amino acid residues in the binding site of CXCR4.18 Like complexes 1 and 2, Pd(II)-cyclam retains a trans-III configuration in both solid state and solution and is inactive. Varying the counterion had no effect on ring configuration and addition of acetate did not induce any configurational change in vanadyl bicyclam.18 Our experiments show that additional axial ligands do not bind or bind only weakly to 1 and 2 (see Supporting Information S5, for solution IR and conductivity measurements). Our studies of these vanadyl cyclam complexes in aqueous solution suggest that they are stable, although it would be interesting to carry our further studies in the presence of potential competitor biomolecules. Our model (Figure 7) suggests that a weak attraction between the vanadyl group oxygen (V=O) and the protein backbone resulting in a new binding position for these cyclam complexes. Another study has shown that Co(III)-cyclam can adopt the trans-III configuration in the solid state with axial acetate ligands, but solution studies show the existence of three different configurations (trans-III, trans-I and a further unidentified trans- configuration).18 This species is also inactive as the axial ligands exchange too slowly. The bicyclam analogue formed intermediate species on reacting with acetate that equilibrated to the trans-III isomer with time.13, 18 In terms of anti-HIV activity, it would appear that for V(IV)=O complexes one exchangeable ligand is enough providing there is a second cyclam linked.
Co-receptor docking
Molecular modelling can offer a representation of possible binding interactions in the CXCR4 active site (see Figure 7). As bicyclam complexes 3 and 4 are active against HIV, their activity may result from binding in an analogous fashion to zinc bicyclam12, through metal-carboxylate interactions involving aspartate residues, Asp171 and Asp272, found in two separate binding sites. However, no crystal structures were available for the oxovanadium(IV)-xylylbicyclam complexes 3,4 and modelling was carried out using the oxovanadium(IV)-cyclam complex 1. Following manual docking and minimization, interactions were found that differed from those in the the Zn2-xylylbicyclam-CXCR4 model studied previously12 (i.e. Zn(II)-carboxylate 2.28 Ǻ compared with V(IV)-carboxylate 2.14 Ǻ in Model 1 and absence of this interaction in Model 2). An additional H-bonding interaction is also seen (Figure 7), specifically a weak attraction between the oxygen of V=O and the ά-carbon proton of Trp195 of CXCR4. A similarly docked oxovanadium(IV)-cyclam in the cis-V configuration in the second binding site could assist in further accounting for the activity of the bicyclam complexes 3 and 4. An attempt was made to crystallize a single oxovanadium(IV)-cyclam attached to a phenyl linker unit in order to investigate the effect of such a side-arm on the macrocycle configuration, but was unsuccessful.
Conclusions
We have reported here the synthesis and characterization of four new vanadyl cyclam complexes. The X-ray structures of the monocyclam complexes 1 and 2 appear to be the first for vanadyl cyclams. The important role of cyclam NH groups in H-bonding is illustrated by the double H-bonds formed in 1 with the uncoordinated oxygens of the axial sulfate ligand, effectively locking the macrocycle in the trans-III configuration. EXAFS data suggest that the xylylbicyclam chlorido complex 4 has a structure in which only one of the vanadyl centers has an axial chlorido ligand or that there is a bridging ligand creating a chlorido-bridged oligomeric species, consistent with difficulties in the synthesis. These structural differences may contribute to the differences in the biological activity of the chlorido and sulfato complexes. EPR data for aqueous solutions revealed two different environments for the vanadium metal center in the xylylbicyclam complexes 3 and 4 and some exchange of axial sulfato and chloride ligands with solvent. Complete exchange of the axial ligands for solvent was seen for the monocyclam complexes 1 and 2. Modelling showed that a weak attraction between the V=O group and the CXCR4 backbone may result in a longer distance between the vanadium center and carboxylate oxygens of Asp171, and consequent loss of a coordination bond, which may account for the inactivity of complexes 1 and 2. For the bicyclam complexes, these data indicate the possibility that one ring is locked in a less-favorable binding mode with the second ring bound in an alternative configuration providing further bonding interactions elsewhere, giving rise to the level of bioactivity seen for complexes 3 and 4. This, in addition to the potential role of the aromatic xylyl-linker in the bicyclam molecule may contribute to the anti-HIV activity.
Supplementary Material
Acknowledgments
We thank the BBSRC for a studentship for A Ross and support for the interdisciplinary center RASOR at the University of Edinburgh. Portions of this research were carried out at the Stanford Synchrotron Radiation laboratory, a National user facility operated by Stanford University of behalf of the US Department of Energy, Office of Basic Energy Sciences. The SSRL Structural Biology Molecular Program is supported by the Department of Energy, Office of Biological and Environmental Research and by the National Institutes of Health, National Centre for Research Resources, Biomedical Technology Programme. We thank Professors Len Lindoy (University of Sydney), David Garner (University of Nottingham) and Dr David Collison for helpful discussion, and Dr Daniel Stone for EPR simulation work at the EPSRC National EPR Centre, University of Manchester, UK.
Footnotes
Supporting Information Available: Experimental protocols for antiviral testing (and results for inactive cyclam complexes), crystallographic parameters, solution IR, conductivity tests and EPR simulations are available. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.De Clercq E. Int J Antimicrob Agents. 2009;33:307–320. doi: 10.1016/j.ijantimicag.2008.10.010. [DOI] [PubMed] [Google Scholar]
- 2.www.pfizer.co.uk/Media/Press/2007/Pages/HIVlaunch.aspx
- 3.Rusconi S, Moonis M, Merrill DP, Pallai PV, Neidhardt EA, Singh SK, Willis KJ, Osburne MS, Profy AT, Jenson JC, Hirsch MS. Antimicrob Agents Chemother. 1996;40:234–236. doi: 10.1128/aac.40.1.234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dalgleish AG, Beverley PCL, Clapham PR, Crawford DH, Greaves MF, Weiss RA. Nature. 1984;312:763–767. doi: 10.1038/312763a0. [DOI] [PubMed] [Google Scholar]
- 5.Klatzmann D, Champagne E, Chamaret S, Gruest J, Guetard D, Hercend T, Gluckman JC, Montagnier L. Nature. 1984;312:767–768. doi: 10.1038/312767a0. [DOI] [PubMed] [Google Scholar]
- 6.(a) De Clercq E. Nature Rev Drug Discov. 2003;2:581–587. doi: 10.1038/nrd1134. [DOI] [PubMed] [Google Scholar]; (b) De Clercq E. Biochem Pharmacol. 2009;77:1655–1664. doi: 10.1016/j.bcp.2008.12.014. [DOI] [PubMed] [Google Scholar]
- 7.Kimura E. Tetrahedron. 1992;48:6175–6217. [Google Scholar]
- 8.Liang X, Sadler PJ. Chem Soc Rev. 2004;33:246–266. doi: 10.1039/b313659k. [DOI] [PubMed] [Google Scholar]
- 9.Valks GC, McRobbie G, Lewis EA, Hubin TJ, Hunter TM, Sadler PJ, Pannecouque C, De Clercq E, Archibald SJ. J Med Chem. 2006;49:6162–6165. doi: 10.1021/jm0607810. [DOI] [PubMed] [Google Scholar]
- 10.Liang X, Weishaupl M, Parkinson JA, Parsons S, McGregor PA, Sadler PJ. Chem Eur J. 2003;9:4709–4717. doi: 10.1002/chem.200304808. [DOI] [PubMed] [Google Scholar]
- 11.Bosnich B, Poon CK, Tobe ML. Inorg Chem. 1965;4:1102–1108. [Google Scholar]
- 12.Liang X, Parkinson JA, Weishaupl M, Gould RO, Paisey SJ, Park Hs, Hunter TM, Blindauer CA, Parsons S, Sadler PJ. J Amer Chem Soc. 2002;124:9105–9112. doi: 10.1021/ja0260723. [DOI] [PubMed] [Google Scholar]
- 13.Paisey S, Sadler PJ. Chem Commun. 2004:306–307. doi: 10.1039/b312752b. [DOI] [PubMed] [Google Scholar]
- 14.Esté JA, Cabrera C, De Clercq E, Struyf S, Van Damme J, Bridger G, Skerlj RT, Abrams MJ, Henson G, Gutierrez A, Clotet B, Schols D. Mol Pharmacol. 1999;55:67–73. doi: 10.1124/mol.55.1.67. [DOI] [PubMed] [Google Scholar]
- 15.Aronne L, Dunn BC, Vyvyan JR, Souvignier CW, Mayer MJ, Howard TA, Salhi CA, Goldie SN, Ochrymowycz LA, Rorabacher DB. Inorg Chem. 1995;34:357–369. [Google Scholar]
- 16.Moriguchi Y, Hashimoto M, Sakata K. Bull Fukuoka Univ Educ. 1990;39:43. [Google Scholar]
- 17.Inouye Y, Kanamori T, Yoshida T, Koike T, Shionoya M, Fujioka H, Kimura E. Biol Pharm Bull. 1996;19:456–458. doi: 10.1248/bpb.19.456. [DOI] [PubMed] [Google Scholar]
- 18.Hunte TM, Paise SJ, Par Hs, Cleggorn L, Parki A, Parson S, Sadle PJ. J Inorg Biochem. 2004;98:713–719. doi: 10.1016/j.jinorgbio.2003.10.018. [DOI] [PubMed] [Google Scholar]
- 19.Rehder D. Angew Chem Int Ed Engl. 1991;30:148. [Google Scholar]
- 20.Thompson KH, McNeill JH, Orvig C. Chem Rev. 1999;99:2561–2571. doi: 10.1021/cr980427c. [DOI] [PubMed] [Google Scholar]
- 21.Crans DC, Smee JJ, Gaidamauskas E, Yang L. Chem Rev. 2004;104:849. doi: 10.1021/cr020607t. [DOI] [PubMed] [Google Scholar]
- 22.Sheldrick GM. SADABS Version 2006-1. University of Gottingen; Gottingen, Germany: 2006. [Google Scholar]
- 23.Altomare A, Cascarano G, Giacovazzo C, Guadliardi A, Burla MC, Polidori G, Camalli M. J Appl Crystallogr. 1994;27:435. [Google Scholar]
- 24.Sheldrick GM. SHELXS-97. University of Gottingen; Gottingen, Germany: 1997. [Google Scholar]
- 25.Betteridge PW, Carruthers JR, Cooper RI, Prout K, Watkin DJ. J Appl Crystallogr. 2003;36:1487. [Google Scholar]
- 26.Choi JH, Oh IG, Linder R, Schonherr T. Chem Phys 2004. 297:7. [Google Scholar]
- 27.Macrae CF, Edgington PR, McCabe P, Pidcock E, Shields GP, Taylor R, Towler M, van de Streek J. J Appl Crystallogr. 2006;39:453–457. [Google Scholar]
- 28.Farrugia LJ. J Appl Crystallogr. 1997;30:565. [Google Scholar]
- 29.Webb SM. Physica Scripta. 2005;T115:1011. [Google Scholar]
- 30.Ravel B, Newville M. J Synchrotron Rad. 2005;12:537. doi: 10.1107/S0909049505012719. [DOI] [PubMed] [Google Scholar]
- 31.Al'Tshuler SA, Kozyrev BM. In: Electron Paramagnetic Resonance. Joole CP Jr, editor. Academic Press; New York: 1964. [Google Scholar]
- 32.Pannecouque C, Daelemans D, De Clercq E. Nat Protocol. 2008;3:427–434. doi: 10.1038/nprot.2007.517. [DOI] [PubMed] [Google Scholar]
- 33.Yang W, Giandomenico CM, Sartori M, Moore DA. Tetrahedron Lett. 2003;44:2481. [Google Scholar]
- 34.Jiang L, Lai L. J Biol Chem. 2002;277:37732–37740. doi: 10.1074/jbc.M204514200. [DOI] [PubMed] [Google Scholar]
- 35.Thirumavalavan M, Martins AM. Inorg Chem Commun. 2006;9:497–499. [Google Scholar]
- 36.Ghosh S, Mukherjee M, Mukherjee AK, Mohanta S, Helliwell M. Acta Cryst. 1994;C50:1204–1207. [Google Scholar]
- 37.Shriver, Atkins . Inorganic Chemistry. 3. Oxford University Press; 1999. p. 296. [Google Scholar]
- 38.Triantafillou GD, Tolis EI, Terzis A, Deligiannakis Y, Raptopoulou CP, Sigalas MP, Kabanos TA. Inorg Chem. 2004;43:79–91. doi: 10.1021/ic034440d. [DOI] [PubMed] [Google Scholar]
- 39.Calviou LJ, Arber JM, Collison D, Garner CD, Clegg W. J Chem Soc Chem Commun. 1992;8:654–656. [Google Scholar]
- 40.Dong Y, Narla RK, Sudbeck E, Uckun FM. J Inorg Biochem. 2000;78:321–330. doi: 10.1016/s0162-0134(00)00060-x. [DOI] [PubMed] [Google Scholar]
- 41.Wong J, Lytle FW, Messmer RP, Maylotte DH. Phys Rev B. 1984;30:5596. [Google Scholar]
- 42.Giorgetti M, Passerini S, Smyrl WH, Mukerjee S, Yang XG, McBreen J. J Electrochem Soc. 1999;146:2387. [Google Scholar]
- 43.Nunes GG, Friedermann GR, Hitchcock PB, de Sa EL, Soares JF. Inorg Chim Acta. 2006;359:1435–1441. [Google Scholar]
- 44.Smith TS, LoBrutto R, Pecoraro VL. Coord Chem Rev. 2002;228:1–18. [Google Scholar]
- 45.Gahan B, Mabbs FE. J Chem Soc Dalton Trans. 1983:1695–1703. [Google Scholar]
- 46.Donnelly MA, Zimmer M. Inorg Chem. 1999;38:1650–1658. doi: 10.1021/ic980918f. [DOI] [PubMed] [Google Scholar]
- 47.Bakaj M, Zimmer M. J Mol Struct. 1999;508:59–72. [Google Scholar]
- 48.Gerlach LO, Jakobsen JS, Jensen KP, Rosenkilde MM, Skerlj RT, Ryde U, Bridger G, Schwartz TW. Biochemistry. 2003;42:710–717. doi: 10.1021/bi0264770. [DOI] [PubMed] [Google Scholar]
- 49.Inouye Y, Kanamori T, Yoshida T, Bu X, Shionoya M, Koike T, Kimura E. Biol Pharm Bull. 1994;17:243–250. doi: 10.1248/bpb.17.243. [DOI] [PubMed] [Google Scholar]
- 50.Hunter TM, McNae IW, Liang X, Bella J, Parsons S, Walkinshaw MD, Sadler PJ. Proc Natl Acad Sci USA. 2005;102:2288–2292. doi: 10.1073/pnas.0407595102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Garman SC, Sechi S, Kinet JP, Jardetzky TS. J Mol Biol. 2001;311:1049–1062. doi: 10.1006/jmbi.2001.4929. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.