

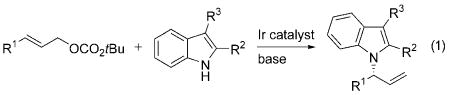
Chiral indole architectures are present in a wide variety of natural products and have been identified as promising lead compounds in medicinal chemistry.[1] Therefore, extensive effort has been dedicated to synthesizing enantioenriched indole derivatives by catalytic, enantioselective reactions,[2] such as 1,2-additions to carbonyl compounds and imines,[3] additions to electron-deficient alkenes,[4] and allylation reactions.[5] However, the indole acts as a carbon nucleophile in each of these reactions; catalytic, enantioselective reactions at the nitrogen atom of an indole are rare, but would provide access to an array of enantioenriched, heterocyclic architectures.
One approach to overcome the greater nucleophilicity of the C3-position of indole, relative to that of N1, is to install an electron-withdrawing substituent at C2. Such a substituent tempers the nucleophilicity at C3, increases the acidity of the N–H bond, and can be used either for additional transformations or removed after reaction at the nitrogen center. Even by following this approach, catalytic, enantioselective reactions of indoles at the nitrogen center are rare. Recent reports by Bandani et al.[6] and Chen et al.[7] have shown that asymmetric alkylation of the indole nitrogen atom occurs with modified cinchona alkaloids, but just one report of enantioselective reactions catalyzed by a transition-metal complex that occur at an indole nitrogen atom has been published. In this work, N-substituted indolopyrrolocarbazole derivatives were synthesized by palladium-catalyzed asymmetric allylic alkylation of bis(indole) lactam pro-aglycons.[8]
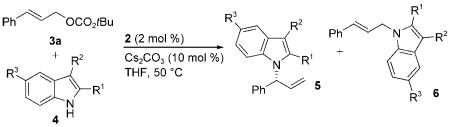
The potential of enantioselective N-allylation of indoles to create an entry into biologically active indole derivatives, particularly with catalysts that preferentially form chiral, branched products from linear allylic esters, led us to investigate iridium-catalyzed, asymmetric, N-allylation of indoles. We report herein the synthesis of enantioenriched, branched N-allylindoles from the reactions of 2-subsituted, 3-substituted, and 2,3-disubstituted indoles with achiral linear allylic carbonates in the presence of a single-component iridium catalyst [Eq. (1)]. The enantioenriched products are readily converted into monoamine reuptake inhibitors,[9] the indole core of integrin αvβ3 inhibitors,[10] and highly substituted dihydropyrrolo[1,2-a]indoles.
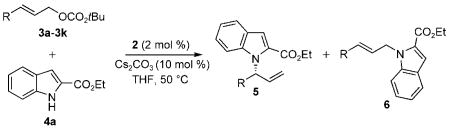
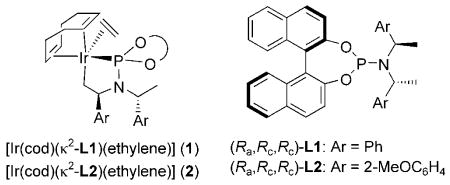
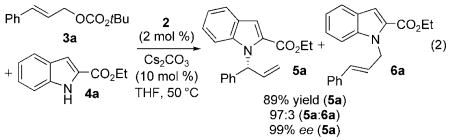
Initial studies to develop catalytic, enantioselective N-allylations of indoles were guided by work from our laboratory on the N-allylation of more acidic and nucleophilic benzimidazoles, imidazoles, and purines catalyzed by the metallacyclic iridium–phosphoramidite complexes [Ir(cod)-(κ2-L1)(ethylene)] (1)[11] and [Ir(cod)(κ2-L2)(ethylene)] (2).[12] To test whether iridium-catalyzed N-allylation of indoles could occur, we studied the reaction of ethyl indole-2-carboxylate (4a) with allylic carbonates. After conducting reactions under various conditions with different catalysts and different carbonate derivatives (see Table S1 in the Supporting Information for details), we found that the allylation of 4a with tert-butylcinnamyl carbonate in the presence of 2 mol% of metallacycle 2 and 10 mol% Cs2CO3 occurred exclusively at N1 with excellent branched-to-linear selectivity (5a/6a = 97:3). This reaction was conducted with the single-component iridium catalyst 2 to eliminate potential complications that would arise from the generation of the metallacyclic catalyst by base[13] in a system containing a weakly basic nucleophile and carbonate base. The branched N-allylindole (R)-5a was isolated in 89% yield and 99% ee [Eq. (2) and Table 1, entry 1]. The absolute configuration of the N-allylindole 5a parallels that of the products from the addition of other nucleophiles that appear to react by backside attack onto the allyl ligand of the intermediate allyliridium complex.[14]
Table 1.
Iridium-catalyzed enantioselective allylation of ethyl indole-2-carboxylate 4a with allylic carbonates 3a–k.[a]
 | |||||
|---|---|---|---|---|---|
| Entry | 3 (R) | 5 | 5/6[b] | Yield [%][c] | ee [%][d] |
| 1 | 3a (Ph) | 5a | 97:3 | 89 | 99 |
| 2 | 3b (4-MeOC6H4) | 5b | 99:1 | 88 | 99 |
| 3 | 3c (4-BrC6H4) | 5c | 97:3 | 87 | 99 |
| 4[e] | 3d (4-F3CC6H4) | 5d | 91:9 | 85 | 99 |
| 5 | 3e (3-MeOC6H4) | 5e | 98:2 | 95 | 98 |
| 6[f] | 3 f (2-MeOC6H4) | 5 f | 95:5 | 72 | 96 |
| 7[f] | 3g (2-furyl) | 5g | 91:9 | 85 | 99 |
| 8[f,g] | 3h ((E)-CH=CHCH3) | 5h | 99:1 | 86 | 99 |
| 9[f] | 3i (n-propyl) | 5i | 94:6 | 92 | 99 |
| 10[f] | 3j (CH2OBn) | 5j | 77:23 | 70 | 98 |
| 11[e] | 3k (cyclohexyl) | 5k | 87:13 | 54 | 99 |
See the Supporting Information for experimental details.
Determined by 1H NMR analysis of the crude reaction mixture.
Yield of isolated 5.
Determined by chiral HPLC methods.
Used 4 mol% 2 as the catalyst.
Reaction was run at room temperature.
Enantiomeric excess was determined after reduction with LiAlH4.
The results of reactions of ethyl indole-2-carboxylate with a series of achiral, linear allylic carbonates under these reaction conditions are shown in Table 1. The reactions of 4a with electron-rich, electron-neutral, and electron-deficient substituted cinnamyl carbonates gave the corresponding N-allylindole products 5b–f in high yields with a range of 91:9 to 99:1 branched-to-linear selectivity and 96–99% enantiomeric excess (Table 1, entries 2–6). The reaction of 4-(trifluoromethyl)cinnamyl tert-butyl carbonate 3d with 4a required a higher loading of catalyst 2 (4 mol%), but gave product 5d in 85% yield with a 91:9 branched-to-linear ratio and 99% ee (Table 1, entry 4). Furthermore, this reaction occurred without isomerization of the chiral 1-arylallyl product into the corresponding achiral 1-arylprop-1-enyl product; an analogous isomerization was observed during the allylation of benzimidazoles and purines with electron-deficient cinnamyl carbonates.[12]
Reactions of 4a with heteroaryl and aliphatic allylic carbonates, as well as a dienyl carbonate, also occurred with moderate to high regioselectivity (5/6=77:23 to 99:1) and excellent enantioselectivity (98–99% ee) (Table 1, entries 7–10). These reactions occurred at room temperature and gave the corresponding N-allylindoles in higher yields with higher regioselectivities at room temperature than at 50°C. The reaction of the benzyloxy-substituted allylic carbonate 3j (R=CH2OBn) occurred with somewhat lower branched-to-linear selectivity, but nevertheless provides a route to hydroxyalkyl-substituted indoles (Table 1, entry 10). The reaction of 4a with the aliphatic carbonate 3k containing branching alpha to the allyl unit (R=cyclohexyl) required more forcing reaction conditions. Although the branched-to-linear ratio was only 87:13, the allylation product 5k was isolated as a single regioisomer in 54% yield and with 99% ee when the reaction was conducted at 50°C in the presence of 4 mol% 2 (Table 1, entry 11).
The range of indoles that undergo the enantioselective N-allylation in the presence of catalyst 2 is summarized in Table 2. Various ethyl indole-2-carboxylates containing electron-donating and electron-withdrawing organic substituents, as well as halogens at the 5-position, were suitable nucleophiles for the allylation reaction with tert-butyl cinnamyl carbonate (Table 2, entries 1–4). The reactions of 5-subsituted ethyl indole-2-carboxylates (4b–e) occurred with high branched-to-linear selectivity (94:6 to 97:3) and enantioselectivity (99% ee), and products 5l–o were isolated in high yields (84–91%). In addition, indole-2-carboxaldehyde (4f) reacted with 3a to form indole 5p in 89% yield with 99% ee (Table 2, entry 5).
Table 2.
Iridium-catalyzed N-allylation of 2-substituted, 3-substituted, and 2,3-disubstituted indoles 4b–p with tert-butyl cinnamyl carbonate 3a.[a]
 | ||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | 4 | R1 | R2 | R3 | 5 | 5/6[b] | Yield [%][c] | ee [%][d] |
| 1 | 4b | CO2Et | H | OMe | 5l | 97:3 | 88 | 99 |
| 2 | 4c | CO2Et | H | F | 5m | 94:6 | 84 | 99 |
| 3 | 4d | CO2Et | H | Cl | 5n | 95:5 | 91 | 99 |
| 4 | 4e | CO2Et | H | NO2 | 5o | 94:6 | 90 | 99 |
| 5 | 4f | CHO | H | H | 5p | 98:2 | 89 | 99 |
| 6 | 4g | H | CO2Me | H | 5q | 96:4 | 84 | 97 |
| 7 | 4h | H | CHO | H | 5r | 94:6 | 87 | 96 |
| 8 | 4i | H | C(O)Me | H | 5 s | 98:2 | 88 | 96 |
| 9 | 4j | H | CN | H | 5t | 96:4 | 93 | 96 |
| 10 | 4k | H | Ph | H | 5u | 99:1 | 21 | 97 |
| 11 | 4 l | CHO | Me | H | 5v | 98:2 | 82 | 99 |
| 12 | 4m | Ph | CHO | H | 5w | 96:4 | 93 | 97 |
| 13 | 4n | Ph | Ph | H | 5x | 98:2 | 95 | 99 |
| 14 | 4o | Ph | Me | H | 5y | 98:2 | 89 | 99 |
| 15 | 4p | -(CH)4- | H | 5z | 98:2 | 88 | 98 | |
See the Supporting Information for experimental details.
Determined by 1H NMR analysis of the crude reaction mixture.
Yield of isolated 5.
Determined by chiral HPLC methods.
Methyl cinnamyl carbonate was used as the electrophile.
3-Substituted indoles also reacted to give N-allylindoles in high yields with excellent selectivities. The reactions of indoles 4g–4j containing a variety of electron-withdrawing substituents at the 3-position (Table 2, entries 6–9) occurred with high branched-to-linear selectivity (94:6 to 98:2) and excellent enantioselectivity (96–97% ee) to form N-allylindoles 5q–t in high yields (84–93%). The reaction of 3-phenylindole (4k) with tert-butyl cinnamyl carbonate (Table 2, entry 10) occurred with excellent selectivities, but low conversion because of deactivation of the catalyst.
Indoles containing substituents in both the 2- and 3-positions also underwent selective iridium-catalyzed N-allylation reactions (Table 2, entries 11–15). For example, reactions of 3a with 2,3-substituted indoles containing an electron-withdrawing substituent at either the 2- or 3-position, such as 3-methylindole-2-carboxaldehyde (4l) or 2-phenylindole-3-carboxaldehyde (4m), gave the corresponding N-allylated products in high yields with excellent regio- and enantioselectivities (Table 2, entries 11 and 12). The presence of a strong electron-withdrawing group at either the 2- or 3-position of a 2,3-disubstituted indole was not even required for N-allylation. The reactions of 2,3-dipheylindole (4n), 3-methyl-2-phenylindole (4o), and carbazole (4p) with 3a occurred with 98:2 branched-to-linear ratio in each case and gave N-allylindoles 5x–z in 88–95% yield with 98–99% ee (Table 2, entries 13–15).
The parent indole and certain indoles containing one phenyl or solely alkyl substituents did not undergo N-allylation, but 7-azaindole did undergo productive N-allylation. Allylations of indole, 2-methylindole, and 2-phenylindole with 3a catalyzed by 1 or 2 and Cs2CO3 at 50°C occurred selectively at the 3-position, whereas no allylation products formed from the analogous reactions of 3-methylindole and 2,3-dimethylindole. However, the reaction of 7-azaindole 7 with tert-butyl cinnamyl carbonate (3a) occurred with greater than 90:10 N1-to-C3 selectivity and 91:9 branched-to-linear selectivity, and the branched N-allylazaindole 8 was isolated from this reaction in 79% yield and 99% ee (Scheme 1).
Scheme 1.

Regioselective and enantioselective N-allylation of 7-azaindole 7 with 3a in the presence of catalyst 2.
Within the past five years, molecular scaffolds containing 3-(1H-indol-1-yl)-3-arylpropan-1-amine and 3-(1H-indol-1-yl)-propanoic acid substructures have been identified as promising lead compounds for medicinal chemistry. 3-(1H-Indol-1-yl)-N-methyl-3-arylpropan-1-amines are potent dual-acting norephinephrine and serotonin reuptake inhibitors,[9a] while 3-(1H-indol-1-yl)-3-arylpropanoic acids and 3-(1H-indol-1-yl)-3-alkylpropanoic acids are constituents of potent inhibitors of the integrin αvβ3.[10] All of these structures contain a stereocenter at the 3-position of the arylpropanamine, arylpropanoic acid, or alkylpropanoic acid subunit that could be formed with control of absolute stereochemistry by iridium-catalyzed allylic substitution. Furthermore, the allyl moieties of the N-allylindole products provide a suitable handle for straightforward elaboration into the requisite propanamine or propanoic acid units.
The syntheses of 3-(1H-indol-1-yl)-N-methyl-3-arylpropan-1-amines 11 and 12a–c are presented in Scheme 2. The known monoamine reuptake inhibitor (R)-3-(1H-indol-1-yl)-N-methyl-3-phenylpropan-1-amine (11) was prepared from 5q by a sequence including removal of the 3-alkoxycarbonyl group by hydrolysis and decarboxylation, and standard conversion of the alkene unit into an aminoethyl group by hydroboration, oxidation, then conversion of the alcohol into the chloride, and finally substitution with methylamine. However, we also developed a more direct route using hydrozirconation. Hydrozirconation of N-allylindoles 5x–z and subsequent reaction of the alkylzirconium intermediate with N-methyl hydroxylamine-O-sulfonic acid gave 3-(1H-indol-1-yl)-N-methyl-3-arylpropan-1-amines 12a–c in a two-step, one-pot sequence from the allylation product in 63–73% yield.
Scheme 2.

Syntheses of enantioenriched 3-(1H-indol-1-yl)-N-methyl-3-arylpropan-1-amines 11 and 12a-c, dihydropyrrolo[1,2-a]indole 13, and indol-1-yl propanonic acid 14 from N-allylindoles. N-Allylindoles: 5p, R1=CHO, R2=H, 99% ee; 5q, R1=H, R2=CO2Me, 97% ee; 5x, R1, R2=Ph, 99% ee; 5y, R1=Ph, R2=Me, 99% ee; 5z, R1, R2=-(CH)4-, 98% ee. Reaction conditions: a) 5q, 9-BBN, THF, −78°C to RT, then H2O2, 3m NaOH, EtOH, 0°C to RT, 97%, 97% ee; b) KOH, MeOH, reflux; c) PhBr, reflux, 68% over two steps; d) PPh3, CCl4, 0°C→RT, 91%; e) MeNH2, EtOH, 90°C, 95%, 97% ee; f) 5x–z, [Cp2Zr(H)Cl], THF, RT, then CH3NHOSO3H, 60°C (see Scheme 2 for results); g) 5p, MeNHOH·HCl, NaOAc, THF, reflux, 83%, 9:1 regioselectivity, >99:1 d.r.; h) Zn, AcOH, H2O, 60°C, 87%, 99% ee; i) PhI(OAc)2, TEMPO, NaHCO3, MeCN/H2O (1:1), 81%, 97% ee. 9-BBN=9-borabicyclo[3.3.1]nonane, TEMPO=2,2,6,6-tetramethyl-1-piperidinyloxy free radical.
We also showed that highly substituted and enantioenriched dihydropyrrolo[1,2-a]indoles, which occur in natural products and other biologically active molecules,[15] are readily synthesized from appropriately substituted N-allylindoles. For example, the reaction of (R)-1-(1-phenylallyl)-1H-indole-2-carboxaldehyde (5p) with N-methylhydroxylamine forms a transient nitrone that undergoes an intramolecular dipolar cycloaddition with the allyl moiety of 5p to form a tricyclic cycloadduct in 83% yield with excellent regio- and diastereoselectivity.[16] Reductive cleavage of the N–O bond contained in the cycloadduct gave the highly substituted dihydropyrrolo[1,2-a]indole 13, a ring-constrained analogue of the 3-(1H-indol-1-yl)-N-methyl-3-arylpropan-1-amines, in 87% yield.
Enantioenriched N-allylindoles are also precursors to indolyl-1-yl propanoic acids, which are key subunits in a class of integrin αvβ3 inhibitors.[10] Sequential hydroboration and oxidation of N-allylindole 5q and then oxidation of the alcohol with iodobenzene diacetate, TEMPO, and sodium bicarbonate led to (R)-3-(3-(methoxycarbonyl)-1H-indol-1-yl)-3-phenylpropanoic acid (14) in 79% yield over two steps.
In summary, we have developed a method for highly regioselective and enantioselective N-allylation of indoles with achiral, acyclic allylic electrophiles in the presence of a metallacyclic iridium catalyst. These reactions encompass a broad range of indole nucleophiles, as well as a variety of unsymmetrical aryl, heteroaryl, and aliphatic allylic carbonates and a dienyl carbonate. The enantioenriched N-allylindole products are formed with consistently high enantioselectivity (96–99% ee) and are readily transformed into 3-(1H-indol-1-yl)-N-methyl-3-arylpropan-1-amines, dihydropyrrolo-[1,2-a]indoles, and indol-1-yl propanoic acids. The use of this reaction to prepare libraries of enantioenriched indole derivatives to identify lead compounds for medicinal chemistry will be the subject of future studies.
Supplementary Material
Footnotes
We thank the NIH for financial support of this work (NIH GM55382 to J.F.H. and GM84584 to L.M.S.) and Johnson-Matthey for gifts of iridium salts.
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/anie.200904338.
References
- 1.For reviews, see: Gupta L, Talwar A, Chauhan PMS. Curr Med Chem. 2007;14:1789. doi: 10.2174/092986707781058904.Cao R, Peng W, Wang Z, Xu A. Curr Med Chem. 2007;14:479. doi: 10.2174/092986707779940998.Lewis SE. Tetrahedron. 2006;62:8655.O'Connor SE, Maresh JJ. Nat Prod Rep. 2006;23:532. doi: 10.1039/b512615k.Gul W, Hamann MT. Life Sci. 2005;78:442. doi: 10.1016/j.lfs.2005.09.007.Aygün A, Pindur U. Curr Med Chem. 2003;10:1113. doi: 10.2174/0929867033457511.
- 2.For reviews, see: Poulsen TB, Jørgensen KA. Chem Rev. 2008;108:2903. doi: 10.1021/cr078372e.Bandini M, Eichholzer A, Umani-Ronchi A. Mini-Rev Org Chem. 2007;4:115.Bandini M, Tommasi S, Umani-Ronchi A. Synlett. 2005:1199.Bandini M, Melloni A, Umani-Ronchi A. Angew Chem. 2004;116:560. doi: 10.1002/anie.200301679.Angew Chem Int Ed. 2004;43:550.You SL, Cai Q, Zeng M. Chem Soc Rev. 2009;38:2190. doi: 10.1039/b817310a.
- 3.For recent examples, see: Jia YX, Zhong J, Zhu SF, Zhang CM, Zhou QL. Angew Chem. 2007;119:5661. doi: 10.1002/anie.200701067.Angew Chem Int Ed. 2007;46:5565.Rowland GB, Rowland EB, Liang Y, Perman JA, Antilla JC. Org Lett. 2007;9:2609. doi: 10.1021/ol0703579.Kang Q, Zhao ZA, You SL. J Am Chem Soc. 2007;129:1484. doi: 10.1021/ja067417a.Terada M, Sorimachi K. J Am Chem Soc. 2007;129:292. doi: 10.1021/ja0678166.Li H, Wang YQ, Deng L. Org Lett. 2006;8:4063. doi: 10.1021/ol061552a.Wang YQ, Song J, Hong R, Li H, Deng L. J Am Chem Soc. 2006;128:8156. doi: 10.1021/ja062700v.
- 4.For recent examples, see: Ganesh M, Seidel D. J Am Chem Soc. 2008;130:16464. doi: 10.1021/ja8063292.Singh PK, Singh VK. Org Lett. 2008;10:4121. doi: 10.1021/ol8016929.Itoh J, Fuchibe K, Akiyama T. Angew Chem. 2008;120:4080. doi: 10.1002/anie.200800770.Angew Chem Int Ed. 2008;47:4016.Rueping M, Nachtsheim BJ, Moreth SA, Bolte M. Angew Chem. 2008;120:603. doi: 10.1002/anie.200703668.Angew Chem Int Ed. 2008;47:593.Evans DA, Fandrick KR, Song HJ, Scheidt KA, Xu R. J Am Chem Soc. 2007;129:10029. doi: 10.1021/ja072976i.Evans DA, Fandrick KR, Song HJ. J Am Chem Soc. 2005;127:8942. doi: 10.1021/ja052433d.
- 5.For recent examples, see: Liu WB, He H, Dai LX, You SL. Synthesis. 2009:2076.Liu WB, He H, Dai LX, You SL. Org Lett. 2008;10:1815. doi: 10.1021/ol800409d.Cheung HY, Yu WY, Lam FL, Au-Yeung TTL, Zhou Z, Chan TH, Chan ASC. Org Lett. 2007;9:4295. doi: 10.1021/ol7018532.Trost BM, Quancard J. J Am Chem Soc. 2006;128:6314. doi: 10.1021/ja0608139.Bandini M, Melloni A, Piccinelli F, Sinisi R, Tommasi S, Umani-Ronchi A. J Am Chem Soc. 2006;128:1424. doi: 10.1021/ja054109o.
- 6.Bandini M, Eichholzer A, Tragni M, Umani-Ronchi A. Angew Chem. 2008;120:3282. doi: 10.1002/anie.200705685. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2008;47:3238. [Google Scholar]
- 7.Cui HL, Feng X, Peng J, Lei J, Jiang K, Chen YC. Angew Chem. 2009;121:5847. doi: 10.1002/anie.200902093. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2009;48:5737. [Google Scholar]
- 8.Trost BM, Krische MJ, Berl V, Grenzer EM. Org Lett. 2002;4:2005. doi: 10.1021/ol020046s. [DOI] [PubMed] [Google Scholar]
- 9.a) Mahaney PE, Vu AT, McComas CC, Zhang P, Nogle LM, Watts WL, Sarkahian A, Leventhal L, Sullivan NR, Uveges AJ, Trybulski EJ. Bioorg Med Chem. 2006;14:8455. doi: 10.1016/j.bmc.2006.08.039. [DOI] [PubMed] [Google Scholar]; b) 3-Amino-1-arylpropyl Indoles as Monoamine Reuptake Inhibitors. Greenhouse R, Jaime-Figueroa S, Raptova L, Reuter DC, Stein KA, Weikert RJ. Int Pat Appl. 2005 WO 2005/118539A1. [Google Scholar]
- 10.Leonard K, Pan W, Anaclerio B, Gushe JM, Guo Z, DesJarlais RL, Chaikin MA, Lattanze J, Crysler C, Manthey CL, Tomczuk BE, Marugan JJ. Bioorg Med Chem Lett. 2005;15:2679. doi: 10.1016/j.bmcl.2005.01.028. [DOI] [PubMed] [Google Scholar]
- 11.Marković D, Hartwig JF. J Am Chem Soc. 2007;129:11680. doi: 10.1021/ja074584h. [DOI] [PubMed] [Google Scholar]
- 12.Stanley LM, Hartwig JF. J Am Chem Soc. 2009;131:8971. doi: 10.1021/ja902243s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kiener CA, Shu C, Incarvito C, Hartwig JF. J Am Chem Soc. 2003;125:14272. doi: 10.1021/ja038319h. [DOI] [PubMed] [Google Scholar]
- 14.Madrahimov ST, Marković D, Hartwig JF. J Am Chem Soc. 2009;131:7228. doi: 10.1021/ja902609g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.a) Wilson RM, Thalji RK, Bergman RG, Ellman JA. Org Lett. 2006;8:1745. doi: 10.1021/ol060485h. [DOI] [PubMed] [Google Scholar]; b) Peters R, Waldmeier P, Joncour A. Org Process Res Dev. 2005;9:508. [Google Scholar]; c) Indole Derivatives and their use as 5-HT2B and 5HT2C Receptor Ligands. Bentley JM, Bickerdike MJ, Hebeisen P, Kennett GA, Lightowler S, Mattei P, Mizrahi J, Morley TJ, Plancher JM, Richter H, Roever S, Taylor S, Vickers SP. Int Pat Appl. 2002 July 4; WO 02/051844A1. [Google Scholar]; d) Pyrroloindoles, Pyridoindoles, and Azopinoindoles as 5-HT2C Agonists. Adams DR, Bentley JM, Roffey JRA, Hamlyn RJ, Gaur S, Duncton MAJ, Davidson JEP, Bickerdike MJ, Cliffe IA, Mansell HL. US Patent 6,433,175. 2002:B1.; e) Johansen MB, Kerr MA. Org Lett. 2008;10:3497. doi: 10.1021/ol8012777. [DOI] [PubMed] [Google Scholar]; f) Kusama H, Takaya J, Iwasawa N. J Am Chem Soc. 2002;124:11592. doi: 10.1021/ja027589h. [DOI] [PubMed] [Google Scholar]
- 16.a) Beccalli EM, Broggini G, Farina A, Malpezzi L, Terraneo A, Zecchi G. Eur J Org Chem. 2002;2080 [Google Scholar]; b) Beccalli EM, Broggini G, La Rosa C, Passarella D, Pilati T, Terraneo A, Zecchi G. J Org Chem. 2000;65:8924. doi: 10.1021/jo000842g. [DOI] [PubMed] [Google Scholar]; c) Baruah B, Prajapati D, Boruah A, Sandhu JS. Synth Commun. 1997;27:2563. [Google Scholar]; d) St. C. Black D, Craig DC, Deb-Das RB, Kumar N. Aust J Chem. 1993;46:603. [Google Scholar]; e) Bhuyan PJ, Boruah RC, Sandhu JS. Tetrahedron Lett. 1989;30:1421. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.