

Chiral phosphines have found widespread use in chemical synthesis as ligands for transition metal catalysis.[1] Along with phosphine oxides and other derivatives, they have also become popular choices as catalysts in organic synthesis.[2] Organophosphorus-based catalysis will undoubtedly benefit from a more diverse range of P-stereogenic phosphines. In response to this demand, metal-catalyzed asymmetric syntheses of P-stereogenic phosphines and their derivatives have recently emerged, with key contributions including alkyne hydrophosphorylation,[3] the alkylation and arylation of secondary phosphines,[4] enantioselective deprotonation,[5] and rhodium-catalyzed [2+2+2] cycloaddition.[6] To date, these catalytic enantioselective routes remain largely outnumbered by well-established methods based on resolutions[7] or on the use of chiral auxiliaries.[8] In spite of recent advances in the area of olefin metathesis, the utility of asymmetric ring-closing metathesis (ARCM)[9] has not been applied to the preparation of P-stereogenic phosphine derivatives.[10] In light of literature precedents, which demonstrate that various P-containing dienes, trienes, and tetraenes are suitable substrates for olefin metathesis,[11] we reasoned that the ARCM of P-templates would be a strategically unique and valuable reaction for the preparation of P-stereogenic compounds. We opted for a catalytic enantioselective desymmetrization process of prochiral P-templates, as this approach offers the opportunity to explore ARCM with the chirality arising from the formation of a stereogenic center other than a carbon atom (Scheme 1). Moreover, the resulting products are structurally novel P-stereogenic scaffolds amenable to rich chemistry further downstream. Herein, we report the first examples of catalytic enantioselective olefin metathesis reactions of phosphinates and phosphine oxides, which lead to the formation of five-, six-, and seven-membered P-heterocycles in up to 98% ee. We also report an unprecedented case of complementary asymmetric induction in reactions promoted by a pair of chiral molybdenum-based complexes differing structurally in their achiral imido ligand.
Scheme 1.

Catalytic ARCM for the generation of P-stereogenic centers.
For the purposes of this investigation, chiral molybdenum catalysts 1–4[12] were selected based on their well-documented ability to promote asymmetric ring-closing metathesis in the context of kinetic resolution or enantioselective desymmetrization (Scheme 2).
Scheme 2.

Chiral and achiral molybdenum-based complexes used for ARCM and RCM reactions.
To initiate our investigations, we examined the ARCM of prochiral phosphinate 6. Catalyst 1b was identified as the optimal catalyst for the ARCM and was established by screening studies involving molybdenum-based chiral complexes 1–4 (Table 1). This optimization study also involved varying the solvent. As indicated, subtle variations in the chiral catalyst impacted significantly on both the conversion and enantioselectivity. The biphenyl-based complex 1b bearing the dimethyl-substituted phenylimido unit delivered 7 with the optimum combination of conversion (63% yield) and optical purity (60% ee) when the reaction was performed in CH2Cl2 (Table 1, entry 4).
Table 1.
Initial catalyst screening for molybdenum-catalyzed ARCM reactions of 6.a
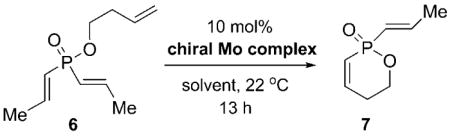 | ||||
|---|---|---|---|---|
| Entry | Chiral Mo Complex | Solvent | Conv. [%]b | ee [%]c |
| 1 | 1 a | C6H6 | <2 | – |
| 2 | 1 a | CH2Cl2 | <2 | – |
| 3 | 1 b | C6H6 | 45 | 51 |
| 4 | 1 b | CH2Cl2 | 63 | 60 |
| 5 | 2 | C6H6 | 15 | −22 |
| 6 | 2 | CH2Cl2 | 49 | 11 |
| 7 | 3 a | C6H6 | 38 | 60 |
| 8 | 3 a | CH2Cl2 | 32 | 53 |
| 9 | 3 b | C6H6 | <2 | – |
| 10 | 3 b | CH2Cl2 | <2 | – |
| 11 | 4 a | C6H6 | 51 | 16 |
| 12 | 4 a | CH2Cl2 | 65 | 11 |
All reactions performed under a nitrogen atmosphere.
Conversion into the desired product was measured by 1H NMR analysis (400 MHz) of the unpurified reaction mixture; traces of homodimer derived from reaction of terminal olefins were present.
Determined by GLC analysis of the purified material; see the Supporting Information for details.
On the basis of these preliminary results, we investigated the molybdenum-catalyzed ARCM of a range of structurally related phosphinates, all of which were synthesized according to literature procedures.[13] The results of these studies are summarized in Table 2. The catalytic ARCM of triene 8 failed to deliver the 5-membered ring-closed product 14 in the presence of all chiral molybdenum-catalysts screened (Table 2, entry 1).[14] The presence of one additional methylene group for the alkenoxy substituent was sufficient to restore reactivity, as demonstrated with the ring-closure of triene 6 and led to the six-membered phosphinate 7 in 60% ee (Table 1 and Table 2, entry 2). The replacement of the propenyl groups of triene 6 by unsubstituted vinyl groups had a beneficial impact. Indeed, the ARCM of triene 9 led to the ring-closed product 15 in higher optical purity (86% ee) and higher yield after purification (54%; Table 2, entry 3). Similarly, the ARCM of trienes 10 and 11 both led to seven-membered products; the vinyl-substituted triene 10 was a superior substrate and delivered 16 in higher yield and enantiomeric purity (73% ee; Table 2, entries 4 and 5). Substrates 12 and 13, substituted with 2-methylallyl groups, were also subjected to ARCM. The ring-closure of phosphinate 12 was not successful, a result consistent with the lack of reactivity observed with 8, which featured the same prop-2-enoxy group. In contrast, triene 13 cyclized efficiently and delivered 19 in 79% yield and 96% ee. The data indicate that the identity of the optimal molybdenum-based catalyst can change as a function of the structural features of the substrate.[10i]
Table 2.
Molybdenum-catalyzed ARCM reactions of phosphinates.a
| Entry | Substance | Product | Chiral Mo Complexb | Solvent; T [°C] | Conv.c; Yield [%] | ee [%]d |
|---|---|---|---|---|---|---|
| 1 |
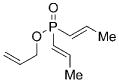 8 |
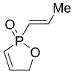 14 |
1–4 | CH2Cl2; 22–60 | <2e | – |
| 2 |
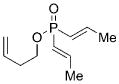 6 |
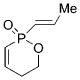 7 |
1 b | CH2Cl2; 22 | 63;43 | 60 |
| 3 |
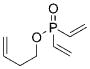 9 |
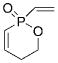 15 |
1 a | C6H6; 22 | 60;54 | 86 |
| 4 |
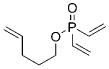 10 |
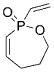 16 |
4 a | C6H6; 22 | 48;42 | 73 |
| 5 |
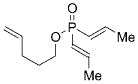 11 |
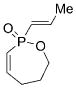 17 |
1 b | CH2Cl2; 22 | 32;30 | 27 |
| 6 |
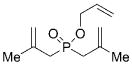 12 |
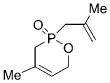 18 |
1–4 | C6H6; 22 | <2e | – |
| 7 |
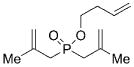 13 |
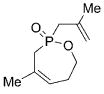 19 |
4 a | C6D6; 60 | 81;79 | 96 |
All reactions performed under a nitrogen atmosphere.
All reactions carried out with 10 mol% of catalyst.
Conversion was measured by 1H NMR analysis (400 MHz) of the unpurified reaction mixture; homodimer was also present (see the Supporting Information).
Determined by GLC analysis of the purified material; see the Supporting Information for details.
Recovered starting material.
The lack of reactivity for 8 and 12 is likely the result of catalyst sequestration and deactivation arising from the formation of an intramolecularly chelated metal-alkylidene complex (Scheme 3).[15] This hypothesis is consistent with the observation that increased distance between the terminal alkene and the Lewis basic phosphinate restores activity. To confirm this hypothesis, a mixture of achiral molybdenum-complex 5 and triene 8 was monitored by 1H NMR spectroscopy (400 MHz). After 20 minutes, 1H NMR analysis showed that initiation of the reaction had taken place and the presence of a molybdenum alkylidyne was observed, as characterized by a broad NH signal at δ = 8.1 ppm along with the absence of an alkylidene resonance.[14] When the reaction was carried out in the presence of a stoichiometric amount of 5, the molybdenum alkylidyne carbon atom resonated at δ = 306.4 ppm as observed by 100 MHz 13C NMR spectroscopy.[17] The above observation may be rationalized by a mechanistic scenario involving the formation of the chelated alkylidene complex 20 and subsequent tautomerization, thus leading to alkylidyne 21 (Scheme 3).
Scheme 3.

Formation of alkylidyne 21.
The second phase of our studies focused on molybdenum-catalyzed ARCM of phosphine oxides. The use of phosphine oxides as Lewis base catalysts highlights the need to expand the range of P-stereogenic phosphine oxides that are available for enantioselective catalysis.[2] We were well aware that, similar to phosphinates, the Lewis basicity of phosphine oxides may result in possible complications resulting from catalyst deactivation.[10i] Substrates 22–24 were subjected to olefin metathesis conditions and the results are summarized in Table 3. All prochiral triene derivatives underwent successful ring closure, including 22 and 23, that could form five-or six-membered chelates between the Lewis basic phosphine oxide and the Lewis acidic molybdenum center—this might be the result of a weak and reversible chelation process. Similar to the ARCM of phosphinates, the identity of the optimal chiral molybdenum-based catalyst can vary.[16] The enantiomeric purities of 25–27 range from 71% to 91% ee. The studies required to optimize the ARCM of the prochiral P-templates examined revealed that the sense of the stereoinduction is reversed upon modulation of the substitution pattern of the catalyst (Table 4).
Table 3.
Molybdenum-catalyzed ARCM reactions of phosphine oxides.a
| Entry | Substrate | Product | Chiral Mo Complexb | Solvent; T [°C] | Conv.c; Yield [%] | ee [%]d |
|---|---|---|---|---|---|---|
| 1 |
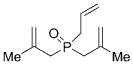 22 |
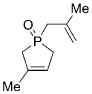 25 |
4 a | CH2Cl2; 60 | 96;88 | 71 |
| 2 |
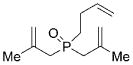 23 |
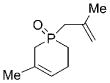 26 |
3 b | CH2Cl2; 22 | 84;80 | 74 |
| 3 |
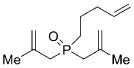 24 |
 27 |
3 b | C6H6; 60 | 81;79 | 91 |
All reactions performed under a nitrogen atmosphere.
All reactions carried out with 10 mol% of catalyst.
Conversion was measured by 1H NMR analysis (400 MHz) of the unpurified reaction mixture; homodimer was also present (see the Supporting Information).
Determined by GLC analysis of the purified material; see the Supporting Information for details.
Table 4.
Formation of the (+) and (−) enantiomers by molybdenum-catalyzed ARCM reactions.a
| Entry | Substrate | Chiral Mo Complexb | Product | Yield [%]c | ee [%]d |
|---|---|---|---|---|---|
| 1 | 13 | 4 a | (−)-19 | 78 | 96 |
| 2 | 13 | 3 b | (+)-19 | 75 | 93 |
| 3 | 24 | 4 a | (−)-27 | 45 | 98 |
| 4 | 24 | 3 b | (+)-27 | 79 | 91 |
| 5 | 13 | 1 a | (−)-19e | 65 | 82 |
| 6 | 13 | 2 | (+)-19e | 40 | 93 |
| 7 | 24 | 1 a | (+)-27f | 61 | 96 |
| 8 | 24 | 2 | (−)-27f | 48 | 73 |
All reactions performed under a nitrogen atmosphere.
All reactions carried out with 10 mol% of catalyst.
Yield of purified product.
Determined by GLC analysis.
Absolute configuration assigned by analogy to 27.
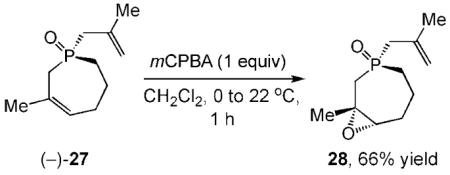
Absolute configuration determined by X-ray analysis of the epoxide derivative 28, see Ref. [18].
The pair of molybdenum-based catalysts 3b and 4a, bearing an identical achiral imido group but differently substituted diol, led to opposite enantiomers upon ARCM of trienes 13 (Table 4, entries 1 and 2). A similar trend was observed with triene 24 (Table 4, entries 3 and 4). For all four reactions, the products were formed in excellent optical purity with enantiomeric excess values ranging from 91% to 98%. In addition, we have established that complexes 1a and 2 promote ARCM reactions with a complementary sense of asymmetric induction. The above molybdenum-based alkylidenes bear identical diolate ligands and are only distinguishable through their achiral imido ligand. The data show that, remarkably, the achiral imido ligand plays a critical role in the observed sense of stereoinduction. We found that the ARCM of 13 led to opposite enantiomers upon treatment with 10 mol% of either catalyst 1a or 2; both product enantiomers were formed in high optical purity (82% and 93% ee) (Table 4, entries 5 and 6). Triene 24 behaved similarly, and led, upon treatment with 10 mol% of 1a or 2, to the two enantiomers (+)-27 and (−)-27 in 96% and 73% ee, respectively (Table 4, entries 7 and 8). The reason why a reversal of stereoinduction is triggered by structural modification of the achiral imido group may be attributed to different reacting alkylidene isomers—possibly the anti isomer for catalyst 1a based on steric repulsion between the alkylidene and the isopropyl groups and the syn isomer for catalyst 2. Although a literature precedent exists, highlighting the disparate reactivity profile of these geometrical isomers,[12] the impact of the alkylidene geometry of chiral molybdenum-based catalysts on the formation of different enantiomers has not been previously observed (Scheme 4).
Scheme 4.

Proposed model for molybdenum-catalyzed ARCM reactions of P-containing trienes, leading to the formation of (+) and (−) enantiomers.
In summary, enantiomerically enriched P-stereogenic phosphinates and phosphine oxides have been prepared in up to 98% ee through molybdenum-catalyzed ARCM reactions. The investigations outlined here represent the first report of ARCM as a route to induce chirality arising from a stereogenic heteroatom, and highlight the importance of the structural features of the reactants on reactivity and of the catalyst on enantiocontrol. Cases where different enantiomers are generated through the use of chiral olefin metathesis catalysts that are structurally distinguishable (imido ligand) are also described. Ongoing efforts in our laboratories are aimed towards expanding the scope of catalytic ARCM of P-templates and investigation of the origin of the complementarity in the enantioselectivity of the aforementioned processes.
Supplementary Material
Footnotes
This research was financially supported by the EPSRC (DTA Award to J.S.H.), the John Fell Fund (062/214), and by the United States National Institutes of Health (GM-59426).
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/anie.200805066.
References
- 1.a) Zhang X. Tetrahedron: Asymmetry. 2004;15:2099–2100. [Google Scholar]; b) Crepy KVL, Imamoto T. Top Curr Chem. 2003;229:1–40. [Google Scholar]; c) Kagan HB, Dang P-D. J Am Chem Soc. 1972;94:6429–6433. [Google Scholar]; d) Kagan HB. Asymmetric Synthesis. 1985;5:1–39. [Google Scholar]; e) Whitesell JK. Chem Rev. 1989;89:1581–1590. [Google Scholar]
- 2.a) Methot JL, Roush WR. Adv Synth Catal. 2004;346:1035–1050. [Google Scholar]; b) Vedejs E, Daugulis O, Diver ST. J Org Chem. 1996;61:430–431. doi: 10.1021/jo951661v. [DOI] [PubMed] [Google Scholar]; c) Seayad J, List B. Org Biomol Chem. 2005;3:719–724. doi: 10.1039/b415217b. [DOI] [PubMed] [Google Scholar]; d) Connon SJ. Angew Chem. 2006;118:4013–4016. [Google Scholar]; Angew Chem Int Ed. 2006;45:3909–3912. doi: 10.1002/anie.200600529. [DOI] [PubMed] [Google Scholar]; e) Ogawa C, Konishi H, Sugiura M, Kobayashi S. Org Biomol Chem. 2004;2:446–448. doi: 10.1039/b315037b. [DOI] [PubMed] [Google Scholar]; f) Kobayashi S, Sugiura M, Ogawa C. Adv Synth Catal. 2004;346:1023–1034. [Google Scholar]; g) Ogawa C, Sugiura M, Kobayashi S. Angew Chem. 2004;116:6653–6655. doi: 10.1002/anie.200461312. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2004;43:6491–6493. doi: 10.1002/anie.200461312. [DOI] [PubMed] [Google Scholar]; h) Douglass MR, Marks TJ. J Am Chem Soc. 2000;122:1824–1825. [Google Scholar]; i) Sadow AD, Togni A. J Am Chem Soc. 2005;127:17012–17024. doi: 10.1021/ja0555163. [DOI] [PubMed] [Google Scholar]
- 3.Join B, Mimeau D, Delacroix O, Gaumont AC. Chem Commun. 2006:3249–3251. doi: 10.1039/b607434k. [DOI] [PubMed] [Google Scholar]
- 4.a) Glueck DS. Chem Eur J. 2008;14:7108–7117. doi: 10.1002/chem.200800267. [DOI] [PubMed] [Google Scholar]; b) Brunker TJ, Anderson BJ, Blank NF, Glueck DS, Rheingold AL. Org Lett. 2007;9:1109–1112. doi: 10.1021/ol0700512. [DOI] [PubMed] [Google Scholar]; c) Scriban C, Glueck DS. J Am Chem Soc. 2006;128:2788–2789. doi: 10.1021/ja058096q. [DOI] [PubMed] [Google Scholar]; d) Moncarz JR, Laritcheva NF, Glueck DS. J Am Chem Soc. 2002;124:13356–13357. doi: 10.1021/ja0267324. [DOI] [PubMed] [Google Scholar]; e) Blank NF, Moncarz JR, Brunker TJ, Scriban C, Anderson BJ, Amir O, Glueck DS, Zakharov LN, Golen JA, Incarvito CD, Rheingold AL. J Am Chem Soc. 2007;129:6847–6858. doi: 10.1021/ja070225a. [DOI] [PubMed] [Google Scholar]; f) Blank NF, McBroom KC, Glueck DS, Kassel WS, Rheingold AL. Organometallics. 2006;25:1742–1748. [Google Scholar]; g) Anderson BJ, Glueck DS, DiPasquale AG, Rheingold AL. Organometallics. 2008;27:4992–5001. [Google Scholar]; h) Chan VS, Stewart IC, Bergman RG, Toste FD. J Am Chem Soc. 2006;128:2786–2787. doi: 10.1021/ja058100y. [DOI] [PubMed] [Google Scholar]; i) Chan VS, Bergman RG, Toste FD. J Am Chem Soc. 2007;129:15122–15123. doi: 10.1021/ja076457r. [DOI] [PubMed] [Google Scholar]; j) Korff C, Helmchen G. Chem Commun. 2004:530–531. doi: 10.1039/b315009g. [DOI] [PubMed] [Google Scholar]
- 5.a) Genet C, Canipa SJ, O’Brien P, Taylor S. J Am Chem Soc. 2006;128:9336–9337. doi: 10.1021/ja062616f. [DOI] [PubMed] [Google Scholar]; b) Gammon JJ, Canipa SJ, O’Brien P, Kelly B, Taylor S. Chem Commun. 2008:3750–3752. doi: 10.1039/b806864j. [DOI] [PubMed] [Google Scholar]
- 6.Nishida G, Noguchi K, Hirano M, Tanaka K. Angew Chem. 2008;120:3458–3461. doi: 10.1002/anie.200800144. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2008;47:3410–3413. doi: 10.1002/anie.200800144. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2008;47:3410–3413. doi: 10.1002/anie.200800144. [DOI] [PubMed] [Google Scholar]
- 7.Pietrusiewicz KM, Zablocka M. Chem Rev. 1994;94:1375–1411. [Google Scholar]
- 8.a) Jugé S. Phosphorus Sulfur Silicon Relat Elem. 2008;183:233–248. [Google Scholar]; b) Jugé S, GenÞt JP. Tetrahedron Lett. 1989;30:2783–2786. [Google Scholar]; c) Juge S, Stephan M, Laffitte JA, Genet JP. Tetrahedron Lett. 1990;31:6357–6360. [Google Scholar]; d) Brown JM, Carey JV, Russell MJH. Tetrahedron. 1990;46:4877–4886. [Google Scholar]; e) Carey JV, Barker MD, Brown JM, Russell MJH. J Chem Soc Perkin Trans 1. 1993:831–839. [Google Scholar]
- 9.a) Hoveyda AH. In: Handbook of Metathesis. Grubbs RH, editor. Vol. 2. Wiley-VCH; Weinheim: 2003. Chapter 3. [Google Scholar]; b) Hoveyda AH, Zhugralin AR. Nature. 2007;450:243–251. doi: 10.1038/nature06351. [DOI] [PubMed] [Google Scholar]
- 10.For molybdenum-catalyzed ARCM reactions, see: Alexander JB, La DS, Cefalo DR, Hoveyda AH, Schrock RR. J Am Chem Soc. 1998;120:4041–4042.La DS, Alexander JB, Cefalo DR, Graf DD, Hoveyda AH, Schrock RR. J Am Chem Soc. 1998;120:9720–9721.Zhu SS, Cefalo DR, La DS, Jamieson JY, Davis WM, Hoveyda AH, Schrock RR. J Am Chem Soc. 1999;121:8251–8259.Weatherhead GS, Houser JH, Ford JG, Jamieson JY, Schrock RR, Hoveyda AH. Tetrahedron Lett. 2000;41:9553–9559.Cefalo DR, Kiely AF, Wuchrer M, Jamieson JY, Schrock RR, Hoveyda AH. J Am Chem Soc. 2001;123:3139–3140.Kiely AF, Jernelius JA, Schrock RR, Hoveyda AH. J Am Chem Soc. 2002;124:2868–2869. doi: 10.1021/ja012679s.Dolman SJ, Schrock RR, Hoveyda AH. Org Lett. 2003;5:4899–4902. doi: 10.1021/ol036026n.Jernelius JA, Schrock RR, Hoveyda AH. Tetrahedron. 2004;60:7345–7351.Sattely ES, Cortez GA, Moebius DC, Schrock RR, Hoveyda AH. J Am Chem Soc. 2005;127:8526–8533. doi: 10.1021/ja051330s.Lee AL, Malcolmson SJ, Puglisi A, Schrock RR, Hoveyda AH. J Am Chem Soc. 2006;128:5153–5157. doi: 10.1021/ja058428r.
- 11.a) Schuman M, Trevitt M, Redd A, Gouverneur V. Angew Chem. 2000;112:2604–2607. [PubMed] [Google Scholar]; Angew Chem Int Ed. 2000;39:2491–2493. [PubMed] [Google Scholar]; b) Slinn CA, Redgrave AJ, Hind SL, Edlin C, Nolan SP, Gouverneur V. Org Biomol Chem. 2003;1:3820–3825. doi: 10.1039/b306940k. [DOI] [PubMed] [Google Scholar]; c) Bisaro F, Gouverneur V. Tetrahedron. 2005;61:2395–2400. [Google Scholar]; d) Dunne KS, Lee SE, Gouverneur V. J Organomet Chem. 2006;691:5246–5259. [Google Scholar]; e) Hanson PR, Stoianova DS. Tetrahedron Lett. 1998;39:3939–3942. [Google Scholar]; f) McReynolds MD, Dougherty JM, Hanson PR. Chem Rev. 2004;104:2239–2258. doi: 10.1021/cr020109k. [DOI] [PubMed] [Google Scholar]; g) Vinokurov N, Michrowska A, Szmigirlska A, Drzaga Z, Wojciuk G, Denchuk OM, Grela K, Pietrusiewicz KM, Butenschon H. Adv Synth Catal. 2006;348:931–938. [Google Scholar]
- 12.a) Schrock RR, Hoveyda AH. Angew Chem. 2003;115:4740–4782. [Google Scholar]; Angew Chem Int Ed. 2003;42:4592–4633. doi: 10.1002/anie.200300576. [DOI] [PubMed] [Google Scholar]; b) Singh R, Czekelius C, Schrock RR, Müller P, Hoveyda AH. Organometallics. 2007;26:2528–2539. doi: 10.1021/om061134+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dunne KS, Bisaro F, Odell B, Paris JM, Gouverneur V. J Org Chem. 2005;70:10803–10809. doi: 10.1021/jo0518708. [DOI] [PubMed] [Google Scholar]
- 14.One possible explanation for why 8 fails to undergo RCM might be that the product is too strained to form. For related precedents, see: BouzBouz S, Boulard L, Cossy J. Org Lett. 2007;9:3765–3768. doi: 10.1021/ol0712632.Kaiser ET, Panar M, Westheimer FM. J Am Chem Soc. 1963;85:602–607.
- 15.For examples of intramolecular Lewis base chelation to molybdenum alkylidene 5, see: Fox HH, Lee JK, Park LY, Schrock RR. Organometallics. 1993;12:759–768.
- 16.a) McCullough LG, Schrock RR, Dewan JC, Murdzek JC. J Am Chem Soc. 1985;107:5987–5998. [Google Scholar]; b) Fox HH, Lee JK, Park LY, Schrock RR. Organometallics. 1993;12:759–768. [Google Scholar]
- 17.For the full optimization study see, the Supporting Information.
-
18.The absolute configuration of (−)-27 was determined by X-ray single crystal structure analysis of the corresponding epoxide 28; see the Supporting Information.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.