

Abstract
Background
The transient outward current Ito is of critical importance in regulating myocardial electrical properties during the very early phase of the action potential. The auxiliary β subunit KCNE2 recently was shown to modulate Ito.
Objective
The purpose of this study was to examine the contributions of KCNE2 and its two published variants (M54T, I57T) to Ito.
Methods
The functional interaction between Kv4.3 (α subunit of human Ito) and wild-type (WT), M54T, and I57T KCNE2, expressed in a heterologous cell line, was studied using patch-clamp techniques.
Results
Compared to expression of Kv4.3 alone, co-expression of WT KCNE2 significantly reduced peak current density, slowed the rate of inactivation, and caused a positive shift of voltage dependence of steady-state inactivation curve. These modifications rendered Kv4.3 channels more similar to native cardiac Ito. Both M54T and I57T variants significantly increased Ito current density and slowed the inactivation rate compared with WT KCNE2. Moreover, both variants accelerated the recovery from inactivation.
Conclusion
The study results suggest that KCNE2 plays a critical role in the normal function of the native Ito channel complex in human heart and that M54T and I57T variants lead to a gain of function of Ito, which may contribute to generating potential arrhythmogeneity and pathogenesis for inherited fatal rhythm disorders.
Keywords: Cardiac arrhythmia, M54T variation, I57T variation, KCNE2, Kv4.3, Sudden cardiac death
Introduction
Classic voltage-gated K+ channels consist of four pore-forming (α) subunits that contain the voltage sensor and ion selectivity filter1,2 and accessory regulating (β) subunits.3 KCNE family genes encode several kinds of β subunits consisting of single transmembrane-domain peptides that co-assemble with α subunits to modulate ion selectivity, gating kinetics, second messenger regulation, and the pharmacology of K+ channels. Association of the KCNE1 product minK with the α subunit Kv7.1 encoding KCNQ1 forms the slowly activating delayed rectifier K+ current IKs in the heart.4,5 In contrast, association of the KCNE2 product MiRP1 with the human ether-a-go-go related gene (HERG) forms the cardiac rapid delayed rectifier K+ current IKr.6
Abbott et al reported that three KCNE2 variants (Q9E, M54T, I57T) caused a loss of function in IKr and thereby were associated with the congenital or drug-induced long QT syndrome.6,7 However, the reported QTc values in two index patients with M54T and I57T variants, both located in the transmembrane segment of MiRP1, were only mildly prolonged (390–500 ms and 470 ms).6 We recently identified the same missense KCNE2 variant, I57T, in which isoleucine was replaced by threonine at codon 57, in three unrelated probands showing a Brugada type 1 ECG. These findings are difficult to explain on the basis of a loss of function in IKr, thus leading us to explore other mechanisms.
Recent studies have demonstrated that interaction between α and β subunits (KCNEs) of voltage-gated K+ channel is more promiscuous; for example, MiRP1 has been shown to interact with Kv7.1,8–10 HCN1,11 Kv2.1,12 and Kv4.2.13 These studies suggest that MiRP1 may also co-associate with Kv4.3 and contribute to the function of transient outward current (Ito) channels.14 Indeed, a recent study reported that Ito is diminished in kcne2 (−/−) mice.15
In the human heart, Ito currents are of critical importance in regulating myocardial electrical properties during the very early phase of the action potential and are thought to be central to the pathogenesis of Brugada-type ECG manifestations.16 Antzelevitch et al demonstrated that a gain of function in Ito secondary to a mutation in KCNE3 contributes to a Brugada phenotype by interacting with Kv4.3 and thereby promoting arrhythmogenicity.14
We hypothesized that mutations in KCNE2 may have similar actions and characterize the functional consequences of interaction of wild-type (WT) and two mutant (I57T, M54T) MiRP1 with Kv4.317,18 using heterologous co-expression of these α and β subunits in Chinese hamster ovary (CHO) cells.
Methods
Heterologous expression of hKv4.3 and β subunits in CHO cells
Full-length cDNA fragment of KCNE2 in pCR3.1 vector10 was subcloned into pIRES-CD8 vector. This expression vector is useful in cell selection for later electrophysiologic study (see below). Two KCNE2 mutants (M54T, I57T) were constructed using a Quick Change II XL site-directed mutagenesis kit according to the manufacturer's instructions (Stratagene, La Jolla, CA, USA) and subcloned to the same vector. Two KCNE2 mutants were fully sequenced (ABI3100×, Applied Biosystems, Foster City, CA, USA) to ensure fidelity. Full-length cDNA encoding the short iso-form of human Kv4.3 subcloned into the pIRES-GFP (Clontech, Palo Alto, CA, USA) expression vector was kindly provided by Dr. G.F. Tomaselli (Johns Hopkins University). Full-length cDNA encoding Kv channel-interacting protein (KCNIP2) subcloned into the PCMV-IRS expression vector was a kind gift from Dr. G.-N. Tseng (Virginia Commonwealth University). KCND3 was transiently transfected into CHO cells together with KCNE2 (or M54T or I57T) cDNA at equimolar ratio (KCND3 1.5 μg, KCNE2 1.5 μg) using Lipofectamine (Invitrogen Life Technologies, Carlsbad, CA, USA) according to the manufacturer's instructions. In one set of experiments, we also co-transfected equimolar levels of KChIP2b (KCND3 1.5 μg, KCNE2 1.5 μg, KCNIP2 1.5 μg). The transfected cells were then cultured in Ham's F-12 medium (Nakalai Tesque, Inc., Kyoto, Japan) supplemented with 10% fetal bovine serum (JRH Biosciences, Inc., Lenexa, KS, USA) and antibiotics (100 international units per milliliter penicillin and 100 μg/mL streptomycin) in a humidified incubator gassed with 5% CO2 and 95% air at 37°C. The cultures were passaged every 4 to 5 days using a brief trypsin-EDTA treatment. The trypsin-EDTA treated cells were seeded onto glass coverslips in a Petri dish for later patch-clamp experiments.
Electrophysiologic recordings and data analysis
After 48 hours of transfection, a coverslip with cells was transferred to a 0.5-mL bath chamber at 25°C on an inverted microscope stage and perfused at 1 to 2 mL/min with extracellular solution containing the following (in mM): 140 NaCl, 5.4 KCl, 1.8 CaCl2, 0.5 MgCl2, 0.33 NaH2PO4, 5.5 glucose, and 5.0 HEPES; pH 7.4 with NaOH. Cells that emitted green fluorescence were chosen for patch-clamp experiments. If co-expressed with KCNE2 (or its mutants), the cells were incubated with polystyrene microbeads pre-coated with anti-CD8 antibody (Dynabeads M450, Dynal, Norway) for 15 minutes. In these cases, cells that emitted green fluorescence and had attached beads were chosen for electrophysiologic recording. Whole-cell membrane currents were recorded with an EPC-8 patch-clamp amplifier (HEKA, Lambrecht, Germany), and data were low-pass filtered at 1 kHz, acquired at 5 kHz through an LIH-1600 analog-to-digital converter (HEKA), and stored on hard disk using PulseFit software (HEKA). Patch pipettes were fabricated from borosilicate glass capillaries (Narishige, Tokyo, Japan) using a horizontal microelectrode puller (P-97, Sutter Instruments, Novato, CA, USA) and the pipette tips fire-polished using a microforge. Patch pipettes had a resistance of 2.5 to 5.0 MΩ when filled with the following pipette solution (in mM): 70 potassium aspartate, 50 KCl, 10 KH2PO4, 1 MgSO4, 3 Na2-ATP (Sigma, Japan, Tokyo), 0.1 Li2-GTP (Roche Diagnostics GmbH, Mannheim, Germany), 5 EGTA, and 5 HEPES (pH 7.2).
Cell membrane capacitance (Cm) was calculated from 5 mV-hyperpolarizing and depolarizing steps (20 ms) applied from a holding potential of −80 mV according to Equation 1 19:
| (1) |
where τc = time constant of capacitance current relaxation, I0 = initial peak current amplitude, ΔVm = amplitude of voltage step, and I∞ = steady-state current value. Whole-cell currents were elicited by a family of depolarizing voltage steps from a holding potential of −80 mV. The difference between the peak current amplitude and the current at the end of a test pulse (1-second duration) was referred to as the transient outward current. To control for cell size variability, currents were expressed as densities (pA/pF).
Steady-state activation curves were obtained by plotting the normalized conductance as a function of peak outward potentials. Steady-state inactivation curves were generated by a standard two-pulse protocol with a conditioning pulse of 500-ms duration and obtained by plotting the normalized current as a function of the test potential. Steady-state inactivation/activation kinetics were fitted to the following Boltzmann equation (Eq. 2):
| (2) |
where Y = normalized conductance or current, V1/2 = potential for half-maximal inactivation or activation, respectively, and k = slope factor.
Data relative to inactivation time constants, time to peak, and mean current levels were obtained by using current data recorded at +50 mV or +20 mV. Recovery from inactivation was assessed by a standard paired-pulse protocol: a 400-ms test pulse to +50 mV (P1) followed by a variable recovery interval at −80 mV and then a second test pulse to +50 mV (P2). Both the inactivation time constants and the time constant for recovery from inactivation were determined by fitting the data to a single exponential (Eq. 3):
| (3) |
where I(t) = current amplitude at time t, A and B = constants, and τ = inactivation time constant or time constant for recovery from inactivation. For measurement of recovery from inactivation, the plot of P2/P1 instead of I(t) was used.
All data were given as mean ± SEM. Statistical comparisons between two groups were analyzed using Student's unpaired t-test. Comparisons among multiple groups were analyzed using analysis of variance followed by Dunnett test. P <.05 was considered significant.
Results
Effects of KCNE2 on Kv4.3 currents and its gating kinetics
WT KCNE2 initially was co-expressed with KCND3, the gene encoding Kv4.3, the α subunit of the Ito channel,17,18 in CHO cells. Figure 1A shows representative whole-cell current traces recorded from cells transfected with KCND3 and co-transfected with (right) or without (left) KCNE2. Cells expressing Kv4.3 channels alone showed rapidly activating and inactivating currents. Co-expression of KCNE2 significantly reduced peak current densities as summarized in the current–voltage relationship curve shown in Figure 1B and slowed both activation and inactivation kinetics (Table 1). Figure 1C (left) shows mean time intervals from the onset of the pulse to maximum current (time to peak), whereas the right panel shows time constants of inactivation (at +20 mV) obtained using Equation 3. Thus, co-transfection of KCNE2 significantly increased both the time to peak and the time constant.
Figure 1.
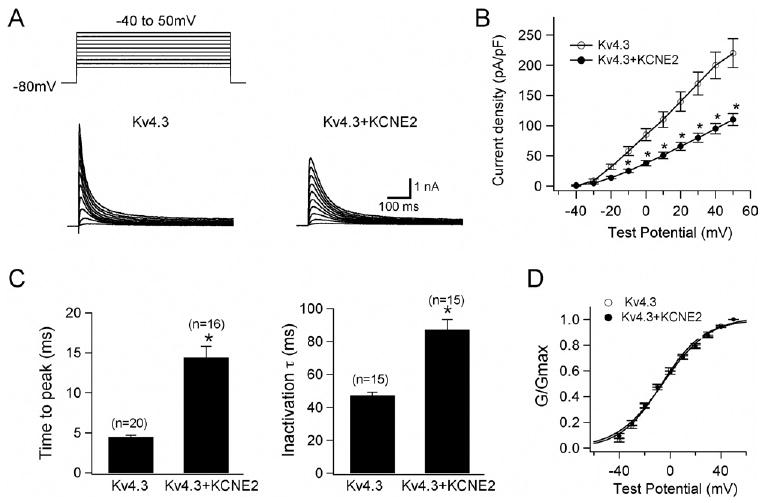
KCNE2 co-expression with Kv4.3 produces smaller Ito-like currents with slower activation/inactivation kinetics. A: Representative current traces recorded from Chinese hamster ovary (CHO) cells expressing Kv4.3 (left) and Kv4.3 + KCNE2 (right). As shown in the inset in panel A, depolarizing step pulses of 1-second duration were introduced from a holding potential of −80 mV to potentials ranging from −40 to +50 mV in 10-mV increments. B: Current–voltage relationship curve showing peak current densities in the absence and presence of co-transfected KCNE2 (*P <.05 vs Kv4.3). C: Bar graphs showing the kinetic properties of reconstituted channel currents: time to peak of activation course (left) and inactivation time constants (right) measured using test potential to +20 mV (*P <.05 vs Kv4.3). Numbers in parentheses indicate numbers of experiments. D: Normalized conductance–voltage relationship for peak outward current of Kv4.3 and Kv4.3 + KCNE2 channels.
Table 1.
Effects of KCNE2 on Kv4.3 and Kv4.3 + KChIP2b
| Parameter | Kv4.3 | Kv4.3 KCNE2 |
Kv4.3 KChIP2b |
Kv4.3 KChIP2b KCNE2 |
|---|---|---|---|---|
| Current density at +20 mV (pA/pF) | 142.0 ± 16.0 (n = 12) |
66.0 ± 6.6* (n = 12) |
191.5 ± 33.8 (n = 15) |
77.8 ± 5.9† (n = 20) |
| Steady-state activation (V0.5 in mV) | −6.5 ± 2.1 (n = 9) |
−5.5 ± 1.7 (n = 11) |
−7.5 ± 1.7 (n = 8) |
−7.4 ± 1.4 (n = 8) |
| Steady-state inactivation (V0.5 in mV) | −46.0 ± 1.3 (n = 10) |
−40.8 ± 1.7* (n = 8) |
−49.8 ± 1.4 (n = 7) |
−44.5 ± 1.9† (n = 7) |
| τ of inactivation at +20 mV (τinact in ms) | 47.3 ± 2.0 (n = 15) |
87.2 ± 6.2* (n = 15) |
47.5 ± 2.2 (n = 15) |
66.6 ± 3.5† (n = 15) |
| Time to peak at +50 mV (TtP in ms) | 4.5 ± 0.2 (n = 20) |
14.4 ± 1.4* (n = 16) |
4.1 ± 0.2 (n = 15) |
6.1 ± 0.5† (n = 21) |
| τ of recovery from inactivation (ms) | 419.6 ± 18.8 (n = 6) |
485.6 ± 74.8 (n = 6) |
89.2 ± 5.3 (n = 6) |
60.2 ± 6.9† (n = 6) |
Significantly different from Kv4.3.
Significantly different from Kv4.3 + KChIP2b.
In contrast, KCNE2 did not affect the voltage dependence of steady-state activation as assessed by plotting the normalized conductance as a function of test potential (Figure 1D). Fitting to the Boltzmann equation (Eq. 2) yielded half-maximal activation potentials of −6.5 ± 2.1 mV for Kv4.3 alone (open circles) and −5.5 ± 1.7 mV for Kv4.3 + KCNE2 channels (filled circles, P = NS; Table 1). These findings are consistent with those previously reported for studies using Xenopus oocytes, CHO cells, and HEK293 cells.20,21
KCNE2 co-expression also caused a positive shift (approximately + 5 mV) of voltage dependence of steady-state inactivation. Steady-state inactivation was assessed using a double-step pulse method (Figure 2A, inset). Peak outward currents recorded at various levels of prepulse (Figure 2A) were normalized by that measured after a 500-ms prepulse at −90 mV and are plotted as a function of prepulse test potentials (Figure 2B). Half-inactivation potentials of steady-state inactivation, determined by fitting data to the Boltzmann equation (Eq. 2), were −46.0 ± 1.3 mV for Kv4.3 (open circles) and −40.8 ± 1.7 mV for Kv4.3 + KCNE2 (filled circles, P <.01), consistent with the observation of Tseng's group.13
Figure 2.
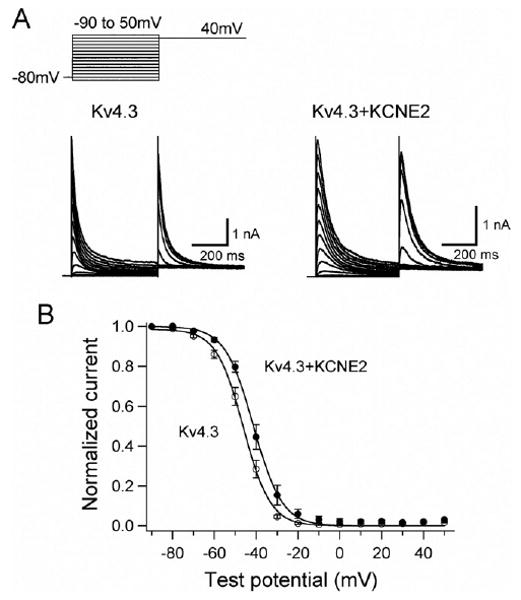
KCNE2 co-expression with Kv4.3 causes a positive shift of voltage dependence of steady-state inactivation. A: Representative Kv4.3 and Kv4.3 + KCNE2 current traces induced by 500-ms pulses (P1) from −90 to +50 mV applied from the holding potential −80 mV in 10-mV steps followed by a second pulse (P2) to +40 mV. B: Steady-state inactivation curves for Kv4.3 (open circles) and Kv4.3 + KCNE2 (closed circles) channels.
A double-pulse protocol (Figure 3A, inset) was used to test the effect of KCNE2 co-expression on the time course for recovery from inactivation. Figure 3A shows the time course of recovery of Kv4.3 alone (open circles) and Kv4.3 + KCNE2 (filled circles). Mean time constants for recovery from inactivation were not significantly different, indicating that co-transfection of KCNE2 did not affect the time course of recovery from inactivation.
Figure 3.
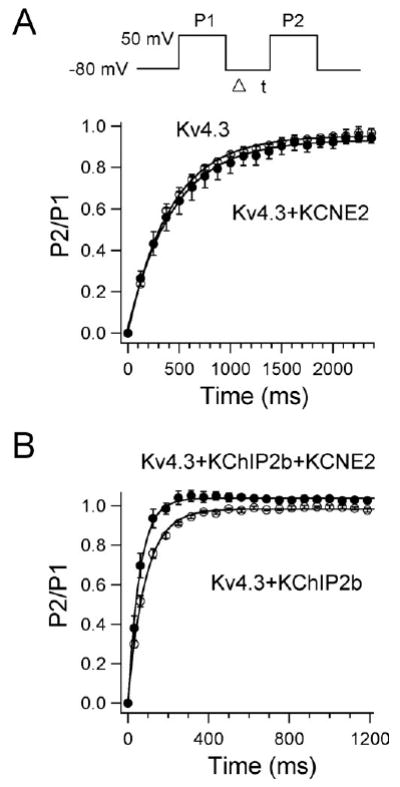
Effects of KCNE2 co-expression on recovery from inactivation of Kv4.3 (A) and Kv4.3 + KChIP2b (B) currents. Recovery from inactivation was assessed by a two-pulse protocol (A, inset): a 400-ms test pulse to +50 mV (P1) followed by a variable interval at −80 mV, then by a second test pulse to +50 mV (P2). Data were fit to a single exponential.
Effects of KCNE2 on Kv4.3 + KChIP2b current and its gating kinetics
For human native cardiac Ito, KChIP2 has been shown to serve as a principal β subunit.22–25 Accordingly, in another series of experiments, we examined the effect of WT and mutant KCNE2 on Kv4.3 + KChIP2b current. Consistent with previous reports, in the presence of KChIP2, Kv4.3 currents showed a significantly faster recovery from inactivation (Figure 3B and Table 1).26,27 Co-expression of WT KCNE2 produced similar changes on Kv4.3 + KChIP2b current as on Kv4.3 current (Table 1). Kv4.3 + KChIP2b current recovery from inactivation was further accelerated: average time constant was 89.2 ± 6.5 ms for Kv4.3 + KChIP2b alone (open circles) and 60.2 ± 8.4 ms for Kv4.3 + KChIP2b + KCNE2 (filled circles, P <.05). In 16 of 21 cells transfected with KCNE2, we observed an “overshoot” phenomenon, which is commonly seen during recording of native Ito in human ventricular myocytes.28
KCNE2 variants increase Kv4.3 + KChIP2b current and alter its gating kinetics
The I57T variant was first identified in an asymptomatic middle-aged woman with very mild QT prolongation.6 In addition to this variant, the authors reported another KCNE2 variant of the transmembrane segment (M54T) that was associated with ventricular fibrillation during exercise in a middle-aged woman. This patient appeared to show a wide range of QTc interval (390–500 ms). Therefore, we tested the functional effects of these two transmembrane KCNE2 variants on Kv4.3 + KChIP2b currents.
The three panels of Figure 4A show three sets of current traces elicited by depolarizing pulses from a holding potential of −80 mV in cells co-transfected with WT (a), I57T (b), or M54T (c) KCNE2. Neither variant caused a significant shift of half-maximal activation voltage: −7.4 ± 1.4 mV (n = 8) for co-expression of WT KCNE2, −6.1 ± 1.5 mV (n = 8) for I57T, and −6.6 ± 1.6 mV (n = 8) for M54T. Both variants significantly increased Ito density: 125.0 ± 10.6 pA/pF in WT KCNE2 (n = 21), 178.1 ± 12.1 pA/pF with I57T (n = 9), and 184.3 ± 27.9 pA/pF with M54T (n = 9, Figure 4C).
Figure 4.
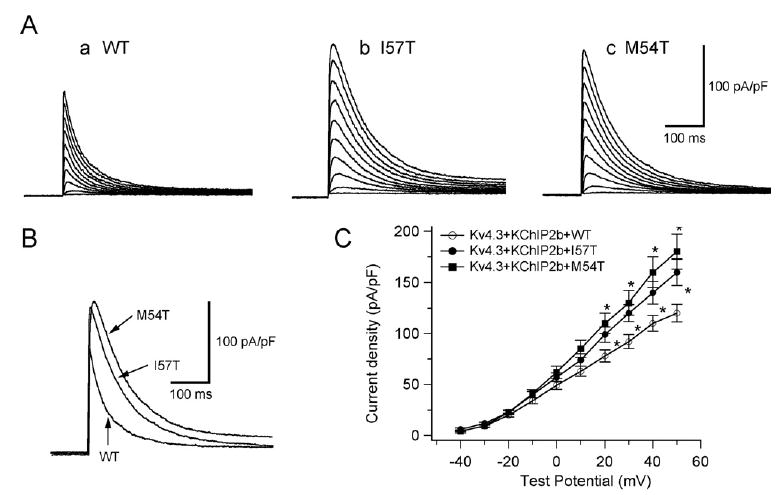
Two KCNE2 transmembrane variants, I57T and M54T, increase the reconstituted Kv4.3 + KChIP2b channel current and slow its inactivation. A: Three sets of current traces elicited by depolarizing pulses for 500 ms from a holding potential of −80 mV to potentials ranging between −40 and +50 mV in 10-mV increments (same protocol as in experiments of Figure 1A). B: Superimposition of three original current traces recorded upon depolarization showing variant-related increase in peak outward current density. C: Current–voltage relationship curve showing average peak outward current densities (*P <.05 vs Kv4.3 + KChIP2b + WT). WT = wild type.
Figure 5A shows the three traces depicted in Figure 4B normalized to their peak current level. This representation shows that the time course of inactivation of the two variant currents is slowed. The current decay was fitted by Equation 3 and the time constants (at + 20 mV) summarized in Figure 5A, panel b. Finally, Figure 5B shows that the time constants of recovery of the two mutant channels from inactivation were significantly reduced. Thus, compared to WT KCNE2, recovery of reconstituted Kv4.3 + KChIP2b channels from inactivation was significantly accelerated with both I57T and M54T mutants.
Figure 5.
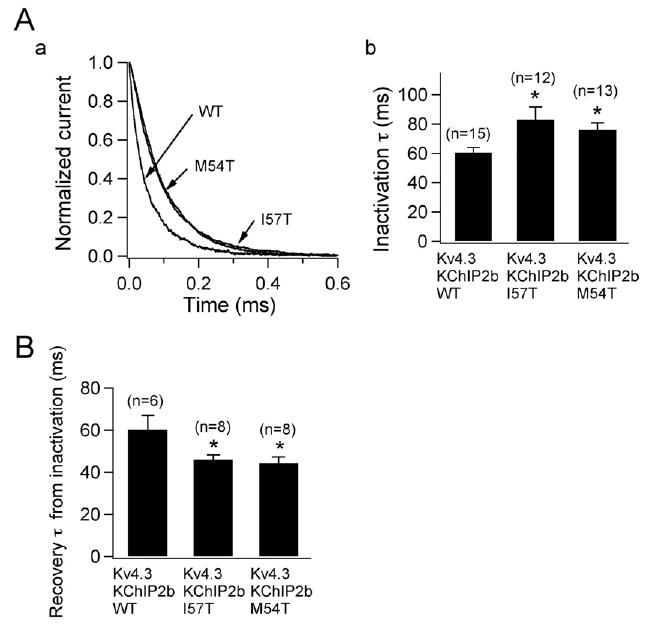
Two KCNE2 variants slow inactivation kinetics and accelerate recovery from inactivation. A, a: Three current traces obtained from Chinese hamster ovary (CHO) cells transfected with wild-type (WT), I57T, and M54T KCNE2 variant co-expressed with Kv4.3 and KChIP2b. Traces, which are normalized and superimposed, show that the variants slow inactivation. A, b: Time constants of decay at +20 mV for WT and variant KCNE2 (*P <.05 vs Kv4.3 + KChIP2b + WT). Numbers in parentheses indicate numbers of observations. B: Time constants of recovery from inactivation recorded using a double-pulse protocol (*P <.05 vs Kv4.3 + KChIP2b + WT). Numbers in parentheses indicate numbers of observations.
Discussion
Kv4.3/KChIP2/MiRP1 complex can recapitulate the native Ito
In the present study, co-expression of WT KCNE2 produced changes in kinetic properties (Figures 1–3 and Table 1) that led to close recapitulation of native cardiac Ito.28,29 Notably, in addition to causing a positive shift of steady-state inactivation (Figure 2), KCNE2 co-expression hastened the recovery of Kv4.3 + KChIP2b channels from inactivation (Figure 3). These modifications rendered Kv4.3 + KChIP2b channels more similar to native cardiac Ito, suggesting that KCNE2 may be an important component of the native Ito channel complex. In contrast to a previous observation in HEK293 cells,21 KCNE2 co-expression decreased the current density of Kv4.3 and Kv4.3 + KChIP2b channel current in the present study, which seems to be a more reasonable result as the native Ito density reportedly was smaller in isolated human heart.28 KCNE2 co-expression has also been shown to reduce the density of Kv7.18,9 and HERG6,7 channels.
Similar to the result of Deschenes and Tomaselli,21 we failed to observe an overshoot during recovery from inactivation when KCNE2 was co-expressed with Kv4.3 (Figure 3A), which is in contrast to the report of another group.13 However, co-expression of KCNE2 with Kv4.3 + KChIP2 channels produced an overshoot (Figure 3B), consistent with the report of Wettwer's group.25 Wettwer et al also found that other KCNE subunits either were ineffective or induced only a small overshoot in CHO cells. Therefore, both MiRP1 and KChIP2 subunits are sufficient and necessary to recapitulate native Ito in the heart. Considering that the overshoot phenomenon has been described only in human ventricular Ito channels of the epicardial but not endocardial region,28 these results may further implicate participation of MiRP1 and KChIP2 in the Ito channel complex in epicardium.
KCNE2 variants may alter the arrhythmogenic substrate by modulating Ito
Heterologous expression in CHO cells was conducted to examine the functional effects of I57T and M54T variants on Kv4.3 + KChIP2 channels. Both I57T and M54T KCNE2 variants significantly (1) increased peak transient outward current density (Figure 4), (2) slowed the decay of the reconstituted Ito (Figure 5A), and (3) accelerated its recovery from inactivation (Figure 5B). Both variants thus caused an important gain of function in human Ito. These sequence changes may play a role in modulating Ito and thereby predispose to some inherited fatal rhythm disorders.
Functional effects on Ito induced by I57T and M54T resemble each other, increasing Ito density and accelerating its recovery from inactivation. The gain of function in Ito opposes the fast inward Na+ currents during phase 0 of the action potential, leading to all or none repolarization at the end of phase 1 and loss of the epicardial action potential dome, thus promoting phase 2 reentry and fatal ventricular arrhythmias.30
Another KCNE2 variant (M54T) associated with fatal arrhythmias was first identified in a woman who had a history of ventricular fibrillation and varied QT intervals.6 It is possible that her arrhythmia was also related to a gain of function in Ito secondary to this variation in KCNE2. Interestingly, the I57T variant has been reported to produce a loss of function of HERG or Kv7.1 channels, thereby predisposing to long QT syndrome,6,8 indicating that the same KCNE2 variant could cause two different cardiac rhythm disorders, similar to long QT syndrome and Brugada syndrome caused by SCN5A mutations.31,32
Acknowledgments
This study was supported by grants from the Ministry of Education, Culture, Sports, Science, Technology Leading Project for Biosimulation to Dr. Horie; Health Sciences Research grants (H18-Research on Human Genome-002) from the Ministry of Health, Labour and Welfare, Japan to Drs. Shimizu and Horie; the National Natural Science Foundation of China (Key Program, No.30930105; General Program, No. 30873058, 30770785) and the National Basic Research Program of China (973 Program, No. 2007CB512005) and CMB Distinguished Professorships Award (No. F510000/G16916404) to Dr. Zang; and National Institutes of Health Grant HL47678 and Free and Accepted Masons of New York State and Florida to Dr. Antzelevitch.
Abbreviations
- CHO
Chinese hamster ovary
- HERG
human ether-a-go-go related gene
- WT
wild type
References
- 1.Kass RS, Freeman LC. Potassium channels in the heart: cellular, molecular, and clinical implications. Trends Cardiovasc Med. 1993;3:149–159. doi: 10.1016/1050-1738(93)90016-Y. [DOI] [PubMed] [Google Scholar]
- 2.MacKinnon R. Determination of the subunit stoichiometry of a voltage-activated potassium channel. Nature. 1991;350:232–235. doi: 10.1038/350232a0. [DOI] [PubMed] [Google Scholar]
- 3.Abbott GW, Goldstein SA. A superfamily of small potassium channel subunits: form and function of the MinK-related peptides (MiRPs) Q Rev Biophys. 1998;31:357–398. doi: 10.1017/s0033583599003467. [DOI] [PubMed] [Google Scholar]
- 4.Barhanin J, Lesage F, Guillemare E, Fink M, Lazdunski M, Romey G. KvLQT1 and IsK (minK) proteins associate to form the IKs cardiac potassium current. Nature. 1996;384:78–80. doi: 10.1038/384078a0. [DOI] [PubMed] [Google Scholar]
- 5.Sanguinetti MC, Curran ME, Zou AR, et al. Coassembly of KvLQT1 and minK (IKs) proteins to form cardiac IKs potassium channel. Nature. 1996;384:80–83. doi: 10.1038/384080a0. [DOI] [PubMed] [Google Scholar]
- 6.Abbott GW, Sesti F, Splawski I, et al. MiRP1 forms IKr potassium channels with HERG and is associated with cardiac arrhythmia. Cell. 1999;97:175–187. doi: 10.1016/s0092-8674(00)80728-x. [DOI] [PubMed] [Google Scholar]
- 7.Sesti F, Abbott GW, Wei J, et al. A common polymorphism associated with antibiotic-induced cardiac arrhythmia. Proc Natl Acad Sci U S A. 2000;97:10613–10618. doi: 10.1073/pnas.180223197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tinel N, Diochot S, Borsotto M, Lazdunski M, Barhanin J. KCNE2 confers background current characteristics to the cardiac KCNQ1 potassium channel. EMBO J. 2000;19:6326–6330. doi: 10.1093/emboj/19.23.6326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wu DM, Jiang M, Zhang M, Liu XS, Korolkova YV, Tseng GN. KCNE2 is colocalized with KCNQ1 and KCNE1 in cardiac myocytes and may function as a negative modulator of I(Ks) current amplitude in the heart. Heart Rhythm. 2006;3:1469–1480. doi: 10.1016/j.hrthm.2006.08.019. [DOI] [PubMed] [Google Scholar]
- 10.Toyoda F, Ueyama H, Ding WG, Matsuura H. Modulation of functional properties of KCNQ1 channel by association of KCNE1 and KCNE2. Biochem Biophys Res Commun. 2006;344:814–820. doi: 10.1016/j.bbrc.2006.03.213. [DOI] [PubMed] [Google Scholar]
- 11.Yu H, Wu J, Potapova I, et al. MinK-related peptide 1: a beta subunit for the HCN ion channel subunit family enhances expression and speeds activation. Circ Res. 2001;88:E84–E87. doi: 10.1161/hh1201.093511. [DOI] [PubMed] [Google Scholar]
- 12.McCrossan ZA, Roepke TK, Lewis A, Panaghie G, Abbott GW. Regulation of the Kv2.1 potassium channel by MinK and MiRP1. J Membr Biol. 2009;228:1–14. doi: 10.1007/s00232-009-9154-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang M, Jiang M, Tseng GN. MinK-related peptide 1 associates with Kv4.2 and modulates its gating function: potential role as beta subunit of cardiac transient outward channel? Circ Res. 2001;88:1012–1019. doi: 10.1161/hh1001.090839. [DOI] [PubMed] [Google Scholar]
- 14.Delpon E, Cordeiro JM, Nunez L, et al. Functional effects of KCNE3 mutation and its role in the development of Brugada syndrome. Circ Arrhythm Electro-physiol. 2008;1:209–218. doi: 10.1161/CIRCEP.107.748103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Roepke TK, Kontogeorgis A, Ovanez C, et al. Targeted deletion of KCNE2 impairs ventricular repolarization via disruption of IK,slow1 and I to,f. FASEB J. 2008;22:3648–3660. doi: 10.1096/fj.08-110171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Calloe K, Cordeiro JM, Di Diego JM, et al. A transient outward potassium current activator recapitulates the electrocardiographic manifestations of Brugada syndrome. Cardiovasc Res. 2009;81:686–694. doi: 10.1093/cvr/cvn339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dixon JE, Shi W, Wang HS, et al. Role of the Kv4.3 K+ channel in ventricular muscle. A molecular correlate for the transient outward current. Circ Res. 1996;79:659–668. doi: 10.1161/01.res.79.4.659. [DOI] [PubMed] [Google Scholar]
- 18.Kääb S, Dixon J, Duc J, et al. Molecular basis of transient outward potassium current downregulation in human heart failure: a decrease in Kv4.3 mRNA correlates with a reduction in current density. Circulation. 1998;98:1383–1393. doi: 10.1161/01.cir.98.14.1383. [DOI] [PubMed] [Google Scholar]
- 19.Benitah JP, Gomez AM, Bailly P, et al. Heterogeneity of the early outward current in ventricular cells isolated from normal and hypertrophied rat hearts. J Physiol. 1993;469:111–138. doi: 10.1113/jphysiol.1993.sp019807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Singleton CB, Valenzuela SM, Walker BD, et al. Blockade by N-3 polyunsaturated fatty acid of the Kv4.3 current stably expressed in Chinese hamster ovary cells. Br J Pharmacol. 1999;127:941–948. doi: 10.1038/sj.bjp.0702638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Deschênes I, Tomaselli GF. Modulation of Kv4.3 current by accessory subunits. FEBS Lett. 2002;528:183–188. doi: 10.1016/s0014-5793(02)03296-9. [DOI] [PubMed] [Google Scholar]
- 22.Wang S, Bondarenko VE, Qu Y, Morales MJ, Rasmusson RL, Strauss HC. Activation properties of Kv4.3 channels: time, voltage and [K+]o dependence. J Physiol. 2004;557:705–717. doi: 10.1113/jphysiol.2003.058578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.An WF, Bowlby MR, Betty M, et al. Modulation of A-type potassium channels by a family of calcium sensors. Nature. 2000;403:553–556. doi: 10.1038/35000592. [DOI] [PubMed] [Google Scholar]
- 24.Decher N, Uyguner O, Scherer CR, et al. hKChIP2b is a functional modifier of hKv4.3 potassium channels: cloning and expression of a short hKChIP2b splice variant. Cardiovasc Res. 2001;52:255–264. doi: 10.1016/s0008-6363(01)00374-1. [DOI] [PubMed] [Google Scholar]
- 25.Radicke S, Cotella D, Graf EM, et al. Functional modulation of the transient outward current Ito by KCNE beta-subunits and regional distribution in human non-failing and failing hearts. Cardiovasc Res. 2006;1:695–703. doi: 10.1016/j.cardiores.2006.06.017. [DOI] [PubMed] [Google Scholar]
- 26.Deschênes I, DiSilvestre D, Juang GJ, Wu RC, An WF, Tomaselli GF. Regulation of Kv4.3 current by KChIP2b splice variants: a component of native cardiac Ito? Circulation. 2002;106:423–429. doi: 10.1161/01.cir.0000025417.65658.b6. [DOI] [PubMed] [Google Scholar]
- 27.Radicke R, Vaquero M, Caballero R, et al. Effects of MiRP1 and DPP6 β-subunits on the blockade induced by flecainide of Kv4.3/KChIP2 channels. Br J Pharmacol. 2008;154:774–786. doi: 10.1038/bjp.2008.134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wettwer E, Amos GJ, Posival H, Ravens U. Transient outward current in human ventricular myocytes of subepicardial and subendocardial origin. Circ Res. 1994;75:473–482. doi: 10.1161/01.res.75.3.473. [DOI] [PubMed] [Google Scholar]
- 29.Patel SP, Campbell DL. Transient outward potassium current, “Ito,” phenotypes in the mammalian left ventricle: underlying molecular, cellular and biophysical mechanisms. J Physiol. 2005;569:7–39. doi: 10.1113/jphysiol.2005.086223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Antzelevitch C. Brugada syndrome. Pacing Clin Electrophysiol. 2006;29:1130–1159. doi: 10.1111/j.1540-8159.2006.00507.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bezzina C, Veldkamp MW, van den Berg MP, et al. A single Na+ channel mutation causing both long-QT and Brugada syndromes. Circ Res. 1999;85:1206–1213. doi: 10.1161/01.res.85.12.1206. [DOI] [PubMed] [Google Scholar]
- 32.Van den Berg MP, Wilde AA, Viersma TJW, et al. Possible bradycardic mode of death and successful pacemaker treatment in a large family with features of long QT syndrome type 3 and Brugada syndrome. J Cardiovasc Electrophysiol. 2001;12:630–636. doi: 10.1046/j.1540-8167.2001.00630.x. [DOI] [PubMed] [Google Scholar]