

Summary
Saccharomyces cerevisiae stimulates dendritic cells (DCs) and represents a promising candidate for cancer vaccine development. Effective cross-presentation of antigen delivered to DCs is necessary for successful induction of cellular immunity. Here, we present a yeast-based vaccine approach that is independent of yeast’s ability to express the chosen antigen, which is instead produced separately and conjugated to the yeast cell wall. The conjugation method is site-specific (based on the SNAP-tag) and designed to facilitate antigen release in the DC phagosome and subsequent translocation for cross-presentation. We demonstrate that nonsite-specific chemical conjugation of the same protein hinders cross-presentation. Phagosomal antigen release was further expedited through the insertion of the invariant chain ectodomain as a linker, which is rapidly cleaved by Cathepsin S. The dose of delivered antigen was increased in several ways: by using yeast strains with higher surface amine densities, by using yeast hulls (cell wall fragments) instead of whole cells, and by conjugating multiple layers of antigen. The novel multilayer conjugation scheme takes advantage of Sfp phosphopantetheinyl transferase and remains site-specific; it enables the antigen dose to grow linearly with the number of layers. We show that whole yeast cells coated with 1 layer of the cancer-testis antigen NY-ESO-1 and yeast hulls bearing 3 layers were able to cross-prime naive CD8+ T cells in vitro, with the latter resulting in higher frequencies of antigen-specific cells after 10 days. This cross-presentation-efficient antigen conjugation scheme is not limited to yeast and can readily be applied toward the development of other particulate vaccines.
Keywords: cross-priming, cross-presentation, cancer vaccine, conjugation, yeast
The goal of cancer vaccination is to elicit a tumor-protective immune response against cancer-associated antigens, to defeat cancer in much the same way as we have defeated many infectious diseases. However, despite the leaps made in understanding of cellular immune function and tumor immunology, objective response rates in cancer vaccine clinical trials have been disappointingly low.1 Many early generation cancer vaccines were simply peptide or protein injections, sometimes paired with a cytokine adjuvant. With respect to infectious diseases, it has been pointed out that modern peptide or protein vaccines are problematic in that they are often considerably less immunogenic than primitive vaccines made from live or dead organisms.2 New, more immunogenic vaccine approaches, including ones that more closely mimic pathogens, are required in the field of cancer immunotherapy.
One such promising new approach is the use of recombinant Saccharomyces cerevisiae yeast expressing antigen in the cytosol3-10 or on the cell wall.11,12 With its fungal cell wall, yeast cells are avidly phagocytosed and induce maturation of dendritic cells (DCs) and the secretion of TH1-type cytokines.3,5-10 S. cerevisiae is not pathogenic and despite its potent effect on the innate immune system, subcutaneous administration of heat-killed yeast did not exhibit dose-limiting toxicity in phase 1 clinical trials.10 The adjuvant properties of yeast are only half the story, as successful vaccine development also requires that a sufficient dose of antigen be delivered to the DCs in a way that facilitates processing and major histocompatibility complex (MHC) class I presentation (ie, cross-presentation).
We have previously shown that timely antigen release from the yeast cell upon phagocytosis by DCs is critical for efficient cross-presentation.12 In that study, the extended peptide antigen was recombinantly expressed on the cell wall as a fusion to the mating adhesion receptor subunit Aga2p, in a technique termed yeast surface display.13 Yeast surface-displayed antigen was cross-presented much more efficiently than antigen expressed in the yeast cytosol, and cross-presentation could be further enhanced by inserting linkers susceptible to Cathepsin S (CatS) cleavage between the antigen and Aga2p.12 Although this model system was useful for studying the impact of phagosomal antigen release kinetics upon cross-presentation, its utility as an immunotherapy vaccine candidate is limited by the reliance on yeast expression levels for antigen delivery. For many reasons, it is often desirable to immunize with a full-length protein as opposed to a known peptide epitope, and yeast surface display levels of the former may not be adequate or optimal. We therefore set out to develop a system for attaching soluble protein antigens to the yeast cell wall, so as to decouple antigen dose from yeast expression levels while retaining a desirable antigen release profile upon DC phagocytosis.
We focused our attentions on the highly immunogenic cancer-testis antigen, NY-ESO-1, which has been widely tested in cancer vaccine clinical trials (reviewed in Refs. 14,15). NY-ESO-1 is surface displayed very poorly by yeast, and although a combination of rational design and directed evolution approaches identified a mutant that has a 100-fold improved display level,16 the number of copies per yeast cell still did not reach peptide display levels. In contrast, NY-ESO-1 can be expressed solubly and at high yield in Escherichia coli when fused to maltose-binding protein (MBP, unpublished observation by A. Piatesi).
Our challenge was to conjugate bacterially expressed NY-ESO-1 to the yeast cell wall, in a manner facilitating release of the antigenic domain in the DC phagosome. Insertion of a CatS-susceptible linker, which worked admirably with yeast surface display, would likely be foiled by conventional chemical conjugation methods where the reaction can occur at multiple amino acid positions along the fusion protein. For example, a reaction scheme based on primary amines might bind a lysine residue of NY-ESO-1 directly to the cell wall, thus preventing release when the CatS-susceptible linker is cleaved. Consequently, we turned to site-specific conjugation methods where the reaction is mediated by a protein and is restricted to a known amino acid residue.
MATERIALS AND METHODS
Reagents
Recombinant human cytokines were obtained from R & D Systems (Minneapolis, MN). Monoclonal α-NY-ESO-1 antibody, clone E978, was obtained from Sigma-Aldrich (St Louis, MO). Dye-conjugated monoclonal antibodies against CD8 and DC markers were from BD Biosciences (San Jose, CA) except for α-CD40 (Calbiochem, Gibbstown, NJ), α-CD86 (Chemicon, Billerica, MA), and α-HLA-DR (Leinco, St Louis, MO).
Cells
Human HLA-A*0201 monocytes purified by negative magnetic cell sorting from leukapheresis packs were purchased from Biological Specialty Corporation (Colmar, PA). The monocytes were cryopreserved in 90% fetal bovine serum (FBS), 10% dimethylsulfoxide, and aliquots were thawed as needed. The monocytes were cultured in C10 medium—RPMI-1640 with 10% FBS, 2 mM l-glutamine, 10 mM N-2-hydroxyl piperazine-N’-2-ethane sulfonic acid, 1 mM sodium pyruvate, 1 nonessential amino acids, 50 μM β-mercaptoethanol, and Primocin (InvivoGen, San Diego, CA)—supplemented with 1000 U/mL each of IL-4 and granulocyte macrophage colony-stimulating factor (GM-CSF) in 6-well plates (4 to 5 × 106 monocytes in 2.5 mL medium per well). Each well was fed with 0.5 mL cytokine-supplemented C10 on days 2 and 4; on day 5 or 6, the floating and loosely adherent immature monocyte-derived DCs were harvested by gentle resuspension.
A human CD8+ T cell line specifically recognizing the cytomegalovirus (CMV)-derived peptide NLVPMVATV (N9V) in the context of HLA-A*0201 was obtained from ProImmune (Oxford, UK). The vials of frozen cells were thawed and cultured overnight in RPMI-1640 with 10% FBS, 5 ng/mL IL-2, and Primocin before use.
Peripheral blood mononuclear cells (PBMCs) were isolated by Ficoll-Paque density gradient centrifugation from HLA-A2 buffy coats (Research Blood Components, Brighton, MA). A portion of the PBMCs from each donor were cryopreserved in 90% FBS, 10% dimethylsulfoxide whereas the rest were subjected to magnetic cell separation by an autoMACS Separator (Miltenyi Biotec, Auburn, CA). CD8 and CD45RO microbeads (Miltenyi) were used sequentially in positive and negative selection modes according to the manufacturer’s protocol to isolate CD8+ CD45RO- cells.
Yeast
S. cerevisiae strain BJ5464α (ATCC, Manassas, VA) was grown in YPD medium (1% yeast extract, 2% peptone, and 2% dextrose) in a 30°C shaker. Plasmids for yeast surface display were constructed based on pCTCON2,17 and the shuttle vector pRS30418 was used to integrate expression cassettes into EBY100,13 a yeast strain that expresses Aga1p under galactose induction. In particular, the strain SWH100 was created from EBY100 by integrating an Aga2p-HA expression cassette, constructed by substituting 2 stop codons between the PstI and BamHI sites in pCTCON2. SWH100 was grown to mid-log phase in SD-CAA medium (2% dextrose, 0.67% yeast nitrogen base, 0.5% casamino acids, 0.1 M sodium phosphate, and pH 6.0) at 30°C and induced for 12 hours at 30°C in YPG (1% yeast extract, 2% peptone, and 2% galactose). Yeast cell densities were estimated by measuring the absorbance at 600 nm, with an optical density (OD) of 1 taken to be 1 × 107 cells/mL.
MBP Fusion Proteins
Plasmids for E. coli expression of MBP fusion proteins were constructed using pMAL-c2x (New England Biolabs, Ipswich, MA) as the vector backbone. The plasmid for expressing MSCE was constructed by inserting sequences encoding the SNAP tag (AvaI/EcoRI; from pSS26b, Covalys, Witterswil, Switzerland), the CatS-susceptible linker (C1)4 and the CMV-derived peptide ARNLVPMVATVQGQN (EcoRI/BamHI; from pCT-(C1)4-N9V12), and the CS variant of NY-ESO-1, with C to S mutations at positions 75, 76, and 78 (BamHI/HindIII; from pCT-NY-ESO-CS16). To express MS74NEY2, further modifications included replacing the (C1)4 coding sequence with that of aa 73 to 207 of human CD74 (EcoRI/NheI; from cDNA clone LIFE-SEQ1001730, Open Biosystems, Huntsville, AL) and C-terminal insertion of a sequence encoding TVQL-(G4S)3-DSLEFIASKLA-H6 (ybbR tag19 is underlined).
Plasmid-bearing BL21(DE3)-RIPL bacteria (Stratagene, La Jolla, CA) in 1 L Luria-Bertani broth were induced at an OD600 of 0.7 with 0.3 mM isopropyl-β-d-thiogalactoside for 4 to 6 hours at 20°C. The cell pellet was frozen, thawed, resuspended in 25 mL of the appropriate column buffer containing Complete Protease Inhibitor Cocktail (Roche Applied Science, Indianapolis, IN) and lysed by sonication. For fusion proteins lacking a His6 tag, the cell lysate was clarified by centrifugation (20,000g, 30 min, 4°C) and loaded onto an amylose resin (New England Biolabs) column equilibrated with amylose resin buffer [antigen retrieval (AR) buffer; 20 mM Tris-HCl, pH 7.4, 200 mM NaCl, 1 mM dithiothreitol (DTT), 1 mM ethylene diaminetetra acetic acid). The column was washed with 50 column volumes (CV) of AR buffer containing 0.1% Triton X-114 (Sigma-Aldrich; to reduce endotoxin content20), followed by 5 CV of AR buffer and eluted with 5 CV of AR buffer containing 10 mM maltose. The eluate was further subjected to size exclusion chromatography (Superdex 200 10/300 GL, GE Healthcare, Piscataway, NJ) to purify full-length protein. For fusion proteins containing a C-terminal His6 tag, the clarified cell lysate was loaded onto a Talon resin (Clontech, Mountain View, CA) column equilibrated with Talon buffer (20 mM Tris-HCl, pH 7.4, 20 mM NaCl, and 5 mM β-mercaptoethanol). The column was washed with 10 CV Talon buffer and eluted with 5 CV Talon buffer with 150 mM imidazole. The eluate was then subjected to amylose resin affinity chromatography as described above. For more thorough endotoxin removal, the purified protein used in the in vitro immunization and DC maturation experiments (see below) were treated with Polymyxin B-agarose (Sigma-Aldrich).
SNAP-tag Conjugation to Yeast
Yeast cells were ultraviolet-irradiated (2 × 1000 J/m2 in a Stratagene Stratalinker), washed 5 × with phosphate-buffered saline (PBS)+0.01% CHAPS (the detergent reduces yeast adherence to tube walls) and once with dimethylformamide (DMF). A pellet of 1 OD/mL or 107 yeast (or the equivalent amount of yeast hulls) was resuspended in 10 μL DMF containing 0.2 mg BG-GLA-NHS (Covalys), shaken at 30°C for 2 hours, and washed 3 × with PBS+0.1% bovine serum albumin (BSA). The benzyl guanine (BG)-modified yeast was then resuspended in 0.4 mL PBS and 0.1 mL of 60 μM SNAP-tagged fusion protein (dissolved in AR buffer with 10 mM maltose) and rotated at 30°C for 5 hours. The yeast was washed at least twice with PBS+0.1% BSA before use. Conjugation levels were monitored by flow cytometry (eg, labeling with α-NY-ESO-1 followed by Alexa Fluor 488-conjugated goat α-mouse antibody).
Conjugation to Yeast by Reductive Amination
Ultraviolet-irradiated BJ5464α yeast (2.5 OD/mL) was washed thrice and resuspended in 450 μL 0.1 M sodium acetate, pH 5.5 buffer. After the addition of 50 μL 100 mM NaIO4 dissolved in the same buffer, the yeast was incubated on ice for 20 minutes and then washed twice with 1 mL sodium acetate buffer. The yeast was resuspended in 200 μL 50 μM MSCE protein that had been buffer-exchanged into PBS+1 mM DTT. NaCNBH3 (2 μL of a 1 M solution in 10 mM NaOH) was added and the tube was rotated at 4°C for 3 days.
N9V Cross-presentation Assay
Immature monocyte-derived DCs were seeded in 96-well round bottom plates with 2 × 105 DCs in 200 μL cytokine-supplemented C10 medium per well. Yeast cells or yeast hulls were added to the DCs at the indicated ratios; after 24 hours, half the medium was replaced by 100 μL of 0.7 to 1 × 106 cells/mL suspension of N9V/HLA-A*0201-specific T cells. Four hours later, the cells in each well were transferred to tubes to perform an interferon gamma (IFN) γ secretion assay (Miltenyi) according to the manufacturer’s protocol. Briefly, the cells were labeled for 5 minutes with a bispecific antibody that captures secreted IFNγ, incubated in medium for 45 minutes at 37°C, and labeled with α-CD8-fluorescein isothiocyanate (FITC) and α-IFNγ-phycoerythrin. The fraction of CD8+ cells that were also IFNγ+ was determined by flow cytometry, with the gate for IFNγ fluorescence set such that the negative control test (no stimulus added to the DCs) gave a result of 0.5% or lower.
ELISpot Assay
Monocytes were isolated by CD14 magnetic cell sorting (Miltenyi) from the PBMCs of a healthy HLA-Cw3 donor and differentiated into DCs by culturing for 6 days in the presence of GM-CSF and IL-4. DCs were pulsed overnight with NY-ESO-192-100 peptide or antigen-coated yeast (20 yeast/DC). The DCs were washed and seeded at 5 × 104 cells/well in a mixed cellulose ester membrane filter plate (Millipore, Billerica, MA) precoated with α-IFNγ (1-D1K, Mabtech, Mariemont, OH) together with the indicated number of C5 cells. C5 is an HLA-Cw3-restricted CD8+ T-cell clone recognizing NY-ESO-192-100.21 After 24 hours of coculture, the plate was developed with biotinylated α-IFNγ (7-B6-1, Mabtech), streptavidin-alkaline phosphatase conjugate (Roche), and BCIP/NBT substrate (Sigma) for evaluation with a CTL ImmunoSpot Analyzer.
CatS Cleavage Time Course
For each time point, 0.2 OD/mL of antigen-conjugated yeast was resuspended in 100 μL AR buffer. Recombinant CatS solution (Calbiochem; 20 ng in 100 μL AR buffer) was added and each tube was incubated at 37°C for the indicated time before the yeast was washed with cold PBS+0.1% BSA. The yeast was labeled with α-NY-ESO-1 followed by Alexa Fluor 488-conjugated goat α-mouse IgG before analysis by flow cytometry.
Multilayer Conjugation
Sfp phosphopantetheinyl transferase was produced as described by Yin et al19 except that BL21(DE3)-RIPL was used as the expression strain and the bacteria were lysed by sonication. BG-CoA was synthesized by reacting 1 mg (2.0 μmoles) of BG-maleimide (Covalys) dissolved in 0.5 mL DMF with an equal volume of PBS containing 1.5 μmoles of Coenzyme A trilithium salt (Sigma-Aldrich). After incubating at 30°C for 24 hours, the presence of the product was confirmed by mass spectrometry. DTT (1 mM) was added to quench the excess BG-maleimide and the BG-CoA solution was used without purification. The initial layer of fusion protein containing both the SNAP and ybbR tags was conjugated to BG-modified yeast (or yeast hulls) as described above. Coated yeast (1 OD/mL) was resuspended in 200 μL PBS+0.1% BSA containing 10 mM MgCl2, 75 μM BG-CoA, and 12.5 μM Sfp. The yeast was incubated at 37°C for 1.5 hours with occasional vortexing and washed twice with PBS+0.1% BSA. The next layer was conjugated by mixing the yeast with 0.4 mL PBS and 0.1 mL 60 μM fusion protein for 2 hours at 30°C. The Sfp/BG-CoA reaction and the SNAP-tag conjugation reaction were alternated to add more layers.
Yeast Hulls
A 1 L culture of induced SWH100 yeast was pelleted, washed, and resuspended in 200 mL PBS to give an OD value of 25. The yeast suspension was passed twice through an M-110EH-30 Microfluidizer processor (Microfluidics, Newton, MA) at 30,000 psi using the G10Z (87 μm) interaction chamber. Lysed yeast suspension (40 mL) was pelleted by centrifugation at 2000g for 5 minutes and then washed once with 100 mL PBS+0.1% CHAPS, once with 100 mL 20% ethanol, and 4 times with 100 mL PBS+0.01% CHAPS. The yeast fragments were resuspended in 100 mL PBS+0.01% CHAPS and were stored in aliquots at -70°C. We assumed that there was no change in the cell wall material concentration during processing and washing, and treated the final yeast hull suspension as being equivalent to 10 OD of yeast.
In Vitro Immunization Experiment
Nine wells of a 24-well plate were each seeded with 5 × 105 immature human monocyte-derived DCs in 1 mL C10 medium containing IL-4 and GM-CSF. Either unmodified yeast hulls, SWH100 whole yeast conjugated with 1 layer of MS74NEY2, or yeast hulls coated 3 times were added to the wells in triplicate, at a dose equivalent to 10 yeast/DC. The next day, CD8+ CD45RO- T cells were isolated from the PBMCs of 3 HLA-A2 donors as described and resuspended in CTL medium (RPMI-1640 with 10% FCS, 50 μM β-mercaptoethanol, 12.5 mM N-2-hydroxyl piperazine-N’-2-ethane sulfonic acid, pH 7.3, and Primocin). The in vitro immunization assay conditions were adapted from a protocol for generating CD8+ T-cell clones.22 For each donor, 5 × 106 naive T cells in 1 mL CTL medium were added directly to 3 DC wells with the 3 test conditions. In addition, 2 × 106 naive T cells from each donor were labeled with 20 μL α-CD8-FITC and 10 μL SLLMWITQV/HLA-A*0201 peptide-MHC pentamer (ProImmune) in a total volume of 100 μL for 30 minutes on ice before analysis by flow cytometry (day 0 results). On day 3, 75% of the medium was replaced by fresh CTL medium containing IL-2 and IL-7 to give final concentrations of 2.5 and 5 ng/mL, respectively. The medium was refreshed in this manner every 2 to 3 days, and on day 7, the cells were transferred to a 12-well plate with the well volume doubling to 4 mL. On day 10, 2 × 106 cells from each well were labeled for CD8 and peptide-MHC pentamer binding as described above. To restimulate the T cells, 3 × 107 PBMCs from each donor were thawed, gamma-irradiated (35 Gy), and resuspended in 3 mL CTL medium containing 40 μM SLLMWITQV peptide (synthesized by AnaSpec, San Jose, CA). The PBMCs were seeded at 1 mL/well of a 12-well plate, and 3 hours later, 5 × 106 day 10 T cells from each test condition were added in 3 mL CTL medium to donor-matched PBMC wells. On day 12 and every 2 to 3 days later, the cells were fed with fresh medium containing IL-2 and IL-7 and split when overcrowded. Flow cytometry analysis was again performed on day 20.
DC Maturation Experiment
Wells of a 24-well plate were each seeded with 4 × 105 immature monocyte-derived DCs in 0.8 mL C10 medium containing IL-4 and GM-CSF. The stimuli (either 1 μg/mL E. coli 055 lipopolysaccharide (LPS), from GE Healthcare, 0.5 μM MS74NEY2 or 10 yeast/DC doses of naked or antigen-coated whole yeast or yeast hulls) were added in duplicate to the wells. After 48 hours, the culture supernatant from each well was set aside to be tested for IL-12p70 content using the OptEIA enzyme-linked immunosorbent assay (ELISA) kit (BD Biosciences). The manufacturer’s protocol was followed and both 2 × and 20 × dilutions of each sample in PBS were tested to cover a wide range of IL-12 concentrations. The DCs from duplicate wells were pooled and then divided into 5 portions; each aliquot was labeled with α-DC-SIGN-FITC and a phycoerythrin-conjugated antibody against 1 of 5 markers of DC maturation (CD40, CD80, CD83, CD86, and HLA-DR). The flow cytometry data were gated by DC-SIGN expression to exclude non-DC events.
RESULTS
Antigen Conjugated to Yeast via the SNAP-tag is Cross-presented
The SNAP-tag is a 20 kd protein domain derived from human O(6)-alkylguanine-DNA alkyltransferase, which reacts with BG moieties resulting in the formation of a covalent thioether bond.23 We anticipated that SNAP-tagged NY-ESO-1 fusion protein could be site-specifically conjugated to yeast cell surfaces modified with BG. Owing to the limited availability of NY-ESO-1-specific T-cell clones, we decided to use a peptide derived from CMV pp65 as a surrogate antigen: ARNLVPMVATVQGQN (HLA-A*0201 epitope N9V is underlined). Cross-presentation of this epitope by DCs stimulated with yeast surface displaying the peptide can be detected using commercially available cognate CD8+ T cells.12
We designed 2 fusion proteins, MSCc-myc and MSCE, both containing (in order from N terminus to C): MBP for yield, solubility, and ease of purification, SNAP-tag for site-specific conjugation, 4 tandem repeats of the CatS-susceptible linker EKARVLAEAAS,12 and the CMV peptide. MSCE further included NY-ESO-1 as the C-terminal component, whereas MSCc-myc had only a final c-myc tag for detection purposes. Both fusion proteins were expressed in E. coli and purified by amylose affinity chromatography (including extensive washes with 0.1% Triton X-114 to greatly reduce endotoxin content)20 and size exclusion chromatography. Wild-type BJ5464α yeast cells (BJ5α for short) were reacted with an aminereactive BG derivative (BG-GLA-NHS) and subsequently incubated with either MSCc-myc or MSCE protein. The SNAP-tag-mediated conjugation was successful as determined by flow cytometry for c-myc and NY-ESO-1, respectively (not shown).
To test for cross-presentation, the antigen-conjugated yeast and the control BG-only yeast were added to immature HLA-A*0201 monocyte-derived DCs. Twenty-four hours later, the DCs were cocultured for 4 hours with an N9V-specific CD8+ T-cell clone. Cross-presentation of the N9V epitope could be assessed by quantifying the percentage of CD8+ cells that had responded by secreting IFNγ. As shown in Figure 1A, yeast coated with either fusion protein induced cross-presentation at levels significantly above the background caused by BG-BJ5α yeast alone. The presence of NY-ESO-1 in the fusion protein did not interfere with cross-presentation of the surrogate CMV epitope, so we continued using MSCE for subsequent experiments.
FIGURE 1.

Antigen site-specifically conjugated to yeast via the SNAP-tag is cross-presented, with yeast expressing Aga1p and Aga2p giving rise to higher conjugation and cross-presentation levels. A to C, Yeast with or without conjugated antigen were added to DCs at a 20:1 ratio. After 24 hours, the DCs were assayed for the ability to stimulate N9V-specific CD8+ T cells to secrete IFNγ. Error bars indicate SDs of duplicate wells. A, BJ5α yeast cells modified with BG were coated via the SNAP-tag reaction with either no protein, MSCc-myc, or MSCE. B, MSCE was conjugated to BJ5α yeast either chemically by reductive amination or site-specifically via the SNAP-tag/BG reaction. The conjugation level of the latter method was matched to the former (within 5%) by reducing the MSCE concentration. C, MSCE was conjugated to either BJ5α yeast or yeast surface displaying 4 to 10 tandem repeats of the sequence GGGGKGS (with X in LysX indicating the repeat number). The negative control was BG-BJ5α lacking MSCE. D, BG and MSCE were conjugated to either BJ5α (grey fill), LysX yeast (where X = 4, 6, 8, or 10; gray lines), or yeast surface displaying a short peptide lacking lysines (black dashed line). The yeast cells were labeled for flow cytometry with α-NY-ESO-1 followed by an Alexa Fluor 488-conjugated secondary antibody. The 4 histograms corresponding to the LysX yeast strains were not distinguished by color as they essentially overlaid each other. E, In an ELISpot assay, HLA-Cw3 DCs were pulsed overnight with antigen and then cultured at 5 × 104 cells/well with the indicated ratio of a HLA-Cw3-restricted CD8+ T-cell clone recognizing NY-ESO-192-100. Lys6 yeast conjugated with MSCE (containing NY-ESO-1) or MSCc-myc (lacking NY-ESO-1) were each added to the DCs at a 20:1 ratio. BG indicates benzyl guanine; DC, dendritic cell; IFNγ, interferon gamma.
Site-specificity of Conjugation is Crucial for Cross-presentation
We had investigated SNAP-tag-mediated conjugation on the premise that conventional chemical conjugation methods create covalent bonds at random sites in the protein and can interfere with antigen release in the DC phagosome and subsequent transport to the cytosol. To test this premise, we conjugated the same fusion protein, MSCE, to the yeast cell wall using either the SNAP-tag to BG reaction or by reductive amination. In the latter chemical conjugation scheme, the sugars on the yeast cell wall were partially oxidized with sodium meta-periodate to generate aldehyde groups, which form covalent bonds with lysine side chains in MSCE in the presence of sodium cyanoborohydride as a reducing agent. To enable a fair comparison, the concentration of MSCE used during SNAP-tag-mediated conjugation was reduced so as to match the conjugation level achieved by reductive amination (resulting in a mere 2% difference as determined by flow cytometry). Figure 1B shows that antigen conjugated by reductive amination was not cross-presented at significant levels, unlike antigen attached via the SNAP-tag.
Increasing Amine Density and Conjugation Levels on Yeast
As BG-GLA-NHS reacts with primary amines on the yeast cell wall, we hypothesized that the antigen dose per yeast cell could be increased by surface-displaying lysine-rich polypeptides. To this end, we constructed yeast strains expressing 4 to 10 tandem repeats of the sequence GGGGKGS fused to Aga2p on the cell wall. These yeast strains, termed LysX, where X indicates the number of added lysines, were reacted with BG-GLA-NHS followed by MSCE and tested for cross-presentation. The results revealed that all the modified yeast strains induced about twice as many CD8+ T cells to secrete IFNγ as compared with wild-type BJ5α, but the number of extra lysine residues seemed to be inconsequential (Fig. 1C). By analyzing the NY-ESO-1 conjugation levels by antibody labeling and flow cytometry, we discovered that a yeast strain surface displaying a lysine-less peptide resulted in the same NY-ESO-1 density as the LysX strains, which was about 6-fold that on BJ5α (Fig. 1D). Similar results were obtained when fluorescently tagged MSCE was conjugated, eliminating antibody access limits as a confounding factor (not shown). It seems that the expression of Aga1p and Aga2p in the yeast surface display system fully accounts for the increase in number of amines available for conjugation. Steric hindrance may impose an upper limit on the number of bulky MSCE molecules that can be attached surrounding a single Aga1p-Aga2p complex. The yeast strain SWH100 with inducible expression of Aga1p and HA-tagged Aga2p was created and used henceforth instead of BJ5α.
An NY-ESO-1 Epitope is Cross-presented
Before investigating further improvements in the antigen conjugation strategy, we wanted to ensure that cross-presentation was not limited to the surrogate epitope. Using an HLA-Cw3-restricted CD8+ T-cell clone specifically recognizing the NY-ESO-1 epitope LAMPFATPM,21 we performed an ELISpot assay to detect IFNγ secretion in response to DCs that had phagocytosed antigen-conjugated yeast. When MSCE fusion protein was conjugated, a significant number of spots were formed, in contrast to when MSCc-myc (with c-myc replacing the NY-ESO-1 domain) was conjugated (Fig. 1E).
CD74 as a CatS-mediated Antigen Release Mechanism
The decision to include (C1)4, a CatS-susceptible linker consisting of 4 repeats of the sequence EKARV-LAEAAS, in MSCE was driven by its effectiveness in boosting cross-presentation efficiency with yeast surface displaying the CMV peptide.12 However, when we deleted (C1)4 from MSCE and conjugated it to SWH100 yeast via the SNAP-tag, the cross-presentation efficiency did not decrease significantly. To shed light on this observation, we digested both proteins in solution with CatS and studied the fragmentation patterns. N-terminal sequencing of one of the earliest fragments revealed that the CMV epitope itself, NLVPMVAITV, can be cleaved by CatS at the indicated position. As competition between antigen release and epitope destruction may be a confounding factor, we sought to eliminate the CatS cleavage site. According to Ruckrich et al,24 only V, L, M, C, and F are found in the P2 position of effective CatS cleavage sites. Mutating the epitope to NLVPMIATV prevented CatS cleavage while maintaining recognition by the CD8+ T-cell clone used in our cross-presentation assays. This mutated CMV epitope was used henceforth, but there remained no clear benefit in cross-presentation when (C1)4 was included versus deleted.
We hypothesized that (C1)4 in the context of MSCE may be inaccessible to phagosomal CatS, perhaps because it assumes a compact instead of extended conformation when sandwiched between 2 sizeable protein domains (SNAP-tag and NY-ESO-1). In contrast, in the yeast surface display model, the linker was more exposed and had only a short peptide C-terminal to it.
Seeking an alternative polypeptide sequence that would not be obscured by the flanking domains and would be rapidly cleaved by CatS, the human invariant chain was considered. CatS-mediated degradation of the invariant chain, also called CD74, is known to be a prerequisite before MHC class II molecules can acquire peptides.25 More than a dozen CatS cleavage sites have been identified in the CD74 sequence.24 We therefore produced a new fusion protein replacing (C1)4 with the ectodomain of CD74 (aa 73 to 207). SWH100 yeast coated with fusion protein containing CD74 as a linker induced more efficient cross-presentation than when either no linker or (C1)4 was used (Fig. 2A). As expected, when yeast cells coated with each of the 3 fusion proteins were incubated with a limiting concentration of CatS, the loss of the NY-ESO-1 domain was dramatically faster when CD74 was used as the linker (Fig. 2B).
FIGURE 2.
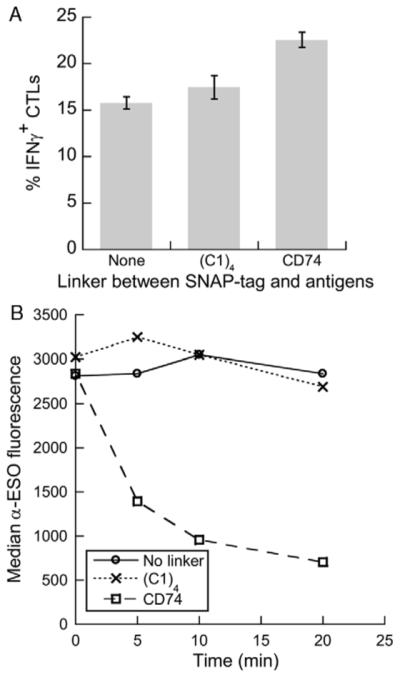
The extracellular domain of CD74 is highly susceptible to CatS cleavage and improves cross-presentation efficiency when used as a linker. Fusion protein variants containing either no linker (C1)4 or CD74 (aa 73 to 207) between the SNAP-tag and the CMV and NY-ESO-1 antigens were conjugated to SWH100 yeast in parallel. A, The conjugated yeast were added at a 15:1 ratio to DCs, which a day later were tested for their ability to activate antigen-specific CD8+ T cells. Error bars indicate SDs of triplicate wells. B, The conjugated yeast (2 × 106 per test) were incubated with 20 ng CatS and incubated at 37°C for different time periods. The loss of the NY-ESO-1 domain was monitored by antibody labeling and flow cytometry. CMV indicates cytomegalovirus.
A Reaction Scheme for Producing Multiple Coats of Antigen
Although SNAP-tag-mediated conjugation to SWH100 resulted in an average of 5 to 7.5 × 105 molecules of antigen per yeast cell, or about 4 to 6 × the typical yeast surface display level for a short peptide, we believed that vaccine potency could be enhanced by further increasing the conjugation level. The experiments with the LysX yeast strains (Figs. 1C, D) suggested that the addition of more primary amine groups to the yeast cell wall would be futile, so we instead pursued the strategy of building up multiple layers of conjugated antigen. To retain rapid phagosomal antigen release by CatS cleavage of the CD74 ectodomain, any new reactions would also need to be site-specific.
Yin et al19 have developed a system for site-specific protein labeling using the enzyme Sfp phosphopantetheinyl transferase (Sfp for short), which covalently transfers small molecule tags linked to Coenzyme A (CoA) to the serine residue of a peptide tag called ybbR (sequence: DSLEFIASKLA). We conjugated BG-maleimide to the free sulfhydryl group of CoA (forming BG-CoA) with the intention of using Sfp to add a fresh layer of BG to yeast coated with ybbR-tagged fusion protein, thus allowing a second coat to be conjugated via the SNAP-tag reaction. Our initial experiments inserting the ybbR tag either between MBP and the SNAP-tag or as a C-terminal fusion to NY-ESO-1 failed to generate a significant second layer. Steric hindrance seemed to be an obstacle, which was solved by the addition of a glycine/serine linker between NY-ESO-1 and the ybbR tag. The final version of the fusion protein, named MS74NEY2, comprised (in order from N terminus to C): MBP, SNAP-tag, CD74 ectodomain, the mutated CMV peptide ASARNLVPMIATV, NY-ESO-1, TVQL (a CatS-sensitive peptide),24 (GGGGS)3 linker, ybbR tag, and His6 tag (for easy purification of the full-length protein).
Under the appropriate reaction conditions, SNAP-tag conjugation and Sfp-mediated BG labeling could be alternated to build up multiple coats of fusion protein. Four coats of MS74NEY2 protein sparsely labeled with FITC (F/P ratio of 0.5) were layered onto SWH100 yeast, and samples from each reaction cycle were analyzed by flow cytometry (Fig. 3A). The increase in fluorescence was essentially linear, suggesting that there was no significant loss in conjugation efficiency as the number of cycles increased. Yeast coated with 0 to 4 layers of unlabeled MS74NEY2 were added to DCs at the low dose of only 2:1. At this dose, yeast coated with a single coat of antigen elicited cross-presentation at nearly background levels, whereas each subsequent antigen layer resulted in an increase in cross-presentation level (Fig. 3B).
FIGURE 3.

Antigen dose and cross-presentation efficiency can be increased by conjugating multiple coats of antigen and by using yeast hulls. A, MS74NEY2 was lightly labeled with FITC and conjugated to SWH100 yeast in multiple layers by alternately performing SNAP-tag/BG and Sfp/BG-CoA reactions. The yeast cells with different number of antigen coats were directly analyzed by flow cytometry. B, SWH100 whole yeast cells with 0 to 4 coats of MS74NEY2 (unlabeled) were added to DCs at a 2:1 ratio. After 24 hours, the DCs were cocultured with N9V-specific T cells and assess for their ability to stimulate IFNγ secretion. Error bars indicate SDs of duplicate wells. C, SWH100 yeast hulls were conjugated with a single layer of MS74NEY2 and added to DCs at varying doses. The same cross-presentation assay was performed. D to F, Confocal microscopy images of yeast hulls coated with 1 (D), 2 (E), or 3 (F) layers of MS74NEY2 lightly tagged with FITC. DC indicates dendritic cell; FITC, fluorescein isothiocyanate; IFNγ, interferon gamma.
Antigen can be Coated Onto Yeast Hulls for Cross-presentation
With this vaccine model, both the functions of antigen delivery and adjuvancy are fulfilled by the cell wall of yeast; presumably, the yeast cytoplasm is dispensable. We anticipated that using yeast cell wall fragments instead of whole yeast cells might hold potential advantages. The antigen density would be increased on a per-volume basis, possibly permitting a higher dose to be delivered to each DC, whereas the variety and quantity of native yeast proteins that are also cross-presented would be reduced. Induced SWH100 yeast cells were disrupted at very high shear rates with a Microfluidizer processor. After 2 passes, the resulting fragments had mean and median diameters of 1.7 and 0.9 μm, respectively, as determined by static light scattering.
After several washes, the fragments, henceforth termed yeast hulls, were subjected to conjugation with BG-GLA-NHS followed by MS74NEY2, using the same reaction conditions as were developed for whole yeast cells. We quantified the yeast hulls in terms of the numbers of originating whole yeast (estimated by measuring OD600), assuming negligible loss during processing and washing.
A dose-response cross-presentation assay was performed by adding antigen-coated yeast hulls to DCs. The coated yeast hulls were surprisingly potent: the lowest dose tested, nominally equivalent to 2 yeast/DC, resulted in a near-maximal response. In contrast, singly coated whole yeast elicited barely detectable levels of cross-presentation at the equivalent dose (Fig. 3B). Increasing the dose of yeast hulls from 5 to 30 yeast equivalents/DC did not result in higher percentages of IFNγ+ T cells (Fig. 3C). The total number of recovered CD8+ T cells dropped with increasing dose (not shown), suggesting a role for T-cell receptor (TCR)-triggered activation-induced cell death in limiting the assay response. We could not accurately quantify the level of antigen conjugation to yeast hulls by flow cytometry (the low forward and side scatter profile of the hulls overlapped with artifactual events), but the levels seemed to be qualitatively higher than achieved with whole yeast despite their smaller size distribution. We speculate that cell wall disruption may have revealed additional primary amine groups for conjugation, perhaps located on the inner face of the cell wall or at fissure sites. The alternating reaction scheme for conjugating multiple layers of antigen was also effective on yeast hulls. Figures 3D-F are confocal microscopy images of yeast hulls coated with 1 to 3 layers of MS74NEY2 lightly labeled with FITC (F/P ratio of 0.1). In addition to the increased fluorescence, the yeast hulls displayed a greater tendency to aggregate with 3 coats of antigen.
NY-ESO-1-specific In Vitro Immunization of Naive CD8+ T Cells
The multiple refinements that we have made to improve our antigen-coated yeast-based vaccine strategy have thus far been tested primarily by detecting cross-presentation of the surrogate CMV epitope using a T-cell clone. We now wished to investigate if the vaccine candidates are capable of priming naive CD8+ T cells to elicit NY-ESO-1-specific immunity. CD8+ CD45RO- T cells were isolated from the PBMCs of 3 HLA-A*0201 donors by magnetic cell sorting. The naive CD8+ T cells were added at a 10:1 ratio to DCs (from a fourth donor) that had been stimulated the previous day with either unconjugated yeast hulls, whole yeast cells carrying 1 coat of MS74NEY2, or yeast hulls with 3 coats of antigen. Before and after 10 days of coculture, the cells were labeled with a SLLMWITQV/HLA-A*0201 peptide-MHC pentamer, with the peptide being the well-characterized NY-ESO-1157-165 epitope with the final Cys residue mutated to Val to avoid oxidation-related issues.26
As shown in Figure 4, the naive precursor frequencies were around 0.02% with all 3 donors, and the frequency of higher avidity T cells (arbitrarily defined as those falling within the rectangular gate) were in the 0.003% to 0.005% range. Ten days after stimulation by the DCs, the frequencies of pentamer-positive CD8+ T cells increased under all 9-test conditions. The proliferation of pentamer-positive T cells after “immunization” with yeast hulls lacking NY-ESO-1 may be a result of extensive TCR degeneracy observed in many experiments,27-33 with theoretical analysis suggesting a TCR ligand repertoire size of 106 to 107 peptide epitopes.34 With all 3 donors, the yeast hulls coated with 3 layers of antigen elicited the highest proportion of pentamer-stained cells, with almost double the frequency of high avidity T cells as compared with the yeast-hull-only controls. The singly coated whole yeast cells, which delivered a smaller dose of antigen as compared with the 3-coat yeast hulls, gave rise to intermediate pentamer-positive T-cell frequencies with 2 of the 3 donors. Although the double-positive events are rare and not clearly distinct from the pentamer-negative cells, we believe that they represent genuine NY-ESO-1-specific T cells because restimulation with peptide-pulsed autologous PBMCs on day 10 expanded the double-positive populations to 5% to 20% by day 20 (Fig. 5). We conclude that DCs fed either whole yeast cells coated with 1 layer of MS74NEY2 or yeast hulls coated 3 times were able to prime naive, NY-ESO-1-recognizing CD8+ T cells, with the latter formulation being initially more potent.
FIGURE 4.

DCs fed MS74NEY2-coated yeast and yeast hulls stimulated the expansion of cells recognizing an NY-ESO-1 peptide/MHC complex from a naive CD8+ pool. CD8+ CD45RO- cells from 3 HLA-A2 donors were added to immature DCs fed either yeast hulls alone, whole yeast coated with 1 layer of MS74NEY2, or yeast hulls with 3 coats (each at a dose equivalent to 10 yeast/DC). The T cells before coculture and after 10 days (IL-2 and IL-7 were added from day 3 onwards) were labeled with α-CD8-FITC and PE-conjugated SLLMWITQV/HLA-A*0201 pentamer. Cells (2 × 106) were labeled per test and 2 × 105 CD8+ events are shown in each dot plot. The proportions of events (out of CD8+ events) falling in the top right quadrant (in bold italics) and in the rectangular gate are shown. DC indicates dendritic cell; FITC, fluorescein isothiocyanate; MHC, major histocompatibility complex; PE, phycoerythrin.
FIGURE 5.

Peptide/MHC pentamer-positive T cells primed by yeast particles continue to multiply after restimulation with peptide-pulsed autologous PBMCs. On day 10 of the experiment described in Figure 4, PBMCs from each donor were thawed, gamma-irradiated, and pulsed with 40 μM SLLMWITQV peptide for 3 hours. Day 10 T cells from each test condition (5 × 106/well) were cocultured with 107 donor-matched PBMCs in 4 mL CTL medium (with 10 μM peptide) in a 12-well plate. The cells were fed and split as necessary. Flow cytometry analysis after labeling with α-CD8-FITC and PE-conjugated SLLMWITQV/HLA-A*0201 pentamer was performed on day 20. FITC indicates fluorescein isothiocyanate; MHC, major histocompatibility complex; PBMC, peripheral blood mononuclear cell; PE, phycoerythrin.
DC Maturation by Yeast Particles
The ability of S. cerevisiae yeast to stimulate inflammatory cytokine secretion by murine splenocytes7,8 and DCs3,9 and to induce maturation of human6 and murine3,9 DCs is well-established. In our preliminary experiments, yeast cells up-regulated various markers of maturation in human monocyte-derived DCs to an extent comparable with 1 μg/mL LPS and induced even more IL-12 secretion than LPS. We now investigated whether the processes of antigen conjugation and cell disruption would alter the immunostimulatory properties of yeast. SWH100 yeast and yeast hulls, each with and without a single layer of conjugated antigen, were added to immature DCs at the nominal dose of 10 yeast/DC. After 48 hours, the DCs were analyzed for the expression levels of CD40, CD80, CD83, CD86, and HLA-DR (all markers of DC maturation), whereas the cell culture supernatants were subjected to an IL-12p70 ELISA assay. As we had observed previously, whole unconjugated yeast up-regulated the maturation markers to levels similar to those caused by LPS (Fig. 6A) and massively induced IL-12 secretion (Fig. 6B). To our surprise, uncoated yeast hulls behaved very differently compared with whole yeast, causing strikingly less marker up-regulation and no detectable IL-12 secretion. Antigen conjugation had opposite effects on whole yeast and yeast hulls, slightly reducing DC maturation and IL-12 secretion by whole yeast while increasing these metrics with yeast hulls. Although some IL-12 was secreted by DCs exposed to antigen-coated yeast hulls, the quantity was 2 orders of magnitude lower than that caused by coated or uncoated whole yeast (Fig. 6B). The effects of antigen conjugation were not simply caused by endotoxin contamination or some intrinsic property of the protein, as soluble fusion protein alone (at 0.5 μM, roughly 100× the level of yeast-conjugated antigen present in a well) had little effect on the DCs (Fig. 6).
FIGURE 6.
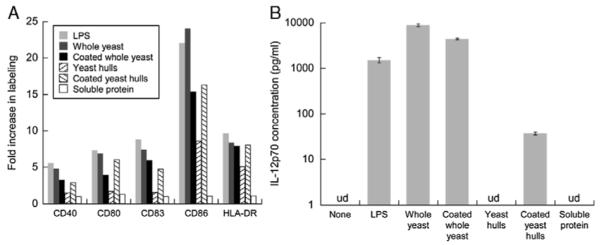
DC maturation in response to yeast is influenced by cell integrity and antigen conjugation. Immature DCs were stimulated with either LPS (1 μg/mL) or soluble fusion protein (0.5 μM) or whole SWH100 yeast or yeast hulls either unmodified or conjugated with 1 layer of antigen (each at a dose equivalent to 10 yeast/DC). A, Two days later, the DCs were labeled with α-DC-SIGN-FITC and a PE-conjugated antibody against 1 of the 5 selected markers of DC maturation. The flow cytometry results were gated by DC-SIGN positivity and the median PE fluorescence in each test was compared against the result with unstimulated DCs. B, The cell culture supernatants after 2 days were tested for IL-12p70 by ELISA. Readings calculated to be lower than 5 pg/mL were considered undetectable (ud). Error bars indicate SDs of duplicate wells. DC indicates dendritic cell; ELISA, enzyme-linked immunosorbent assay; FITC, fluorescein isothiocyanate; LPS, lipopolysaccharide; PE, phycoerythrin.
DISCUSSION
We present here a flexible vaccine-oriented scheme for conjugating antigen to yeast particles that has undergone multiple refinements with the aim of maximizing cross-presentation. The fusion protein MS74NEY2 represents the latest embodiment, consisting of the following functional elements: MBP, SNAP-tag, CD74 ectodomain, 15-mer CMV-derived surrogate antigen, NY-ESO-1 cancer-testis antigen, glycine/serine linker, ybbR tag, and His6 tag. We have demonstrated cross-presentation of both the surrogate antigen and an NY-ESO-1 HLA-A2 epitope; these antigens can readily be exchanged to suit different immunotherapy applications. The highly hydrophobic NY-ESO-1 protein used in clinical trials is purified from E. coli inclusion bodies,35 but insertion of MBP as the N-terminal domain enabled our fusion proteins to be expressed solubly and also facilitated purification. For other antigens, MBP may be omitted or replaced if desired. A key feature of our conjugation scheme is its site-specific nature, which ensures that antigen can be released in the DC phagosome to translocate to the cytosol for entry into the MHC class I processing pathway. The initial layer of fusion protein is conjugated to BG-derivatized particles through the formation of a stable thioether bond with the SNAP-tag domain. We showed that cross-presentation is highly inefficient when a nonsite-specific chemical conjugation method was used to attach the same protein to yeast. The lack of efficacy is especially noteworthy considering that there were no reactive lysine residues within the CMV peptide and only one in the C-terminal direction. This suggests that merely “tethering” an epitope on both ends with no intervening phagosomal protease sites can impede cross-presentation. To expedite phagosomal antigen release in our scheme, we inserted the extremely CatS-susceptible CD74 ectodomain between the SNAP-tag tether and the antigens, resulting in improved cross-presentation efficiency.
In addition to developing the conjugation scheme with antigen release kinetics in mind, we have also made several improvements to increase antigen dose delivery. The first refinement was to use a yeast strain expressing Aga1p and Aga2p, which provided more free amines on the cell wall for BG and fusion protein conjugation compared with the wild type. Fusing lysine-containing polypeptides to Aga2p did not further increase the antigen density in this first layer, so we devised a novel multilayer conjugation strategy that retains site-specificity. Sfp enzyme is used to attach BG-linked CoA to the ybbR tags of 1 antigen layer, thus permitting the next layer to be added via the SNAP-tag/BG reaction. We experienced no loss in conjugation efficiency in building up to 4 antigen layers (suggesting that further coats can be added almost indefinitely) and demonstrated that cross-presentation levels rose with the number of layers. Finally, breaking up the cell wall into fragments before conjugation further increased the amount of antigen delivered per yeast cell equivalent, probably because the surfaces that were revealed provided additional free amines for BG attachment.
Although the use of yeast hulls instead of whole yeast improves antigen delivery and reduces native yeast protein content, it is at present unclear whether yeast hulls will ultimately be more effective in the setting of immunotherapy. A major reason behind the attractiveness of yeast-based vaccines is their ability to mature DCs and stimulate the secretion of proinflammatory cytokines, obviating the need for additional adjuvants.5 To our surprise, yeast hulls were significantly inferior to whole yeast cells in inducing DC maturation, and more importantly, they elicited little or no IL-12 secretion. In retrospect, this observation is not without precedent as several groups have reported that Zymosan, a preparation of S. cerevisiae cell wall particles, is a poor inducer of DC IL-12 secretion despite its long-standing reputation as a proinflammatory agent.36-38 IL-12 secretion is likely to be important to vaccine efficacy because it plays a role in enhancing the clonal expansion of primed CD8+ T cells,39,40 perhaps by increasing resistance to activation-induced cell death.41
The disparity in DC reactions to whole yeast cells and yeast cell wall fragments is puzzling and warrants further investigation. One possibility is that an immunostimulatory component is lacking in yeast hulls, which was either removed with the yeast cytoplasm or washed away from the cell wall fragments. Reevaluation of the wash steps may be helpful in the latter case. In the former case, a possible candidate is double-stranded RNA from viruses that ubiquitously infect yeast cells.42 It has been observed that a combination of Zymosan and poly-I:C (a synthetic double-stranded RNA analog) stimulates far greater levels of IL-12 secretion than either agent alone,38 raising the intriguing possibility that whole yeast cells induce massive IL-12 secretion through a similar synergism. If this is the case, conjugating poly-I:C to yeast hulls may present a simple solution. An alternative explanation stems from the complex structure of the yeast cell wall, with fragmentation resulting in the exposure of different molecular patterns leading to altered signaling cascades in DCs. To elaborate, the outer layer of the S. cerevisiae cell wall is composed primarily of mannan (hypermannosylated proteins), which in undamaged yeast cells largely masks the inner layer of β1,3-glucan, β1,6-glucan, and chitin.43 Our observation that antigen conjugation (and thus partial disruption of the molecular patterns) had opposite effects on whole yeast cells and yeast hulls could be an indication that DC recognition of the inner cell wall layer partially counteracts the maturation signals derived from ligands in the outer layer.
Vaccination experiments in vivo will be required to discover the effects of yeast particle choice on T-cell memory and protective immunity and to develop an optimal formulation (possibly a mixture of uncoated whole yeast and coated yeast hulls). At the same time, the conjugation scheme presented here is not restricted to yeast and can easily be applied to any particle that is or can be amine-functionalized, including heat-killed bacteria and bacterial ghosts, polymer and inorganic microspheres, ribosomes, and liposomes. Although the ability of antigen-coated particles to induce protective tumor immunity was demonstrated almost 15 years ago,44 particulate antigen delivery vehicles (with the exception of viruslike particles) have received little attention in the field of cancer immunotherapy. Codelivery of adjuvant and antigen in particles holds several advantages over injection of soluble agents, including a high local concentration that does not quickly dissipate, assurance that both adjuvant and a minimum bolus of antigen are delivered to the same cell, and inherent targeting for uptake by phagocytic cells (besides DCs, macrophages45 and neutrophils46 are also capable of cross-priming). Poor phagosomal escape of antigen represents one barrier to the development of particulate vaccines, and the conjugation strategy described here presents a novel way to break this barrier.
ACKNOWLEDGMENTS
The authors thank Yuhua Hu for her help with the confocal microscope, and Jason Auer and Steve Mesite for their technical assistance with the Microfluidizer.
Funded in part by NIH Grant AI065824 and the Ludwig Foundation. Shanshan W. Howland is supported by Singapore’s Agency for Science, Technology, and Research.
Footnotes
Financial Disclosure: The authors have declared there are no financial conflicts of interest in regards to this work.
REFERENCES
- 1.Rosenberg SA, Yang JC, Restifo NP. Cancer immunotherapy: moving beyond current vaccines. 2004;10:909–915. doi: 10.1038/nm1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Petrovsky N, Aguilar JC. Vaccine adjuvants: current state and future trends. 2004;82:488–496. doi: 10.1111/j.0818-9641.2004.01272.x. [DOI] [PubMed] [Google Scholar]
- 3.Stubbs AC, Martin KS, Coeshott C, et al. Whole recombinant yeast vaccine activates dendritic cells and elicits protective cell-mediated immunity. Nat Med. 2001;7:625–629. doi: 10.1038/87974. [DOI] [PubMed] [Google Scholar]
- 4.Lu Y, Bellgrau D, Dwyer-Nield LD, et al. Mutation-selective tumor remission with Ras-targeted, whole yeast-based immunotherapy. Cancer Res. 2004;64:5084–5088. doi: 10.1158/0008-5472.CAN-04-1487. [DOI] [PubMed] [Google Scholar]
- 5.Franzusoff A, Duke RC, King TH, et al. Yeasts encoding tumour antigens in cancer immunotherapy. Expert Opinion Biol Ther. 2005;5:565–575. doi: 10.1517/14712598.5.4.565. [DOI] [PubMed] [Google Scholar]
- 6.Barron MA, Blyveis N, Pan SC, et al. Human dendritic cell interactions with whole recombinant yeast: implications for HIV-1 vaccine development. J Clin Immunol. 2006;26:251–264. doi: 10.1007/s10875-006-9020-8. [DOI] [PubMed] [Google Scholar]
- 7.Haller AA, Lauer GM, King TH, et al. Whole recombinant yeast-based immunotherapy induces potent T cell responses targeting HCV NS3 and Core proteins. Vaccine. 2007;25:1452–1463. doi: 10.1016/j.vaccine.2006.10.035. [DOI] [PubMed] [Google Scholar]
- 8.Riemann H, Takao J, Shellman YG, et al. Generation of a prophylactic melanoma vaccine using whole recombinant yeast expressing MART-1. Exp Dermatol. 2007;16:814–822. doi: 10.1111/j.1600-0625.2007.00599.x. [DOI] [PubMed] [Google Scholar]
- 9.Bernstein MB, Chakraborty M, Wansley EK, et al. Recombinant Saccharomyces cerevisiae (yeast-CEA) as a potent activator of murine dendritic cells. Vaccine. 2008;26:509–521. doi: 10.1016/j.vaccine.2007.11.033. [DOI] [PubMed] [Google Scholar]
- 10.Munson S, Parker J, King TH, et al. Coupling innate and adaptive immunity with yeast-based cancer immunotherapy. In: Orentas R, Hodge JW, Johnson BD, editors. Cancer Vaccines and Tumor Immunity. Wiley-Liss; New York: 2008. pp. 131–149. [Google Scholar]
- 11.Wadle A, Held G, Neumann F, et al. Cross-presentation of HLA class I epitopes from influenza matrix protein produced in Saccharomyces cerevisiae. Vaccine. 2006;24:6272–6281. doi: 10.1016/j.vaccine.2006.05.096. [DOI] [PubMed] [Google Scholar]
- 12.Howland SW, Wittrup KD. Antigen release kinetics in the phagosome are critical to cross-presentation efficiency. J Immunol. 2008;180:1576–1583. doi: 10.4049/jimmunol.180.3.1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Boder ET, Wittrup KD. Yeast surface display for screening combinatorial polypeptide libraries. Nat Biotechnol. 1997;15:553–557. doi: 10.1038/nbt0697-553. [DOI] [PubMed] [Google Scholar]
- 14.Gnjatic S, Nishikawa H, Jungbluth AA, et al. NY-ESO-1: review of an immunogenic tumor antigen. Adv Cancer Res. 2006;95:1–30. doi: 10.1016/S0065-230X(06)95001-5. [DOI] [PubMed] [Google Scholar]
- 15.Nicholaou T, Ebert L, Davis ID, et al. Directions in the immune targeting of cancer: lessons learned from the cancer-testis Ag NY-ESO-1. 2006;84:303–317. doi: 10.1111/j.1440-1711.2006.01446.x. [DOI] [PubMed] [Google Scholar]
- 16.Piatesi A, Howland SW, Rakestraw JA, et al. Directed evolution for improved secretion of cancer-testis antigen NY-ESO-1 from yeast. Protein Exp Purif. 2006;48:232–242. doi: 10.1016/j.pep.2006.01.026. [DOI] [PubMed] [Google Scholar]
- 17.Chao G, Lau WL, Hackel BJ, et al. Isolating and engineering human antibodies using yeast surface display. 2006;1:755–768. doi: 10.1038/nprot.2006.94. [DOI] [PubMed] [Google Scholar]
- 18.Sikorski RS, Hieter P. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics. 1989;122:19–27. doi: 10.1093/genetics/122.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yin J, Lin AJ, Golan DE, et al. Site-specific protein labeling by Sfp phosphopantetheinyl transferase. Nat Protoc. 2006;1:280–285. doi: 10.1038/nprot.2006.43. [DOI] [PubMed] [Google Scholar]
- 20.Reichelt P, Schwarz C, Donzeau M. Single step protocol to purify recombinant proteins with low endotoxin contents. Protein Exp Purif. 2006;46:483–488. doi: 10.1016/j.pep.2005.09.027. [DOI] [PubMed] [Google Scholar]
- 21.Gnjatic S, Nagata Y, Jager E, et al. Strategy for monitoring T cell responses to NY-ESO-1 in patients with any HLA class I allele. Proc Natl Acad Sci. 2000;97:10917–10922. doi: 10.1073/pnas.97.20.10917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ho WY, Nguyen HN, Wolfl M, et al. In vitro methods for generating CD8+ T-cell clones for immunotherapy from the naive repertoire. J Immunol Methods. 2006;310:40–52. doi: 10.1016/j.jim.2005.11.023. [DOI] [PubMed] [Google Scholar]
- 23.Keppler A, Kindermann M, Gendreizig S, et al. Labeling of fusion proteins of O6-alkylguanine-DNA alkyltransferase with small molecules in vivo and in vitro. Methods. 2004;32:437–444. doi: 10.1016/j.ymeth.2003.10.007. [DOI] [PubMed] [Google Scholar]
- 24.Ruckrich T, Brandenburg J, Cansier A, et al. Specificity of human cathepsin S determined by processing of peptide substrates and MHC class II-associated invariant chain. Biol Chem. 2006;387:1503–1511. doi: 10.1515/BC.2006.188. [DOI] [PubMed] [Google Scholar]
- 25.Riese RJ, Wolf PR, Bromme D, et al. Essential role for cathepsin S in MHC class II-associated invariant chain processing and peptide loading. Immunity. 1996;4:357–366. doi: 10.1016/s1074-7613(00)80249-6. [DOI] [PubMed] [Google Scholar]
- 26.Chen J-L, Dunbar PR, Gileadi U, et al. Identification of NY-ESO-1 peptide analogues capable of improved stimulation of tumor-reactive CTL. J Immunol. 2000;165:948–955. doi: 10.4049/jimmunol.165.2.948. [DOI] [PubMed] [Google Scholar]
- 27.Wucherpfennig KW, Strominger JL. Molecular mimicry in T cell-mediated autoimmunity: viral peptides activate human T cell clones specific for myelin basic protein. Cell. 1995;80:695–705. doi: 10.1016/0092-8674(95)90348-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kersh G, Allen P. Structural basis for T cell recognition of altered peptide ligands: a single T cell receptor can productively recognize a large continuum of related ligands. J Exp Med. 1996;184:1259–1268. doi: 10.1084/jem.184.4.1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hemmer B, Vergelli M, Gran B, et al. Cutting edge: predictable TCR antigen recognition based on peptide scans leads to the identification of agonist ligands with no sequence homology. J Immunol. 1998;160:3631–3636. [PubMed] [Google Scholar]
- 30.Hemmer B, Vergelli M, Pinilla C, et al. Probing degeneracy in T-cell recognition using peptide combinatorial libraries. Immunol Today. 1998;19:163–168. doi: 10.1016/s0167-5699(97)01217-6. [DOI] [PubMed] [Google Scholar]
- 31.Misko IS, Cross SM, Khanna R, et al. Crossreactive recognition of viral, self, and bacterial peptide ligands by human class I-restricted cytotoxic T lymphocyte clonotypes: implications for molecular mimicry in autoimmune disease. Proc Natl Acad Sci. 1999;96:2279–2284. doi: 10.1073/pnas.96.5.2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen J, Eisen HN, Kranz DM. A model T-cell receptor system for studying memory T-cell development. Microbes Infect. 2003;5:233–240. doi: 10.1016/s1286-4579(03)00016-9. [DOI] [PubMed] [Google Scholar]
- 33.Wucherpfennig KW. T cell receptor crossreactivity as a general property of T cell recognition. Molecular immunology degeneracy of T cell recognition and its relationship to molecular mimicry. 2004;40:1009–1017. doi: 10.1016/j.molimm.2003.11.003. [DOI] [PubMed] [Google Scholar]
- 34.Mason D. A very high level of crossreactivity is an essential feature of the T-cell receptor. Immunol Today. 1998;19:395–404. doi: 10.1016/s0167-5699(98)01299-7. [DOI] [PubMed] [Google Scholar]
- 35.Murphy R, Green S, Ritter G, et al. Recombinant NY-ESO-1 cancer antigen: production and purification under cGMP conditions. Prep Biochem Biotechnol. 2005;35:119–134. doi: 10.1081/pb-200054732. [DOI] [PubMed] [Google Scholar]
- 36.Edwards AD, Manickasingham SP, Sporri R, et al. Microbial recognition via toll-like receptor-dependent and -independent path-ways determines the cytokine response of murine dendritic cell subsets to CD40 triggering. J Immunol. 2002;169:3652–3660. doi: 10.4049/jimmunol.169.7.3652. [DOI] [PubMed] [Google Scholar]
- 37.Dillon S, Agrawal S, Banerjee K, et al. Yeast zymosan, a stimulus for TLR2 and dectin-1, induces regulatory antigen-presenting cells and immunological tolerance. J Clin Invest. 2006;116:916–928. doi: 10.1172/JCI27203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bohnenkamp HR, Papazisis KT, Burchell JM, et al. Synergism of toll-like receptor-induced interleukin-12p70 secretion by monocyte-derived dendritic cells is mediated through p38 MAPK and lowers the threshold of T-helper cell type I responses. Cell Immunol. 2007;247:72–84. doi: 10.1016/j.cellimm.2007.07.008. [DOI] [PubMed] [Google Scholar]
- 39.Valenzuela J, Schmidt C, Mescher M. The roles of IL-12 in providing a third signal for clonal expansion of naive CD8 T cells. J Immunol. 2002;169:6842–6849. doi: 10.4049/jimmunol.169.12.6842. [DOI] [PubMed] [Google Scholar]
- 40.Chang J, Cho J-H, Lee S-W, et al. IL-12 priming during in vitro antigenic stimulation changes properties of CD8 T cells and increases generation of effector and memory cells. J Immunol. 2004;172:2818–2826. doi: 10.4049/jimmunol.172.5.2818. [DOI] [PubMed] [Google Scholar]
- 41.Díaz-Montero C, El Naggar S, Al Khami A, et al. Priming of naive CD8+ T cells in the presence of IL-12 selectively enhances the survival of CD8+CD62Lhi cells and results in superior anti-tumor activity in a tolerogenic murine model. Cancer Immunol, Immunother. 2008;57:563–572. doi: 10.1007/s00262-007-0394-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wickner RB. Prions and RNA viruses of Saccharomyces cerevisiae. Annu Rev Genet. 1996;30:109–139. doi: 10.1146/annurev.genet.30.1.109. [DOI] [PubMed] [Google Scholar]
- 43.Wheeler RT, Fink GR. A drug-sensitive genetic network masks fungi from the immune system. PLoS Pathogens. 2006;2:e35. doi: 10.1371/journal.ppat.0020035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Falo LD, Kovacsovics-Bankowski M, Thompson K, et al. Targeting antigen into the phagocytic pathway in vivo induces protective tumour immunity. 1995;1:649–653. doi: 10.1038/nm0795-649. [DOI] [PubMed] [Google Scholar]
- 45.Pozzi L-AM, Maciaszek JW, Rock KL. Both dendritic cells and macrophages can stimulate naive CD8 T cells in vivo to proliferate, develop effector function, and differentiate into memory cells. J Immunol. 2005;175:2071–2081. doi: 10.4049/jimmunol.175.4.2071. [DOI] [PubMed] [Google Scholar]
- 46.Beauvillain C, Delneste Y, Scotet M, et al. Neutrophils efficiently cross-prime naive T cells in vivo. Blood. 2007;110:2965–2973. doi: 10.1182/blood-2006-12-063826. [DOI] [PubMed] [Google Scholar]