

Abstract
Silent synapses abound in the young brain, representing an early step in the pathway of experience-dependent synaptic development. Discovered amidst the debate over whether long-term potentiation reflects a presynaptic or a postsynaptic modification, silent synapses — which in the hippocampal CA1 subfield are characterized by the presence of NMDA receptors but not AMPA receptors — have stirred some mechanistic controversy of their own. Out of this literature has emerged a model for synapse unsilencing that highlights the central role for postsynaptic AMPA-receptor trafficking in the expression of excitatory synaptic plasticity.
Given the pace of research in LTP during the past few years, we may not have to wait long for the answers.
Malenka et al. (1989)1
The synapse, that richly modifiable point where one neuron passes information to another, is the basic unit of information storage in the brain. The human brain contains roughly 104 times as many synapses as it does neurons — more, perhaps, than we use. Especially during development, some synapses are incapable of neurotransmission under basal conditions and wait in a reserve capacity until an appropriate trigger activates them. In the CA1 subfield of the hippocampus, one of the most heavily studied areas in the mammalian brain, a silent synapse is defined as a synapse in which an excitatory postsynaptic current (EPSC) is absent at the resting membrane potential but becomes apparent on depolarization (FIG. 1). Silent synapses are thought to reflect the functional presence of NMDARs but not AMPARs. Because only AMPARs can conduct current at the resting membrane potential (BOX 1; FIG. 1), the absence of functional postsynaptic AMPARs renders a synapse ‘silent’ — unable to mediate synaptic transmission under physiological conditions.
Figure 1. Electrophysiological demonstration of silent synapses and their unsilencing.

A silent synapse is defined as a synapse in which an excitatory postsynaptic current (EPSC) is absent at the resting membrane potential but becomes apparent on depolarization. The traces here were obtained during whole-cell recordings from CA1 pyramidal neurons from acute rat hippocampal slices. a | (From left to right.) High-intensity stimulation evoked a fast, AMPAR-mediated EPSC at a holding potential of −60 mV. When the stimulus intensity was reduced to below the threshold for triggering EPSCs, as shown in a series of superimposed traces from repeated trials, no evoked current appeared at −60 mV. However, using the same stimulus intensity, a slow EPSC did appear at a holding potential of +30 mV; this EPSC disappeared on application of the NMDAR antagonist D-APV, indicating that it was purely mediated by NMDARs. On returning the holding potential to −60 mV, the lower-intensity stimulus again did not evoke a current. The flat traces in this series indicate failures (see BOX 2). b | The left-hand panel shows an EPSC appearing at baseline at a holding potential of +55 mV but not at −65 mV. However, after a long-term potentiation protocol, in which stimulation was paired with postsynaptic depolarization to 0 mV, EPSCs appeared at −65 mV (middle panel). As illustrated in the time course graph (right-hand panel), the number of failures diminished markedly after pairing. Part a reproduced, with permission, from REF. 3 © (1995) Elsevier Science. Part b reproduced, with permission, from REF. 2 © (1995) Macmillan Publishers Ltd. All rights reserved.
Box 1. Hippocampal neurotransmission.
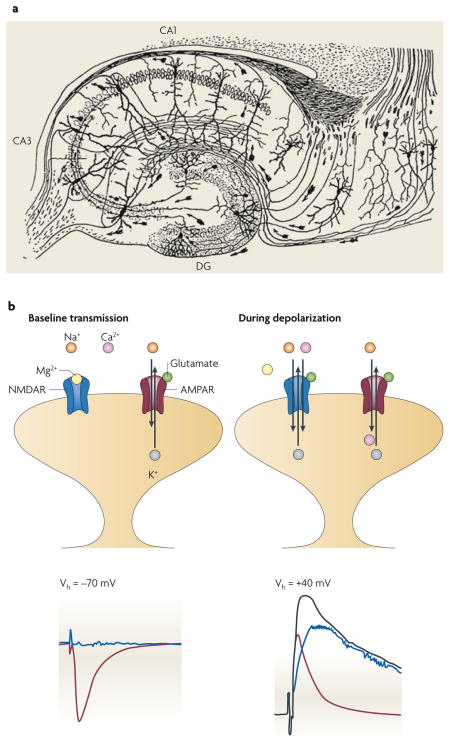
For decades the hippocampus has been a favourite brain structure of neurophysiologists. In part this is because of its central role in the consolidation of new episodic memories, and the hope that studying it will reveal the cellular and molecular mechanisms that underlie learning and memory. Another attractive feature of the hippocampus is its simple, consistent circuitry. Santiago Ramón y Cajal revealed some details of this circuitry in his classic drawing, pictured here in part a of the figure121.
There are three main types of excitatory neurons in the hippocampus: dentate gyrus (DG) granule cells project their axons (mossy fibres) to CA3 pyramidal cells. These CA3 neurons synapse recurrently onto other CA3 neurons and project axons (Schaffer collaterals) to the CA1, where they synapse onto the pyramidal cells there. These CA1 neurons convey the main output of the hippocampus proper.
Nowhere else in the brain has excitatory neurotransmission been more thoroughly described than at the synapses between Schaffer collaterals and the apical dendrites of CA1 pyramidal neurons. At these synapses, multiple types of glutamate receptors coexist (see figure, part b, upper panels), including AMPARs and NMDARs. Both are permeable to Na+ and K+, with reversal potentials close to 0 mV. NMDARs additionally exhibit important interactions with divalent cations: whereas Ca2+ is highly permeant, mediating the important signalling functions of these receptors, Mg2+ gets stuck in the pore, producing voltage-dependent NMDAR blockade at negative membrane potentials (see figure, part b, blue trace in bottom left panel). Thus, at the resting membrane potential (left-hand panels in part b), synaptic glutamate will evoke an excitatory postsynaptic current (EPSC) that is mediated almost entirely by AMPARs (bottom left panel in part b, red trace). Depolarized potentials will relieve the Mg2+ blockade, and EPSCs will subsequently contain contributions from both AMPARs and NMDARs (bottom right panel in part b, black trace). Using selective antagonists to one or the other receptor, these components can be isolated: the AMPAR component is represented by the red trace in the bottom right panel of part b; the NMDAR component is represented by the blue trace in this panel. These traces also highlight the important kinetic differences between the two receptor subtypes: whereas AMPAR-mediated currents activate quickly and decay within milliseconds, NMDAR-mediated currents activate more slowly and decay over hundreds of milliseconds. It should be noted that under physiological conditions the membrane potential of these dendritic spines never reaches positive values. Part a of the figure modified from REF. 121.
Over a decade has elapsed since the first direct observation of silent synapses in the hippocampus2,3. More exciting than the fact that they exist here — researchers have since found silent synapses almost everywhere in the brain where they have looked (FIG. 2) — was the demonstration that manipulations that were used to trigger long-term potentiation (LTP) in the hippocampus could also ‘unsilence’ these silent CA1 synapses. This finding spawned a rich body of literature on the mechanisms that underlie silent synapses and their unsilencing. In addition to providing, at last, conclusive evidence that LTP is mainly expressed postsynaptically, this literature has provided insight into the workings of the postsynaptic density, including the critical role for AMPAR trafficking in determining synaptic strength. In this Review, we describe the discovery of silent synapses, the mechanism that underlies their silence, and their role in synaptic plasticity.
Figure 2. Silent synapses throughout the brain.

Since the original discovery of silent synapses in the hippocampus2,3,54, such synapses have now been described all over the CNS. In rats, silent synapses are abundant at thalamocortical connections during postnatal days (P) 2–5 and are markedly reduced by P8–9 (REF. 122). Silent synapses exhibit a similar developmental gradient in the rat visual cortex, being easily detectable at P3–5 and significantly reduced by P9–11 (REFS 123,124). At retinocollicular connections in mice125, and at cerebellar granule cells126, nucleus tractus solitarii neurons127 and the dorsal horn of the spinal cord4,8 in rats, the prevalence of silent synapses declines over the first two postnatal weeks. Interestingly, in rats, synapses in the principal sensory nucleus of the trigeminal nerve have matured by birth, but they can be silenced by postnatal deafferentation128. At all of these anatomical locations, as in the hippocampal CA1 subfield, silent synapses were signified by the presence of an NMDAR-mediated excitatory postsynaptic current (EPSC) and the absence of AMPAR-mediated EPSCs. Silent synapses and their unsilencing seem to be pervasive features of excitatory transmission, contributing to synaptic development and plasticity.
Discovery of silent synapses
Patrick Wall and his student, Eugene Merrill, were the first to propose the existence of “ineffective synapses” between primary afferent sensory fibres and the spinal cord dorsal horn, after they found that some presynaptic stimuli could not trigger postsynaptic firing in spinal-cord neurons4. Further research showed that after transection and subsequent degeneration of a subset of presynaptic fibres, previously ineffective synapses became reliable signal conductors5. In these studies, the authors could not conclude whether the inability to trigger postsynaptic action potentials reflected a failure of presynaptic transmitter release or a subthreshold or absent postsynaptic response6–8.
In part to address this issue, Faber and colleagues studied silent synapses on the goldfish Mauthner cell9. These giant brainstem neurons mediate the escape reflex in fish and receive inhibitory inputs from local glycinergic interneurons and excitatory inputs from vestibulocochlear (VIIIth) nerve afferent fibres. Paired recordings from presynaptic interneurons and postsynaptic Mauthner cells revealed that transmission failed to occur in a large percentage of cases, despite there being morphological evidence of synapse formation between the recorded cells; this suggested the presence of silent synapses10. Synapse unsilencing occurred after postsynaptic injection of cyclic AMP11, but not in response to direct stimulation of presynaptic fibres by injection of Ca2+ or of a K+ channel antagonist10. Together, these data suggested that glycinergic silent synapses comprise a functional presynaptic bouton and a non-functional postsynaptic membrane. Many of the VIIIth-nerve excitatory synapses onto Mauthner cells were also silent. Presynaptic injection of K+ channel antagonists unsilenced these synapses, presumably by enhancing Ca2+ influx and amplifying the triggers for vesicle release; this indicated that, in contrast to the postsynaptically silent glycinergic synapses, these glutamatergic synapses were presynaptically silent12,13. At approximately the same time that this research was being carried out, other researchers described presynaptically silent synapses at the crayfish neuromuscular junction. At these synapses, prolonged high-frequency stimulation resulted in the creation of new sites of vesicle release, as revealed by both quantal analysis and ultrastructural study14,15. These studies established two themes: first, that although silent synapses are common among different species and different areas of the nervous system, the mechanisms that underlie synaptic silence might vary6; and second, that synaptic activity is the trigger for unsilencing.
LTP and silent synapses in the hippocampus
The discovery of silent synapses in hippocampal CA1 pyramidal neurons took place amidst a debate over whether the locus of LTP expression is pre- or postsynaptic. LTP is the prototypical model of synaptic plasticity, in which coincident presynaptic activity and postsynaptic depolarization trigger enhanced synaptic transmission; it could potentially be explained by a presynaptic increase in glutamate release16 or by a postsynaptic increase in responsiveness to glutamate17. This pre- versus postsynaptic controversy, which so dominated the hippocampal physiology literature of the 1980s and 1990s, persisted despite the multitude of physiological techniques that competing research groups applied to the question.
The problem was that these techniques yielded what seemed to be contradictory results. Many researchers had observed that LTP involves an augmentation of AMPAR-mediated EPSCs (A-EPSCs), with little or no increase in NMDAR-mediated EPSCs (N-EPSCs)18–21. Because both receptor subtypes colocalize at the post-synaptic membrane22 and bind synaptic glutamate, this finding suggested that some postsynaptic modification favouring the activation of AMPARs must underlie LTP. Countering this argument was the theoretical notion that weak presynaptic glutamate release should engage only NMDARs, because they bind glutamate with a higher affinity than AMPARs23, whereas a more robust glutamate signal should activate both types of receptor. However, the synaptic glutamate concentration peaks quickly after vesicle release, and by the time glutamate dissipates by diffusion and uptake, it has not had time to achieve equilibrium with postsynaptic receptors. Taking into account the fact that NMDARs have much slower activation kinetics than AMPARs (BOX 1), the apparent affinities of these two receptor types for glutamate converge in these non-equilibrium conditions24,25 (but see REF. 26), making it less likely that there is a physiological scenario in which glutamate would activate NMDARs without any trace of AMPAR activation. Moreover, manipulations that selectively affect presynaptic activity — such as increasing stimulus intensity or applying the GABARB (γ-aminobutyric acid type B receptor) agonist baclofen, the adenosine antagonist theophylline or phorbol esters — produced parallel changes in A-EPSCs and N-EPSCs over a range of amplitudes that were even broader than the amplitudes that are typically encountered during LTP20,24,27,28. In addition, LTP did not trigger a change in paired pulse facilitation and therefore did not seem to involve any change in the probability of transmitter release ( p)20,28 (BOX 2). These results argued against a presynaptic mechanism for LTP.
Box 2. Quantal theory.
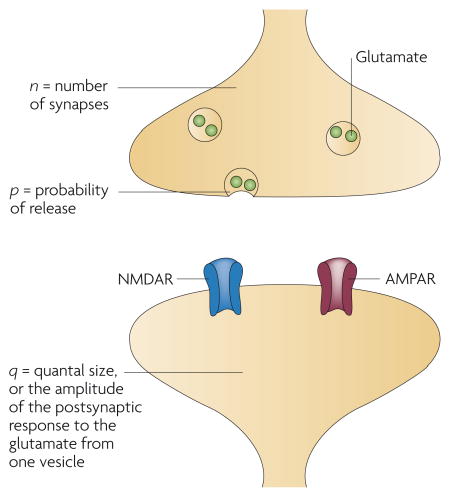
The minimal magnitude of neurotransmission that can occur at a synapse is the release of a single vesicle of neurotransmitter. Because the amount of neurotransmitter (for example, glutamate) is roughly the same from vesicle to vesicle, the postsynaptic response is graded by quantal steps, the biological principle on which ‘quantal analysis’ is based. Quantal analysis can be performed most directly by isolating the postsynaptic response to a single quantum of neurotransmitter, typically by blocking action-potential firing with tetrodotoxin (TTX) and waiting for spontaneous, non-action-potential-initiated vesicle release to occur onto a recorded neuron. In this scenario the amplitude, or quantal size (q), of a so-called miniature excitatory postsynaptic current (mEPSC) corresponds to the glutamate sensitivity of the postsynaptic membrane (that is, the number and conductance of its AMPARs), whereas the frequency with which mEPSCs occur is thought to reflect the probability of vesicle release from the presynaptic terminal (p) and the number of release sites (n). (n is probably equivalent to the number of synapses, because most central synapses contain only one release site each.) Quantal content, or n·p, should represent the total presynaptic output.
Quantal analysis can also be performed on stimulus-evoked EPSCs in the absence of TTX, on the premise that the EPSC is the summation of multiple quanta. If trial-to-trial fluctuations in EPSC amplitude reflect stochastic differences in the number of quanta that are released, then these amplitudes should distribute in a multimodal, quantal manner. According to this simple model, the coefficient of variation (CV) (which is the standard deviation of the EPSC amplitudes normalized to the mean amplitude) varies oppositely with quantal content, and the inverse square,CV−2, is directly proportional to quantal content. As discussed in the text, quantal theory was originally developed in studies of the neuromuscular junction. Because central synapses differ from those at the neuromuscular junction in a number of fundamental ways that go beyond the scope of this Review, some of the assumptions on which quantal theory is based might not apply to glutamatergic synapses (see the references cited in the main text).
At CA1 synapses transmission is quite unreliable, with a p of approximately 0.3; this means that most action potentials do not result in glutamate release (they are ‘synaptic failures’) and that the ones that are successful rarely result in the release of more than one vesicle. Paired pulse facilitation occurs, in theory, when the Ca2+ influx at a presynaptic bouton has not entirely cleared before the next action potential arrives, resulting in a higher peak presynaptic Ca2+ concentration after this second stimulus than was achieved by the first; a higher Ca2+ concentration increases p and results in the facilitation of the EPSC. Presynaptic Ca2+ concentrations quickly return to baseline values, accounting for the limited time window between the first and second stimulus during which paired pulse facilitation can be observed.
Quantal content provides a direct read-out of p in the simplest experimental scenario, in which only one synapse is activated and n = 1 (see figure). But quantal content is not always a faithful presynaptic parameter: in a scenario in which more synapses are sampled in one phase of an experiment than in another (for example, after synapse unsilencing by a long-term potentiation protocol), n will seem to rise, resulting in an increased quantal content even if p remains constant. This increase in n could reflect a true increase in the number of release sites (presynaptic unsilencing) or an increase in the number of functional postsynaptic units that respond to a constant number of release sites (postsynaptic unsilencing); because n does not distinguish between these two possibilities, a change in quantal content — and consequently in CV−2 — cannot be used in isolation as a means to determine whether a change in synaptic strength has a pre- or postsynaptic locus. To take this reasoning one step further, in the scenario in which multiple synapses are activated, some containing both AMPARs and NMDARs and some containing only NMDARs, the quantal content (and thus the CV−2) for AMPAR-mediated EPSCs will seem to be smaller than that for NMDAR-mediated EPSCs21 — a result that would be confusing if these variables were wrongly assumed to reflect only the state of the presynaptic terminal.
As convincing as the above arguments for a post-synaptic mechanism for LTP may have been, contemporary sentiments clearly lay on the other side of the synapse. What made a presynaptic mechanism for LTP so persuasive? It boiled down to a single observation, namely that the trial-to-trial variance of EPSC amplitudes declined after LTP. According to quantal theory (BOX 2), this reduced variance — manifested as a decrease in the frequency of synaptic failures (that is, instances in which stimulation does not result in transmission) and an increase in the index CV−2 (the inverse square of the coefficient of variation of EPSC amplitudes) — should correlate with presynaptic changes but not with postsynaptic sensitivity29,30 (see also REFS 31–33). Other research groups also observed LTP-triggered increases in quantal content (n·p; in which n is the number of release sites), either alone or in concert with increases in quantal size (q; see BOX 2)34–37. There were concerns over the validity of using classical methods of quantal analysis at central synapses — relating to, among other aspects, the applicability of binomial analysis and the influence of dendritic filtering on q7,16,25,38–41 (but see REFS 42–44) — but these concerns alone could not completely invalidate all of the arguments for a presynaptic mechanism for LTP. For instance, it was difficult to dispute the fact that the frequency of synaptic failures dropped dramatically after LTP, which in theory reflects an increased p29,34,35,45.
This apparent contradiction — that different measures of quantal content produced opposite results — was highlighted by the conflicting observations that CV−2 increased after LTP in the same experiments in which paired pulse facilitation remained constant46. Moreover, CV−2 seemed to change for A-EPSCs but not for N-EPSCs21 (but see REF. 47). Such observations were crucial to the realization that some unmeasured phenomenon must confound the perception of ‘quantal content’ (BOX 2).
Silent synapses came to the rescue. Several research groups21,34–36,39,48,49 speculated that the activation of silent synapses could explain all of the prior findings: by increasing the number of active synapses that contribute to EPSCs after LTP has been triggered, synapse unsilencing mimics both an increase in n, leading to an apparent increase in quantal content, and a decrease in the failure rate (BOX 2). The rise in quantal content would occur with no change in p, accounting for the absence of any change in paired pulse facilitation. This theory was supported by the observation that the only experimental manipulation that could recreate the disparate effects on CV−2 and paired pulse facilitation was increasing the stimulus intensity, which causes additional synapses to be recruited46.
Indeed, at stimulation intensities that were just below the threshold that is necessary to elicit EPSCs in CA1 neurons at the resting membrane potential (which would indicate an absence of an AMPAR-mediated response), switching to a positive holding potential revealed an EPSC that could be blocked entirely by NMDAR antagonists2,3 (FIG. 1). In other words, the synapse(s) that was activated by this low-level stimulation seemed to contain only NMDARs. In the same series of experiments, pairing repetitive presynaptic stimulation with postsynap-tic depolarization, a protocol that reliably induces LTP in conventional experiments, resulted in the recruitment of A-EPSCs that persisted stably for the rest of the recording. Such activity-induced unsilencing of silent synapses follows Hebb’s rule of association, requires NMDAR activation and calcium/calmodulin-dependent protein kinase II (CaMKII)50,51 and seems to be identical to the phenomenon of LTP itself.
Silent synapses in neuronal development
One pervasive feature of silent synapses is that there is a developmental gradient to their expression. During the first few postnatal days, virtually all Schaffer-collateral-to-CA1 synapses are silent; by the second to third post-natal week, roughly half have become activated2,3,52–54. These findings, which are supported by ultrastructural analysis (see below), suggest that during spine maturation NMDAR expression occurs first and recruitment of AMPARs occurs later. However, differential timing in receptor expression is not the only mechanism that could drive the phenomenon of synaptic silence (as we discuss below), and the process of synaptic maturation might be more complex. For instance, in the neonatal visual cortex, silent synapses abound in layer VI pyramidal neurons and gradually become active during the first postnatal week, much as they do in the hippocampus55. By contrast, pyramidal neurons in layer I/II, which are formed later in development, bear synapses that are fully functional at birth and that then become silent before going on to reactivate55. It is not known whether this bimodal pattern of AMPAR expression is a special case, or whether a similar pattern would emerge in other cell types if they could be studied early enough in development. There is evidence that NMDARs play an active part in keeping AMPARs away from immature spines until an appropriate trigger signals for their recruitment56–59. For example, conditional deletion of NMDARs from hippocampal neurons before and during synaptogenesis, by mosaic expression of Cre recombinase in floxed NR1 mice, resulted in a massive increase in AMPAR activity and a profound decrease in the incidence of silent synapses, compared with neighbouring control neurons56. Taken together with the findings in the visual cortex, one could speculate that excitatory synapses progress through three developmental stages, the timing of which might differ between different cell populations: first, an early period of AMPAR and NMDAR co-expression; second, the emergence of silent synapses after active removal of AMPARs — triggered, perhaps, by low-level NMDAR activation; and third, synapse unsilencing by the recruitment of functional AMPARs in response to more-robust synaptic activity that is sufficient to trigger LTP. Rigorous examination of synaptic physiology from embryogenesis through the early postnatal period would be helpful in elucidating this issue.
One group suggested that silent synapses do not occur naturally but appear as a result of experimental manipulation60,61. They found that after obtaining an EPSC using a fixed, suprathreshold stimulation intensity, ongoing regular stimulation caused the EPSC amplitude to decline (and plateau after approximately 40 pulses), in a manner that required postsynaptic Ca2+, through NMDAR- and voltage-gated-Ca2+-channel-independent means. This synaptic depression reversed after an LTP pairing protocol, and exhibited a developmental gradient that resembled that of silent synapses in the hippocampus. The authors proposed that silent synapses result from the sort of repetitive, low-frequency electrical stimulation that is commonly used during slice physiology experiments. Although they typically saw only depression — not silencing — they did observe that depression could lead to complete synaptic silence in a few recordings; in the example that they give60, the longest period of consecutive synaptic failure was thirteen sweeps. However, in most other studies silent synapses had been defined using a different criterion — that a subthreshold stimulus produced failures for at least 100 consecutive traces — and such synapses were not as hard to find2,3. Thus, synaptic depression at functional synapses does not preclude, and is probably functionally distinct from, the phenomenon of natively silent synapses62.
Presynaptic silence models
Although the discovery of silent synapses in the hippocampus reconciled previously contradictory findings, it did not quell the debate over whether LTP expression has a pre- or a postsynaptic locus. Although the simplest explanation for silent synapses is that they lack AMPARs on the postsynaptic membrane, another possibility is that AMPARs are present but not activated, and that synaptic silence is a state of the presynaptic terminal. All of the proposed presynaptic models emanate (perhaps tenuously24,25) from the different glutamate affinities of AMPARs and NMDARs (see above). Specifically, a low synaptic glutamate concentration ([Glu]) might activate NMDARs but fail to activate AMPARs; after LTP induction, enhanced glutamate release would unmask the A-EPSC component. Indirect evidence for this model — including the apparent emergence of A-EPSCs in response to paired pulse facilitation63,64, blockade of AMPAR desensitization26,63 or a rise in temperature (which is thought to increase p)63 — has not withstood more rigorous experimental testing (see below)65. Here we will examine the remaining evidence for a presynaptic mechanism of synaptic silence.
Kullmann proposed that a silent synapse consists of an incompetent presynaptic terminal and a mature postsynaptic membrane, and that isolated NMDA responses occur when glutamate spills over, slowly and at a low concentration, from a nearby functional synapse47,66. Contrary to prior findings21,24,67, he found that pairing-induced LTP resulted in a small potentiation of N-EPSCs, with a corresponding increase in CV−2 (REF. 47). In addition, the NMDAR antagonist MK-801, which occupies the channel pore only after glutamate binding, blocked the receptor slightly more rapidly after pairing, which implied a greater availability of synaptic glutamate during LTP (but see REF. 68). Furthermore, potentiation of N-EPSCs occurred after tetanic stimulation even when Ca2+ chelation and hyperpolarization of the voltage-clamped postsynaptic cell prevented A-EPSC potentiation. Presumably the tetanus triggered enhanced glutamate release from nearby potentiated synapses, thereby enhancing spillover onto the recorded cell. There were no recordings from silent synapses in these experiments, and the question of whether spillover could account for unsilencing was not experimentally addressed. Importantly, no studies have directly demonstrated that spillover accounts for synapse unsilencing. Such a scenario would be troubling, because it would violate the input specificity of LTP (that is, the notion that heightened activity at one synapse should lead to potentiation of that synapse alone, without the side effect of potentiating bystander synapses). It is more likely that the low concentration of glutamate that reaches neighbouring synapses will cause a low level of NMDAR-mediated Ca2+ influx, triggering depression rather than potentiation and thus creating a ‘centre on, surround off’ phenomenon that in fact amplifies input specificity69,70. Given the density of excitatory synapses in CA1, spillover probably does occur — to a limited extent because glial glutamate transporters efficiently curb the amount of glutamate diffusion69,71,72 — but there are no data to support the hypothesis that spillover accounts for silent synapses.
The spillover model supposes that the presynaptic terminal at a silent synapse is non-functional. Another possibility is that the presynaptic terminal is handicapped or only partly functional, so that glutamate release occurs in such a limited quantity or over such a long time course that there is a severe reduction in the concentration that ultimately reaches the postsynaptic membrane. One study demonstrated that a low concentration (250 μM) of the rapidly reversible NMDAR antagonist L-AP5 was sufficient to block N-EPSCs at silent synapses (somewhat surprising, given its low affinity25) but failed to block N-EPSCs after pairing26. This indicates that after pairing there was greater competition for NMDAR binding between the antagonist and glutamate, owing to a higher [Glu]. Cyclothiazide, which by itself did not change synaptic [Glu] as assessed by glial glutamate-transporter currents, revealed that AMPARs were present at silent synapses and that a faster rise time and a slower decay of A-EPSCs occurred after pairing, consistent with increased [Glu] and slower transmitter clearance, respectively. Notably, in the presence of cyclothiazide the A-EPSC failure rate was identical before and after pairing, as was the N-EPSC failure rate. The authors concluded that pairing improves the efficiency of transmitter delivery into the cleft without actually changing p; specifically, they proposed that the bouton converts from a mode that allows only partial vesicle fusion events to one that permits full vesicle collapse. Notably, the authors did not report what happened to glutamate-transporter currents after pairing; in fact, two prior reports73,74 showed no change (see below), contradicting the hypothesis that there can be any change in [Glu] after LTP26,63,64. Of note, the findings with cyclothiazide could not subsequently be reproduced65.
Another study supported the notion that silent synapses exist in conditions of slow glutamate release. In cultured hippocampal neurons, individual synapses were visualized by FM1-43 dye labelling and stimulated by focal electrical stimulation or iontophoretic glutamate application75. At ‘silent’ synapses that exhibited no A-EPSC with electrical stimulation or slow glutamate iontophoresis (10 nA for 10 ms), faster glutamate iontophoresis (100 nA for 1 ms) did reveal functional AMPARs. The authors concluded that the rate of glutamate delivery, rather than the absolute amount, dictates whether or not AMPARs will be activated. Support for this hypothesis came from studies in which cultures were treated with tetanus toxin for 1 to 3 hours, which was not long enough to cleave all presynaptic vesicle-fusion proteins and shut down evoked transmission completely; presumably by partially handicapping vesicle release, this manipulation boosted the number of ‘silent’ synapses that exhibited the differential responses to slow and fast iontophoresis described above75. One caveat to this work is that some aspects of iontophoresis (namely leakage, the difficulty of passing a square-wave current through a high-resistance pipette and placement with respect to the tiny patch of postsynaptic membrane) prevent the tight control over agonist delivery that is needed for a detailed consideration of transmitter kinetics. Nevertheless, despite their limitations, these two studies26,75 come to the same conclusion using different techniques — that synapse unsilencing results from enhanced presynaptic glutamate release, without any change in postsynaptic AMPAR expression.
Models of presynaptic silence suffer from the same flaw that models of presynaptic LTP suffer from: they invoke a role for a retrograde messenger. Like LTP, synapse unsilencing in CA1 requires Ca2+ influx through postsynaptic NMDARs3,54. If synaptic silence occurs through any of the presynaptic mechanisms discussed above, then there must be a means by which the postsynaptic signalling cascade communicates with the presynaptic effectors of unsilencing — whether to activate a non-functional release machinery or to enhance its efficiency. Advocates of presynaptic LTP proposed that nitric oxide might be the retrograde messenger76–78, but many groups dispute this claim73,74,79–81. It would be interesting to know whether, in the experiments described above, pharmacological inhibition or genetic deletion of nitric oxide synthase can prevent the observed changes in presynaptic glutamate release.
There is evidence for presynaptically silent synapses at the connections between hippocampal mossy fibres and CA3 pyramidal cells. Mossy-fibre synapses exhibit a presynaptic form of LTP that involves an increase in release probability82,83. From experiments that used MK-801, it seems that this form of LTP also involves presynaptic unsilencing84. MK-801 was applied during baseline transmission to completely block NMDARs at functional synapses, and was then removed before an LTP-inducing tetanus was delivered; because MK-801 is poorly reversible, any NMDARs that were activated during the baseline period would be expected to remain blocked after the tetanus. The observation that N-EPSCs appeared immediately after the tetanus, and persisted stably for the rest of the experiment, indicates the recruitment of new sites of transmitter release — that is, the unsilencing of presynaptic terminals. These presynaptically silent synapses are a special case that is physiologically distinct from most silent synapses in the brain (which exhibit N-EPSCs but no A-EPSCs) (FIG. 2); nevertheless, they remind us of the goldfish Mauthner cell and of the fact that the mechanism of synaptic silence might differ from synapse to synapse.
Postsynaptic silence models
A preponderance of evidence suggests that the main mode of synaptic silence in the mammalian brain results from an absence of postsynaptic AMPARs rather than from impaired presynaptic glutamate release. Part of the challenge in defining the mechanism of synaptic silence has been that conventional hippocampal synaptic physiology experiments do not permit absolute control over the population of synapses that is being activated. Indeed, during recordings of somatic electrical events that are triggered by extracellular stimulation, one cannot be sure that the combination of pre- and post-synaptic elements that is being sampled remains stable from sweep to sweep — in other words, the technique does not permit the definitive isolation of a population of synapses. Given that a pyramidal cell contains a mixture of synapses — some silent and some not — such a lack of control introduces problems that could confound data interpretation, potentially leading to wrong conclusions. For instance, some experimental manipulations, such as raising the temperature63, could conceivably affect the way that the electrical field that emanates from a stimulating electrode interacts with the many axons that pass nearby. It might therefore be wrong to interpret the emergence of A-EPSCs at silent synapses in warm solutions as necessarily reflecting presynaptic unsilencing63, as it could instead reflect a change in the population of synapses that is being activated by the electrode.
One study addressed this concern by making paired recordings from synaptically connected CA3 neurons65. Unlike the mossy-fibre-to-CA3 synapses discussed above, recurrent CA3 synapses seem to be functionally identical to CA3–CA1 synapses83,85. By studying only the EPSCs that were elicited by current injection and action-potential generation in the presynaptic neuron, the authors isolated a single, precisely defined population of synapses. In these experiments, recordings from some cell pairs revealed all-silent synapses that became unsilenced after a pairing protocol. Raising the temperature, delivering paired pulses or applying cyclothiazide all failed to reveal A-EPSCs at silent synapses; by contrast, each manipulation did facilitate A-EPSCs at functional synapses. In addition, N-EPSCs seemed to be identical in every respect before and after pairing, suggesting that unsilencing was not associated with a change in [Glu] or in the speed of glutamate diffusion. These carefully controlled experiments refuted many of the arguments in favour of a presynaptic mechanism for synaptic silence (see above) and made a compelling case for a lack of functional postsynaptic AMPARs at silent synapses.
Recordings from synapses that were isolated by other means provided similar evidence of a postsynaptic mechanism for synaptic silence. For example, in a study of single cultured neurons (which permitted simultaneous control of the pre- and postsynaptic cell, as the only possible synapses were those made by the neuron onto itself), a proportion of autaptic events exhibited N-EPSCs but no A-EPSCs86; keeping in mind the limitations of such artificial conditions, one could conclude that this observation specifically refutes any role for glutamate spillover from a functional synapse onto a presynaptically silent synapse, as the A-EPSC from a functional synapse should always appear before the N-EPSC at a silent synapse. In another study, confocal fluorescent Ca2+ imaging and simultaneous electrophysiological recordings of CA1 neurons in hippocampal slice cultures allowed visualization of individual silent synaptic spines87. At these spines, using fluorescent Ca2+ transients as a surrogate for N-EPSCs, no Ca2+ transient occurred in response to presynaptic stimulation. After delivery of a tetanus, evoked Ca2+ transients did occur. The authors demonstrated that NMDARs were present both before and after the tetanus, and that the reason that they could not elicit Ca2+ transients (N-EPSCs) at silent synapses was that the spine never depolarized enough to relieve the voltage-dependent Mg2+ blockade of NMDARs (see BOX 1). After the tetanus spine depolarization could occur, presumably through the activity of newly recruited AMPARs; NMDAR activation was thus permitted. Together these studies provide further support for the notion that synapse unsilencing results from the recruitment of functional AMPARs to the postsynaptic membrane.
Because it is theoretically possible that presynaptic (for example, changes in [Glu]) and postsynaptic modifications could occur simultaneously when silent synapses are unsilenced, it is important to note the studies that have argued against LTP-induced changes in [Glu]. Glial glutamate-transporter currents provide a reliable read-out of [Glu], changing predictably in response to pharmacological and physiological manipulations that are known to alter the probability of presynaptic glutamate release73,74; tetanus-induced LTP triggered no change in glial transporter currents. In addition, nitric oxide, which had been proposed to be the retrograde messenger that links postsynaptic NMDAR activation to enhanced presynaptic vesicle release (see above), also failed to alter transporter currents73. Similarly, visualization of presynaptic vesicles using FM1-43 revealed no change in vesicle turnover rates in response to conventional methods of LTP induction88; only extreme induction protocols (a 200 Hz tetanus or global slice depolarization with the K+ channel blocker tetraethylammonium (TEA)) triggered an enhancement of presynaptic release. Although direct proof that [Glu] never changes during standard LTP induction or synapse unsilencing is lacking, these studies provide compelling indirect evidence in support of this notion.
If a large population of hippocampal synapses does in fact express postsynaptic NMDARs but not AMPARs, then there should be a simple way to visualize this. Indeed, anatomical studies confirmed that surface AMPARs are absent at some spines (FIG. 3). In studies of cultured neurons, a subpopulation of synaptic spines was immunopositive for surface NMDARs but not for AMPARs86,89–91, and activity triggered the rapid appearance of AMPAR immunoreactivity at such spines in an NMDAR-dependent manner92–94, consistent with the hypothesis that synapse unsilencing involves the physical recruitment of AMPARs. Electron microscopy of intact tissue similarly revealed CA1 spines with immunogold labelling for NMDARs but not AMPARs95–98. Although a lack of AMPAR labelling does not necessarily imply a complete absence of AMPARs (given concerns over antibody sensitivity), it is notable that no AMPAR-negative mossy-fibre-to-CA3 synapse appeared95,98. Moreover, the prevalence of AMPAR-negative synapses in CA1 exhibited a developmental gradient95,96, similar to what was predicted by electrophysiological experiments2,3,54. Thus, there seems to be an anatomical basis for a postsynaptic mechanism of synaptic silence. The data summarized in this section, which were obtained using various experimental techniques, provide compelling evidence that silent synapses lack AMPARs and that synaptic unsilencing occurs when AMPARs arrive.
Figure 3. Electron microscopy of developing glutamatergic synapses.

In hippocampal tissue obtained from young rats, postembedding immunogold labelling was performed using an antibody raised against the carboxyl terminus of the AMPAR subunit GluR1 (a,d,g) or using antibodies that bind to both the GluR2 and the GluR3 subunits (b,c,e,f,h,i); both GluR1 and GluR2 contribute to virtually every tetrameric AMPAR complex in CA1 pyramidal cells. The age at which the tissue was obtained increases from left to right, from postnatal day 2 (P2) to 5 weeks. In each part of the figure, the presynaptic terminals, with glutamate-containing vesicles, are labelled P; opposite each of these terminals is the postsynaptic membrane, marked by the presence of an electron-dense band (the postsynaptic density) where glutamate receptors (black dots) crowd alongside associated anchoring and signalling proteins. A large increase in AMPAR-subunit labelling, relative to the postnatal period, is evident at 5 weeks. Figure reproduced, with permission, from REF. 96 © (1999) Macmillan Publishers Ltd. All rights reserved.
Silencing the debate
The proof of postsynaptic silence came from experiments that used laser photolysis of caged glutamate onto single dendritic spines99–103. In such studies, an experimental preparation is bathed with an inert ‘caged’ glutamate derivative; subsequently, one- or two-photon laser stimulation that is focused on a dedritic spine head in a fluorescently labelled neuron (visualized under confocal or two-photon microscopy) uncages glutamate at a spatial resolution that, at its best (~0.43 μm99), approaches the size of a presynaptic bouton. Such stimulation evokes a single-synapse, uncaging-evoked EPSC (uEPSC); no other means of glutamate application, including iontophoresis, can approach this level of precision — one of the key strengths of this technique. Another compelling aspect of this technique is that glutamate uncaging effectively removes the presynaptic terminal from consideration, thus permitting a discrete, unequivocal investigation into the dynamics of the postsynaptic membrane.
Initially, this technique was used to demonstrate conclusively that LTP can occur postsynaptically, in the absence of any change in [Glu]. The first such study revealed that pairing postsynaptic depolarization with repetitive glutamate uncaging at single spines potentiated A-uEPSCs in an NMDAR- and CaMKII-dependent manner103. These authors also described a relationship between spine size and A-uEPSC amplitude, with thin spines and filopodia exhibiting the smallest responses and large mushroom spines producing the largest102; spines undergoing LTP enlarged after the pairing protocol103, supporting the authors’ hypothesis that spine size reflects AMPAR content. In their hands, LTP occurred preferentially at small spines with small baseline A-uEPSCs; larger spines exhibited only transient potentiation. This group proposed, but did not observe directly, that silent synapses might occur at thin spines that are devoid of AMPARs. Other groups also triggered LTP by pairing glutamate uncaging at single spines with postsynaptic depolarization, although they disagreed on whether there is a relationship between spine size and A-uEPSC amplitude99,101,104. This disagreement, which mirrors the debate over whether silent spines are smaller than their functional counterparts87,95,98,100, could reflect different imaging sensitivities, different preparations or different developmental ages. It does not detract from the incontrovertible demonstration that LTP involves an increase in postsynaptic glutamate sensitivity.
A recent publication carried this line of investigation to its logical conclusion100. In acute slices of neonatal rat somatosensory cortex, some spines exhibited N-uEPSCs but not A-uEPSCs in response to glutamate uncaging (FIG. 4). Focal application of a hypertonic solution to such silent spines, which triggered vesicle fusion at the associated presynaptic bouton, likewise revealed N-EPSCs but not A-EPSCs, indicating that an ‘AMPAR-silent’ postsynaptic membrane sits opposite a fully competent presynaptic terminal. As expected, the prevalence of silent synapses exhibited a developmental gradient, with silent synapses disappearing after postnatal day 12. Although this study does not prove that the presynaptic bouton always matures before the postsynaptic density, it leaves little room to argue that the phenomenon of synaptic silence, as defined at the beginning of this article, reflects anything other than an absence of functional AMPARs on the postsynaptic membrane (FIG. 5).
Figure 4. Postsynaptic silence demonstrated by two-photon laser glutamate uncaging.

a | The left-hand panel shows a spine and its parent dendrite, with sites of glutamate uncaging indicated by asterisks; the show the average| AMPAR-mediated unitary excitatory postsynaptic current (uEPSC) at each site. On each spine tested, an area of maximal response (in this case the site indicated by the lower asterisk) could be located, illustrating the precision of the technique of glutamate uncaging. The right-hand panel shows a lower-magnification view of the basal dendrite of a layer II/III cortical pyramidal neuron from a postnatal day (P) 9 rat, showing examples of mature spines (indicated by the white arrowheads) and a filopodium (indicated by the red arrowhead). b | At P9, glutamate uncaging at a spine revealed no uEPSC at −70 mV but did reveal a slow, NMDAR-mediated uEPSC at +40 mV; five individual trials (grey traces) and the corresponding averages (blue traces) are shown. This is a silent synapse that clearly lacks functional AMPARs in the postsynaptic membrane. c | NMDAR-mediated uEPSC amplitudes versus AMPAR-mediated uEPSC amplitudes are plotted in control conditions (n = 72 spines, P9–13) or in the presence of the AMPAR antagonist NBQX (n = 13 spines, P9–15). The dashed line marks the expected position for silent spines lacking AMPARs. Many control spines fall along this line, exhibiting AMPAR-mediated uEPSCs similar to those recorded in the presence of NBQX, illustrating the large number of silent synapses in these young animals. During the second postnatal week, silent synapses became increasingly hard to find and had virtually disappeared by P14–17 (not shown). Parts a and b reproduced and modified (respectively), with permission, from REF. 100 © (2008) Cambridge University Press.
Figure 5. Silent synapses.

A CA1 pyramidal neuron projects its large apical dendrite down into the stratum radiatum, where Schaffer collateral axons en passant synapses onto dendritic spines. The spine pictured on the left is mature, with a full complement of both AMPARs and NMDARs. By contrast, the spine on the right possesses only NMDARs and therefore cannot conduct current in response to presynaptic glutamate release. The expanded view of this silent synapse illustrates how an LTP-induction protocol will cause AMPARs to migrate towards the postsynaptic density, either through lateral diffusion along the synaptic membrane107 or through the fusion of AMPAR-containing endosomes105,106,129.
The simplest mechanism for the rapid appearance of AMPARs at the postsynaptic membrane following an LTP protocol is a physical movement of heteromeric receptor complexes to the postsynaptic density105,106, either from intracellular stores105,106,129 or through lateral diffusion along the plasma membrane107. As discussed in the previous section, immunohistochemical and ultrastructural studies indeed support this simple mechanism. However, the other formal possibility is that AMPARs are present on the postsynaptic membrane but fail to activate on binding glutamate; activation of NMDARs during LTP would then convert these AMPARs from a non-conducting to a conducting state. In fact, this hypothesis dates to before the discovery of silent synapses108. Although there has never been evidence for non-conducting AMPARs in mammals, such receptors were recently observed in the roundworm Caenorhabditis elegans109. In these animals, AMPARs are composed of glutamate-receptor-channel-forming subunits that are coupled to auxiliary transmembrane AMPAR regulatory proteins (TARPs), which include two homologous members, STG-1 and STG-2 (REFS 109,110). In mutant worms lacking both of these TARP proteins, AMPARs were present on the surface but could not conduct current109. In mammals, TARPs also influence AMPAR gating111, but there is no evidence that AMPARs lacking associated TARPs fail to gate. Nevertheless, these new findings in C. elegans indicate that it might be premature to dismiss the notion that TARPs could represent a switch that converts ‘silent’ AMPARs into functional ones.
Implications for plasticity
Is synapse unsilencing the long-sought-after mechanism that underlies LTP? The reverse is certainly true: unsilencing is qualitatively and mechanistically indistinguishable from LTP2,50,54,103. However, LTP can also occur at synapses that are already functional. The first evidence that synapses might not need to be silent in order to be potentiated was provided by measures of the amplitude of the postsynaptic response to a single quantum of transmitter (BOX 2). One way to measure q is to use minimal stimulation and select only those trials in which a postsynaptic response occurs; if a single synapse is responsible for these responses, then the average size of the EPSCs is a measure of q. Using this approach, some studies found no change in q following LTP32,33, implying that LTP occurs solely by recruiting new synapses (not by making changes to existing ones). Other studies, however, have routinely observed an increase in q34–37,112,113. Some of the confusion resulting from these apparently conflicting results could arise from the uncertainty over whether only a single fibre is being stimulated.
The second, more traditional way of studying quantal events is to record miniature EPSCs (mEPSCs) in the presence of tetrodotoxin (BOX 2). Whereas mEPSCs are elicited in the recorded cell by many thousands of presynaptic elements, synapses undergoing potentiation represent a very small proportion of all sampled synapses, and any changes in quantal parameters that affect only the potentiated synapses might therefore be obscured. In the presence of strontium (Sr2+) instead of Ca2+, stimulus-evoked EPSCs devolve into a series of asynchronous quantal events, each of which is identical in size to an mEPSC; this permits quantal analysis of the subset of synapses that is activated by the stimulating electrode114. In these conditions, LTP reliably triggered an increase in mEPSC amplitude. Based on what we know now, this observation implies that functional synapses can recruit additional AMPARs during LTP. The modern demonstration of this phenomenon came from the glutamate uncaging work discussed above, which showed an increase in A-uEPSC amplitude at functional synapses after LTP99,101,103. Although enhanced single-AMPAR conductance might partly account for this increase in post-synaptic glutamate sensitivity115, a boost in the number of receptors is likely to be the dominant mechanism116. Since the initial visualization of green fluorescent protein (GFP)-tagged AMPARs moving into synapses during LTP105,106, and in light of much other work reviewed elsewhere117–119, it has become generally accepted that movement of AMPARs into and out of synapses accounts for excitatory synaptic potentiation and depression.
Therefore, LTP is the recruitment of new AMPARs to synapses — either to synapses that had previously been silent or to synapses that already possessed some functional AMPARs. The relative contributions of these different scenarios to LTP could depend on age, previous activity at a synapse or other factors. In addition, some research groups still advocate a role for the presynaptic terminal in LTP. For example, in the Ca2+-imaging experiments described above, even though p did not change after synapse unsilencing, it did increase after LTP at functional120 or previously unsilenced87 synapses. The authors of these reports used closely spaced trains of 100 Hz stimulation as the trigger for LTP; as mentioned above, others found that such intense activity enhances presynaptic release, but that more-typical triggers — which are still fully capable of producing postsynaptic LTP — do not88. To achieve maximal potentiation in response to extreme levels of activity, perhaps synapses must draw on both pre- and postsynaptic resources. Finally, little is known about what happens after the first hour following LTP induction, because recordings usually do not last that long. What is known is that, despite there being enormous overall heterogeneity in the size and shape of synapses, in individual synapses the pre-synaptic active zone and the postsynaptic density exhibit remarkable congruence49. Thus, it would not be surprising if, after AMPARs have had a chance to settle into a potentiated postsynaptic membrane, the presynaptic terminal expands proportionately.
The effort to understand the nature of LTP drove the discovery of silent synapses. In turn, the characterization of silent synapses has highlighted the central role of AMPAR trafficking in synaptic plasticity. Shuttling AMPARs into synapses requires the coordinated effort of many signalling and structural molecules, with NMDAR activation and AMPAR recruitment representing merely the beginning and end of a sequence of events that is still only partly understood. We are making progress towards understanding LTP — slower progress, perhaps, than some might have predicted. Moving beyond the old controversies, the focus of future research should be on identifying the minimal molecular requirements that link NMDAR activation to AMPAR trafficking.
Acknowledgments
We thank A. Jackson, W. Lu, A. Milstein and A. Tzingounis for their thoughtful comments on the manuscript. R.A.N. is supported by grants from the US National Institutes of Health and G.A.K. was supported by the Larry L. Hillblom Foundation.
- CA1
The area of the hippocampus that lies at the end of the hippocampal trisynaptic circuit
- Silent synapse
Aside from the exceptions indicated in this Review, silent synapses are synapses that exhibit an NMDA-receptor-mediated response but no AMPA-receptor-mediated response
- Excitatory postsynaptic current
(EPSC). The current that is recorded in a voltage-clamped neuron in response to release of synaptic glutamate
- NMDAR
(N-methyl-D-aspartate receptor). The subtype of ionotropic glutamate receptor, containing the subunit NR1 complexed with some combination of the subunits NR2A–NR2D and NR3A or NR3B, that responds selectively to NMDA by gating a cationic channel that is distinguished both by its permeability to Ca2+ and by its voltage-dependent blockade by Mg2+ at the resting membrane potential
- AMPAR
(α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid receptor). The subtype of ionotropic glutamate receptor that contains a tetramer of some combination of the subunits GluR1–GluR4, responds selectively to AMPA by gating a monovalent cation current, and mediates most fast excitatory neurotransmission in the brain
- LTP
(Long-term potentiation). A form of synaptic plasticity that is mostly studied in hippocampal CA1 pyramidal neurons but that is found in many other areas of the brain. It is characterized by a persistent enhancement of neurotransmission following an appropriate stimulus (see Hebb’s rule)
- Pyramidal neurons
The principal cells of the hippocampus and the neocortex. These large cells use glutamate as their neurotransmitter
- Paired pulse facilitation
The phenomenon whereby the amplitude of an EPSC that is triggered shortly (for example, 50 ms) after a prior EPSC is increased relative to that of the prior EPSC, reflecting an increased probability of presynaptic vesicle release and probably resulting from an increased presynaptic Ca2+ concentration
- Pairing
Delivering low-frequency presynaptic stimulation and simultaneously depolarizing the postsynaptic cell. This triggers LTP in a manner that is consistent with Hebb’s rule
- Hebb’s rule
(or Hebb’s postulate). The notion that after a presynaptic neuron and a postsynaptic neuron fire in unison, the efficiency of neurotransmission between them improves
- MK-801
An NMDAR antagonist that is use-dependent (that is, it binds inside the open channel pore only after agonist binding) and poorly reversible (that is, once it is in the pore it binds tightly with a very slow unbinding rate)
- Tetanic stimulation
The delivery of a rapid train of presynaptic stimuli through an extracellular stimulating electrode positioned near a bundle of axons, typically to trigger synaptic plasticity
- Glutamate transporters
Transmembrane proteins that bind extracellular glutamate and transport it into astrocytes and neurons, thereby contributing to the cessation of excitatory synaptic transmission after presynaptic glutamate release
- Cyclothiazide
A drug that binds AMPARs, blocking the normal fast desensitization of these receptors to glutamate
- FM1-43
An ampiphilic styryl dye that reversibly partitions into lipid bilayers; after exposing neurons to this dye, inducing neuronal activity and then washing off the dye, recycled presynaptic vesicles retain a fluorescent signal, which allows visualization of presynaptic boutons under light microscopy
- Iontophoresis
Ejection of a charged chemical from a high-resistance glass microelectrode through the delivery of a current pulse
- Autapse
A synapse formed by a neuron onto itself, usually in the context of isolated single-neuron microisland cultures
Footnotes
FURTHER INFORMATION
Roger Nicoll's homepage: http://keck.ucsf.edu/neurograd/faculty/nicoll.html
References
- 1.Malenka RC, Kauer JA, perkel DJ, Nicoll RA. The impact of postsynaptic calcium on synaptic transmission — its role in long-term potentiation. Trends Neurosci. 1989;12:444–450. doi: 10.1016/0166-2236(89)90094-5. [DOI] [PubMed] [Google Scholar]
- 2.Liao D, Hessler NA, Malinow R. Activation of postsynaptically silent synapses during pairing-induced LTP in CA1 region of hippocampal slice. Nature. 1995;375:400–404. doi: 10.1038/375400a0. This paper and reference 3, which was published two months later, first established the presence of hippocampal CA1 silent synapses. Both groups used a minimal-stimulation technique to isolate synapses exhibiting the (now classic) physiological signature of a silent synapse — the presence of N-EPSCs but not A-EPSCs — and both groups went on to show that LTP triggered the rapid recruitment of an AMPAR-mediated response. [DOI] [PubMed] [Google Scholar]
- 3.Isaac JT, Nicoll RA, Malenka RC. Evidence for silent synapses: implications for the expression of LTP. Neuron. 1995;15:427–434. doi: 10.1016/0896-6273(95)90046-2. [DOI] [PubMed] [Google Scholar]
- 4.Merrill EG, Wall PD. Factors forming the edge of a receptive field: the presence of relatively ineffective afferent terminals. J Physiol. 1972;226:825–846. doi: 10.1113/jphysiol.1972.sp010012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wall PD. The presence of ineffective synapses and the circumstances which unmask them. Philos Trans R Soc Lond B Biol Sci. 1977;278:361–372. doi: 10.1098/rstb.1977.0048. [DOI] [PubMed] [Google Scholar]
- 6.Malenka RC, Nicoll RA. Silent synapses speak up. Neuron. 1997;19:473–476. doi: 10.1016/s0896-6273(00)80362-1. [DOI] [PubMed] [Google Scholar]
- 7.Redman S. Quantal analysis of synaptic potentials in neurons of the central nervous system. Physiol Rev. 1990;70:165–198. doi: 10.1152/physrev.1990.70.1.165. [DOI] [PubMed] [Google Scholar]
- 8.Kerchner GA, Li P, Zhuo M. Speaking out of turn: a role for silent synapses in pain. IUBMB Life. 1999;48:251–256. doi: 10.1080/713803505. [DOI] [PubMed] [Google Scholar]
- 9.Faber DS, Lin JW, Korn H. Silent synaptic connections and their modifiability. Ann NY Acad Sci. 1991;627:151–164. doi: 10.1111/j.1749-6632.1991.tb25920.x. [DOI] [PubMed] [Google Scholar]
- 10.Charpier S, Behrends JC, Triller A, Faber DS, Korn H. “Latent” inhibitory connections become functional during activity-dependent plasticity. Proc Natl Acad Sci USA. 1995;92:117–120. doi: 10.1073/pnas.92.1.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wolszon L, Faber D. The effects of postsynaptic levels of cyclic AMP on excitatory and inhibitory responses of an identified central neuron. J Neurosci. 1989;9:784–797. doi: 10.1523/JNEUROSCI.09-03-00784.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lin JW, Faber DS. Synaptic transmission mediated by single club endings on the goldfish Mauthner cell. II Plasticity of excitatory postsynaptic potentials. J Neurosci. 1988;8:1313–1325. doi: 10.1523/JNEUROSCI.08-04-01313.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lin JW, Faber DS. Synaptic transmission mediated by single club endings on the goldfish Mauthner cell. I Characteristics of electrotonic and chemical postsynaptic potentials. J Neurosci. 1988;8:1302–1312. doi: 10.1523/JNEUROSCI.08-04-01302.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wojtowicz JM, Marin L, Atwood HL. Activity-induced changes in synaptic release sites at the crayfish neuromuscular junction. J Neurosci. 1994;14:3688–3703. doi: 10.1523/JNEUROSCI.14-06-03688.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wojtowicz JM, Smith BR, Atwood HL. Activity-dependent recruitment of silent synapses. Ann NY Acad Sci. 1991;627:169–179. doi: 10.1111/j.1749-6632.1991.tb25922.x. [DOI] [PubMed] [Google Scholar]
- 16.Stevens CF. Quantal release of neurotransmitter and long-term potentiation. Cell. 1993;72:55–63. doi: 10.1016/s0092-8674(05)80028-5. [DOI] [PubMed] [Google Scholar]
- 17.Nicoll RA, Malenka RC. Contrasting properties of two forms of long-term potentiation in the hippocampus. Nature. 1995;377:115–118. doi: 10.1038/377115a0. [DOI] [PubMed] [Google Scholar]
- 18.Kauer JA, Malenka RC, Nicoll RA. A persistent postsynaptic modification mediates long-term potentiation in the hippocampus. Neuron. 1988;1:911–917. doi: 10.1016/0896-6273(88)90148-1. [DOI] [PubMed] [Google Scholar]
- 19.Muller D, Joly M, Lynch G. Contributions of quisqualate and NMDA receptors to the induction and expression of LTP. Science. 1988;242:1694–1697. doi: 10.1126/science.2904701. [DOI] [PubMed] [Google Scholar]
- 20.Muller D, Lynch G. Long-term potentiation differentially affects two components of synaptic responses in hippocampus. Proc Natl Acad Sci USA. 1988;85:9346–9350. doi: 10.1073/pnas.85.23.9346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kullmann DM. Amplitude fluctuations of dual-component EPSCs in hippocampal pyramidal cells: implications for long-term potentiation. Neuron. 1994;12:1111–1120. doi: 10.1016/0896-6273(94)90318-2. As one of the last milestones in the journey that ultimately led to the discovery of silent synapses in the hippocampus, this study elegantly illustrated that the quantal content for N-EPSCs is paradoxically different from the quantal content for A-EPSCs, and that only the latter changes during LTP. Kullmann proposed that LTP-induced uncovering of latent AMPAR clusters could explain this finding, in the absence of any change in presynaptic glutamate release. [DOI] [PubMed] [Google Scholar]
- 22.Bekkers JM, Stevens CF. NMDA and non-NMDA receptors are co-localized at individual excitatory synapses in cultured rat hippocampus. Nature. 1989;341:230–233. doi: 10.1038/341230a0. [DOI] [PubMed] [Google Scholar]
- 23.Patneau DK, Mayer ML. Structure-activity relationships for amino acid transmitter candidates acting at N-methyl-D-aspartate and quisqualate receptors. J Neurosci. 1990;10:2385–2399. doi: 10.1523/JNEUROSCI.10-07-02385.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Perkel DJ, Nicoll RA. Evidence for all-or-none regulation of neurotransmitter release: implications for long-term potentiation. J Physiol. 1993;471:481–500. doi: 10.1113/jphysiol.1993.sp019911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tong G, Jahr CE. Multivesicular release from excitatory synapses of cultured hippocampal neurons. Neuron. 1994;12:51–59. doi: 10.1016/0896-6273(94)90151-1. [DOI] [PubMed] [Google Scholar]
- 26.Choi S, Klingauf J, Tsien RW. Postfusional regulation of cleft glutamate concentration during LTP at ‘silent synapses’. Nature Neurosci. 2000;3:330–336. doi: 10.1038/73895. [DOI] [PubMed] [Google Scholar]
- 27.Asztely F, Wigstrom H, Gustafsson B. The relative contribution of NMDA receptor channels in the expression of long-term potentiation in the hippocampal CA1 region. Eur J Neurosci. 1992;4:681–690. doi: 10.1111/j.1460-9568.1992.tb00177.x. [DOI] [PubMed] [Google Scholar]
- 28.Gustafsson B, Huang YY, Wigstrom H. Phorbol ester-induced synaptic potentiation differs from long-term potentiation in the guinea pig hippocampus in vitro. Neurosci Lett. 1988;85:77–81. doi: 10.1016/0304-3940(88)90432-6. [DOI] [PubMed] [Google Scholar]
- 29.Malinow R, Tsien RW. Presynaptic enhancement shown by whole-cell recordings of long-term potentiation in hippocampal slices. Nature. 1990;346:177–180. doi: 10.1038/346177a0. This paper was groundbreaking for two reasons: first, it marked the first time that LTP was induced in a hippocampal slice by a pairing protocol during whole-cell patch-clamp recording; and second, because the signal-to-noise ratio of this technique is superior to that of intracellular sharp-electrode recordings, the authors could perform quantal analysis on EPSCs evoked by minimal stimulation and demonstrate an increase in quantal content and a decrease in synaptic failures following LTP. [DOI] [PubMed] [Google Scholar]
- 30.Bekkers JM, Stevens CF. Presynaptic mechanism for long-term potentiation in the hippocampus. Nature. 1990;346:724–729. doi: 10.1038/346724a0. Published a month after reference 29, this paper yielded similar findings. [DOI] [PubMed] [Google Scholar]
- 31.Voronin LL. Long-term potentiation in the hippocampus. Neuroscience. 1983;10:1051–1069. doi: 10.1016/0306-4522(83)90099-4. [DOI] [PubMed] [Google Scholar]
- 32.Bolshakov VY, Siegelbaum SA. Regulation of hippocampal transmitter release during development and long-term potentiation. Science. 1995;269:1730–1734. doi: 10.1126/science.7569903. [DOI] [PubMed] [Google Scholar]
- 33.Stevens CF, Wang Y. Changes in reliability of synaptic function as a mechanism for plasticity. Nature. 1994;371:704–707. doi: 10.1038/371704a0. [DOI] [PubMed] [Google Scholar]
- 34.Kullmann DM, Nicoll RA. Long-term potentiation is associated with increases in quantal content and quantal amplitude. Nature. 1992;357:240–244. doi: 10.1038/357240a0. [DOI] [PubMed] [Google Scholar]
- 35.Liao D, Jones A, Malinow R. Direct measurement of quantal changes underlying long-term potentiation in CA1 hippocampus. Neuron. 1992;9:1089–1097. doi: 10.1016/0896-6273(92)90068-o. [DOI] [PubMed] [Google Scholar]
- 36.Larkman A, Hannay T, Stratford K, Jack J. Presynaptic release probability influences the locus of long-term potentiation. Nature. 1992;360:70–73. doi: 10.1038/360070a0. [DOI] [PubMed] [Google Scholar]
- 37.Stricker C, Field AC, Redman SJ. Changes in quantal parameters of EPSCs in rat CA1 neurones in vitro after the induction of long-term potentiation. J Physiol. 1996;490:443–454. doi: 10.1113/jphysiol.1996.sp021156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Edwards FA, Konnerth A, Sakmann B. Quantal analysis of inhibitory synaptic transmission in the dentate gyrus of rat hippocampal slices: a patch-clamp study. J Physiol. 1990;430:213–249. doi: 10.1113/jphysiol.1990.sp018289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Edwards F. LTP is a long term problem. Nature. 1991;350:271–272. doi: 10.1038/350271a0. [DOI] [PubMed] [Google Scholar]
- 40.Larkman A, Stratford K, Jack J. Quantal analysis of excitatory synaptic action and depression in hippocampal slices. Nature. 1991;350:344–347. doi: 10.1038/350344a0. [DOI] [PubMed] [Google Scholar]
- 41.Faber DS, Korn H. Applicability of the coefficient of variation method for analyzing synaptic plasticity. Biophys J. 1991;60:1288–1294. doi: 10.1016/S0006-3495(91)82162-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liu G, Choi S, Tsien RW. Variability of neurotransmitter concentration and nonsaturation of postsynaptic AMPA receptors at synapses in hippocampal cultures and slices. Neuron. 1999;22:395–409. doi: 10.1016/s0896-6273(00)81099-5. [DOI] [PubMed] [Google Scholar]
- 43.Mainen ZF, Malinow R, Svoboda K. Synaptic calcium transients in single spines indicate that NMDA receptors are not saturated. Nature. 1999;399:151–155. doi: 10.1038/20187. [DOI] [PubMed] [Google Scholar]
- 44.Pankratov YV, Krishtal OA. Distinct quantal features of AMPA and NMDA synaptic currents in hippocampal neurons: implication of glutamate spillover and receptor saturation. Biophys J. 2003;85:3375–3387. doi: 10.1016/S0006-3495(03)74757-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Malinow R. Transmission between pairs of hippocampal slice neurons: quantal levels, oscillations, and LTP. Science. 1991;252:722–724. doi: 10.1126/science.1850871. [DOI] [PubMed] [Google Scholar]
- 46.Manabe T, Wyllie DJ, Perkel DJ, Nicoll RA. Modulation of synaptic transmission and long-term potentiation: effects on paired pulse facilitation and EPSC variance in the CA1 region of the hippocampus. J Neurophysiol. 1993;70:1451–1459. doi: 10.1152/jn.1993.70.4.1451. [DOI] [PubMed] [Google Scholar]
- 47.Kullmann DM, Erdemli G, Asztély F. LTP of AMPA and NMDA receptor-mediated signals: evidence for presynaptic expression and extrasynaptic glutamate spill-over. Neuron. 1996;17:461–474. doi: 10.1016/s0896-6273(00)80178-6. [DOI] [PubMed] [Google Scholar]
- 48.Collingridge GL. Long-term potentiation. A question of reliability. Nature. 1994;371:652–653. doi: 10.1038/371652a0. [DOI] [PubMed] [Google Scholar]
- 49.Lisman JE, Harris KM. Quantal analysis and synaptic anatomy — integrating two views of hippocampal plasticity. Trends Neurosci. 1993;16:141–147. doi: 10.1016/0166-2236(93)90122-3. [DOI] [PubMed] [Google Scholar]
- 50.Wu G, Malinow R, Cline HT. Maturation of a central glutamatergic synapse. Science. 1996;274:972–976. doi: 10.1126/science.274.5289.972. [DOI] [PubMed] [Google Scholar]
- 51.Lledo PM, et al. Calcium/calmodulin-dependent kinase II and long-term potentiation enhance synaptic transmission by the same mechanism. Proc Natl Acad Sci USA. 1995;92:11175–11179. doi: 10.1073/pnas.92.24.11175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hsia AY, Malenka RC, Nicoll RA. Development of excitatory circuitry in the hippocampus. J Neurophysiol. 1998;79:2013–2024. doi: 10.1152/jn.1998.79.4.2013. [DOI] [PubMed] [Google Scholar]
- 53.Liao D, Malinow R. Deficiency in induction but not expression of LTP in hippocampal slices from young rats. Learn Mem. 1996;3:138–149. doi: 10.1101/lm.3.2-3.138. [DOI] [PubMed] [Google Scholar]
- 54.Durand GM, Kovalchuk Y, Konnerth A. Long-term potentiation and functional synapse induction in developing hippocampus. Nature. 1996;381:71–75. doi: 10.1038/381071a0. Another early demonstration of silent CA1 synapses (see also references 2 and 3), this paper was the first to provide an explicit analysis of the developmental time course of their expression. [DOI] [PubMed] [Google Scholar]
- 55.Rumpel S, Kattenstroth G, Gottmann K. Silent synapses in the immature visual cortex: layer-specific developmental regulation. J Neurophysiol. 2004;91:1097–1101. doi: 10.1152/jn.00443.2003. [DOI] [PubMed] [Google Scholar]
- 56.Adesnik H, Li G, During MJ, Pleasure SJ, Nicoll RA. NMDA receptors inhibit synapse unsilencing during brain development. Proc Natl Acad Sci USA. 2008;105:5597–5602. doi: 10.1073/pnas.0800946105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hall BJ, Ghosh A. Regulation of AMPA receptor recruitment at developing synapses. Trends Neurosci. 2008;31:82–89. doi: 10.1016/j.tins.2007.11.010. [DOI] [PubMed] [Google Scholar]
- 58.Hall BJ, Ripley B, Ghosh A. NR2B signaling regulates the development of synaptic AMPA receptor current. J Neurosci. 2007;27:13446–13456. doi: 10.1523/JNEUROSCI.3793-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Luthi A, Schwyzer L, Mateos JM, Gahwiler BH, McKinney RA. NMDA receptor activation limits the number of synaptic connections during hippocampal development. Nature Neurosci. 2001;4:1102–1107. doi: 10.1038/nn744. [DOI] [PubMed] [Google Scholar]
- 60.Xiao MY, Wasling P, Hanse E, Gustafsson B. Creation of AMPA-silent synapses in the neonatal hippocampus. Nature Neurosci. 2004;7:236–243. doi: 10.1038/nn1196. [DOI] [PubMed] [Google Scholar]
- 61.Abrahamsson T, Gustafsson B, Hanse E. Reversible synaptic depression in developing rat CA3–CA1 synapses explained by a novel cycle of AMPA silencing-unsilencing. J Neurophysiol. 2007;98:2604–2611. doi: 10.1152/jn.00602.2007. [DOI] [PubMed] [Google Scholar]
- 62.Montgomery JM, Madison DV. State-dependent heterogeneity in synaptic depression between pyramidal cell pairs. Neuron. 2002;33:765–777. doi: 10.1016/s0896-6273(02)00606-2. [DOI] [PubMed] [Google Scholar]
- 63.Gasparini S, Saviane C, Voronin LL, Cherubini E. Silent synapses in the developing hippocampus: lack of functional AMPA receptors or low probability of glutamate release? Proc Natl Acad Sci USA. 2000;97:9741–9746. doi: 10.1073/pnas.170032297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Maggi L, Le Magueresse C, Changeux JP, Cherubini E. Nicotine activates immature “silent” connections in the developing hippocampus. Proc Natl Acad Sci USA. 2003;100:2059–2064. doi: 10.1073/pnas.0437947100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Montgomery JM, Pavlidis P, Madison DV. Pair recordings reveal all-silent synaptic connections and the postsynaptic expression of long-term potentiation. Neuron. 2001;29:691–701. doi: 10.1016/s0896-6273(01)00244-6. By simultaneously patch-clamping both the pre- and the postsynaptic neurons during recordings of silent synapses, these authors achieved a level of experimental control superior to contemporary methods of minimal extracellular stimulation and refuted many of the arguments for a presynaptic mechanism for synaptic silence. [DOI] [PubMed] [Google Scholar]
- 66.Kullmann DM, Asztely F. Extrasynaptic glutamate spillover in the hippocampus: evidence and implications. Trends Neurosci. 1998;21:8–14. doi: 10.1016/s0166-2236(97)01150-8. [DOI] [PubMed] [Google Scholar]
- 67.Selig DK, Hjelmstad GO, Herron C, Nicoll RA, Malenka RC. Independent mechanisms for long-term depression of AMPA and NMDA responses. Neuron. 1995;15:417–426. doi: 10.1016/0896-6273(95)90045-4. [DOI] [PubMed] [Google Scholar]
- 68.Manabe T, Nicoll RA. Long-term potentiation: evidence against an increase in transmitter release probability in the CA1 region of the hippocampus. Science. 1994;265:1888–1892. doi: 10.1126/science.7916483. [DOI] [PubMed] [Google Scholar]
- 69.Diamond JS. Neuronal glutamate transporters limit activation of NMDA receptors by neurotransmitter spillover on CA1 pyramidal cells. J Neurosci. 2001;21:8328–8338. doi: 10.1523/JNEUROSCI.21-21-08328.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Diamond JS. A broad view of glutamate spillover. Nature Neurosci. 2002;5:291–292. doi: 10.1038/nn0402-291. [DOI] [PubMed] [Google Scholar]
- 71.Arnth-Jensen N, Jabaudon D, Scanziani M. Cooperation between independent hippocampal synapses is controlled by glutamate uptake. Nature Neurosci. 2002;5:325–331. doi: 10.1038/nn825. [DOI] [PubMed] [Google Scholar]
- 72.Lozovaya NA, Kopanitsa MV, Boychuk YA, Krishtal OA. Enhancement of glutamate release uncovers spillover-mediated transmission by N-methyl-D-aspartate receptors in the rat hippocampus. Neuroscience. 1999;91:1321–1330. doi: 10.1016/s0306-4522(98)00638-1. [DOI] [PubMed] [Google Scholar]
- 73.Diamond JS, Bergles DE, Jahr CE. Glutamate release monitored with astrocyte transporter currents during LTP. Neuron. 1998;21:425–433. doi: 10.1016/s0896-6273(00)80551-6. This paper, together with reference 74 (published back-to-back in the same issue), established that the magnitude of a glutamate-transporter current in CA1 astrocytes is a reliable surrogate for synaptic glutamate concentration, and that this concentration does not change after LTP. [DOI] [PubMed] [Google Scholar]
- 74.Lüscher C, Malenka RC, Nicoll RA. Monitoring glutamate release during LTP with glial transporter currents. Neuron. 1998;21:435–441. doi: 10.1016/s0896-6273(00)80552-8. [DOI] [PubMed] [Google Scholar]
- 75.Renger JJ, Egles C, Liu G. A developmental switch in neurotransmitter flux enhances synaptic efficacy by affecting AMPA receptor activation. Neuron. 2001;29:469–484. doi: 10.1016/s0896-6273(01)00219-7. [DOI] [PubMed] [Google Scholar]
- 76.Arancio O, et al. Nitric oxide acts directly in the presynaptic neuron to produce long-term potentiation in cultured hippocampal neurons. Cell. 1996;87:1025–1035. doi: 10.1016/s0092-8674(00)81797-3. [DOI] [PubMed] [Google Scholar]
- 77.Schuman EM, Madison DV. A requirement for the intercellular messenger nitric oxide in long-term potentiation. Science. 1991;254:1503–1506. doi: 10.1126/science.1720572. [DOI] [PubMed] [Google Scholar]
- 78.Son H, et al. Long-term potentiation is reduced in mice that are doubly mutant in endothelial and neuronal nitric oxide synthase. Cell. 1996;87:1015–1023. doi: 10.1016/s0092-8674(00)81796-1. [DOI] [PubMed] [Google Scholar]
- 79.Cummings JA, Nicola SM, Malenka RC. Induction in the rat hippocampus of long-term potentiation (LTP) and long-term depression (LTD) in the presence of a nitric oxide synthase inhibitor. Neurosci Lett. 1994;176:110–114. doi: 10.1016/0304-3940(94)90883-4. [DOI] [PubMed] [Google Scholar]
- 80.Selig DK, et al. Examination of the role of cGMP in long-term potentiation in the CA1 region of the hippocampus. Learn Mem. 1996;3:42–48. doi: 10.1101/lm.3.1.42. [DOI] [PubMed] [Google Scholar]
- 81.Serulle Y, et al. A GluR1-cGKII interaction regulates AMPA receptor trafficking. Neuron. 2007;56:670–688. doi: 10.1016/j.neuron.2007.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Nicoll RA, Schmitz D. Synaptic plasticity at hippocampal mossy fibre synapses. Nature Rev Neurosci. 2005;6:863–876. doi: 10.1038/nrn1786. [DOI] [PubMed] [Google Scholar]
- 83.Zalutsky RA, Nicoll RA. Comparison of two forms of long-term potentiation in single hippocampal neurons. Science. 1990;248:1619–1624. doi: 10.1126/science.2114039. [DOI] [PubMed] [Google Scholar]
- 84.Tong G, Malenka RC, Nicoll RA. Long-term potentiation in cultures of single hippocampal granule cells: a presynaptic form of plasticity. Neuron. 1996;16:1147–1157. doi: 10.1016/s0896-6273(00)80141-5. [DOI] [PubMed] [Google Scholar]
- 85.Debanne D, Gahwiler BH, Thompson SM. Long-term synaptic plasticity between pairs of individual CA3 pyramidal cells in rat hippocampal slice cultures. J Physiol. 1998;507:237–247. doi: 10.1111/j.1469-7793.1998.237bu.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Gomperts SN, Rao A, Craig AM, Malenka RC, Nicoll RA. Postsynaptically silent synapses in single neuron cultures. Neuron. 1998;21:1443–1451. doi: 10.1016/s0896-6273(00)80662-5. [DOI] [PubMed] [Google Scholar]
- 87.Ward B, et al. State-dependent mechanisms of LTP expression revealed by optical quantal analysis. Neuron. 2006;52:649–661. doi: 10.1016/j.neuron.2006.10.007. [DOI] [PubMed] [Google Scholar]
- 88.Zakharenko SS, Zablow L, Siegelbaum SA. Visualization of changes in presynaptic function during long-term synaptic plasticity. Nature Neurosci. 2001;4:711–717. doi: 10.1038/89498. [DOI] [PubMed] [Google Scholar]
- 89.Liao D, Zhang X, O’Brien R, Ehlers MD, Huganir RL. Regulation of morphological postsynaptic silent synapses in developing hippocampal neurons. Nature Neurosci. 1999;2:37–43. doi: 10.1038/4540. [DOI] [PubMed] [Google Scholar]
- 90.Richmond SA, et al. Localization of the glutamate receptor subunit GluR1 on the surface of living and within cultured hippocampal neurons. Neuroscience. 1996;75:69–82. doi: 10.1016/0306-4522(96)00217-5. [DOI] [PubMed] [Google Scholar]
- 91.Pickard L, Noel J, Henley JM, Collingridge GL, Molnar E. Developmental changes in synaptic AMPA and NMDA receptor distribution and AMPA receptor subunit composition in living hippocampal neurons. J Neurosci. 2000;20:7922–7931. doi: 10.1523/JNEUROSCI.20-21-07922.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Liao D, Scannevin RH, Huganir R. Activation of silent synapses by rapid activity-dependent synaptic recruitment of AMPA receptors. J Neurosci. 2001;21:6008–6017. doi: 10.1523/JNEUROSCI.21-16-06008.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Lu WY, et al. Activation of synaptic NMDA receptors induces membrane insertion of new AMPA receptors and LTP in cultured hippocampal neurons. Neuron. 2001;29:243–254. doi: 10.1016/s0896-6273(01)00194-5. [DOI] [PubMed] [Google Scholar]
- 94.Pickard L, et al. Transient synaptic activation of NMDA receptors leads to the insertion of native AMPA receptors at hippocampal neuronal plasma membranes. Neuropharmacology. 2001;41:700–713. doi: 10.1016/s0028-3908(01)00127-7. [DOI] [PubMed] [Google Scholar]
- 95.Nusser Z, et al. Cell type and pathway dependence of synaptic AMPA receptor number and variability in the hippocampus. Neuron. 1998;21:545–559. doi: 10.1016/s0896-6273(00)80565-6. [DOI] [PubMed] [Google Scholar]
- 96.Petralia RS, et al. Selective acquisition of AMPA receptors over postnatal development suggests a molecular basis for silent synapses. Nature Neurosci. 1999;2:31–36. doi: 10.1038/4532. [DOI] [PubMed] [Google Scholar]
- 97.Racca C, Stephenson FA, Streit P, Roberts JDB, Somogyi P. NMDA receptor content of synapses in stratum radiatum of the hippocampal CA1 area. J Neurosci. 2000;20:2512–2522. doi: 10.1523/JNEUROSCI.20-07-02512.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Takumi Y, Ramirez-Leon V, Laake P, Rinvik E, Ottersen OP. Different modes of expression of AMPA and NMDA receptors in hippocampal synapses. Nature Neurosci. 1999;2:618–624. doi: 10.1038/10172. [DOI] [PubMed] [Google Scholar]
- 99.Bagal AA, Kao JP, Tang CM, Thompson SM. Long-term potentiation of exogenous glutamate responses at single dendritic spines. Proc Natl Acad Sci USA. 2005;102:14434–14439. doi: 10.1073/pnas.0501956102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Busetto G, Higley MJ, Sabatini BL. Developmental presence and disappearance of postsynaptically silent synapses on dendritic spines of rat layer 2/3 pyramidal neurons. J Physiol. 2008;586:1519–1527. doi: 10.1113/jphysiol.2007.149336. By using glutamate uncaging to trigger uEPSCs and thus to bypass the presynaptic terminal, this paper was the first to demonstrate unequivocally that silent synapses express no functional AMPARs in the postsynaptic membrane. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Harvey CD, Svoboda K. Locally dynamic synaptic learning rules in pyramidal neuron dendrites. Nature. 2007;450:1195–1200. doi: 10.1038/nature06416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Matsuzaki M, et al. Dendritic spine geometry is critical for AMPA receptor expression in hippocampal CA1 pyramidal neurons. Nature Neurosci. 2001;4:1086–1092. doi: 10.1038/nn736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Matsuzaki M, Honkura N, Ellis-Davies GCR, Kasai H. Structural basis of long-term potentiation in single dendritic spines. Nature. 2004;429:761–766. doi: 10.1038/nature02617. This paper was the first to induce LTP at single spines through repetitive glutamate uncaging. It demonstrated conclusively that LTP could occur by a purely postsynaptic mechanism. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Zhang YP, Holbro N, Oertner TG. Optical induction of plasticity at single synapses reveals input-specific accumulation of αCaMKII. Proc Natl Acad Sci USA. 2008;105:12039–12044. doi: 10.1073/pnas.0802940105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Hayashi Y, et al. Driving AMPA receptors into synapses by LTP and CaMKII: requirement for GluR1 and PDZ domain interaction. Science. 2000;287:2262–2267. doi: 10.1126/science.287.5461.2262. [DOI] [PubMed] [Google Scholar]
- 106.Shi S-H, et al. Rapid spine delivery and redistribution of AMPA receptors after synaptic NMDA receptor activation. Science. 1999;284:1811–1816. doi: 10.1126/science.284.5421.1811. In hippocampal neurons that expressed a recombinant GluR1 AMPAR subunit tagged with GFP, these authors observed rapid delivery of tagged receptors to the postsynaptic membrane from intracellular stores following LTP induction, providing evidence that LTP occurs by trafficking AMPARs to synapses. [DOI] [PubMed] [Google Scholar]
- 107.Adesnik H, Nicoll RA, England PM. Photoinactivation of native AMPA receptors reveals their real-time trafficking. Neuron. 2005;48:977–985. doi: 10.1016/j.neuron.2005.11.030. [DOI] [PubMed] [Google Scholar]
- 108.Lynch G, Baudry M. The biochemistry of memory: a new and specific hypothesis. Science. 1984;224:1057–1063. doi: 10.1126/science.6144182. [DOI] [PubMed] [Google Scholar]
- 109.Wang R, et al. TARP proteins have fundamental roles in the gating of glutamate receptors and the tuning of synaptic function. Neuron. doi: 10.1016/j.neuron.2008.07.023. (in the press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Walker CS, et al. Reconstitution of invertebrate glutamate receptor function depends on stargazin-like proteins. Proc Natl Acad Sci USA. 2006;103:10781–10786. doi: 10.1073/pnas.0604482103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Nicoll RA, Tomita S, Bredt DS. Auxiliary subunits assist AMPA-type glutamate receptors. Science. 2006;311:1253–1256. doi: 10.1126/science.1123339. [DOI] [PubMed] [Google Scholar]
- 112.Manabe T, Renner P, Nicoll RA. Postsynaptic contribution to long-term potentiation revealed by the analysis of miniature synaptic currents. Nature. 1992;355:50–55. doi: 10.1038/355050a0. [DOI] [PubMed] [Google Scholar]
- 113.Isaac JT, Hjelmstad GO, Nicoll RA, Malenka RC. Long-term potentiation at single fiber inputs to hippocampal CA1 pyramidal cells. Proc Natl Acad Sci USA. 1996;93:8710–8715. doi: 10.1073/pnas.93.16.8710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Oliet SH, Malenka RC, Nicoll RA. Bidirectional control of quantal size by synaptic activity in the hippocampus. Science. 1996;271:1294–1297. doi: 10.1126/science.271.5253.1294. [DOI] [PubMed] [Google Scholar]
- 115.Benke TA, Luthi A, Isaac JTR, Collingridge GL. Modulation of AMPA receptor unitary conductance by synaptic activity. Nature. 1998;393:793–797. doi: 10.1038/31709. [DOI] [PubMed] [Google Scholar]
- 116.Andrásfalvy BK, Magee JC. Changes in AMPA receptor currents following LTP induction on rat CA1 pyramidal neurons. J Physiol. 2004;559:543–554. doi: 10.1113/jphysiol.2004.065219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Bredt DS, Nicoll RA. AMPA receptor trafficking at excitatory synapses. Neuron. 2003;40:361–379. doi: 10.1016/s0896-6273(03)00640-8. [DOI] [PubMed] [Google Scholar]
- 118.Malinow R, Malenka RC. AMPA receptor trafficking and synaptic plasticity. Annu Rev Neurosci. 2002;25:103–126. doi: 10.1146/annurev.neuro.25.112701.142758. [DOI] [PubMed] [Google Scholar]
- 119.Song I, Huganir RL. Regulation of AMPA receptors during synaptic plasticity. Trends Neurosci. 2002;25:578–588. doi: 10.1016/s0166-2236(02)02270-1. [DOI] [PubMed] [Google Scholar]
- 120.Emptage NJ, Reid CA, Fine A, Bliss TVP. Optical quantal analysis reveals a presynaptic component of LTP at hippocampal Schaffer-associational synapses. Neuron. 2003;38:797–804. doi: 10.1016/s0896-6273(03)00325-8. [DOI] [PubMed] [Google Scholar]
- 121.Ramón y Cajal S. Histologie du Système Nerveux de l’Homme et des vertébrés. Maloine; paris: 1909. [Google Scholar]
- 122.Isaac JT, Crair MC, Nicoll RA, Malenka RC. Silent synapses during development of thalamocortical inputs. Neuron. 1997;18:269–280. doi: 10.1016/s0896-6273(00)80267-6. [DOI] [PubMed] [Google Scholar]
- 123.Blakemore C, Hillman P. An attempt to assess the effects of monocular deprivation and strabismus on synaptic efficiency in the kitten’s visual cortex. Exp Brain Res. 1977;30:187–202. doi: 10.1007/BF00237250. [DOI] [PubMed] [Google Scholar]
- 124.Rumpel S, Hatt H, Gottmann K. Silent synapses in the developing rat visual cortex: evidence for postsynaptic expression of synaptic plasticity. J Neurosci. 1998;18:8863–8874. doi: 10.1523/JNEUROSCI.18-21-08863.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Shah RD, Crair MC. Retinocollicular synapse maturation and plasticity are regulated by correlated retinal waves. J Neurosci. 2008;28:292–303. doi: 10.1523/JNEUROSCI.4276-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Losi G, Prybylowski K, Fu Z, Luo JH, Vicini S. Silent synapses in developing cerebellar granule neurons. J Neurophysiol. 2002;87:1263–1270. doi: 10.1152/jn.00633.2001. [DOI] [PubMed] [Google Scholar]
- 127.Balland B, Lachamp P, Kessler JP, Tell F. Silent synapses in developing rat nucleus tractus solitarii have AMPA receptors. J Neurosci. 2008;28:4624–4634. doi: 10.1523/JNEUROSCI.5355-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Lo FS, Erzurumlu RS. Conversion of functional synapses into silent synapses in the trigeminal brainstem after neonatal peripheral nerve transection. J Neurosci. 2007;27:4929–4934. doi: 10.1523/JNEUROSCI.5342-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Park M, Penick EC, Edwards JG, Kauer JA, Ehlers MD. Recycling endosomes supply AMPA receptors for LTP. Science. 2000;305:1972–1975. doi: 10.1126/science.1102026. [DOI] [PubMed] [Google Scholar]