

Abstract
The ends of eukaryotic chromosomes have the potential to be mistaken for damaged or broken DNA and must therefore be protected from cellular DNA damage response pathways. Otherwise, cells might permanently arrest in the cell cycle, and attempts to “repair” the chromosome ends would have devastating consequences for genome integrity. This end-protection problem is solved by protein-DNA complexes called telomeres. Studies of mammalian cells have recently uncovered the mechanism by which telomeres disguise the chromosome ends. Comparison to unicellular eukaryotes reveals key differences in the DNA damage response systems that inadvertently threaten chromosome ends. Telomeres appear to be tailored to these variations, explaining their variable structure and composition.
Of the three major questions in telomere biology, two were solved in the 1980s. First, the nature of the DNA sequences that confer telomere function onto chromosome ends was revealed when Blackburn and Szostak showed that the short G-rich repeats from the ends of yeast chromosomes were sufficient to stabilize a linear plasmid (1, 2). Since then it has become clear that G-rich repeats cap the ends of most eukaryotic chromosomes, including mammalian chromosomes that end in TTAGGG repeats.
Second, the mechanism by which telomeric DNA is maintained was resolved when Blackburn and Greider showed that telomeric DNA is synthesized by telomerase. Telomerase is a reverse transcriptase that adds telomeric repeats to the 3′ ends of each chromosome (3). In doing so, telomerase makes up for the shortcomings of semiconservative DNA replication, which cannot complete the synthesis of chromosome ends. Other solutions to this end-replication problem exist, notably in Drosophila and other dipterans, but it is now clear that telomerase is the main method by which eukaryotes avoid sequence loss at the ends of their chromosomes.
It has been suggested that early eukaryotes used a primitive form of telomeres without telomerase to solve the end-replication problem (4). The later acquisition of telomerase not only solved the end-replication problem but ensured the presence of the same sequence at all chromosome ends. Once all telomeres in the cell had the same sequence, telomeric DNA binding factors could evolve, thereby enabling cells to distinguish natural chromosome ends from sites of DNA damage.
The End-Protection Problem
Research on the third major issue in telomere biology, how telomeres solve the end-protection problem, stagnated until the 1990s. The end-protection problem first surfaced early last century, when Muller and McClintock observed a critical distinction between the behavior of broken chromosome ends and telomeres. Muller found that chromosomes lacking their natural ends were unstable; McClintock documented the propensity of broken ends, but not telomeres, to fuse. However, the full extent of the end-protection problem remained obscure until the principles of the DNA damage response were revealed in the 1980s.
The first insight came when Szostak, Rothstein, and Orr-Weaver found that linear DNA introduced into eukaryotic cells is unstable because the DNA ends recombine with the genome (5). It is now clear that introduced linear DNA falls victim to two important DNA repair pathways that mend broken chromosomes: homology-directed repair (HDR) and nonhomologous end joining (NHEJ). The observation that DNA ends (also known as double-strand breaks) are processed by these DNA repair reactions raised the question of whether the natural ends of chromosomes are also attacked by HDR and NHEJ, and if not, why not.
A second question arose from the work of Hartwell and Weinert, who found that budding yeast lacking the RAD9 gene failed to arrest the cell division cycle in response to double-strand breaks (6). This experiment, and earlier observations on fission yeast and mammalian cells (7), revealed that the cell cycle arrest normally associated with DNA damage is not due to the DNA damage itself. Rather, cells arrest because of the activation of a pathway that detects DNA damage and blocks cell cycle progression in response. Why, then, are these pathways not activated by the natural ends of linear chromosomes?
These findings on how eukaryotes respond to DNA damage shaped the current molecular definition of the end-protection problem: How do telomeres prevent the activation of the DNA damage signaling pathways, and why are they resistant to the repair pathways that act on DNA ends?
In the context of mammalian cells, the end-protection problem can be rephrased in more precise terms, based on current knowledge of the molecular pathways that recognize and repair double-strand breaks (Fig. 1). Mammalian cells have two independent signaling pathways that are activated by double-strand breaks: (i) the ATM (ataxia telangiectasia mutated) kinase pathway, which is activated directly by DNA ends, and (ii) the ATR (ataxia telangiectasia and Rad3 related) kinase pathway, which is activated by the single-stranded DNA formed when the 5′ end of a double-strand break gets trimmed back, or resected. Solutions to the end-protection problem must include mechanisms that keep both kinases dormant at telomeres, because mammalian telomeres have features (both a DNA end and a constitutive region of single-stranded DNA) that could activate ATM and ATR. A second set of reactions also needs to be blocked at telomeres: Mammalian cells can repair double-strand breaks via either HDR or NHEJ, and therefore a mechanism must exist that allows telomeres to avoid these reactions.
Fig. 1.
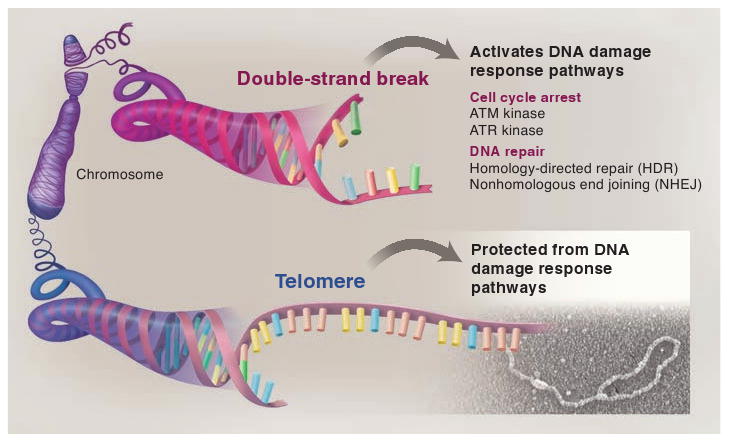
The end-protection problem. When a mammalian chromosome breaks (top), the exposed DNA ends can activate two signaling pathways (the ATM and ATR kinase pathways) that arrest the cell division cycle and can induce cell death. The broken chromosome is usually repaired by one of two different DNA repair pathways (NHEJ and HDR), allowing cells to continue their divisions with an intact genome. The presence of these DNA damage response pathways poses a problem for the ends of linear chromosomes (telomeres, bottom) because activation of DNA damage signaling or DNA repair at telomeres would be disastrous. Mammalian telomeres solve this end-protection problem through the use of a telomere-specific protein complex (shelterin) and an altered structure (the t-loop) that together ensure that all four pathways remain blocked.
The end-protection problem of mammalian chromosomes thus involves escaping the potential harmful effects of four different pathways (Fig. 1). Failure to do so will result in cell cycle arrest (under the command of ATM and/or ATR), chromosome end-to-end fusions (a product of NHEJ), or sequence exchanges (mediated by HDR) that involve two telomeres or a telomere and another part of the genome.
How Shelterin Solves the End-Protection Problem in Mammals
Mammalian telomeres solve the end-protection problem through the agency of a six-subunit protein complex called shelterin (8) (Fig. 2). Shelterin is endowed with specificity for telomeres through the DNA sequence preference of several DNA binding proteins in the complex. Two shelterin subunits, TRF1 and TRF2, bind to the TTAGGG sequences in double-stranded DNA, and one subunit, POT1, binds to these sequences in single-stranded form. Because these three proteins are held together by TIN2 and TPP1, the selectivity of shelterin for telomeric DNA is exquisite.
Fig. 2.

Mammalian telomeres. Human and mouse telomeres are composed of long stretches of the repetitive sequence TTAGGG and a telomere-specific protein complex, shelterin (upper left). Shelterin derives its specificity for telomeric DNA from three DNA binding proteins (lower left). TRF1 and TRF2 are two similar proteins that bind to the double-stranded telomeric repeats while POT1 interacts with TTAGGG repeats in single-stranded form. TIN2 and TPP1 connect POT1 to TRF1 and TRF2. Rap1 is bound to TRF2. Telomeres are found in a lariat conformation (upper right), the t-loop, which results from the strand invasion of the 3′ single-stranded overhang into the double-stranded telomeric DNA. Shelterin is sufficiently abundant to cover most of the double-stranded telomeric DNA, and there is sufficient POT1 to cover single-stranded telomeric DNA either in the 3′ overhang or in the D loop. Telomeres also contain nucleosomes and numerous shelterin-associated proteins (not shown).
The logic of the mammalian telomere system is that the repeats synthesized by telomerase function as binding sites for shelterin. As a consequence, shelterin accumulates at all natural chromosome ends, where it prevents the activation of the DNA damage response. In turn, shelterin is thought to be required for the recruitment of telomerase (9), ensuring that this enzyme does not add telomeric DNA to broken ends that lack shelterin binding sites. The sequence specificity of shelterin is critical: If it accumulated at chromosome-internal sites, it could interfere with the normal steps of the DNA damage response in case of local damage, and it might promote inappropriate “healing” of the broken ends by telomerase.
The repression of the ATM kinase pathway at telomeres is the assignment of the TRF2 subunit (Fig. 3A). Loss of TRF2 leads to activation of the ATM kinase at the natural ends of mouse or human chromosomes (10, 11). The consequences of ATM activation can be directly visualized at chromosome ends in the form of DNA damage foci containing DNA damage response factors such as γ-H2AX, MDC1, and 53BP1 (12, 13). Cells lacking TRF2 at their telomeres arrest in the cell cycle because of up-regulation of p53 and show other hallmarks of ATM signaling, including the phosphorylation of ATM and Chk2. The DNA damage response at these dysfunctional telomeres is not only completely dependent on ATM, but also requires a DNA end binding complex [;the MRN (Mre11/Rad50/Nbs1) complex] that senses double-strand breaks and activates ATM (14–16).
Fig. 3.
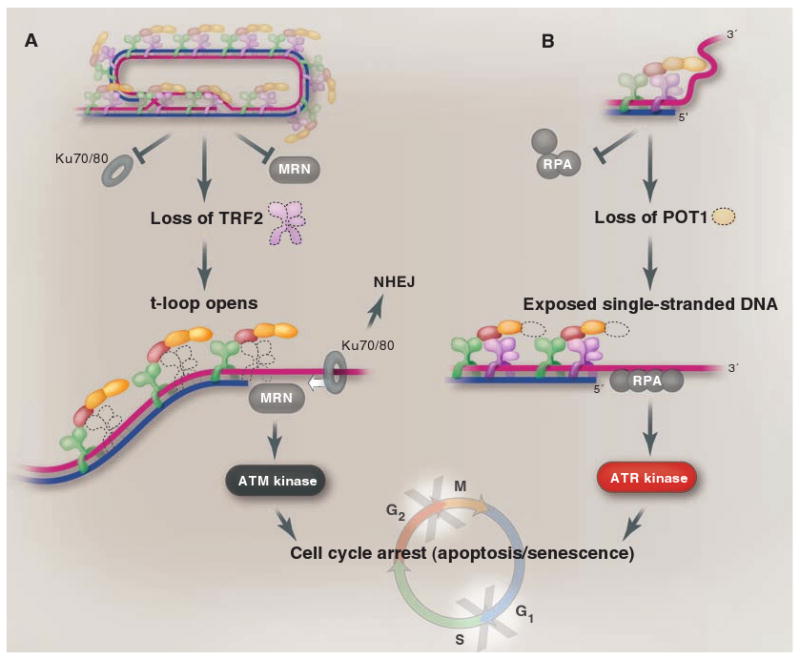
Different components of shelterin are dedicated to different aspects of the end-protection problem. TRF2 represses the ATM kinase signaling pathway (A), whereas POT1 ensures that the ATR kinase is not activated (B). In addition, TRF2 is the main repressor of NHEJ at telomeres, although POT1 contributes to the repression of NHEJ, especially after DNA replication. Both TRF2 and POT1 function to block HDR at telomeres (not shown). TRF2 is proposed to block NHEJ and ATM kinase signaling by forming the t-loop. In the t-loop structure (A), the telomere end is hidden from the DNA end sensor MRN that activates the ATM kinase pathway (MRN), and the Ku70/80 ring (which initiates NHEJ) will not be able to load onto the chromosome end. In (B), POT1 is proposed to block ATR kinase signaling by preventing the binding of RPA, the single-stranded DNA binding protein that activates the ATR kinase pathway. POT1 could block RPA from binding to the single-stranded telomeric DNA either when present at the telomere terminus (as shown) or when exposed in the D loop of the t-loop configuration.
The threat of the ATR signaling pathway is handled by POT1 (17) (Fig. 3B). Deletion of the two mouse POT1 genes results in a telomere damage response, as evidenced by DNA damage foci at telomeres and phosphorylation of the ATR target Chk1 (18, 19). The response to loss of POT1 is dependent on the ATR kinase but not on ATM. The ATM kinase remains repressed when POT1 is removed, because TRF2 is still associated with the telomeres. Thus, two different shelterin subunits independently repress the two main DNA damage signaling pathways. Together, TRF2 and POT1 distinguish telomeres from the chromosome-internal double-strand breaks that require DNA repair and modulation of cell cycle transitions.
The TRF2 and POT1 subunits are also instrumental in blocking the two DNA repair pathways that could harm telomeres (Figs. 3 and 4). The NHEJ pathway is a major threat to telomeres because it could create dicentric chromosomes when two telomeres fuse. Dicentric chromosomes are unstable in mitosis, the time when cells segregate their chromosomes during cell division, and thereby promote genome instability. In the G1 phase of the cell cycle, before DNA replication starts, TRF2 is the main repressor of NHEJ at telomeres (11, 20), whereas in the G2 phase, after DNA replication, both TRF2 and POT1 contribute to blocking this type of repair (18, 21). In addition, TRF2 and POT1 inhibit the processing of telomeres by HDR (21–24). However, HDR at telomeres can also be repressed by the Ku70/80 heterodimer, a DNA repair factor that binds to DNA ends. Thus, HDR between telomeres is only fully unleashed when both Ku70/80 is absent and either TRF2 or POT1 is deleted (22, 23).
Fig. 4.
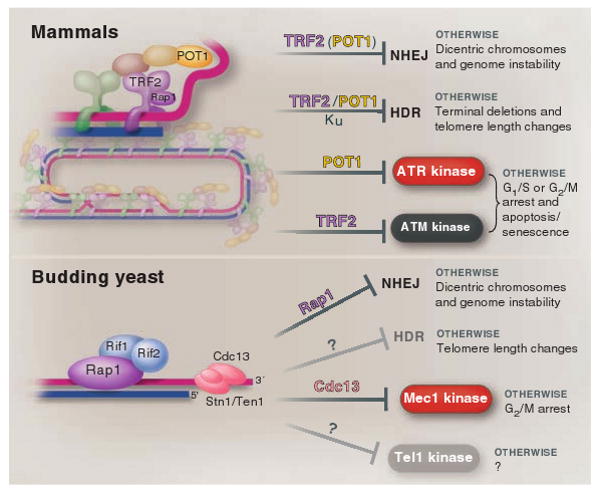
Different solutions to the end-protection problem. At mammalian telomeres, the presence of shelterin and the t-loop structure together ensure the repression of the four pathways that threaten telomeres throughout the cell cycle (top). The DNA damage response in budding yeast is not the same as in mammalian cells, hence budding yeast telomeres face a different set of threats (bottom). Whereas Mec1 (ATR equivalent) is a major threat, Tel1 (ATM-like) is not, and HDR is less stringently repressed at budding yeast telomeres than in mammals. Budding yeast telomeres appear tailored to cope with this simpler set of problems, which may explain why none of the shelterin components, except for Rap1, are conserved (bottom). Shelterin is at telomeres throughout the cell cycle, whereas Cdc13/Stn1/Ten1 is not at telomeres before the initiation of DNA replication (not shown).
The protective role of Ku70/80 at telomeres brings up a dilemma that has fascinated the field, because Ku70/80 is a component of the NHEJ pathway. Ku70/80 forms a ring-shaped complex that loads onto DNA ends and promotes the association of double-strand breaks in preparation for their ligation (25). Given its role in NHEJ, Ku70/80 could reasonably be expected to be barred from telomeres. Yet this factor binds to telomeres, probably not by loading onto the end, but rather through an interaction with shelterin (26). The current interpretation of this paradox is that shelterin might curb the actions of Ku70/80 in such a way that it becomes helpful in repressing HDR without being able to initiate NHEJ. Ku70/80 is one of several DNA repair factors that seem to have been “tamed” by shelterin to protect telomeres without engaging in activities that could pose a threat to telomeres (27).
How TRF2 and POT1 Hide the End
How does a DNA binding protein such as TRF2 prevent activation of the ATM kinase at telomere termini? A possible answer arose from an analysis of the structure of telomeric DNA in human and mouse cells, which revealed that the telomere terminus can be hidden in a configuration termed the t-loop (28) (Fig. 2). T-loops appear to form through strand invasion of the 3′ telomeric overhang into the duplex part of the telomere. Consistent with such a strand invasion, there is a short segment of single-stranded telomeric DNA at the base of the loop (the D loop). T-loops have also been found in chickens, Caenorhabditis elegans, plants, and protozoa (29–32).
Given that t-loops hide the telomere terminus, their formation and maintenance is expected to block DNA end-binding factors from gaining access to the chromosome end (Fig. 3A). In particular, t-loops could provide an architectural solution to the repression of the ATM kinase pathway, which relies on a sensor (the MRN complex) with DNA end-binding activity. In addition, t-loops could prevent the Ku70/80 heterodimer from loading onto the telomere terminus, thereby blocking the initiation of the NHEJ pathway (Fig. 3A). TRF2, which is dedicated to the repression of ATM and is a key factor for the repression of NHEJ, has the unusual ability to generate t-loop like structures in vitro (28, 33, 34). Thus, a model can be proposed wherein TRF2, through its ability to remodel telomeres into the t-loop configuration, takes the telomere terminus into custody, sheltering it from the potentially ruinous actions of MRN/ATM and the NHEJ pathways (Fig. 3A).
The above t-loop model does not explain how telomeres deter the ATR kinase, which is activated by replication protein A (RPA), which binds to single-stranded DNA. Although the t-loop sequesters the telomere terminus, binding of RPA to the single-stranded D loop could lead to the activation of the ATR kinase at telomeres.
A likely model for how telomeres block the activation of ATR is based on competition between POT1 and RPA for single-stranded DNA (17) (Fig. 3B). POT1 has the advantage over RPA that, as a component of shelterin, it can accumulate in excess of its single-stranded target sequences at telomeres. In agreement with this competition model, RPA is not normally observed at mammalian telomeres but becomes readily detectable when POT1 is impaired (35). Furthermore, POT1 can only repress the ATR kinase pathway when linked to the rest of shelterin. Inhibition of TPP1, the tether between POT1 and the rest of shelterin, also activates the ATR kinase pathway (19, 36, 17). The ability of POT1 to compete with single-strand DNA binding proteins might also play an important role in the repression of HDR, which involves binding of both RPA and the HDR factor Rad51 to single-stranded DNA.
These are speculative models that are influenced by the current understanding of the earliest steps in ATM and ATR signaling and NHEJ, and many issues remain to be addressed (Box 1). As insights into the mammalian DNA damage signaling pathways deepen, more sophisticated models and accompanying tests will emerge. Similarly, future insights into the initiation of HDR in mammalian cells will help to elucidate at which step this pathway is blocked by shelterin and the Ku70/80 heterodimer.
Box 1. Outstanding issues concerning the mammalian end-protection problem.
• What are the molecular mechanisms by which TRF2 and POT1 control ATM and ATR signaling and prevent repair by NHEJ and HDR?
• How do t-loops contribute to end protection? Are the loops lost when shelterin is impaired? Are they resolved by passage of the replication fork in the S phase of the cell cycle, or are they present throughout the cycle?
• What is the role of the many shelterin accessory factors that also function in DNA damage signaling and DNA repair? How are their potentially harmful actions repressed at telomeres?
• How is the 3′ overhang of mammalian telomeres generated? Is this process responsible for the high rate of telomere shortening in mammalian cells?
• Does telomeric repeat–containing RNA (TERRA) contribute to end protection? (TERRA has recently been observed in several eukaryotes. Its function and regulation are of potential interest in all aspects of telomere biology.)
• What happens at human telomeres during replicative senescence and crisis? What type of DNA damage response takes place at critically shortened telomeres? Which signal transducers enforce arrest? What repair pathways act on short telomeres? What are the key differences between a critically short telomere and a functional one?
• How is the length of mammalian telomeres regulated, and how is telomerase recruited? Does temporary loss of end protection (for example, in the S phase of the cell cycle) contribute to these pathways?
• How do human ALT cells bypass the repression of HDR at their telomeres? (Alternative lengthening of telomeres, or ALT, is a mechanism of telomere maintenance that relies on HDR.)
True for an Elephant, but Is It True for…?
In the context of the eukaryotic genomes, the essence of Jacques Monod's dictum (“what is true for E. coli is true for an elephant”) clearly pertains to one aspect of telomeres: the end-replication problem. Both the problem itself and its telomerase-based solution have been highly conserved during eukaryotic evolution. In contrast, the manner in which telomeres solve the end-protection problem appears to be much less conserved—most likely because the problem itself is not identical in different eukaryotes (Fig. 4).
Telomeres have been studied extensively in two types of single-celled eukaryotes to which we owe much of our current knowledge of telomerase: ciliates and yeast. These organisms also provided early hints about telomere binding proteins, the first of which was found in Oxytricha, a hypotrichous ciliate. In their vegetative nucleus (the macronucleus), these ciliates have very short telomeres (one-thousandth the length of mammalian telomeres) that are capped by a single protein dimer, TEBPα/β. TEBPα/β is distantly related to POT1 and its binding partner TPP1 in the shelterin complex (37, 38), although little is known about its function.
Two yeasts (budding yeast and fission yeast), on the other hand, have delivered both the proteins that bind to their telomeres and the phenotypes associated with their functional impairment. Fission yeast telomeres associate with a protein complex that bears similarity to shelterin (39). In this complex, a TRF-like module, Taz1, connects to a TPP1/POT1-like dimer, Tpz1/Pot1, through protein-protein interactions. The Rap1 subunit is also conserved and, as in shelterin, it binds to the TRF module, Taz1. Like mammalian TRF2, Taz1 represses the NHEJ pathway at telomeres and also acts with Ku70/80 to inhibit telomere recombination, specifically in cells lacking telomerase (40, 41). NHEJ threatens fission yeast primarily when cells are nitrogen-starved and arrest in the G1 phase of the cell cycle; when growing in rich medium, fission yeast spends most of its time in the G2 phase, where HDR dominates (42). A corollary of lingering in the G2 phase is the constant threat of telomere resection, which is blocked by Pot1 (37). Cells lacking Pot1 rapidly lose all telomeric DNA, a disastrous phenotype not (yet?) observed at mammalian chromosome ends. How fission yeast avoids the activation of Rad3 (the ATR homolog) and Tel1 (related to ATM) at its chromosome ends is not yet clear. Given that the details of the DNA damage signaling pathways are well-defined in this system, it will be particularly informative to understand at which steps fission yeast telomeres intervene.
The most extensively studied telomeres are those of budding yeast (Fig. 4). These telomeres contain two distinct telomeric complexes, one on the double-stranded telomeric DNA and one at the telomere terminus; neither of them resemble shelterin. The double-stranded DNA binding complex is formed by Rap1, the only shelterin component conserved in budding yeast (43–45), and its interacting partners Rif1 and Rif2. Unlike mammalian and fission yeast Rap1, however, budding yeast Rap1 binds directly to telomeric DNA (46, 47). Furthermore, budding yeast Rap1 has a prominent nontelomeric function in regulating transcription of numerous genes (48). At telomeres, the Rap1 complex has a well-described and highly conserved role in the regulation of telomere length (49) and contributes to the repression of NHEJ (50), a function it shares with fission yeast Rap1 (51). Whether mammalian Rap1 also inhibits NHEJ of chromosome ends remains to be determined (52).
The complex that binds to the termini of budding yeast telomeres is composed of three subunits: Cdc13, Stn1, and Ten1 (53–56). This complex binds to single-stranded telomeric DNA and appears to be a telomere-specific version of RPA, rather than being related to TPP1/POT1 (57). The Cdc13 complex is prominent at telomeres during DNA replication, when it has a role in the telomerase pathway (49) and—relevant to the end-protection problem—it limits resection of the telomere end, preventing formation of a region of single-stranded DNA. Without the Cdc13 complex, exonucleolytic attack on the 5′ end generates long regions of single-stranded DNA that activate the Mec1 kinase (related to ATR), resulting in arrest after DNA synthesis in the G2 phase (58). It is not yet clear whether Cdc13 prevents Mec1 activation primarily through limiting end resection or whether it also blocks RPA binding to the single-stranded telomeric DNA (as proposed for ATR inhibition by POT1 in mammals).
Unlike TRF2 and POT1, which are required to repress ATM and ATR throughout the mammalian cell cycle (20), budding yeast Cdc13 complex is not needed for the protection of telomeres in the G1 phase (59). How then do yeast telomeres prevent the activation of the DNA damage signaling pathways during G1? One pertinent consideration is that the budding yeast version of ATM, Tel1, like its fission yeast counterpart, has a very limited ability to enforce a G1 arrest; it may thus not pose a threat to cell cycle progression (and hence viability) when it is activated at telomeres. The task for budding yeast telomeres in the G1 phase is therefore primarily to prevent the activation of Mec1.
The dependence of Mec1 activation on single-stranded DNA may have given budding yeast a reasonable way to avoid its activation in the G1 phase: limiting the single-stranded DNA at chromosome ends. Indeed, before their replication, budding yeast telomeres do not contain enough single-stranded DNA for RPA binding and hence avoid activation of Mec1 (60). Furthermore, end-resection activities are minimal in the G1 phase, so telomeres may not be at risk in terms of activating Mec1 even when the Cdc13 complex is not bound. According to this logic, the Cdc13 complex is only needed to prevent the activation of Mec1 in the S/G2 phases, when end-resection activities rise and telomeres gain transient long 3′ overhangs (61).
How budding yeast represses HDR at its telomeres is not yet clear, but it appears that the repression is weaker than in mammalian cells. HDR events can occur spontaneously at budding yeast telomeres (62), and telomere-telomere recombination can readily compensate for telomerase deficiency (63). HDR at mammalian telomeres is more tightly restricted, and mammalian cells are consequently poor at escaping replicative senescence without telomerase (64). Unlike the large vertebrate genomes, the budding yeast chromosomes lack substantial chromosome-internal telomeric DNA; therefore, disastrous recombination events between telomeres and chromosome-internal sites should be rare, perhaps obviating the need for stringent HDR control.
These insights suggest interesting differences between budding yeast and vertebrates (and fission yeast) with regard to the end-protection problem and its solutions. Consistent with this divergence, the composition of the telomeric protein complex is distinct. TRF1 and TRF2 have been lost in budding yeast, and part of the role of POT1 has been taken over by the Cdc13 complex. The telomeric DNA itself is also quite different. Although t-loops have been demonstrated directly and indirectly in budding yeasts with unusual telomere length (65, 66), t-loops are unlikely to occur in the wild-type cells. Budding yeast telomeres have three features that may restrict the formation of t-loops: They are short (∼300 base pairs), lack a long 3′ overhang in most of the cell cycle, and are made up of imprecise repeats, limiting the options for strand invasion. It could also be argued that the advantages the t-loop has to offer as a solution to the budding yeast end-protection problem are minimal, because the t-loop structure would block the weak Tel1 pathway but is helpless against the bigger threat of the Mec1 pathway.
In addition to ciliates, yeast, and mammals, telomeres are being analyzed in chickens, Xenopus, Drosophila, C. elegans, plants, and assorted protozoa. The resulting comparative telomere biology should lead to a deeper understanding of the spectrum of challenges faced by chromosome ends and how these end-protection problems are solved by variations on the themes observed so far.
Tackling Human Telomeres
Several of the outstanding questions about mammalian telomeres (Box 1) are currently being addressed using genetic tools in the mouse. Although mouse genetics is the only way of assessing null phenotypes in the context of different genetic backgrounds (a prerequisite for understanding how telomeres work), mice have the drawback that they are not human. Human and mouse telomeres, although the same in broad strokes, differ in some detailed aspects that should caution against facile extrapolations from one system to another. For instance, whereas TRF2 and POT1 appear to work very similarly in human and mouse cells, the single POT1 gene in human cells combines the two distinct functions of the two mouse POT1 genes (18, 23), and whereas mice survive without Ku70/80, human cells perish without Ku70/80 because of deletion of their telomeres (67). These examples imply that subtle differences in the DNA damage response of human and mouse cells may dictate variations in how telomeres solve the end-protection problem.
Acknowledgments
I thank M. Wellinger, J. Petrini, J. Haber, V. Lundblad, and M. Godinho Ferreira for helpful discussion; Y. Doksani, P. Wu, F. Lottersberger, and A. Sfeir for their comments on this manuscript; and J. Griffith for providing the t-loop EM in Fig. 1. Supported by NIH grants CA076027, GM049046, and AG016642.
References and Notes
- 1.Szostak JW, Blackburn EH. Cell. 1982;29:245. doi: 10.1016/0092-8674(82)90109-x. [DOI] [PubMed] [Google Scholar]
- 2.Shampay J, Szostak JW, Blackburn EH. Nature. 1984;310:154. doi: 10.1038/310154a0. [DOI] [PubMed] [Google Scholar]
- 3.Greider CW, Blackburn EH. Cell. 1985;43:405. doi: 10.1016/0092-8674(85)90170-9. [DOI] [PubMed] [Google Scholar]
- 4.de Lange T. Nat Rev Mol Cell Biol. 2004;5:323. doi: 10.1038/nrm1359. [DOI] [PubMed] [Google Scholar]
- 5.Orr-Weaver TL, Szostak JW, Rothstein RJ. Proc Natl Acad Sci USA. 1981;78:6354. doi: 10.1073/pnas.78.10.6354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Weinert TA, Hartwell LH. Science. 1988;241:317. doi: 10.1126/science.3291120. [DOI] [PubMed] [Google Scholar]
- 7.Callegari AJ, Kelly TJ. Cell Cycle. 2007;6:660. doi: 10.4161/cc.6.6.3984. [DOI] [PubMed] [Google Scholar]
- 8.de Lange T. Genes Dev. 2005;19:2100. doi: 10.1101/gad.1346005. [DOI] [PubMed] [Google Scholar]
- 9.Stern JL, Bryan TM. Cytogenet Genome Res. 2008;122:243. doi: 10.1159/000167810. [DOI] [PubMed] [Google Scholar]
- 10.Karlseder J, Broccoli D, Dai Y, Hardy S, de Lange T. Science. 1999;283:1321. doi: 10.1126/science.283.5406.1321. [DOI] [PubMed] [Google Scholar]
- 11.Celli GB, de Lange T. Nat Cell Biol. 2005;7:712. doi: 10.1038/ncb1275. [DOI] [PubMed] [Google Scholar]
- 12.d'Adda di Fagagna F, et al. Nature. 2003;426:194. doi: 10.1038/nature02118. [DOI] [PubMed] [Google Scholar]
- 13.Takai H, Smogorzewska A, de Lange T. Curr Biol. 2003;13:1549. doi: 10.1016/s0960-9822(03)00542-6. [DOI] [PubMed] [Google Scholar]
- 14.Attwooll CL, Akpinar M, Petrini JHJ. Mol Cell Biol. 2009;29:5540. doi: 10.1128/MCB.00479-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dimitrova N, de Lange T. Mol Cell Biol. 2009;29:5552. doi: 10.1128/MCB.00476-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Deng Y, Guo X, Ferguson DO, Chang S. Nature. 2009;460:914. doi: 10.1038/nature08196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lazzerini Denchi E, de Lange T. Nature. 2007;448:1068. doi: 10.1038/nature06065. [DOI] [PubMed] [Google Scholar]
- 18.Hockemeyer D, Daniels JP, Takai H, de Lange T. Cell. 2006;126:63. doi: 10.1016/j.cell.2006.04.044. [DOI] [PubMed] [Google Scholar]
- 19.Guo X, et al. EMBO J. 2007;26:4709. doi: 10.1038/sj.emboj.7601893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Konishi A, de Lange T. Genes Dev. 2008;22:1221. doi: 10.1101/gad.1634008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wu L, et al. Cell. 2006;126:49. doi: 10.1016/j.cell.2006.05.037. [DOI] [PubMed] [Google Scholar]
- 22.Celli GB, Lazzerini Denchi E, de Lange T. Nat Cell Biol. 2006;8:885. doi: 10.1038/ncb1444. [DOI] [PubMed] [Google Scholar]
- 23.Palm W, Hockemeyer D, Kibe T, de Lange T. Mol Cell Biol. 2009;29:471. doi: 10.1128/MCB.01352-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang RC, Smogorzewska A, de Lange T. Cell. 2004;119:355. doi: 10.1016/j.cell.2004.10.011. [DOI] [PubMed] [Google Scholar]
- 25.Lieber MR, Ma Y, Pannicke U, Schwarz K. Nat Rev Mol Cell Biol. 2003;4:712. doi: 10.1038/nrm1202. [DOI] [PubMed] [Google Scholar]
- 26.Hsu HL, et al. Genes Dev. 2000;14:2807. doi: 10.1101/gad.844000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Palm W, de Lange T. Annu Rev Genet. 2008;42:301. doi: 10.1146/annurev.genet.41.110306.130350. [DOI] [PubMed] [Google Scholar]
- 28.Griffith JD, et al. Cell. 1999;97:503. doi: 10.1016/s0092-8674(00)80760-6. [DOI] [PubMed] [Google Scholar]
- 29.Cesare AJ, Quinney N, Willcox S, Subramanian D, Griffith JD. Plant J. 2003;36:271. doi: 10.1046/j.1365-313x.2003.01882.x. [DOI] [PubMed] [Google Scholar]
- 30.Munoz-Jordan JL, Cross GA, de Lange T, Griffith JD. EMBO J. 2001;20:579. doi: 10.1093/emboj/20.3.579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Murti KG, Prescott DM. Proc Natl Acad Sci USA. 1999;96:14436. doi: 10.1073/pnas.96.25.14436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Raices M, et al. Cell. 2008;132:745. doi: 10.1016/j.cell.2007.12.039. [DOI] [PubMed] [Google Scholar]
- 33.Stansel RM, de Lange T, Griffith JD. EMBO J. 2001;20:5532. doi: 10.1093/emboj/20.19.5532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Poulet A, et al. EMBO J. 2009;28:641. doi: 10.1038/emboj.2009.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Barrientos KS, et al. Mol Cell Biol. 2008;28:5251. doi: 10.1128/MCB.00048-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hockemeyer D, et al. Nat Struct Mol Biol. 2007;14:754. doi: 10.1038/nsmb1270. [DOI] [PubMed] [Google Scholar]
- 37.Baumann P, Cech TR. Science. 2001;292:1171. doi: 10.1126/science.1060036. [DOI] [PubMed] [Google Scholar]
- 38.Wang F, et al. Nature. 2007;445:506. doi: 10.1038/nature05454. [DOI] [PubMed] [Google Scholar]
- 39.Miyoshi T, Kanoh J, Saito M, Ishikawa F. Science. 2008;320:1341. doi: 10.1126/science.1154819. [DOI] [PubMed] [Google Scholar]
- 40.Ferreira MG, Cooper JP. Mol Cell. 2001;7:55. doi: 10.1016/s1097-2765(01)00154-x. [DOI] [PubMed] [Google Scholar]
- 41.Subramanian L, Moser BA, Nakamura TM. Mol Cell Biol. 2008;28:1443. doi: 10.1128/MCB.01614-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ferreira MG, Cooper JP. Genes Dev. 2004;18:2249. doi: 10.1101/gad.315804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Longtine MS, Wilson NM, Petracek ME, Berman J. Curr Genet. 1989;16:225. doi: 10.1007/BF00422108. [DOI] [PubMed] [Google Scholar]
- 44.Conrad MN, Wright JH, Wolf AJ, Zakian VA. Cell. 1990;63:739. doi: 10.1016/0092-8674(90)90140-a. [DOI] [PubMed] [Google Scholar]
- 45.Lustig AJ, Kurtz S, Shore D. Science. 1990;250:549. doi: 10.1126/science.2237406. [DOI] [PubMed] [Google Scholar]
- 46.Buchman AR, Kimmerly WJ, Rine J, Kornberg RD. Mol Cell Biol. 1988;8:210. doi: 10.1128/mcb.8.1.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li B, Oestreich S, de Lange T. Cell. 2000;101:471. doi: 10.1016/s0092-8674(00)80858-2. [DOI] [PubMed] [Google Scholar]
- 48.Shore D, Nasmyth K. Cell. 1987;51:721. doi: 10.1016/0092-8674(87)90095-x. [DOI] [PubMed] [Google Scholar]
- 49.Bianchi A, Shore D. Mol Cell. 2008;31:153. doi: 10.1016/j.molcel.2008.06.013. [DOI] [PubMed] [Google Scholar]
- 50.Marcand S, Pardo B, Gratias A, Cahun S, Callebaut I. Genes Dev. 2008;22:1153. doi: 10.1101/gad.455108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Miller KM, Ferreira MG, Cooper JP. EMBO J. 2005;24:3128. doi: 10.1038/sj.emboj.7600779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bae NS, Baumann P. Mol Cell. 2007;26:323. doi: 10.1016/j.molcel.2007.03.023. [DOI] [PubMed] [Google Scholar]
- 53.Lin JJ, Zakian VA. Proc Natl Acad Sci USA. 1996;93:13760. doi: 10.1073/pnas.93.24.13760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nugent CI, Hughes TR, Lue NF, Lundblad V. Science. 1996;274:249. doi: 10.1126/science.274.5285.249. [DOI] [PubMed] [Google Scholar]
- 55.Grandin N, Reed SI, Charbonneau M. Genes Dev. 1997;11:512. doi: 10.1101/gad.11.4.512. [DOI] [PubMed] [Google Scholar]
- 56.Grandin N, Damon C, Charbonneau M. EMBO J. 2001;20:1173. doi: 10.1093/emboj/20.5.1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gao H, Cervantes RB, Mandell EK, Otero JH, Lundblad V. Nat Struct Mol Biol. 2007;14:208. doi: 10.1038/nsmb1205. [DOI] [PubMed] [Google Scholar]
- 58.Garvik B, Carson M, Hartwell L. Mol Cell Biol. 1995;15:6128. doi: 10.1128/mcb.15.11.6128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Vodenicharov MD, Wellinger RJ. Cell Cycle. 2007;6:1161. doi: 10.4161/cc.6.10.4224. [DOI] [PubMed] [Google Scholar]
- 60.Vodenicharov MD, Wellinger RJ. Mol Cell. 2006;24:127. doi: 10.1016/j.molcel.2006.07.035. [DOI] [PubMed] [Google Scholar]
- 61.Wellinger RJ, Wolf AJ, Zakian VA. Cell. 1993;72:51. doi: 10.1016/0092-8674(93)90049-v. [DOI] [PubMed] [Google Scholar]
- 62.Teixeira MT, Arneric M, Sperisen P, Lingner J. Cell. 2004;117:323. doi: 10.1016/s0092-8674(04)00334-4. [DOI] [PubMed] [Google Scholar]
- 63.Lundblad V, Blackburn EH. Cell. 1993;73:347. doi: 10.1016/0092-8674(93)90234-h. [DOI] [PubMed] [Google Scholar]
- 64.Bodnar AG, et al. Science. 1998;279:349. doi: 10.1126/science.279.5349.349. [DOI] [PubMed] [Google Scholar]
- 65.Li B, Lustig AJ. Genes Dev. 1996;10:1310. doi: 10.1101/gad.10.11.1310. [DOI] [PubMed] [Google Scholar]
- 66.Cesare AJ, Groff-Vindman C, Compton SA, McEachern MJ, Griffith JD. Mol Cell Biol. 2008;28:20. doi: 10.1128/MCB.01122-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wang Y, Ghosh G, Hendrickson EA. Proc Natl Acad Sci USA. 2009;106:12430. doi: 10.1073/pnas.0903362106. [DOI] [PMC free article] [PubMed] [Google Scholar]