

Abstract
Purpose
To test the ability of a new IGF-IR tyrosine kinase inhibitor BMS-536924 to reverse the ability of constitutively active IGF-IR (CD8-IGF-IR) to transform MCF10A cells, and to examine the effect of the inhibitor on a range of human breast cancer cell lines.
Experimental Design
CD8-IGF-IR-MCF10A cells were grown in monolayer culture, three-dimensional (3D) culture, and as xenografts, and treated with BMS-536924. Proliferation, cell-cycle, polarity, and apoptosis were measured. Twenty three human breast cancer cell lines were treated in monolayer culture with BMS-536924 and cell viability was measured. MCF7, MDA-MB-231, and MDA-MB-435 were treated with BMS-536924 in monolayer and 3D culture and proliferation, migration, polarity, and apoptosis were measured.
Results
Treatment of CD8-IGF-IR-MCF10A cells grown in 3D culture with BMS-536924 caused a blockade of proliferation, restoration of apical-basal polarity, and enhanced apoptosis, resulting in a partial phenotypic reversion to normal acini. In monolayer culture, BMS-536924 induced a dose-dependent inhibition of proliferation, with an accumulation of cells in G0/G1, and completely blocked CD8-IGF-IR-induced migration, invasion, and anchorage-independent growth. CD8-IGF-IR-MCF10A xenografts treated with BMS-536924 (100mg/kg/day) showed a 76% reduction in xenograft volume. In a series of twenty three human breast cancer cell lines, BMS-536924 inhibited monolayer proliferation of sixteen cell lines. Most strikingly, treatment of MCF7 cells grown in 3D culture with BMS-536924 caused blockade of proliferation, and resulted in the formation of hollow polarized lumen.
Conclusions
These results demonstrate that the new small molecule BMS-536924 is an effective inhibitor of IGF-IR, causing a reversion of an IGF-IR-mediated transformed phenotype.
Keywords: IGF-IR, growth transformation, mammary epithelial cell, breast cancer, small molecule inhibitor
Introduction
The insulin-like growth factor (IGF) family consists of two ligands (IGF-I and IGF-II), two receptors (IGF-IR and IGF-IIR), six high-affinity binding proteins (IGFBP 1-6), and several IGFBP-related proteins (IGFBPrP) [1, 2]. IGF-I mediates cell signaling through the IGF-IR, which upon binding IGF-I undergoes conformational changes resulting in activation of the intrinsic tyrosine kinase domain and subsequent multi-site auto-phosphorylation [2, 3] which permits activation of downstream signaling pathways such as Erk1/2 and Akt that lead to cell proliferation and survival [4].
Expression of IGF family members are often altered in numerous neoplasms, including breast cancer [5]. IGF-IR is present in human breast cancer cell lines and is frequently overexpressed and auto-phosphorylated in breast cancer [6, 7]. IGF-IR is a potent oncogene, with overexpression in NIH-3T3 fibroblasts causing transformation and tumor growth in vivo [8]. Indeed, IGF-IR is actually required for transformation, with fibroblasts derived from IGF-IR null mice (R-cells) being resistant to transformation by a variety of viral and cellular oncogenes [9-11].
Recent studies have shown that IGF-IR is a critical regulator of mammary epithelial transformation, and that overexpression in the mouse mammary gland is sufficient to induce mammary epithelial hyperplasia and tumor formation [12, 13]. Consistent with this, two studies found that overexpression of IGF-IR in human mammary epithelial cells (MCF10A) disrupted acini formation due to hyper-proliferation and increased survival [14, 15]. We have recently shown that overexpression of a constitutively active IGF-IR (CD8-IGF-IR) in MCF10A cells likewise disrupted mammary acini formation, but also caused an epithelial to mesenchymal transition and full transformation, with growth as xenografts in immunocompromised mice, an uncommon feature following transformation of MCF10A cells with a single oncogene [16].
Due to IGF-IR's critical role in proliferation and survival, the receptor has become a major focus for anti-cancer therapy [17, 18]. Various strategies, including antisense technology [19], dominant-negative IGF-IR [20], inhibitory antibodies [21-25], and small molecule inhibitors [26-29] have been used to inhibit breast cancer cell growth in vitro or in vivo by disrupting IGF-IR function. Indeed, clinical trials of antibodies and small molecule inhibitors are currently ongoing as an approach for the therapeutic blockade of the IGF-IR in human cancer [18, 30].
Here we report the ability of a new small molecule inhibitor targeting IGF-IR, BMS-536924, to reverse IGF-IR mediated transformation of mammary epithelial cells and affect breast cancer cell growth and migration. BMS-536924 completely reversed all measures of CD8-IGF-IR induced transformation in vitro, and caused a partial phenotypic reversion to more normal acini formation in Matrigel™ culture. Furthermore, the inhibitor blocked proliferation of a wide range of breast cancer cells. Most strikingly, treatment of MCF7 cells grown in three dimensional cultures with BMS-536924 caused blockade of proliferation and restoration of apical-basal polarity, resulting in a partial phenotypic reversion to normal acini similar to MCF10A cells. These results demonstrate that the new small molecule BMS-536924 is an effective inhibitor of IGF-IR, and may be effective in the treatment of IGF-IR-stimulated human breast cancer.
Materials and Methods
Cell culture
MCF10A cells were cultured in complete growth medium consisting of DMEM/F12 supplemented with 5% horse serum (Sigma, St. Louis, MO), 20ng/ml epidermal growth factor (Sigma), 10μg/ml insulin (Sigma), 0.5μg/ml hydrocortisone (Sigma), 100ng/ml cholera toxin (Sigma), and 100 units/ml penicillin-streptomycin (Invitrogen, Carlsbad, CA). CD8-IGF-IR-MCF10A and pBabe-MCF10A were generated by infection of retrovirus containing the CD8-IGF-IR-pBabe-puro or pBabe-puro as previously described [16]. All breast cancer cell lines were obtained from American Type Culture Collection (Manassas, VA), except H3396 which was obtained from the Pacific Northwest Institute (Seattle, WA). Breast tumor cell lines were maintained in RPMI1640 media, 10% heat inactivated fetal bovine serum, 100 μg/mL penicillin, and 100 μg/mL streptomycin (Invitrogen). The MCF7/Her2 cell line was made by stable transfection of the HER2 gene into MCF7 cells. MDA-MB-435 cells were originally classified as a breast cancer cell line, but have since been shown to be derived from the M14 melanoma cancer cell line and thus must be considered to be of melanoma origin [31].
IGF-I and BMS-536924 treatment and preparation of total cell lysates
For testing the inhibition of the IGF-IR pathway in pBabe-MCF10A control cells, 1×106 pBabe-MCF10A cells were seeded onto 60mm dishes. After 24 hours, the medium was changed to serum-free medium (SFM) and incubated overnight at 37°C for 24 hours. Cells were then preincubated with or without 1μM BMS-536924 for 1 hour in SFM followed by stimulation with IGF-I (50ng/ml) for 10 min. Cell monolayers were washed twice with PBS and harvested for immunoblot analysis. For dose-dependent inhibitor treatment, 1×106 CD8-IGF-IR-MCF10A cells were plated onto 60 mm cell culture dishes and grown in complete medium for 24 hours. The medium was replaced with complete growth medium plus BMS-536924 diluted in DMSO at the following concentrations: 0, 0.01, 0.1, 1, and 10 μM. After 24h cells were harvested for immunoblot analysis. For time-dependent inhibitor treatment 1×106 CD8-IGF-IR-MCF10A cells were plated onto 60 mm cell culture dishes and grown in complete medium for 24h. Medium was changed to complete growth media plus 1 μM BMS-536924. Cells were harvested after 10 min and 1, 8, 24, and 48 hours. For studies assessing the effect of the inhibitor on epidermal growth factor receptor (EGFR), 1×106 pBabe-MCF10A cells were seeded onto 60 mm dishes. After 24 hours, the medium was changed to serum-free medium (SFM) and incubated overnight at 37°C for 24 hours. Cells were then pre-incubated with increasing concentrations of BMS-536924 for 1 hour in serum-free medium (SFM) followed by stimulation with epidermal growth factor (EGF) (20ng/ml) for 10 min. Cell monolayers were washed twice with PBS and harvested for immunoblot analysis.
3D Matrigel™ culture
Three-dimensional (3D) cultures were grown in 8-well chamber slides (Falcon) as described previously [16] using growth factor reduced Matrigel™ (BD Biosciences). Cells were treated with assay medium containing 2% Matrigel™ and 1μM BMS-536924. Assay medium containing 2% Matrigel™ was replaced every 4 days. For BMS-536924 treatment day 0 was designated the next day after plating cells. Acini structures were harvested and immunostained with antibodies to Ki-67 (Dako), laminin-V (BD Biosource), activated cleaved caspase 3 (Cell signaling) and GM130 (BD Biosource) as described previously [16].
Immunoblot analysis
To evaluate phosphorylation levels of the selected IGF-IR signaling molecules, monolayer cultures were lysed with 200μl 5% SDS. Total protein extract (50-75μg) was resuspended in denaturing sample loading buffer, separated by 8% SDS-PAGE, and electrophoretically transferred to a nitrocellulose membrane overnight at 4°C. The membrane was blocked with phosphate buffered saline plus 0.05% Tween-20 (PBST) containing 5% nonfat milk for 1 h. The following antibodies were utilized: anti-pY1162/1163-IGF-IR (#44-804G, Biosource, Camarillo, CA, 1:500), anti-IGF-IRß (#sc-713, Santa Cruz Biotechnology, Santa Cruz, CA, 1:500), antiphospho-Erk1/2 (#9101, Cell Signaling Technology, Danvers, MA, 1:1000), anti-Erk1/2 (#9102, Cell Signaling Technology: 1:1000), anti-pS473-AKT (#9271,Cell Signaling, 1:1000), anti-AKT (#9272, Cell Signaling, 1:1000), anti-pS21/9-GSK3 alpha/beta (#9315, Cell Signaling, 1:1000), anti-GSK3 alpha/beta (#9316, Cell Signaling, 1:1000) and anti-β-actin (#A1978, Sigma, 1:4000). Antibodies were incubated in blocking solution overnight at 4°C. Subsequently, the membrane was washed three times for 5 min each with PBST and then incubated with a horseradish peroxidase-linked secondary antibody (Amersham Pharmacia Biotech, Piscataway, NJ) at a dilution of 1:4000 in blocking solution. After the membrane was washed three times for 5 min each with PBST, bands were visualized by enhanced chemiluminescence according to the manufacturer's protocol (Pierce Biotechnology, Rockford, Il) and captured using an Alpha Innotech 7000 (Alpha Innotech, San Leandro, CA).
Cell cycle analysis
5×105 CD8-IGF-IR-MCF10A cells and pBabe-MCF10A control cells were seeded in duplicate onto 60 mm dishes. Day 0 was designated as the day after plating cells. Cells were then incubated overnight with SFM. At day 1, medium was replaced either by complete medium or SFM containing 0, 0.1 μM, 0.5 μM, and 1μM BMS-536924 and incubated for 24 hours. Cells were washed with PBS, trypsinized, and resuspended in 2 ml 0.9% NaCl. 90% ice cold ethanol was added drop wise to fix cells for 30 minutes at room temperature. Cells were centrifuged at 1000 rpm, and stained with 50μg/ml in H2O propidium iodide and 1mg/ml RNase A (Sigma) was added. Cells were incubated for 30 min at 37°C in the dark. Single color fluorescent flow cytometry was performed with a FACScalibur flow cytometer (Becton Dickinson, USA). The histograms were analyzed with ModFit LT™ software (Verify Software, Topsham, ME).
Monolayer growth, soft agar, wound healing, and invasion assays
All assays were preformed as described previously [16]. Cells were cultured with the increasing concentrations of BMS-536924 in complete growth medium or SFM and cell number counted at day 4 using a Beckman Coulter Z Series (Beckman Coulter, Fullerton, CA). For breast cancer cell line studies, cells were seeded at 1,000 to 12,000 cells per well depending on the cell line in 96 well microtiter plates and incubated overnight. Compound was serially diluted and added. After 72 hr exposure, a cytotoxicity evaluation was performed using an MTS tetrazolium dye conversion assay. The IC50 values were calculated and expressed graphically for each cell line exposed to the compound. Anchorage-independent colony formation was assessed as described previously. Every 4 days growth medium (200μl) containing 0, 0.1μM, 0.5μM, and 1μM BMS-536924, respectively, was added. Colonies were stained with 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) (5mg/ml in PBS) by incubation for 4 hrs at 37°C and photographed using an Alpha Imager 7000 (Alpha Innotech, San Leandro, CA) and then counted. The wound healing assay was performed as described previously with proliferation inhibited with 5μM mitomycin C. The invasion assay was performed as described previously. The lower compartment was filled with 800μl complete medium or complete medium containing 1μM BMS-536924.
Xenograft studies
All procedures were conducted in accordance with the NIH Guide for the Care and Use of Laboratory Animals, and were approved by the IACUC of Baylor College of Medicine. Mice were maintained on a 12-hr light, 12-hr dark schedule with ad libitum access to laboratory chow (Pico Lab Rodent Diet 20, Lab Diet 5053, PMI Nutrition International Inc., Brentwood, MO) and water. 5×106 pBabe-MCF10A or CD8-IGF-IRMCF10A cells were injected orthotopically into the thoracic mammary gland of athymic nude mice (with or without Matrigel™). pBabe-MCF10A cells did not grow. When CD8-IGF-IR-MCF10A cells were between 100-200mm3 they were randomized to receive either vehicle or BMS-536924 (100mg/kg) daily by gavage. Tumor size was measured by digital caliper and tumor volumes were estimated according to the formula for an ellipse (short dimension)2 × (long dimension)/2. Mice didn't lost weight during treatment, and there were no outward signs of toxicity.
Immunohistochemistry
Tumor sections were cut at intervals of 100 μm and stained by hematoxylin and eosin (H&E) and then examined microscopically. Serial sections (5 μm thick) were placed on Superfost Plus slides (Fisher Scientific, Fair Lawn, NJ), deparaffinized, and gradually hydrated. Immunohistochemistry was performed for mouse cleaved caspase 3 (CC3) (1:100 Covance, Berkeley, CA) using a Vectastain ABC peroxidase immunodetection kit (rat immunoglobulin G [IgG] as a negative control) and a mouse on mouse (M.O.M.) immunodetection kit (both purchased from Vector Laboratories, Burlingame, CA).
Results
BMS-536924 inhibits IGF-I-stimulated IGF-IR signaling in MCF10A cells and blocks constitutive IGF-IR activity in CD8-IGF-IR-MCF10A
BMS-536924 was identified in a research program aimed at development of small molecule ATP-competitive inhibitors of the IGF-IR. BMS-536924 inhibits IGF-IR kinase activity with an IC50 of 80nM, but also inhibits a small number of other kinases including insulin receptor [32]. To investigate the ability of BMS-536924 to inhibit IGF-IR activity, tyrosine phosphorylation of the IGF-IR was examined in the presence or absence of the inhibitor and IGF-I (Fig. 1). MCF10A cells showed no detectable phosphorylation of the IGF-IR when cells were cultured in serum-free medium (SFM) without addition of growth factors. However, in the presence of IGF-I, key tyrosine residues in the kinase domain of the β-subunit of the IGF-IR became phosphorylated (Fig. 1A). Pre-incubation of cells with 1μM BMS-536924 completely blocked the ability of IGF-I to stimulate IGFIR phosphorylation. There was no change in the amount of total IGF-IR between those different treatment groups as determined by immunoblotting for total receptor levels. We next determined the effect of BMS-536924 on key signaling molecules downstream of IGF-IR. MCF10A cells in SFM showed weak phosphorylation of ERK1/2 and GSK3β, but p-AKT was not detectable. IGF-I stimulation resulted in increased phosphorylation of ERK1/2, GSK3β, and AKT. BMS-536924 inhibited this ligand-induced phosphorylation, consistent with the blockade of IGF-IR phosphorylation and activation. Basal levels of GSK3β and ERK1/2 phosphorylation were reduced by BMS-536924 potentially due either to low-level autocrine (IGF-I or IGF-II) activation of IGF-IR or due to BMS-536924 inhibition of other signaling pathways. We did not observe any appreciable change in the level of total ERK1/2, AKT, or GSK3β among the various treatment groups.
Figure 1. BMS-536924 inhibits IGF-IR signaling in pBabe-MCF10A cells and inhibits phosphorylation of CD8-IGF-IR.

A. pBabe-MCF10A cells in SFM were pretreated with BMS-536924, and then incubated with or without BMS-536924 in the presence or absence of IGF-I (50ng/ml). Cells were lysed and analyzed by immunoblot as described in Materials and Methods and probed using IGF-IRβ-specific antibodies as well as total/phospho-specific antibodies for AKT, ERK1/2, and GSK3β. β-actin was used as a loading control. B. CD8-IGF-IR-MCF10A cells were incubated in SFM and then exposed to increasing concentrations of BMS-536924 for 24 hours and harvested for immunoblot analysis. C. CD8-IGF-IR-MCF10A cells were cultured in SFM and then incubated with 1μM BMS-536924 and harvested after 10min, 1 hour, 8 hours, 24 hours, or 48 hours for immunoblot analysis. D. pBabe-MCF10A cells were incubated in SFM and pretreated for 1hour with increasing concentrations of BMS-536924 (0.01, 0.1 0.25, 0.5, 0.75, 1μM), followed by stimulation with EGF (20ng/ml) for 15min. Cells were lysed and analyzed by immunoblot.
CD8-IGF-IR is a chimeric receptor consisting of the extracellular domain of human CD8α fused to the β-subunit of IGF-IR. Disulfide bonding between CD8α subunits is believed to cause the IGF-IR kinase domains to have constitutive activity [12]. To study the role of IGF-IR in mammary epithelial cell transformation, and the ability of BMS-536924 to reverse this, MCF10A human immortalized mammary epithelial cells were previously infected with a retrovirus encoding CD8-IGF-IR and stable clones isolated (thereafter called CD8-IGF-IR-MCF10A) [16]. Treatment of the CD8-IGF-IRMCF10A cells with BMS-536924 resulted in a dose-dependent inhibition of phosphorylation with partial inhibition at 0.01μM and 0.1μM, but complete receptor inhibition at a concentration of 1μM (Fig. 1B). Maximal inhibition of phosphorylated IGF-IR was observed as early as 10 min following incubation (Fig. 1C). BMS-536924 retained its ability to inhibit IGF-IR phosphorylation for up to 48hr. Furthermore, addition of BMS-536924 time-dependently inhibited AKT phosphorylation starting at 1 hour. By 48 hours, AKT activation was completely blocked.
MCF10A cells are dependent upon EGF for proliferation, and typically require EGF in the culture medium for growth [33]. To assure that any further work with the inhibitor was not due non-specific blockade of EGFR, we examined EGF activation of EGFR in the presence of increasing concentrations of BMS-536924. BMS-536924 up to a concentration of 1μM (which was maximal for blocking IGF-IR – see Fig. 1A and B) had no effect upon EGF-mediated activation of EGFR (Fig. 1D). EGFR-mediated activation of AKT and ERK1/2 was also not affected by BMS-536924.
CD8-IGF-IR disrupts normal mammary acini formation in three-dimensional Matrigel™, which is partially reversed by BMS-536924
Previous studies have shown that abnormal expression of oncogenes can induce hyperproliferation, disrupt apico-basal polarity, and suppress apoptosis to alter normal mammary acinar morphogenesis of MCF10A cells in 3D Matrigel™ [34]. Similarly, transformed breast epithelial cells grown in 3D Matrigel™ show phenotypes similar to those of transformed MCF10A cells, with uncontrolled proliferation and lack of polarity. Targeting β1 integrin can lead to a reversion of the malignant phenotype and formation of normal acini [35, 36]. In our previous study we reported that CD8-IGF-IR-MCF10A cells grown in Matrigel™ formed large, multilobulated structures with enhanced proliferation and luminal filling [16]. Herein, we further analyzed CD8-IGF-IR-mediated disruption of MCF10A acini morphogenesis and found that pBabe-MCF10A cells underwent growth arrest similar to that described by others [14, 15, 34], whereas in stark contrast, CD8-IGF-IR-MCF10A cells were highly proliferative with the high rate of proliferation over an extended period of time (Supplementary Figure 1). This high degree of proliferation likely contributed to expansion and overgrowth of the acini. Consistent with IGF-IR stimulating survival and luminal filling, CD8-IGF-IR showed little or no apoptosis in the luminal space as assessed by CC3 staining (data not shown and Fig 2C). Additionally, pBabe-MCF10A cells showed a polarized layer of cells as described by others [37], whereas CD8-IGF-IR-MCF10A cells lost polarity as indicated by aberrant deposition of LamininV in the luminal space (Supplementary Figure 1).
Figure 2. BMS-536924 blocks acinar proliferation, partially restores polarization, and induces apoptosis in CD8-IGF-IR-MCF10A acini.

A. A. pBabe-MCF10A and CD8-IGF-IR-MCF10A acini were plated in Matrigel™ and treated every four days with or without 1μM BMS-536924. Acini were cultured for 12 days and stained with antibodies against Ki67 (green) or laminin V (red). Nuclei were counterstained with ToPro3. Confocal microscopy visualized cross-sections through the middle of the acini with 40x magnification. Pictures show representative acini. B. Quantification of the number of cells per acinus was performed by image analysis. Data represent means ± SE from ten different acini. The percentage of Ki67 positive cells within acini was scored after the indicated number of days in culture. Values represent the mean ± the SE of 10 acini. C. pBabe-MCF10A and CD8-IGF-IR-MCF10A were cultured for 12 days and then treated with 1μM BMS-536924. After 4 days treatment, acini were stained as in A. D. The percentage of CC3 positive cells within the acini was scored after the indicated number of days in culture. Values represent the mean ± SE of 10 acini.
These studies showed that overexpressed IGF-IR caused dramatic alterations in mammary acinar morphogenesis involving enhanced proliferation, suppressed apoptosis, and a loss of polarization. To investigate the effect of BMS-536924 on proliferation and apicobasal polarization, pBabe-MCF10A and CD8-IGF-IR-MCF10A cells were cultured in Matrigel and treated with 1μM BMS-536924 every 4 days. BMS-536924 inhibited growth with both pBabe-MCF10A and CD8-IGF-IR-MCF10A acini being smaller in size and a reduction of more than 80% of Ki67-positive cells (Fig. 2A and quantified in 2B) The size reduction was more dramatic in CD8-IGF-IR-overexpressing cells but most strikingly, BMS-536924 treatment of CD8-IGF-IR-MCF10A cells resulted in the formation of acini that resembled the MCF10A acini, and we didn't observe any large multilobular structures (Fig. 2A). BMS-536924 treatment of CD8-IGF-IR-MCF10A cells resulted in acini with distinct polarization and lamininV deposition to the basal surface of the acini and a disappearance of lamininV from the lumen (Fig. 2A). Therefore, BMS-536924 is able to partially reverse IGF-IR mediated disruption of proliferation and polarization and results in the formation of acini that resemble normal MCF10A cells. These results are similar to those results originally observed with targeting β1-integrin that led to a reversion of the malignant phenotype and formation of normal acini [35, 36].
BMS-536924 induces apoptosis in CD8-IGF-IR-MCF10A acini
IGF-IR induces filling of the luminal space, in part, by blocking apoptosis [14, 15, 34]. We therefore tested whether BMS-536924 had the ability to enhance apoptosis in the center of the acini. CD8-IGF-IR-MCF10A and pBabe-MCF10A were cultured in matrigel for 12 days, and then treated for 4 days with 1μM BMS-536924. At day 16 acini were harvested, fixed, and stained for cleaved caspase 3 (CC3) as a marker for apoptosis, and Ki67 as a marker of proliferation. At day 16, pBabe-MCF10A acini showed little or no proliferation either in the presence or absence of BMS-536924 (Fig. 2C). In contrast, CD8-IGF-IR-MCF10A acini were large and hyperproliferative, and treatment with BMS-536924 caused a dramatic reduction in Ki67 staining (Fig. 2C). CC3 was not detectable in untreated pBabe-MCF10A acini whereas treatment with BMS-536924 resulted in the detection of cells positive for CC3, indicating a role for IGF-IR in survival of MCF10A cells in culture. More importantly, treatment of CD8-IGF-IR-MCF10A cells with BMS-536924 resulted in a dramatic induction of apoptosis, with up to 40% of CC3 positive cells (Fig. 2C and quantified in 2D). Interestingly, the majority of the staining was in the center of the largely misshapen acini, but we also noted apoptotic cells in the outer layer. Despite this large increase in apoptosis, we didn't observe a complete elimination of cells in the center of the lumen. Even a longer incubation with BMS-536924 for 6 days after 12 days of culturing CD8-IGF-IR-MCF10A cells in matrigel didn't allow luminal clearance (data not shown), suggesting either that the increased apoptosis is not sufficient alone to clear the luminal space or that a longer incubation is needed to see this effect.
BMS-536924 inhibits growth of CD8-IGF-IR-MCF10A cells and causes a G0/G1 block
MCF10A cells require serum and EGF for proliferation [38]. In SFM MCF10A cells undergo growth arrest. To assess the anti-proliferative ability of BMS-536924, we performed a monolayer growth assay on pBabe-MCF10A and CD8-IGF-IR-MCF10A cells. In SFM, pBabe-MCF10A cells were mostly refractory to BMS-536924, while CD8-IGF-IR-MCF10A cell growth was inhibited in a dose-dependent manner with a complete blockade of proliferation at an IC50 of 0.48μM BMS-536924 (Fig. 3A). The lack of response of pBabe-MCF10A cells to BMS-536924 was due to the fact that in SFM very few cells (~1.5-2.0%) were in S-phase (Fig. 3B); most were found in G0/G1 (data not shown). In contrast, a large fraction of CD8-IGF-IR-MCF10A cells were found in S-phase (~24%), and this fraction was reduced in a dose-dependent manner with BMS-536924, with 1μM causing an 89% reduction in S-phase fraction (Fig. 3B). At the same time there was a dramatic increase (from 66% to 91.53%) of cells in G0/G1 after BMS-536924 treatment (Fig. 3C). Interestingly, when the same assays were performed in cells in complete growth medium, BMS-536924 had little effect on proliferation or S-phase fraction (data not shown), showing only a 9% decrease in S-phase cells. This indicates that IGF-I or the CD8-IGF-IR is not sufficient to induce proliferation in this setting and that in complete growth medium, other growth factors are sufficient for cell growth, e.g. EGF. This data is also consistent with data from Fig. 1D showing that BMS-536924 (1μM) is selective for IGF-IR and doesn't block EGFR. We noted that during the FACS experiments, cell death was negligible as nearly 100% of cells were gated for analysis, and we didn't didn't detect a subG1 population indicative of apoptosis (Fig. 3C). This is in contrast to the ability of BMS-536924 to induce apoptosis in 3D culture and suggests that CD8-IGF-IR may be required for survival signals in 3D but not 2D culture. This has previously been reported by Dr. Baserga's group [39].
Figure 3. BMS-536924 induces a dose-dependent inhibition of proliferation with a decrease in S-phase cells, and inhibits IGF-IR-induced migration and invasion.
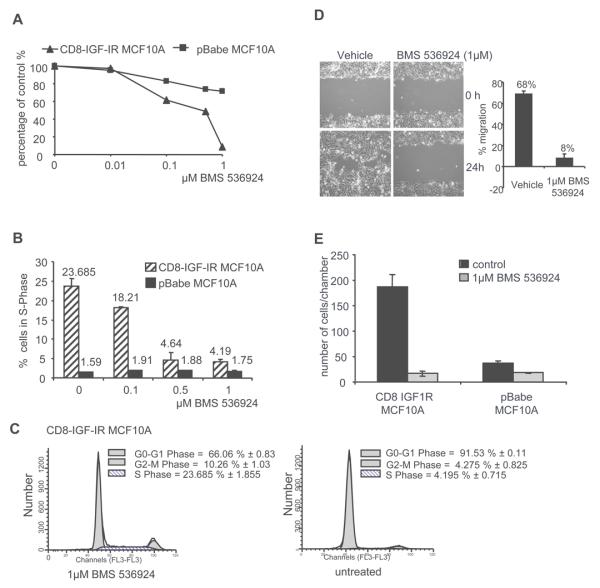
A. Monolayer growth assay. pBabe-MCF10A and CD8-IGF-IR-MCF10A were exposed to increasing concentrations of BMS-536924 in SFM for 4 days. The panel displays percentage of growth inhibition with an IC50 of 0.4μM BMS-536924 for CD8-IGF-IR MCF10A. B. Cell cycle analysis of CD8-IGF-IR-MCF10A and pBabe-MCF10A in SFM with increasing concentrations of BMS-536924. C. Single-parameter histogram of DNA shows the discrimination of cell populations existing in G0/G1, S, and G2/M phases of the cell cycle. D. Confluent monolayers of CD8-IGF-IR-MCF10A cells were incubated overnight in either full medium or SFM. One hour before treatment proliferation was inhibited by 1μM mitomycin C. Monolayers were lightly scratched and cultured in SFM or complete medium containing 1μM mitomycin C or 1μM mitomycin C and 1μM BMS-536924. Photographs were taken immediately (0h) and 24 hour post-scratch at 10x magnification and the percentage of wound closure was quantified. E. Matrigel invasion assay showed that BMS-536924 blocks CD8-IGF-IR-MCF10A cells from penetrating the membrane. Cells were counted in a minimum of 4 high-powered fields (HPF). Data are presented as number of cells per chamber (number of cells/HPF), and represent mean ± SE of duplicate experiments.
BMS-536924 blocks CD8-IGF-IR-induced migration and invasion
To measure the effect of BMS-536924 on cellular migration in CD8-IGF-IRMCF10A cells, a scratch-in assay was performed. In the absence of BMS-536924, CD8-IGF-IR-MCF10A cells were able to migrate into a scratch in either SFM (68% of scratch filled and Fig. 3D) or complete growth medium (58% of scratch filled, data not shown). Treatment with 1μM BMS-536924 completely blocked migration in both SFM (Fig. 3D) and complete growth medium (data not shown). In contrast to the proliferation assays (Fig. 3A), where BMS-536924 had little effect on growth of CD8-IGF-IR MCF10A cells grown in complete medium, the complete blockade of migration in cells grown in complete medium suggests that CD8-IGF-IR alone is both sufficient and required for migration even in the presence of serum or other growth factors. We also tested the effect of BMS-536924 on CD8-IGF-IR-MCF10A cell invasion using a modified Boyden chamber assay with Matrigel™-coated wells (Fig. 3E). pBabe-MCF10A cells showed a poor ability to invade through the Matrigel™-coated chamber whereas CD8-IGF-IRMCF10A cells were highly invasive. BMS-536924 completely blocked invasion of CD8-IGFIR-MCF10A through Matrigel™-coated wells.
BMS-536924 blocks CD8-IGF-IR-MCF10A soft-agar growth and causes regression of xenografts in vivo
CD8-IGF-IR-MCF10A cells exhibit several hallmarks of transformation. Consistent with this, CD8-IGF-IR-MCF10A cells were able to grow in soft agar assays, whereas pBabe-MCF10A cells failed to grow in soft agar consistent with their non-transformed phenotype (Fig. 4A). Increasing doses of BMS-536924 caused a dose dependent reduction in CD8-IGF-IR-MCF10A colony formation, with a low concentration of BMS-536924 (0.1μM BMS) causing significantly smaller colonies (Fig. 4A), and 0.5μM BMS-536924 completely inhibiting colony formation. We next examined the effects of BMS-536924 in vivo. Injection of CD8-IGF-IR-MCF10A resulted in the appearance of palpable tumors that rapidly grew to ~100-200mm3 in size. Interestingly, despite the in vitro transformed phenotype of CD8-IGF-IR-MCF10A cells, xenografts stopped growing once they reached 100-200mm3. Histological analysis of the xenografts showed them to be poorly differentiated carcinomas that were relatively benign and non-invasive. Similar results following transformation of human immortalized mammary epithelial cells have been previously reported [40]. Despite this lack of growth past 100-200mm3, we tested the ability of BMS-536924 to cause regression of these xenografts. Therefore, when xenografts had reached 100-200mm3, mice were treated daily with either vehicle or BMS-536924 (100mg/kg) and tumor volume measured. Within 1 week of treatment xenograft volume was reduced and after 2 weeks an average reduction of 76% tumor volume was noted (Fig. 4B). Treated xenografts showed large areas of necrosis with apoptotic cells as assessed by CC3 staining (Fig. 4C).
Figure 4. BMS-536924 inhibits transformation in vitro and in vivo.
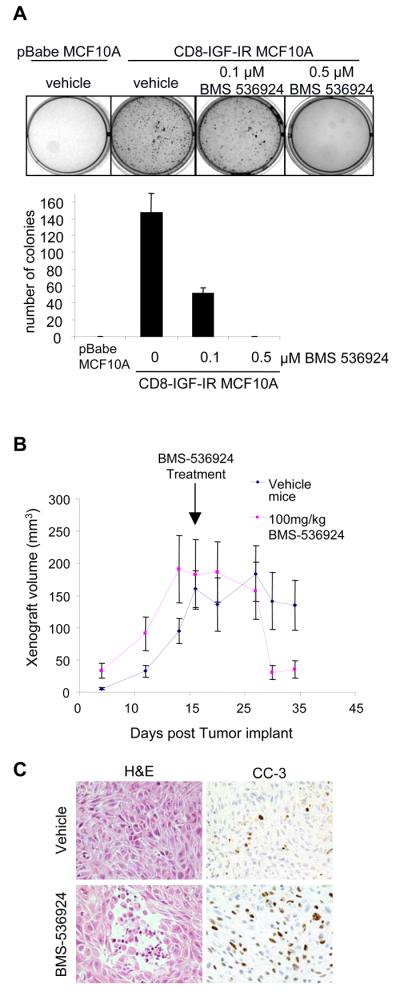
A. For anchorage-independent growth assay, 1×104 cells were suspended in their growth medium containing 0.35% agarose and increasing concentrations of BMS-536924 and seeded in 6 well plates coated with a basal layer of complete medium containing 0.7% agarose. Colony assay was set up in triplicate. 3 weeks later, colonies were stained with 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT), counted, and plotted against increasing concentrations of BMS-536924. B. 6 week-old mice were injected with CD8-IGF-IR-MCF10A cells in the #3 mammary gland. Once xenografts reached a volume of 100-200 mm3 they were randomized and treated once daily with 100 mg/kg BMS-536924. Data represents the mean ± SE of xenograft volumes. C. Treated and untreated CD8-IGF-IR-MCF10A xenografts were harvested after 3 weeks of treatment and embedded in paraffin. 5μm tumor sections were stained by H&E or processed for IHC using an anti-CC3 antibody. Pictures show representative tumor cross-sections at 40x magnification.
BMS-536924 inhibits growth of a wide range of breast cancer cell lines
To assess the ability of BMS-536924 to inhibit growth of human breast cancer cell lines, the minimal and maximal IC50 were determined on 23 different cell lines (Fig. 5A). MCF7 cells were the most sensitive breast cancer cell line with an IC50 of 1.2 µM BMS-536924. To confirm the ability of BMS-536924 to inhibit IGF-IR activity of MCF7 cells, tyrosine phosphorylation of the IGF-IR was examined in the presence or absence of the inhibitor and IGF-I (Fig. 5B). MCF7 cells showed no detectable phosphorylation of the IGF-IR when cultured in SFM. However, in the presence of IGF-I the IGF-IR receptor was phosphorylated (Fig. 5B). Pre-incubation of cells with 1μM BMS-536924 completely blocked the ability of IGF-I to stimulate IGF-IR phosphorylation. Furthermore, addition of BMS-536924 inhibited downstream activation of AKT (Fig. 5B). In stark contrast to the sensitivity of MCF7 cells to BMS-536824, MCF7 cells that overexpressed HER2/ErbB2 were completely resistant (IC50 >9.9 μM, Fig. 5A). Among the different tumor cell lines sensitivity varied widely, of the 23 cell lines, 17 responded with an average IC50 of 5.24 μM, while 7 were resistant to BMS-536924 with IC50 of >9.9 μM. Many of these resistant cell lines overexpressed ErbB2/HER2 (such as MCF7/HER2), consistent with other reports that Her2 can cause resistance to IGF-IR inhibition and favoring the notion of co-targeting of these pathways in breast cancer.
Figure 5. BMS-536924 inhibits growth in a panel of breast cancer cell lines.

A. Breast cancer cells were seeded at 1,000 to 12,000 cells per well depending on the cell line in 96 well microtiter plates and incubated overnight. BMS-536924 was serially diluted and added. After 72 hr exposure, a cytotoxicity evaluation was performed using an MTS tetrazolium dye conversion assay. Bars represent the average IC50 (μM) of each breast cancer cell line. For 7 cells lines the IC50 was not reached (>9.9μM). B. MCF7 cells were pretreated with BMS-536924, and then exposed to BMS-536924 in the presence or absence of IGF-I (50ng/ml). Cells were analyzed by immunoblot as described in Materials and Methods and probed using IGF-IRβ-specific antibodies as well as total/phospho-specific antibodies for AKT. C. The relative IGF-IR level (densitometry value from immunoblot) was determined for cells with an IC50 below or above the mean of the group of cells (5.89μM). Bars represent the average IGF-IR level for cells with an IC50 <6μM (n=7) and >6μM (n=6) +/− S.E.M. Statistical significance was determined by the Student t test; *, P < 0.05. D. Scratch-in assay of MDA-MB-231 cells was performed as described in Materials and Methods. Photographs were taken immediately (0h) and 24 hour post-scratch at 10x magnification and the percentage of wound closure was quantified. E. For anchorage-independent growth assay, cells were cultured in their growth medium containing 0.35% agarose and increasing concentrations of BMS-536924. Colony assay was set up in triplicate. After 3 weeks, colonies were stained with 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT), counted, and plotted against increasing concentrations of BMS-536924.
To determine the correlation between the sensitivity of cell lines to BMS-536924 and level of IGF-I receptor protein expression we examined publicly available data from Neve et al. [41]. IGF-IR protein levels (derived from densitometric quantitation of immunoblots) were available on 13 cell lines for which we had the IC50 for BMS-536924. Dichotomization of the cell lines based upon the mean IC50 (5.89μM) showed significant higher (p<0.05) IGF-IR levels were correlated sensitivity to the drug (Fig. 5C).
BMS-536924 inhibits migration of MDA-MB-231 cells and blocks MCF7 soft agar growth
To assess whether BMS-536924 may affect other biological properties of breast cancer cells, we examined the influence of the inhibitor upon migration and anchorage-independent growth. MDA-MB-231 cell migration was measured using a scratch-in assay (Fig. 5D). After 24 hours, 42% of cells migrated into the gap. BMS-536924 (2μM) completely inhibited migration of MDA-MB-231 cells in the scratch-in assay (Fig. 5D). Of note, that maximal concentration for inhibition of migration was still lower than the IC50 required for inhibition of monolayer proliferation (IC50 4.82 μM, Fig. 5A). In soft agar assays, MCF7 cells showed a dose-dependent decrease in colony number and colony size (Fig. 5E) which is consistent with the results seen in CD8-IGF-IR-MCF10A. Effects on colony formation were observed at concentrations as low as 0.1μM BMS-536924, with a complete blockade at 1μM BMS-536924. Similar to results in MDA-MB-231 cells, the IC50 for growth inhibition of MCF7 cells in soft agar growth was lower (approximately 10-fold) than for monolayer growth.
BMS-536924 decreases proliferation and cell number in MCF7 and MDA-MB-435 cells grown in 3D culture, and in MCF7 cells causes lumen formation
In 3D culture, breast cancer cells show uncontrolled proliferation, suppression of apoptosis, and a lack of polarity that result in the cells forming clumps that have little or no resemblance to MCF10A acini. To investigate the effect of BMS-536924 on proliferation and apicobasal polarization, we analyzed 3D cultures of MCF7 cells and MDA-MB-435 cells cultured in Matrigel™ and treated with 1μM BMS-536924 every 4 days. BMS-536924 inhibited growth with more than a 27% reduction compared to vehicle control. (Fig. 6A and B). To determine the effect of treatment on cell number as a whole, we counted the total number of cells per acinus and compared untreated and treated acini. We found that IGF-IR inhibition resulted in a significant decrease in total cell number (Fig. 6B). Most strikingly, however, BMS-536924 treatment of MCF7 cells resulted in the formation of acini that resembled the MCF10A acini with hollow lumen (Fig. 6A). Staining with an antibody for GM130, a matrix protein in the cis-Golgi compartment [42] showed distinct acinar polarization. Similar to the results observed with targeting CD8-IGF-IR in MCF10A cells, BMS-536924 was able to reverse disruption of polarization, allowing formation of acini that resemble normal MCF10A cells. We next examined the effects of BMS-536924 on MDA-MB-435 acini formation. BMS-536924 was able to suppress proliferation indicated by a 41% reduction compared to vehicle (Fig. 6C, D). However, treatment didn't cause polarization in MDA-MB-435 acini, as GM130 was dispersed. In both cell lines we didn't observed no change in apoptosis as indicated by CC3 staining (Fig. 6A and C); perhaps because apoptosis occurred at an earlier stage of acinar morphogenesis.
Figure 6. BMS-536924 decreases proliferation and cell number in MCF7 and MDAMB-435 acini and reverses MCF7 acini formation.

A. MCF7 cells were placed in morphogenesis assays, treated every four days with BMS-536924, and then fixed and immunostained at day 12 with antibodies to Ki67, active caspase-3 or GM130. Nuclei were counterstained with TOPRO-3. B. The number of cells per acinus was quantified. Values represent the mean ± the SE of 10 acini. C. MDA-MB-435 acini were treated every four days with BMS-536924 and immunostained with antibodies to Ki67, activated caspase-3, or GM130. Nuclei were counterstained with TOPRO-3 (blue). D. The number of cells per acinus was quantified. Values represent the mean ± the SE of 10 acini.
Discussion
In the present study we showed several significant findings. First, BMS-536924 fulfills the key characteristics expected from an IGF-IR inhibitor, completely blocking IGF-IR tyrosine kinase activity. Second, treatment of CD8-IGF-IR-MCF10A acini with BMS-536924 caused a partial phenotypic reversion to normal acini with blockade of proliferation, restoration of apico-basal polarization, and enhanced luminal apoptosis. Third, BMS-536924 completely blocked and/or reversed all aspects of IGF-IR-induced transformation including IGF-IR-mediated migration, invasion, anchorage-independent growth, and xenograft growth. Finally, BMS-536924 was effective at inhibiting growth of a wide range of breast cancer cell lines, and caused MCF7 cells in 3D culture to form polarized hollow lumen.
Previously, Bissell's group showed that phenotypic reversion of malignant T4-2 cells was possible by inhibition with antibodies directed against EGFR or β1-integrin [43]. Different antibodies reversed invasive and metastatic breast cancer cells to a near normal phenotype of growth-arrested acini structures, or caused cell death. Phenotypic reversion of T4-2 cells was also achieved by treatment with a PI3 kinase (PI3K) inhibitor [44]. Similar to the studies by Bissell, we found that BMS-536924 caused a phenotypic reversion of CD8-IGF-IR acini structures to near-normal polarized MCF10A acini. Most strikingly, multi-acini structures didn't develop after treatment with BMS-536924. Consistent with this, BMS-536924 was able to inhibit proliferation and partially restore polarization. In contrast to the partial reversion in 3D culture, BMS-536924 completely reversed all measures of transformation in vitro.
BMS-536924 showed a lower IC50 against CD8-IGF-IR-MCF10A and MCF7 cells grown in anchorage-independent versus monolayer cultures. This is consistent with studies showing that IGF-IR is not an essential requirement for monolayer growth [45, 46] but that IGF-IR is a strict requirement for anchorage-independent growth [39]. Presumably there are IGF-IR-dependent survival pathways that are selectively active in anchorage-independent conditions and are inhibited by BMS-536924. In addition, BMS-536924 was effective at blocking migration of MDA-MB-231 cells at a concentration below the IC50 for monolayer proliferation. Our results thus reaffirm the need to test inhibitors in multiple settings and not only in monolayer proliferation assays.
Overexpression of a dominant-acting oncogene can cause cells to become ‘addicted’ to the oncogene [47]. This is in part revealed by overexpression of the CD8-IGF-IR in MCF10A cells. Thus, CD8-IGF-IR allows MCF10A cells to grow in SFM, but they are now completely reliant upon IGF-IR for proliferation, since blockade causes a complete loss of cells in S-phase. Interestingly, BMS-536924 did not block proliferation or S-phase fraction of CD8-IGF-IR-MCF10A cells grown in complete medium, indicating that in this setting other factors confer proliferation. In contrast to this, BMS-536924 completely inhibited migration in both SFM and complete medium, indicating that active IGF-IR is both sufficient and required for migration in both conditions.
CD8-IGF-IR-MCF10A xenografts showed an unusual growth curve, with rapid growth until 100-200mm3 but then slow growth and a stable size for over 3 weeks. This is consistent with other reports showing that transformation of mammary epithelial cells results in the formation of undifferentiated carcinomas that are benign, relatively noninvasive, and do not metastasize [40]. It has recently been reported that a special serum-free medium allows the isolation of breast primary epithelial cells (BPECs) that when transformed result in highly tumorigenic adenocarcinomas [48]. It seems that the mammary epithelial precursor of the tumorigenic cells affect the tumor cell type including its metastatic potential. Despite the relative benign nature of our transformed CD8-IGF-IR-MCF10A cells, we were still able to show that BMS-536924 caused regression and reduction in tumor volume.
One exciting aspect of this study is the finding that IGF-IR inhibition not only reduces proliferation but induces apoptosis. Preclinical studies by other groups have found similar observations. The small molecule inhibitor ADW742 inhibits proliferation not only by interfering with cell cycle progression but also by inducing apoptosis [27]. Similarly an antibody against IGF-IR (A12, Imclone) also acts to block MCF7 xenograft growth via an induction of apoptosis rather than a blockade of proliferation [21]. This dual mechanism is of great importance as it suggests that IGF-IR inhibitors may act as both cytostatic and cytotoxic agents. Additionally, it was recently shown that the combination of an anti-IGF-IR antibody with chemotherapy enhances anti-tumor activity [49].
In summary, this is the first study to show that a small molecule inhibitor reverses IGF-IR-mediated transformation of mammary epithelial cells in vitro and in vivo. In addition, BMS 536924 causes a phenotypic reversion of CD8-IGF-IR-induced transformed acini structures, accompanied by suppression of proliferation, induction of apoptosis, and a partial restoration of polarization. Finally BMS-536924 can restore polarization in MCF7 cells, and affects growth of a range of breast cancer cells lines, highlighting a possible role in breast cancer therapy.
Statement of Translational Relevance.
The insulin-like growth factor receptor (IGF-IR) is critical for cell transformation, and within the last few years several drugs targeting IGF-IR have entered clinical trials and are showing promising early results. In this report we show that a new IGF-IR tyrosine kinase inhibitor BMS-536924 is a potent inhibitor of IGF-IR-induced transformation of human mammary epithelial cells, being able to completely reverse all measures of transformation in vitro and reducing xenograft tumor burden in vivo. Supporting these proof-of-concept studies, BMS-536924 also affects cell viability of a range of human breast cancer cell lines. This work suggests that targeting IGF-IR may be an effective strategy for the treatment of human breast cancer.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Dr. Xiaojiang Cui (John Wayne Cancer Institute, Santa Monica, CA) and others in the Breast Center at Baylor College of Medicine for helpful suggestions and technical assistance, and Dr. Gary Chamness for critical reading of the article. The study was supported in part by a research grants from the National Cancer Institute, P01CA30195, R01CA94118, and P50CA58183. Beate Litzenburger is a recipient of the German Academic Exchange Service Award and a Department of Defense Predoctoral Traineeship Award (DAMD-W81XWH-08-1-0220).
Literature Cited
- 1.Baserga R, Peruzzi F, Reiss K. The IGF-1 receptor in cancer biology. Int J Cancer. 2003;107:873–7. doi: 10.1002/ijc.11487. [DOI] [PubMed] [Google Scholar]
- 2.LeRoith D, Roberts JCT. The insulin-like growth factor system and cancer. Cancer Letters. 2003;195:127. doi: 10.1016/s0304-3835(03)00159-9. [DOI] [PubMed] [Google Scholar]
- 3.O'Connor R. Regulation of IGF-I Receptor Signaling in Tumor Cells. Hormone and Metabolic Research. 2003:771. doi: 10.1055/s-2004-814166. [DOI] [PubMed] [Google Scholar]
- 4.Van Obberghen E, Baron V, Delahaye L, et al. Surfing the insulin signaling web. European Journal of Clinical Investigation. 2001;31:966–77. doi: 10.1046/j.1365-2362.2001.00896.x. [DOI] [PubMed] [Google Scholar]
- 5.Pollak MN, Schernhammer ES, Hankinson SE. INSULIN-LIKE GROWTH FACTORS AND NEOPLASIA. Nature Reviews Cancer. 2004;4:505. doi: 10.1038/nrc1387. [DOI] [PubMed] [Google Scholar]
- 6.Resnik JL, Reichart DB, Huey K, Webster NJ, Seely BL. Elevated insulin-like growth factor I receptor autophosphorylation and kinase activity in human breast cancer. Cancer Res. 1998;58:1159–64. [PubMed] [Google Scholar]
- 7.Surmacz E. Function of the IGF-I receptor in breast cancer. J Mammary Gland Biol Neoplasia. 2000;5:95–105. doi: 10.1023/a:1009523501499. [DOI] [PubMed] [Google Scholar]
- 8.Kaleko M, Rutter WJ, Miller AD. Overexpression of the human insulinlike growth factor I receptor promotes ligand-dependent neoplastic transformation. Mol Cell Biol. 1990;10:464–73. doi: 10.1128/mcb.10.2.464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Baserga R. The contradictions of the insulin-like growth factor 1 receptor. Oncogene. 2000;19:5574–81. doi: 10.1038/sj.onc.1203854. [DOI] [PubMed] [Google Scholar]
- 10.Sell C, Rubini M, Rubin R, Liu JP, Efstratiadis A, Baserga R. Simian virus 40 large tumor antigen is unable to transform mouse embryonic fibroblasts lacking type 1 insulin-like growth factor receptor. Proc Natl Acad Sci U S A. 1993;90:11217–21. doi: 10.1073/pnas.90.23.11217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sell C, Dumenil G, Deveaud C, et al. Effect of a null mutation of the insulin-like growth factor I receptor gene on growth and transformation of mouse embryo fibroblasts. Mol Cell Biol. 1994;14:3604–12. doi: 10.1128/mcb.14.6.3604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Carboni JM, Lee AV, Hadsell DL, et al. Tumor development by transgenic expression of a constitutively active insulin-like growth factor I receptor. Cancer Res. 2005;65:3781–7. doi: 10.1158/0008-5472.CAN-04-4602. [DOI] [PubMed] [Google Scholar]
- 13.Jones RA, Campbell CI, Gunther EJ, et al. Transgenic overexpression of IGF-IR disrupts mammary ductal morphogenesis and induces tumor formation. Oncogene. 2006 doi: 10.1038/sj.onc.1209955. [DOI] [PubMed] [Google Scholar]
- 14.Irie HY, Pearline RV, Grueneberg D, et al. Distinct roles of Akt1 and Akt2 in regulating cell migration and epithelial-mesenchymal transition. J. Cell Biol. 2005;171:1023–34. doi: 10.1083/jcb.200505087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yanochko GM, Eckhart W. Type I insulin-like growth factor receptor overexpression induces proliferation and anti-apoptotic signaling in a three-dimensional culture model of breast epithelial cells. Breast Cancer Res. 2006;8:R18. doi: 10.1186/bcr1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kim HJ, Litzenburger BC, Cui X, et al. Constitutively active type I insulin-like growth factor receptor causes transformation and xenograft growth of immortalized mammary epithelial cells and is accompanied by an epithelial-to-mesenchymal transition mediated by NF-kappaB and snail. Mol Cell Biol. 2007;27:3165–75. doi: 10.1128/MCB.01315-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Haddad T, Yee D. Targeting the insulin-like growth factor axis as a cancer therapy. Future Oncol. 2006;2:101–10. doi: 10.2217/14796694.2.1.101. [DOI] [PubMed] [Google Scholar]
- 18.Sachdev D, Yee D. Inhibitors of insulin-like growth factor signaling: a therapeutic approach for breast cancer. J Mammary Gland Biol Neoplasia. 2006;11:27–39. doi: 10.1007/s10911-006-9010-8. [DOI] [PubMed] [Google Scholar]
- 19.Schillaci R, Salatino M, Cassataro J, et al. Immunization with murine breast cancer cells treated with antisense oligodeoxynucleotides to type I insulin-like growth factor receptor induced an antitumoral effect mediated by a CD8+ response involving Fas/Fas ligand cytotoxic pathway. J Immunol. 2006;176:3426–37. doi: 10.4049/jimmunol.176.6.3426. [DOI] [PubMed] [Google Scholar]
- 20.Sachdev D, Hartell JS, Lee AV, Zhang X, Yee D. A dominant negative type I insulin-like growth factor receptor inhibits metastasis of human cancer cells. J Biol Chem. 2004;279:5017–24. doi: 10.1074/jbc.M305403200. [DOI] [PubMed] [Google Scholar]
- 21.Burtrum D, Zhu Z, Lu D, et al. A Fully Human Monoclonal Antibody to the Insulin-Like Growth Factor I Receptor Blocks Ligand-Dependent Signaling and Inhibits Human Tumor Growth in Vivo. Cancer Res. 2003;63:8912–21. [PubMed] [Google Scholar]
- 22.Cohen BD, Baker DA, Soderstrom C, et al. Combination Therapy Enhances the Inhibition of Tumor Growth with the Fully Human Anti-Type 1 Insulin-Like Growth Factor Receptor Monoclonal Antibody CP-751,871. Clin Cancer Res. 2005;11:2063–73. doi: 10.1158/1078-0432.CCR-04-1070. [DOI] [PubMed] [Google Scholar]
- 23.Li SL, Liang SJ, Guo N, Wu AM, Fujita-Yamaguchi Y. Single-chain antibodies against human insulin-like growth factor I receptor: expression, purification, and effect on tumor growth. Cancer Immunol Immunother. 2000;49:243–52. doi: 10.1007/s002620000115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Maloney EK, McLaughlin JL, Dagdigian NE, et al. An Anti-Insulin-like Growth Factor I Receptor Antibody That Is a Potent Inhibitor of Cancer Cell Proliferation. Cancer Res. 2003;63:5073–83. [PubMed] [Google Scholar]
- 25.Sachdev D, Li S-L, Hartell JS, Fujita-Yamaguchi Y, Miller JS, Yee D. A Chimeric Humanized Single-Chain Antibody against the Type I Insulin-like Growth Factor (IGF) Receptor Renders Breast Cancer Cells Refractory to the Mitogenic Effects of IGF-I. Cancer Res. 2003;63:627–35. [PubMed] [Google Scholar]
- 26.Blum G, Gazit A, Levitzki A. Development of New Insulin-like Growth Factor-1 Receptor Kinase Inhibitors Using Catechol Mimics. J Biol Chem. 2003;278:40442–54. doi: 10.1074/jbc.M305490200. [DOI] [PubMed] [Google Scholar]
- 27.Garcia-Echeverria C, Pearson MA, Marti A, et al. In vivo antitumor activity of NVPAEW541--A novel, potent, and selective inhibitor of the IGF-IR kinase. Cancer Cell. 2004;5:231. doi: 10.1016/s1535-6108(04)00051-0. [DOI] [PubMed] [Google Scholar]
- 28.Haluska P, Carboni JM, Loegering DA, et al. In vitro and in vivo antitumor effects of the dual insulin-like growth factor-I/insulin receptor inhibitor, BMS-554417. Cancer Res. 2006;66:362–71. doi: 10.1158/0008-5472.CAN-05-1107. [DOI] [PubMed] [Google Scholar]
- 29.Parrizas M, Gazit A, Levitzki A, Wertheimer E, LeRoith D. Specific Inhibition of Insulin-Like Growth Factor-1 and Insulin Receptor Tyrosine Kinase Activity and Biological Function by Tyrphostins. Endocrinology. 1997;138:1427–33. doi: 10.1210/endo.138.4.5092. [DOI] [PubMed] [Google Scholar]
- 30.Garber K. IGF-1: Old Growth Factor Shines as New Drug Target. J Natl Cancer Inst. 2005;97:790–2. doi: 10.1093/jnci/97.11.790. [DOI] [PubMed] [Google Scholar]
- 31.Rae JM, Creighton CJ, Meck JM, Haddad BR, Johnson MD. MDA-MB-435 cells are derived from M14 melanoma cells--a loss for breast cancer, but a boon for melanoma research. Breast Cancer Res Treat. 2007;104:13–9. doi: 10.1007/s10549-006-9392-8. [DOI] [PubMed] [Google Scholar]
- 32.Wittman M, Carboni J, Attar R, et al. Discovery of a 1H-Benzoimidazol-2-yl)-1H-pyridin-2-one (BMS-536924) Inhibitor of Insulin-like Growth Factor I Receptor Kinase with in Vivo Antitumor Activity. J. Med. Chem. 2005;48:5639–43. doi: 10.1021/jm050392q. [DOI] [PubMed] [Google Scholar]
- 33.LeVea CM, Reeder JE, Mooney RA. EGF-dependent cell cycle progression is controlled by density-dependent regulation of Akt activation. Experimental Cell Research. 2004;297:272. doi: 10.1016/j.yexcr.2004.03.026. [DOI] [PubMed] [Google Scholar]
- 34.Debnath J, Muthuswamy SK, Brugge JS. Morphogenesis and oncogenesis of MCF10A mammary epithelial acini grown in three-dimensional basement membrane cultures. Methods. 2003;30:256. doi: 10.1016/s1046-2023(03)00032-x. [DOI] [PubMed] [Google Scholar]
- 35.Bissell MJ, Rizki A, Mian IS. Tissue architecture: the ultimate regulator of breast epithelial function. Current Opinion in Cell Biology. 2003;15:753. doi: 10.1016/j.ceb.2003.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Park CC, Zhang H, Pallavicini M, et al. Beta1 integrin inhibitory antibody induces apoptosis of breast cancer cells, inhibits growth, and distinguishes malignant from normal phenotype in three dimensional cultures and in vivo. Cancer Res. 2006;66:1526–35. doi: 10.1158/0008-5472.CAN-05-3071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Arthur MM, Robin EB, Jun C, et al. Integrin Laminin Receptors and Breast Carcinoma Progression. Journal of Mammary Gland Biology and Neoplasia. 2001;6:299. doi: 10.1023/a:1011323608064. [DOI] [PubMed] [Google Scholar]
- 38.Soule HD, Maloney TM, Wolman SR, et al. Isolation and characterization of a spontaneously immortalized human breast epithelial cell line, MCF-10. Cancer Res. 1990;50:6075–86. [PubMed] [Google Scholar]
- 39.Baserga R. The price of independence. Exp Cell Res. 1997;236:1–3. doi: 10.1006/excr.1997.3732. [DOI] [PubMed] [Google Scholar]
- 40.Elenbaas B, Spirio L, Koerner F, et al. Human breast cancer cells generated by oncogenic transformation of primary mammary epithelial cells. Genes Dev. 2001;15:50–65. doi: 10.1101/gad.828901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Neve RM, Chin K, Fridlyand J, et al. A collection of breast cancer cell lines for the study of functionally distinct cancer subtypes. Cancer Cell. 2006;10:515–27. doi: 10.1016/j.ccr.2006.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Matsumoto H, Noguchi S, Sugie K, et al. Subcellular Localization of Fukutin and Fukutin-Related Protein in Muscle Cells. J Biochem (Tokyo) 2004;135:709–12. doi: 10.1093/jb/mvh086. [DOI] [PubMed] [Google Scholar]
- 43.Wang F, Hansen RK, Radisky D, et al. Phenotypic Reversion or Death of Cancer Cells by Altering Signaling Pathways in Three-Dimensional Contexts. J Natl Cancer Inst. 2002;94:1494–503. doi: 10.1093/jnci/94.19.1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liu H, Radisky DC, Wang F, Bissell MJ. Polarity and proliferation are controlled by distinct signaling pathways downstream of PI3-kinase in breast epithelial tumor cells. J Cell Biol. 2004;164:603–12. doi: 10.1083/jcb.200306090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ludwig T, Eggenschwiler J, Fisher P, D'Ercole AJ, Davenport ML, Efstratiadis A. Mouse mutants lacking the type 2 IGF receptor (IGF2R) are rescued from perinatal lethality in Igf2 and Igf1r null backgrounds. Dev Biol. 1996;177:517–35. doi: 10.1006/dbio.1996.0182. [DOI] [PubMed] [Google Scholar]
- 46.Surmacz E, Sell C, Swantek J, et al. Dissociation of mitogenesis and transforming activity by C-terminal truncation of the insulin-like growth factor-I receptor. Exp Cell Res. 1995;218:370–80. doi: 10.1006/excr.1995.1168. [DOI] [PubMed] [Google Scholar]
- 47.Sharma SV, Fischbach MA, Haber DA, Settleman J. “Oncogenic Shock”: Explaining Oncogene Addiction through Differential Signal Attenuation. Clin Cancer Res. 2006;12:4392s–5s. doi: 10.1158/1078-0432.CCR-06-0096. [DOI] [PubMed] [Google Scholar]
- 48.Ince TA, Richardson AL, Bell GW, et al. Transformation of Different Human Breast Epithelial Cell Types Leads to Distinct Tumor Phenotypes. Cancer Cell. 2007;12:160–70. doi: 10.1016/j.ccr.2007.06.013. [DOI] [PubMed] [Google Scholar]
- 49.Goetsch Liliane. AGOLABPJPJFHNC: A recombinant humanized anti-insulin-like growth factor receptor type I antibody (h7C10) enhances the antitumor activity of vinorelbine and anti-epidermal growth factor receptor therapy against human cancer xenografts. International Journal of Cancer. 2005;113:316–28. doi: 10.1002/ijc.20543. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.