

Abstract
The fruit fly Drosophila melanogaster has brought significant advances to research in neurodegenerative disease, notably in the identification of genes that are required to maintain the structural integrity of the brain, defined by recessive mutations that cause adult-onset neurodegeneration. Here, we survey these genes in the fly and classify them according to five key cell biological processes. Over half of these genes have counterparts in mouse or human that are also associated with neurodegeneration. Fly genetics continues to be instrumental in the analysis of degenerative disease, with notable recent advances in our understanding of several inherited disorders, as well as Parkinson’s Disease and the central role of mitochondria in neuronal maintenance.
Introduction
Typically afflicting adults in mid-life, neurodegenerative diseases are characterized by motor or cognitive symptoms that get progressively worse with age and that usually reduce life expectancy. Human neurodegenerative disease can result from a variety of environmental and genetic causes. Genetic factors in particular have been instrumental in developing our understanding of the etiology and progression of such diseases, and can range from mutations that increase the risk for a particular disorder, to mutations that are the sole, direct cause of a disease. As with another collection of related diseases—cancer—neurodegeneration can result from dominant or recessive mutations. In this review, we focus on the role of Drosophila melanogaster in characterizing “neurodegeneration suppressor” genes, analogous to cancer tumor suppressor genes. Defined by recessive loss of function mutations that cause neurodegeneration, such genes are required for maintaining the integrity of the adult central nervous system.
Drosophila has been a key tool in much of the work to identify genes involved in neuronal integrity and to discover their functions. Gene discovery is quick and straightforward in the fly, as is the analysis of how separate genes function together, two points which we expand on in the following section. Other useful Drosophila techniques are also addressed below: the expression of transgenes, including RNAi constructs, that can be directed precisely in space and time; and genetically mosaic flies, useful for identifying the location of gene function.
Although superficially different, humans and flies are remarkably similar in key respects. Critical signaling pathways in development, cancer and innate immunity are conserved between the fly and human1,2. The central nervous systems of invertebrates and vertebrates share a common evolutionary origin3, and the fly has been used successfully for the genetic analysis of complex behaviors ranging from sleep4, to learning and memory5 and aggression6. Of human protein sequences associated with disease in the Online Mendelian Inheritance in Man (OMIM) database, 74% have highly related sequences in the fly7 (see also the Homophila web site). Moreover, a number of dominantly inherited human neurodegenerative diseases, such as those caused by polyglutamine repeat expansions, have been successfully modeled in Drosophila by transgenic expression of the human disease genes (reviewed in reference 8). Subsequent screens for fly genes that modify the effects of toxic human proteins have been enormously successful, leading to new insight into these diseases and demonstrating the parallels between human and fly neurodegeneration.
Here we focus on a complementary approach, distinct from transgenic modeling of human neurodegenerative diseases: the identification of loss of function mutations in endogenous fly genes that cause brain degeneration. More than half of such genes either have human orthologs linked to disease or otherwise provide insight into conserved processes required for maintaining the structural integrity of the central nervous system, a tissue with complex cell types that for the most part undergo no cell division or renewal during the lifetime of the animal. The last few years in particular have seen rapid advances in this field with screens to identify new fly neurodegeneration genes, and new insight into the roles of genes previously known to be critical in human neurodegeneration. Below, we highlight examples showing how fly genetics can be used to address human disease. From a survey of currently known fly neurodegeneration genes, five key cell biological processes stand out as critical for maintaining CNS (central nervous system) integrity. Finally, we address insights from Drosophila mutants into the role of glia and cell-cell interactions in neuronal integrity. Parallels between the fly, mouse and human underscore the conservation of gene function in maintaining the nervous system, and suggest that further investigations in the fly will reveal additional insight relevant to the entire field.
Of screens and genes
Genes required for the maintenance of the adult fly brain have been discovered by three approaches: screens, candidate genes, and fortuitous mutations. In screens to identify neurodegeneration genes, the classic approach has been to select viable adult mutant fly lines with a behavioral defect, and then screen by histology for CNS degeneration. Interestingly, the first assay devised for the genetic analysis of fly behavior was instrumental in the first discovery of a mutation that causes neurodegeneration in the adult fly, in the gene drop-dead8,9. Later screens in Seymour Benzer’s lab focused on mutants with shortened lifespans10,11. In the late 1970s, Heisenberg and Bohl screened for fly mutants defective in phototaxis and then performed a type of high-throughput mass histology on fly heads12. Recent screens have looked for degeneration in mutant flies that become paralyzed with a change in temperature13 or mechanical stress14, or that have altered circadian rhythms15. The candidate gene approach is an alternative to screens, in which fly orthologs of mouse or human genes known to cause neurodegeneration have been identified and characterized. Finally, many fortuitous mutations isolated in unrelated studies have shown unexpected loss of integrity of the brain.
Currently, 44 genes involved in CNS integrity in Drosophila have been characterized (Supplementary Information 1, Table). A subset of these genes, those discussed most extensively in this Review, is shown in Table 1. A gene is included in SI TABLE 1 if the recessive, loss-of-function mutation causes progressive, adult-onset histological abnormalities in the fly brain. Retinal degeneration, a related topic, is addressed in BOX 1. With two exceptions, the genes in S1 TABLE 1 have readily identifiable orthologs in mouse and human, and over half are related to mouse or human genes also associated with neurodegeneration. Below, we discuss three genes from the table that illustrate advantages of Drosophila genetics that are crucial in addressing neurodegenerative disease.
TABLE 1. Neurodegeneration Due to Loss-of-Function Mutations.
A subset of the Drosophila genes associated with progressive, adult-onset histological defects in the fly brain, shown in full in SUPPLEMENTARY TABLE 1, Mouse and human genes are orthologs of the respective fly genes, called by Flybase/InParanoid or NCBI/Homologene, and are in  if they are known to be associated with neurodegeneration. Mouse knockout phenotypes are from the Mouse Genome Informatics web site; human diseases from Online Mendelian Inheritance in Man. Mutations in fly, mouse, and human genes are recessive loss-of-function alleles unless otherwise noted. An expanded, updated version of this table with links is at the Bonini Lab web site.
if they are known to be associated with neurodegeneration. Mouse knockout phenotypes are from the Mouse Genome Informatics web site; human diseases from Online Mendelian Inheritance in Man. Mutations in fly, mouse, and human genes are recessive loss-of-function alleles unless otherwise noted. An expanded, updated version of this table with links is at the Bonini Lab web site.
| FLY GENE | PROTEIN | MOUSE GENE: KNOCKOUT | HUMAN GENE: DISEASE | NOTES |
|---|---|---|---|---|
| Ace | acetylcholinesterase | Ache: delayed postnatal development, organophosphate sensitivity, death by 3 weeks. | ACHE | Fly: Null allele is lethal. Large mutant brain clones induced in embryogenesis show degeneration62. Temperature sensitive allele at 32°causes paralysis and quick death; at 29° for 11 days causes abnormal neuropil63. |
| ATPα | Na+/K+ ATPase α subunit | Atp1a3: neonatal lethal. Heterozygous mice: learning defects, hyperactivity. |
 : dystonia 12 : dystonia 12 |
Fly: Degeneration with both recessive and dominant alleles65. |
Mouse: β subunit gene  knockout causes neurodegeneration and death at 17–18d93. knockout causes neurodegeneration and death at 17–18d93. | ||||
| Human diseases caused by dominant allele. | ||||
| ATP6 | F1F0-ATP synthase subunit | mt-Atp6 |
 : Leigh syndrome : Leigh syndrome |
Fly: Subtle thoracic ganglia defects; no gross histological defects in aged brains. Enhances sesB phenotype. Thoracic muscle degeneration. Mechanical stress sensitivity Abnormal mitochondrial ultrastructure68. |
| bubblegum (bgm) | fatty acid CoA synthetase | Acsbg1 | ACSBG2 | Fly: Histological defects in optic lobe only11. |
Human; Adrenoleukodystrophy is caused by a recessive X-linked  allele, affecting a peroxisomal transporter involved in importation or anchoring of the synthetase. allele, affecting a peroxisomal transporter involved in importation or anchoring of the synthetase. | ||||
| Cystein string protein (Csp) | Hsp40-like; component of synaptic vesicles | Dnajc5b | DNAJC5B | Fly: Subtle synaptic degeneration seen at TEM level58. |
Mouse: Knockout of paralog  dies within 3 months, with progressive neuromuscular junction degeneration and behavioral abnormalities59. dies within 3 months, with progressive neuromuscular junction degeneration and behavioral abnormalities59. | ||||
| dare | ferredoxin reductase | Fdxr | FDXR | Fly: Hypomorphic allele; null alleles are larval lethal. Severe uncoordination43. |
| drop-dead (drd) | membrane | no significantly similar gene | no significantly similar gene | Fly: Abnormal glial morphology in young adults76. |
| EAAT1 | glutamate transporter | Slc1a3: ataxia, abnormal Purkinje cell innervation by climbing fibers94. | SLC1A3 | Fly: Loss of function via RNAi. Behavioral defects. Increased sensitivity to paraquat64. |
Mouse: Knockout of neuronally-expressed paralog  causes neurodegeneration95. causes neurodegeneration95. | ||||
| easily shocked (eas) | ethanolamine kinase | Etnk1 | ETNK1 | Fly: Mechanical shock causes brief hyperactivity and then paralysis; possible epilepsy model. Electrophysiological defects in giant fiber pathway33,96. |
| fumble (fbl) | pantothenate kinase | Pank1 | PANK1 | Fly: Hypomorphic alleles. Flight and climbing defects. Sensitive to paraquat36. |
Mouse paralog  knockout causes retinal degeneration. knockout causes retinal degeneration. | ||||
Human: Mutation of paralog  causes antothenate kinase-associated neurodegeneration (a.k.a. Hallervorden-Spatz syndrome). causes antothenate kinase-associated neurodegeneration (a.k.a. Hallervorden-Spatz syndrome). | ||||
| futsch | microtubule-associated protein 1B | Mtap1b: CNS developmental defects | MAP1B | Fly: Hypomorphic alleles; null alleles are lethal. Learning defect observed before brain histological defects. Partial rescue by Tau97. |
Human: Dominant splicing mutation in the paralog 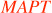 (Tau) causes frontotemporal dementia with parkinsonism. (Tau) causes frontotemporal dementia with parkinsonism. | ||||
| levy | cytochrome C oxidase (COX) subunit VIa | Cox6a1 | COX6A1 | Fly: Histological defects limited to retina & optic lobes98. |
Human Leigh syndromes can be caused by mutation of  or or 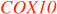 (see Conclusion in text) (see Conclusion in text) | ||||
| parkin (park) | E3 ubiquitin ligase |
 : behavioral defects, dopaminergic neuron loss in some but not all knockout alleles : behavioral defects, dopaminergic neuron loss in some but not all knockout alleles |
 : Parkinson disease 2 : Parkinson disease 2 |
Fly: Principal pathology is in mitochondria of sperm & flight muscles. Dopaminergic neurons are smaller in size99,100. |
| pink1 | mitochondrial protein kinase | Pink1: normal number of dopaminergic neurons; mitochondrial defects |
 : Parkinson disease 6 : Parkinson disease 6 |
Fly: Conflicting data on dopaminergic neuron loss. Male sterility and wing, muscle, and mitochondrial defects24–26. |
| reverse polarity (repo) | homeodomain transcription factor | Alx4:developmental skeletal defects. | ALX4: parietal foramina 2 | Fly: Hypomorphic allele. Apoptotic loss of both neurons and glia limited to optic lobe75. |
| SNF4Aγ a.k.a. löchrig (loe) | AMP-activated protein kinase γ subunit | Prkag2 | PRKAG2: Wolff-Parkinson-White syndrome | Fly: Mutation affects 1 of 3 isoforms39,101. |
| Human disease caused by a dominant allele. | ||||
| swiss cheese (sws) | membrane lipid esterase; protein kinase A regulatory subunit |
 : Hippocampus, thalamus, & cerebellar degeneration21. : Hippocampus, thalamus, & cerebellar degeneration21. |
 (a.k.a NTE): spastic paraplegia-39 (a.k.a NTE): spastic paraplegia-39 |
Fly: Increased apoptosis. Both neuronal and glial death12,16,19. |
| Mouse: neurodegeneration due to conditional brain-specific knockout | ||||
Retinal Degeneration in Drosophila
Phototransduction—the conversion of light into neural signals—occurs through a G protein-coupled pathway. Mutations in many genes in this pathway in the fly typically induce light-dependent retinal degeneration but not neuronal loss elsewhere in the fly82. However, since vision in the fly is not essential for viability or reproduction, the eye is an attractive tissue in which to model general neurodegeneration. A number of tools are available for such purposes. Genetic tricks include transgenic RNAi constructs targeted specifically to the developing eye or to mature photoreceptor neurons, and the generation of homozygous mutant eyes in an otherwise heterozygous animal83. Either method allows perturbation of the eye without affecting viability of the animal as a whole. Methods for assaying photoreceptor degeneration include a simple behavioral assay, such as phototaxis (healthy flies run robustly towards light), or the electroretinogram (ERG), which measures the amplitude of phototransduction and efficacy of the synaptic output of photoreceptor neurons. Photoreceptor loss can also be seen directly by histology or, more easily, by optical neutralization, commonly called the pseudopupil preparation. This visualization of the light-gathering organelles of the photoreceptors can be performed on unfixed, intact heads, allowing easy quantification84. The figure shows pseudopupil preparations of a normal retina (a) and a degenerate retina (b). The table highlights six genes associated with retinal degeneration that are unlikely to be directly involved in phototransduction. Roles for these genes in maintaining CNS integrity in the fly have not yet been addressed, but are likely, due to their widespread expression or association of orthologs with neurodegenerative disease.

swiss cheese
One of the mutations discovered in the Heisenberg and Bohl screen12 was named swiss cheese (sws), after the holes discovered in sections of mutant brains16 (see BOX 2 for a discussion of techniques). Characterization of sws in 1997 revealed what was then a novel protein16, but one year later the human ortholog was determined to be neuropathy target esterase17 (NTE, or PNPLA6). In mammalian cells, the phospholipase activity of NTE breaks down the membrane lipid phosphotidylcholine to glycerophosphocholine18; sws mutant flies have excess phosphatidylcholine19, which may affect membrane properties in a deleterious manner. SWS and NTE also share a conserved domain that acts as a noncanonical regulatory subunit of cAMP-dependent protein kinase (PKA)20. Inactive PKA is a tetramer of two regulatory subunits and two catalytic subunits; when the regulatory subunits bind cAMP, they release the catalytic subunits, which then become active. An N-terminal transmembrane domain anchors SWS/NTE to the cytoplasmic face of endoplasmic reticulum membranes18 and thus may sequester PKA catalytic subunits there. The PKA regulatory activity of SWS (discussed further in a later section) has at least a partial role in neurodegeneration, as exogenously expressed SWS with a mutation in the binding domain cannot fully rescue the sws mutant defect20. Presumably, the partial rescue observed with this transgene is mediated through the intact phospolipase activity.
Techniques used to study neurodegeneration in Drosophila
Histology
The most common approach used to asses neurodegeneration in the fly is examination of sections of the brain, usually embedded in paraffin or plastic, typically treated with a non-specific stain or contrast such as toluidine blue or autofluorescence (see the Flybrain website). The most striking lesions in these histological sections are the vacuoles or holes in the central neuropil or outer rind where most neuron cell bodies reside, as shown in the figure in a 20-day old swiss cheese mutant brain (b), compared to an age-matched normal brain (a). Arrows indicate dying glial cells. An advantage of this method is that it is a direct assay of CNS defect; a disadvantage is its low resolution as commonly practiced, as one cannot determine which neurons are dying.
Electron Microscopy (EM)
A specialized form of histology, EM affords unparalleled resolution of sub-cellular structures and is the definitive technique for identifying autophagic defects. A disadvantage is that it is labor-intensive, and antibody labeling can be difficult.
Electrophysiology
Many preparations are commonly used for electrophysiological recordings of neural activity. The giant fiber system – illustrated in the figure, panel c - mediates the fly’s escape response. With a mix of electrical, cholinergic and glutaminergic synapses, this prep is particularly useful for testing CNS function in the adult92. The giant fiber neuron (blue) is located in the brain and, in the thoracic ganglion, synapses with a motor neuron (green) that directs the jump muscle, and with an interneuron (black) which in turn synapses with five motor neurons (one only shown for clarity) that direct a set of flight muscles. Neurons and muscles are bilaterally symmetrical and are shown on one side only for simplicity. Recordings can be made from the muscles, or intracellularly from the giant fiber axon or from the motor neurons. An advantage of this technique is that it provides a direct assay of neuronal dysfunction; a disadvantage is that is labor intensive.
Behavior
Well-characterized behaviors in the fly range from mating to aggression. Climbing (sometimes called “negative geotaxis”) is the most commonly assayed behavior with respect to fly neurodegeneration for three reasons: as a test of mobility, it may reflect the ataxia that is common in human neurodegenerative diseases; large numbers of flies can be tested relatively quickly; and the behavior is robust (for most strains, ~90% of flies will immediately start climbing the walls of a container after they have been tapped to the bottom). An advantage of this techniques is that it correlates with human disease; disadvantages are that a loss of climbing can be due to factors other than neurodegeneration, and the behavior can vary significantly with genetic background.
Lifespan
Not all flies that die early do so from brain degeneration, but all neurodegenerative mutants have lifespans that are shortened to some degree. Therefore a straightforward first look at a mutant can be obtained with a survival curve, which can be coupled with feeding oxidative stressors such as paraquat. Advantages of this technique are that it is easy to perform and correlates with human disease; disadvantages are that it is time-consuming, there are possible causes other than neurodegeneration, it can vary significantly with genetic background, and it provides little insight into pathology.
See BOX1 for techniques associated specifically with the eye.

Biochemical activities of SWS and NTE appear to be conserved, but what about the role of NTE in maintaining CNS integrity with age? Knockout of Pnpla6/Nte in the mouse is lethal, but if loss of the gene is restricted to the CNS, then the mouse survives to adulthood but suffers from neurodegeneration21. Underscoring functional conservation, mutations in human PNPLA6 cause spastic paraplegia 39, a hereditary motor neuron degenerative disease22. Furthermore, NTE is a target of organophosphates, a class of compounds that includes many pesticides and the neurotoxin sarin. Upon long-term exposure, these compounds bind to the catalytic serine residue of NTE and cause axonal degeneration18. Thus, characterization of the sws mutation in flies led to a series of studies that defined biochemical activities of the SWS/NTE protein and revealed its role in disease and in response to environmental toxins.
The example of swiss cheese demonstrates a key advantage of studies of neurodegeneration in the fly: the unbiased, forward-genetic screen is a tried-and-true, classic method in Drosophila that has led directly to the identification of new neurodegeneration genes in mouse and human.
pink1 and park
At least three inherited forms of parkinsonism are caused by recessive mutations; in two of these, loss of PINK1(PTEN induced putative kinase 1) or PARK2 (Parkinson disease 2, parkin) function causes familial, juvenile-onset parkinsonism23. Several groups have taken a candidate gene approach to studying the roles of these Parkinson’s disease (PD) genes in Drosophila. One defect in pink1 or park mutant flies is abnormal wing posture. Ostensibly unrelated to loss of brain integrity, the wing effect has been revelatory, since it is caused by problems in underlying muscles with a high demand for energy, thus focusing attention on the role of mitochondria in PD.
Loss of either pink1 or park in flies results in remarkably similar effects, including mitochondrial structural defects, muscle degeneration, shortened lifespan, and male infertility in addition to the wing defect24–26. A breakthrough in the understanding of these genes came from a simple genetic experiment in the fly: the forced expression of the normal park gene in a pink1 mutant background. This demonstrated that park gene function can rescue all pink1 mutant defects24–26. This is a classic example of an epistasis experiment —the study of combinations of gene activities, through either recessive loss-of-function or dominant gain-of-function mutations— and demonstrates that PARK functions downstream of PINK1. The result has been confirmed in human cells: expression of PARK2 in cells derived from two patients with different PINK1 mutations can rescue the mitochondrial defect27.
Pink1 is a protein kinase principally localized to mitochondria, and PARK is principally cytoplasmic, suggesting the intriguing possibility that aberrant signaling between the mitochondria and cytoplasm may play a role in disease. Mitochondria are not static structures but are in a constant flux of fusion and fission in response to cellular conditions. Further evidence that PINK1 and PARK work together to regulate mitochondrial structure comes from genetic manipulation of mitochondrial dynamics in flies: pink1 and park mitochondrial defects can be rescued by either upregulating a mitochondrial fission protein (Drp1), or knocking down mitochondrial fusion proteins (OPA1 or MARF/MFN2)28,29. In humans, MFN2 (mitofusin 2) mutations cause the neurodegenerative disease Charcot-Marie-Tooth type 2A, and in the mouse, loss of Mfn2 in the cerebellum causes aberrant mitochondrial structure and function in Purkinje cells, resulting in their degeneration30.
The work with pink1 and park demonstrates a second advantage of neurodegeneration studies in the fly: epistasis experiments are routine in Drosophila and are a powerful approach to defining functional gene order in pathways conserved in the mouse and human.
Processes critical to CNS integrity
The genes in S1 TABLE 1 can be classified according to five cell biological processes, although many of these genes play multiple roles (FIG. 1). While this analysis stems from Drosophila neurodegeneration mutants, an important theme is the commonality of these processes between flies and mammals. The Protein homeostasis process includes genes that function in both the ubiquitin-proteasome system and in autophagy/lysosomal degradation, a topic reviewed elsewhere31,32. Genes classified in the Cytoskeleton process have roles both in the function of microtubules, the principal structural element of long neurites like axons, and in the actin-based cytoskeleton, which is critical for synapse formation and plasticity. Below, we further highlight recent work on aspects of the other three processes.
FIGURE 1. Cellular processes implicated by neurodegeneration genes.

A Venn diagram showing the relationships between five cellular processes and a suggested classification of the neurodegeneration genes from SI TABLE 1, many of which have multiple roles.
Lipid homeostasis
Lipids serve as the key constituent of membranes, as a source of energy, and as signaling molecules. A number of Drosophila neurodegeneration mutants have been linked to pathways of lipid homeostasis. Ethanolamine kinase, encoded by eas33, catalyzes the first step in one pathway for synthesizing phosphatidylethanolamine, a phospholipid that in mammals is enriched in neuronal and mitochondrial membranes34. Exactly how phospholipid composition could affect degeneration is unclear, but one possibility involves membrane properties such as fluidity, which could influence channel or neurotransmitter functions. Furthermore, different organelles can have characteristic phospholipids within their membranes, and so altering phospholipid composition could affect, for example, organelle trafficking35.
Three other genes linked to neurodegeneration are involved in fatty acid catabolism (FIG. 2). fbl (fumble) and PPCS (phosphopantothenoylcysteine synthetase) have recently been characterized and encode enzymes in the pathway for synthesis of coenzyme A, required for the first step in the degradation of fatty acids36. bgm encodes a fatty acid coenzyme A ligase that appears to be specific for very long chain fatty acids. Degeneration in bgm mutants can be partially rescued by feeding the flies a fatty-acid component of “Lorenzo’s Oil,” a putative preventive treatment for adrenoleukodystrophy, perhaps by inhibiting the synthesis of very long chain fatty acids11. Together, fbl, PPCS and bgm could affect neuronal membrane properties or energy availability. If the latter function is critical, then an interaction would be predicted between these genes and those encoding the mitochondrial fatty acyl translocation apparatus (FIG. 2), consisting of a translocase and enzymes which attach and remove fatty acids from the carrier molecule carnitine37.
FIGURE 2. Neurodegeneration proteins associated with the mitochondrion.

Gene products from SI TABLE 1 are highlighted in orange. In black in the same font are proteins not in the table that genetically interact (or are predicted to interact) with the neurodegeneration proteins. At the bottom left, transport of mitochondria into neurites depends on the (+) end-directed motor Kinesin and on microtubules (pink tubule), which are stabilized by FUTSCH. The dare product transfers electrons from NADPH to adrenodoxin, which in turn transfers them to a cytochrome P450, which catalyzes the first step in steroid hormone synthesis. At left, fatty acids are activated by a number of steps before importation (black dashed arrow; see text) into the matrix for oxidation. Together with the citric acid cycle, these pathways generate the electron carriers NADH and FADH2, which in turn power the complexes of the electron transport chain (blue, top). Proton flux across the inner membrane is indicated by dashed arrows. Incoming protons drive synthesis of ATP, which is transported out of the matrix (dashed arrow) by the ATP/ADP translocator (green). Low ATP generation results in activation of AMP-activated protein kinase (AMPK, orange). At the bottom right, PINK1 regulates TRAP1 and the localization of PARK (see text); pink1 and park interact with genes that regulate mitochondrial fission and fusion, consistent with these gene products acting downstream of PARK, as shown here, although there is no direct evidence for this yet. Pink1 also regulates another parkinsonism protein, HTRA2, and a Na+/Ca2+ antiporter activity (see text). High Ca2+ levels within the matrix will trigger formation of the permeability transition pore (PTP), through which cytochrome c can be released, activating caspases and apoptosis. (Not shown are apoptosis inducing factor and members of the BCL2 family, proteins that also regulate apoptosis and translocate between the cytoplasm and the outer surface of mitochondria. See ref. 72)
Cholesterol is an essential constituent of animal membranes and has been linked to Alzheimer’s disease (AD). Deposition of amyloid-β (Aβ) peptide into amyloid plaques, the hallmark of AD, is enhanced by high cholesterol through unknown mechanisms, perhaps changes in membrane properties38 (APP, the Aβ precursor, is a transmembrane protein). In the fly, SNF4Aγ mutants have a 40% reduction in cholesteryl ester levels, which may increase free cholesterol, and loss of appl, the fly version of APP, enhances degeneration in SNF4Aγ mutants39. SNF4Aγ encodes a subunit of AMP-activated protein kinase (AMPK), discussed further below. Although flies do not synthesize cholesterol de novo, they do have HMG CoA reductase, the enzyme which catalyzes the rate-limiting step in cholesterol synthesis in vertebrates and which is directly phosphorylated and inhibited by AMPK40. Loss of one copy of the gene for HMG CoA reductase (Hmgcr) partially rescues the degeneration in SNF4Aγ mutant flies, and overexpression of Hmgcr enhances degeneration39. Statins, a class of compounds that target Hmgcr and are used to treat high blood cholesterol levels, appear to reduce the risk of AD38, and feeding a statin to SNF4Aγ mutant flies decreases degeneration39. Cholesterol is a versatile lipid: it is the precursor of hormones (see below), an adduct of signaling proteins41, and a regulator of membrane fluidity with a key role in synapse function42. The relative importance of each of these roles in neurodegeneration remains to be determined.
In flies, dare activity is required in mitochondria for the synthesis of ecdysteroids from cholesterol (FIG. 2). Feeding larvae ecdysone rescues an early developmental defect caused by null dare mutations43, and since neuronal expression of dare can rescue adult neurodegeneration44, there may be a role for ecdysteroid synthesis in CNS maintenance. The mammalian gene NPC1/Npc1 presents an interesting parallel to the dare story. Niemann-Pick disease can be caused by loss of NPC1 and is characterized by misregulated cholesterol and sterol trafficking. Mice lacking Npc1 have many of the same neurodegenerative symptoms as Niemann-Pick patients, as well as deficits in the neuron-specific synthesis of steroid hormones45. Strikingly, these effects can be rescued by feeding the mouse a brain-specific steroid45. The dare and Npc results raise the intriguing possibility that steroid hormone signaling in the adult brain plays a role in maintaining morphological integrity.
Signal transduction
A number of the genes classified in Figure 1 implicate known signal transduction pathways in neurodegeneration, but at present few molecular details are known on how dysfunction of these pathways causes degeneration. alc and SNF4Aγ, encoding the β and γ subunits of AMPK, respectively, are the only genes that can be classified into three of the processes depicted in FIG. 1, thus appearing to be at a nexus of cellular pathways involved in neurodegeneration39,46. AMPK is a crucial control point for metabolism and is composed of the α catalytic subunit, the β subunit (which appears to be a scaffold), and the γ regulatory subunit (which binds AMP)40. Rising AMP levels indicates an energy deficit, which activates the kinase40 (see FIG. 2). The sws mutation suggests a role for protein kinase A in maintaining CNS integrity (discussed above); PKA functions in a well-characterized pathway that is required during development and for learning and memory47, and that plays a role in axon regeneration48. Both AMPK and PKA signaling are essential for the development and function of many cell types, and so uncovering the details of how these specific pathways affect brain integrity will require the use of tools for investigating their roles specifically in mature neurons.
Recent mammalian cell culture experiments that have grown out of the pink1-park epistasis experiments have shed further light on this mitochondrial pathway. While PARK is typically cytoplasmic, coexpression of PINK1 causes translocation of PARK to mitochondria; translocation depends on PINK1 kinase activity49. Further experiments have suggested that Pink1 may directly phosphorylate PARK49. In mammalian cells, PARK can mediate the autophagic engulfment of dysfunctional mitochondria50. How this is done is unclear, although PARK has alternative ubiquitin ligase activities51 that are associated not with proteasomal degradation but rather with other processes such as protein trafficking52. A likely direct substrate of PINK1 was recently shown to be the serine protease HTRA253, which is released from mitochondria during apoptosis and has proapoptotic activity54. However, loss-of-function mutations of HTRA2 can cause PD, and knockout of Htra2 in mice causes parkinsonism-like defects54. Pro-apoptotic activity for Drosophila HtrA2 has been shown recently55,56, but a role for the fly gene in neurodegeneration is unknown at this point, and the mechanism of HTRA2’s role in CNS integrity remains unclear.
Many of the neurodegeneration-associated signal transduction genes play a direct role in neuronal activity, either by maintaining membrane excitability or by mediating signals across the synapse. For example, CSP is a synaptic vesicle protein that is required for proper neurotransmitter release57. Since its initial characterization in flies58, loss of CSP has been shown to cause neurodegeneration in the mouse59 and, surprisingly, this can be rescued by upregulation of α-synuclein60 (dominant mutations of which, including increased copy number of the normal gene, are a cause of PD61). These studies reveal a potential overlap in biological function between CSP and α-synuclein at the synapse, and the susceptibility of neurons to toxicity from abnormal synaptic function.
Ace encodes acetylcholinesterase, which degrades the neurotransmitter acetylcholine; work on Ace in the fly 30 years ago thus implicated excitotoxicity as a mechanism for neurodegeneration62,63. Similar to acetylcholinesterase, the transporter encoded by EAAT1 buffers an excitatory neurotransmitter, in this case glutamate64. Further, excessive neuronal firing is observed in neurodegenerative, dominant mutations of ATP α 65, which encodes the α subunit of the Na+/ K+ ATPase. Collectively these studies highlight the role of excitotoxicity in neural integrity. In contrast, other electrophysiological studies demonstrate decreased transmitter release and reduced phototransduction and synaptic transmission in spin mutants66,67. Perhaps CNS integrity in the adult requires that neurons be maintained in a “Goldilocks state” of neither abnormally high nor abnormally low levels of activity, a phenomenon parallel to vertebrate neural development, where excess neurons are trimmed dependent on their level of activity.
Mitochondrial function
Mitochondria are central to the processes affecting neurodegeneration (FIG. 1): the set of genes affecting mitochondria overlaps each of the other four sets described. FIG. 2 illustrates the functions of gene products from TABLE 1 that are associated with mitochondria. The synthesis of the cellular energy currency ATP is the principle purpose of mitochondria and is directly implicated in neuronal integrity by mutants in levy and ATP6, which encode components of the electron transport chain, and by the sesB product, which transports newly-synthesized ATP to the cytoplasm. ATP6 is the only gene implicated so far in Drosophila neurodegeneration that resides in the mitochondrial genome. The ATP6 mutation arose spontaneously in a sesB mutant background and has a mild effect on neuronal integrity by itself68. Interestingly, patients with the human disease associated with the sesB ortholog (progressive external opthalmoplegia 2) also have multiple, varying mitochondrial DNA deletions, raising the possibility that nuclear gene mutations that cause mitochondrial dysfunction also leave mitochondrial DNA in a state more vulnerable to lesions69 (see also OMIM record #609283). Reactive oxygen species such as superoxide (O2−) are toxic products of mitochondrial respiration, are associated with DNA damage, and have long been suspected as a cause of aging and neurodegeneration70. For example, the Sod product superoxide dismutase scavenges O2− (BOX 1). TRAP1, a mitochondrial chaperone that is phosphorylated in response to oxidative stress, is a recently described in vivo substrate of PINK1 in human cells71. PINK1 can protect these cells against oxidative stress, an activity that depends on TRAP171.
Mitochondria are not simply passive passengers in eukaryotic cells. They regulate their morphology in response to specific cellular needs and their own level of functionality, as discussed above with respect to PINK1 and PARK. Further, mitochondria integrate apoptotic signals and facilitate apoptotic cell death. A key event to initiate apoptosis is the opening of the permeability transition pore72; this allows release into the cytoplasm of cytochrome c, Apoptosis inducing factor, and perhaps HTRA2 (see above). The pore is opened upon increased Ca2+ concentration within the mitochondrial matrix. Recently, a Na+/Ca2+ antiporter activity (which has not yet been molecularly characterized in mammals or in flies) was shown to be blocked in cultured mammalian Pink1 mutant neurons, resulting in increased mitochondrial Ca2+ levels73. Thus, misregulated apoptosis may be a contributing cause of neuronal death in pink1-dependent parkinsonism. Indeed, the proximal cause of neuronal death for many of the mutants in TABLE 1 is apoptosis, although in many cases apoptosis may be triggered simply in response to an underlying pathology.
Glia and cellular interactions
The discussion above is largely focused on cell-autonomous ways that neurons can die, that is, from intrinsic problems such as dysfunctional mitochondria. But neurodegeneration can occur through aberrant cell-cell signaling as well, implicated by genes that play a role in synaptic transmission and that suggest a role for neuronal steroid hormones. Glial interactions represent a clear case of cells that are necessary for the proper maintenance of neighboring neurons. Below we consider fly genes that have implicated glia in neurodegeneration and how genetically mosaic flies can uncover roles for cell-cell signaling in neuronal maintenance.
Glia perform essential functions in vertebrates, such as modulation of synapses, formation of the blood-brain barrier, basic immune system duties, and protection of long axonal tracts. All of these roles have clear parallels in the fly74. For example, while flies do not have a vascular circulatory system, glia form a blood-brain barrier analog by sealing off neurons from the surrounding hemolymph. Flies do not have an adaptive immune system, but fly glia perform some of the immune tasks of vertebrate microglia, such as engulfing the corpses of dead neurons. Finally, although flies do not have myelin, they do have a glial cell type that ensheaths axons, comparable to Schwann cells which myelinate long axonal tracts in mammals. Thus, glia are essential for proper neuronal function in flies, and indeed four genes in TABLE 1 implicate glial function in the maintenance of the brain.
The expression of two of these genes— repo and EAAT1 —is restricted to glia. The Repo protein is a transcriptional regulator that is required for glial development; null alleles are lethal, reflecting the essential role for glia in the animal. A partial loss-of-function allele of repo reveals a role of the gene in maintaining the adult CNS75, although the specific defects in these mutant glia that cause neuronal loss are unclear. EAAT1, a transporter mentioned above, likely scavenges excess glutamate, a neurotransmitter. The transporter is enriched in glial membranes at synaptic clefts, and thus this gene implicates Drosophila glia in modulating synaptic activity64.
Flies mutant for drop-dead (drd) appear to be normal for about a week, then suddenly become uncoordinated, dying within hours with holes throughout the brain. Glia in drd mutants have structural defects, and there are subtle glial abnormalities in young adult mutants, apparent before the onset of behavioral defects76. The location of drd function was addressed in the early 70s in genetically mosaic flies that are partly mutant for drd and partly normal8. Strikingly, most flies with heads that were half-mutant and half-normal behaved like normal flies and had normal brain morphology. That is, in these animals brain histology was normal on the side of the head that was genotypically mutant. Therefore the function of drd mutant tissue can be rescued by adjacent normal tissue, implying the existence of drd-dependent diffusible signals.
Young adult sws mutants also have an early glial defect: multiple glial membranes wrap adjacent neurites which, normally, are wrapped by only a single layer16. Eventually glia die along with neurons19. In contrast to the non-autonomy of drd mosaics, however, degeneration in mosaic sws flies corresponds to genotypically mutant parts of the brain16, ruling out a longrange, sws-dependent signal. The method used to construct the mosaic flies was the same used to analyze drd, producing broad swatches of mutant brain that can shed little light on a possible requirement for sws activity in glia, which are intermingled with neurons. Other methods of making mosaic flies, such as looking at sws mutant glia situated adjacent to normal neurons, could address questions such as sws-dependent glial-neuronal interactions. The reciprocal experiment – determining the degree of rescue in a mutant animal upon targeted expression of the wild type gene – is also informative. For example, neuronal expression of normal npc1a can rescue npc1a mutant adults, and interestingly, glial expression of the normal gene also rescues, although not as efficiently77. In summary, as in higher organisms, glial function is critical for neuronal integrity in the fly, and the repertoire of tools for fly genetics should allow for both glial function and interneuronal signaling to be addressed systematically.
Conclusions
The genes discussed in this review share a number of themes. First, most are widely expressed: either ubiquitously or enriched throughout the CNS (interesting exceptions being EAAT1 and repo, which are restricted to glial cells). Second, behavioral defects are common in flies mutant for these genes. Third, all these Drosophila neurodegeneration mutants have shortened lifespans, which correspond with the severity and timecourse of degeneration (see below). These aspects of fly neurodegeneration mutants mirror observations of neurodegenerative diseases in humans: widespread expression of causative genes, neurological symptoms such as ataxia or tremor, and early death.
A variation among the genes is the spatial extent of degeneration that occurs in the brains of mutant flies. In many cases, lesions are seen throughout the brain and even in the thoracic ganglion (the large ganglion that is a thickening of the ventral nerve cord). In other mutants, lesions are seen only in specific subregions of the brain, typically the optic lobes. The timecourse of degeneration can also vary widely among the mutants: from hours in the case of dare43 to about a month in the case of Adar78. Moreover, whereas adult dare mutant flies die within a few days, Adar mutants can live nearly as long as wild-type flies. Such variation is probably due only partly to the absolute requirement for a particular gene in maintaining CNS integrity. Other factors include possible earlier roles in development and the strength of the mutant allele.
Undiscovered neurodegeneration genes
Identifying new genes in the fly with a role in maintaining adult CNS integrity is perhaps the fastest way to identify them in humans, given the ease of genetic screens for brain integrity mutants and range of genetic tools available for assessing gene function. There are almost certainly many as-yet undiscovered genes that are required for maintaining the CNS Drosophila screens specifically designed to identify neurodegeneration genes have not been any more successful than other methods in flies: 14 genes in SI TABLE 1 were identified in such screens; 16 were candidate genes; and 14 were fortuitous mutations. Probably due to their labor-intensive nature, none of the previous screens achieved saturation, since most of the relevant genes are represented by single alleles. Genes with a narrow role in maintaining a specific neuronal subtype may be underrepresented due to technical limitations (see below); most surveyed here cause broad neurodegeneration when mutated. Genes essential for development are also underrepresented: all those discussed here that are characterized by complete loss-of-function mutations are viable. This limitation could be overcome by the study of gene activity in differentiated adult neurons using transgenic RNAi constructs, and if necessary, methods to control expression of the RNAi construct temporally79. Developmentally essential genes can also be analyzed in genetically mosaic flies.
Fly genes and human neurodegeneration
In a number of cases the mouse or human ortholog of a fly gene in SI TABLE 1 is directly associated with neurodegeneration. There are three other possible relationships between a fly gene and mammalian brain integrity. The mouse or human ortholog may be closely related to another disease-associated gene through gene duplication (for example, futsch in TABLE 1). Second, a fly gene may encode a protein that is part of a complex, a separate subunit of which has been shown to cause neurodegeneration in mouse or human (for example, ATPα ). Finally, a fly gene may function in a pathway in which another gene is linked to neurodegeneration. For example, fly levy encodes a component of the COX complex, whereas COX10 and LRPPRC are required for proper assembly and expression of this complex, but the mutations are functionally equivalent. Taken together, 52% of the Drosophila genes in SI TABLE 1 are currently linked to mouse or human genes in pathways associated with neurodegeneration. There remain fly genes in SI TABLE 1 for which, at present, no mammalian genes related by sequence or function are associated with neurodegeneration. Such genes may have functions in maintaining adult brain integrity in mammals that are still awaiting discovery.
Open questions
As discussed above, there are probably many more genes associated with neurodegeneration that are not yet identified, and it is likely that other processes will be added to the five highlighted in this Review. Further, there are a number of issues that have been yet to be systematically addressed for fly neurodegeneration genes. For example, how are the neurons dying? Apoptosis is involved in many but not all of the degeneration mutants. How widespread is necrotic cell loss? Do neurons die due to defective intrinsic processes or from interactions with sick neighboring cells? For widely expressed genes, tools that enable one to look at requirements for gene activity in glia or in specific neurons are available and might answer this latter question: RNAi can be restricted to only glial cells, or to only small subsets of neurons80. Further, genetically mosaic flies can be analyzed with marked mutant clones as small as a single cell81. What types of neurons are dying? For the most part this question has been addressed only for PD candidate genes. Cell type-specific or brain structure-specific markers combined with confocal/3-D reconstruction are likely to become more prevalent in the study of the brain degeneration; commonly used histological techniques at present (BOX 2) cannot differentiate between affected neuronal types. Finally, in humans, age is the single largest risk factor for diseases such as PD and AD. A challenge for the future will be to use the advantages of the fly to tease apart the role of aging in neurodegeneration.
Although an excellent system for investigating neurodegenerative disease, flies are not humans. Lacking an adaptive immune system, for example, Drosophila may not be well-suited for addressing the role of inflammation in neurodegeneration, although flies will allow detailed study of the role of innate immunity. However, striking similarities between fly and human nervous systems mean that studies in flies and mammals complement each other well. Moreover, identification of genes and pathways critical for brain maintenance is relatively straightforward in the fly and serves as a springboard for further investigation in mice and humans. Manipulation of such pathways in the fly enhances approaches to human diseases associated with loss of brain integrity and cognitive function.
Links
Flybase: http://flybase.org/
Flybrain: http://flybrain.neurobio.arizona.edu/
Homophila: http://superfly.ucsd.edu/homophila/
OMIM: http://www.ncbi.nlm.nih.gov/sites/entrez?db=omim&TabCmd=Limits
Mouse Genome Informatics: http://www.informatics.jax.org/
Bonini Lab: http://bonini.bio.upenn.edu/index.html
ONLINE SUMMARY
More than 40 Drosophila melanogaster genes have been discovered for which recessive, loss-of-function mutations cause adult onset degeneration of the central nervous system (CNS). A table presenting these genes is provided, and an expanded, updated version can be found at the Bonini Laboratory homepage.
Almost all of these genes have easily identifiable orthologues in the mouse and human. Over half have mouse or human orthologues that are also associated with neurodegeneration.
The swiss cheese (sws) gene demonstrates the value of unbiased screens in the fly. Since its discovery, two biochemical functions have been characterized for the protein; loss of the mouse orthologue in the brain has been shown to cause neurodegeneration, and loss-of-function mutations in the human orthologue have been discovered as the cause of spastic paraplegia 39.
Pink1 and park are associated with Parkinson’s disease, and they demonstrate the value of epistasis experiments in the fly, which have shown that these two genes function together in a pathway that regulates mitochondrial fusion and fission.
Fly neurodegeneration genes can be grouped into the following cellular processes: mitochondrial function, signal transduction, lipid homeostasis, protein homeostasis and the cytoskeleton. Many of the genes have roles in more than one of these processes.
Some glial-specific genes have been shown to be required for maintaining neurons in the adult. Mutations in other, more widely expressed genes have defective glia, underscoring the importance of glia in CNS integrity.
Many genetic tricks are possible in the fly, such as: the precise control in space and time of the expression of transgenes, including through RNAi constructs; and the possibility of making marked homozygous mutant clones as small as a single neuron in otherwise heterozygous animals. These techniques, and the ease of forward genetics screens for identifying new neurodegeneration mutants, ensure that D. melanogaster will remain a key tool for the analysis of genes required for CNS integrity.
Supplementary Material
ACKNOWLEDGMENTS
We thank M. Bland, N. Liu, Z. Yu, L.-Y. Hao, and C. J. Thut for comments on the manuscript. N.M.B. receives funding from the National Institute of Aging and the National Institute of Neurological Disorders and Stroke, and she is an Investigator of the Howard Hughes Medical Institute.
GLOSSARY TERMS
- RNAi
RNA interference in the adult fly is achieved with transgenic constructs expressing an inverted repeat sequence targeted to the mRNA of interest. The expressed double-stranded RNA is processed in vivo into short interfering RNAs (siRNAs), which lead to degradation of the target gene transcripts for a loss of function mutant effect.
- Mosaic
Animal comprised of tissue of different genotypes. In flies, mosaics are generated by site-specific recombination, to yield homozygous mutant tissue or cells in an otherwise heterozygous animal.
- Autophagy
More precisely, macroautophagy–the engulfment of protein aggregates or organelles by vesicles with double-bilayer membranes, which then fuse with lysosomes for degradation of their contents.
- Ubiquitin-proteasome system
Members of a large family of E3 ubiquitin ligases recognize specific substrate proteins, tagging them by polyubiquitination for degradation in the proteasome, a large cylindrical protein complex.
- Glia
Support cells for neurons.
- Phototaxis
Movement towards a light source. A behavior often used in flies to test locomotor activity and eye function.
- Parkinsonism
Symptoms characteristic of Parkinson’s Disease (tremor, rigidity, slowing of movement, postural instability, shuffling gait) that respond to treatment with dopamine.
- Purkinje cells
Vertebrate neurons with huge, dense dendrites that integrate complex inputs in the cerebellum and project axons to the deep motor nuclei of the brain.
- Neurites
General term for axons and dendrites.
- Amyloid
Protein aggregates that accumulate as fibers of 7–10nm in diameter with common structural features including β-pleated sheet conformation and resistance to detergents and proteases.
- Ecdysone
Steroid hormones found in arthropods. In insects, 20-hydroxyecdysone stimulates moulting and metamorphosis.
- Ecdysteroids
Steroids that are similar in structure to ecdysones, found in arthropods and some plants.
- Excitotoxicity
The over-stimulation of excitatory neurotransmitter receptors, which causes an influx of calcium in the postsynaptic neuron.
- Hemolymph
The interstitial fluid in insects, which have an open circulatory system. Unlike blood, hemolymph plays a very small role in carrying O2/CO2, which is principally done by the tracheal system.
- Optic lobes
Large, bilaterally symmetric structures of the fly brain that process visual input.
Biographies
Derek Lessing is a senior research scientist working with Nancy M. Bonini. He received his Ph.D. from the Department of Developmental Biology at Stanford University, Palo Alto, California, in the laboratory of Roel Nusse, and did a postdoctoral fellowship with John Carlson at Yale University, New Haven, Connecticut. His research interests are characterizing endogenous fly genes that cause neurodegeneration and that modify polyglutamine disease.
Nancy M. Bonini is the Lucille B. Williams Term Professor of Biology and an Investigator of the Howard Hughes Medical Institute. She received her Ph.D. in Neuroscience from the University of Wisconsin–Madison in the laboratory of David Nelson, and did a postdoctoral fellowship with Seymour Benzer at California Institute of Technology (Caltech), Pasadena. She has been at the University of Pennsylvania, Philadelphia, since 1994, and her interests are genes and mechanisms that modulate neurodegenerative disease and neural trauma.
Footnotes
TABLE OF CONTENTS BLURB
Identifying genes that are essential for maintaining neuronal integrity provides significant insight into the mechanisms underlying neurodegenerative disorders. Recessive mutants in the fly have proven invaluable for finding such genes and for highlighting key biological processes that contribute to neurodegeneration.
References
- 1.Pires-daSilva A, Sommer RJ. The evolution of signalling pathways in animal development. Nat. Rev. Genet. 2003;4:39–49. doi: 10.1038/nrg977. [DOI] [PubMed] [Google Scholar]
- 2.Ben-Shlomo I, Hsu SY, Rauch R, Kowalski HW, Hsueh AJW. Signaling Receptome: A Genomic and Evolutionary Perspective of Plasma Membrane Receptors Involved in Signal Transduction. Science STKE. 2003;2003:re9. doi: 10.1126/stke.2003.187.re9. [DOI] [PubMed] [Google Scholar]
- 3.Hirth F, Reichert H. Conserved genetic programs in insect and mammalian brain development. Bioessays. 1999;21:677–684. doi: 10.1002/(SICI)1521-1878(199908)21:8<677::AID-BIES7>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 4.Sehgal A, et al. Molecular analysis of sleep: wake cycles in Drosophila. Cold Spring Harb. Symp. Quant. Biol. 2007;72:557–564. doi: 10.1101/sqb.2007.72.018. [DOI] [PubMed] [Google Scholar]
- 5.Roman G, Davis RL. Molecular biology and anatomy of Drosophila olfactory associative learning. Bioessays. 2001;23:571–581. doi: 10.1002/bies.1083. [DOI] [PubMed] [Google Scholar]
- 6.Dierick HA. Fly Fighting: Octopamine Modulates Aggression. Curr. Biol. 2008;18:R161–R163. doi: 10.1016/j.cub.2007.12.026. [DOI] [PubMed] [Google Scholar]
- 7.Chien S, Reiter LT, Bier E, Gribskov M. Homophila: human disease gene cognates in Drosophila. Nucleic Acids Res. 2002;30:149–151. doi: 10.1093/nar/30.1.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hotta Y, Benzer S. Mapping of behaviour in Drosophila mosaics. Nature. 1972;240:527–535. doi: 10.1038/240527a0. [DOI] [PubMed] [Google Scholar]
- 9.Bonini NM. A Tribute to Seymour Benzer, 1921–2007. Genetics. 2008;180:1265–1273. doi: 10.1534/genetics.104.97782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Min KT, Benzer S. Spongecake and eggroll: two hereditary diseases in Drosophila resemble patterns of human brain degeneration. Curr. Biol. 1997;7:885–888. doi: 10.1016/s0960-9822(06)00378-2. [DOI] [PubMed] [Google Scholar]
- 11.Min KT, Benzer S. Preventing neurodegeneration in the Drosophila mutant bubblegum. Science. 1999;284:1985–1988. doi: 10.1126/science.284.5422.1985. [DOI] [PubMed] [Google Scholar]
- 12.Heisenberg M, Bohl K. Isolation of anatomical brain mutants of Drosophila by histological means. Z. Naturforsch. B. 1979;34:143–147. [Google Scholar]
- 13.Palladino MJ, Hadley TJ, Ganetzky B. Temperature-sensitive paralytic mutants are enriched for those causing neurodegeneration in Drosophila. Genetics. 2002;161:1197–1208. doi: 10.1093/genetics/161.3.1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fergestad T, Bostwick B, Ganetzky B. Metabolic disruption in Drosophila bang-sensitive seizure mutants. Genetics. 2006;173:1357–1364. doi: 10.1534/genetics.106.057463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rezaval C, et al. A functional misexpression screen uncovers a role for enabled in progressive neurodegeneration. PLoS ONE. 2008;3:e3332. doi: 10.1371/journal.pone.0003332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kretzschmar D, Hasan G, Sharma S, Heisenberg M, Benzer S. The swiss cheese mutant causes glial hyperwrapping and brain degeneration in Drosophila. J. Neurosci. 1997;17:7425–7432. doi: 10.1523/JNEUROSCI.17-19-07425.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lush MJ, Li Y, Read DJ, Willis AC, Glynn P. Neuropathy target esterase and a homologous Drosophila neurodegeneration-associated mutant protein contain a novel domain conserved from bacteria to man. Biochem. J. 1998;332:1–4. doi: 10.1042/bj3320001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Glynn P. Neuropathy target esterase and phospholipid deacylation. Biochim. Biophys. Acta. 2005;1736:87–93. doi: 10.1016/j.bbalip.2005.08.002. [DOI] [PubMed] [Google Scholar]
- 19.Muhlig-Versen M, et al. Loss of Swiss cheese/neuropathy target esterase activity causes disruption of phosphatidylcholine homeostasis and neuronal and glial death in adult Drosophila. J. Neurosci. 2005;25:2865–2873. doi: 10.1523/JNEUROSCI.5097-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bettencourt da Cruz A, Wentzell J, Kretzschmar D. Swiss Cheese, a Protein Involved in Progressive Neurodegeneration, Acts as a Noncanonical Regulatory Subunit for PKA-C3. J. Neurosci. 2008;28:10885–10892. doi: 10.1523/JNEUROSCI.3015-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Akassoglou K, et al. Brain-specific deletion of neuropathy target esterase/swisscheese results in neurodegeneration. Proc. Natl. Acad. Sci. U.S.A. 2004;101:5075–5080. doi: 10.1073/pnas.0401030101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rainier S, et al. Neuropathy target esterase gene mutations cause motor neuron disease. Am. J. Hum. Genet. 2008;82:780–785. doi: 10.1016/j.ajhg.2007.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Thomas B, Beal MF. Parkinson's disease. Hum. Mol. Genet. 2007;16:R183–R194. doi: 10.1093/hmg/ddm159. [DOI] [PubMed] [Google Scholar]
- 24.Clark IE, et al. Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature. 2006;441:1162–1166. doi: 10.1038/nature04779. [DOI] [PubMed] [Google Scholar]
- 25.Park J, et al. Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by parkin. Nature. 2006;441:1157–1161. doi: 10.1038/nature04788. [DOI] [PubMed] [Google Scholar]
- 26.Yang Y, et al. Mitochondrial pathology and muscle and dopaminergic neuron degeneration caused by inactivation of Drosophila Pink1 is rescued by Parkin. Proc. Natl. Acad. Sci. U.S.A. 2006;103:10793–10798. doi: 10.1073/pnas.0602493103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Exner N, et al. Loss-of-function of human PINK1 results in mitochondrial pathology and can be rescued by parkin. J. Neurosci. 2007;27:12413–12418. doi: 10.1523/JNEUROSCI.0719-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Poole AC, et al. The PINK1/Parkin pathway regulates mitochondrial morphology. Proc. Natl. Acad. Sci. U.S.A. 2008;105:1638–1643. doi: 10.1073/pnas.0709336105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Deng H, Dodson MW, Huang H, Guo M. The Parkinson's disease genes pink1 and parkin promote mitochondrial fission and/or inhibit fusion in Drosophila. Proc. Natl. Acad. Sci. U.S.A. 2008;105:14503–14508. doi: 10.1073/pnas.0803998105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen H, McCaffery JM, Chan DC. Mitochondrial Fusion Protects against Neurodegeneration in the Cerebellum. Cell. 2007;130:548–562. doi: 10.1016/j.cell.2007.06.026. [DOI] [PubMed] [Google Scholar]
- 31.Opal P, Zoghbi HY. The role of chaperones in polyglutamine disease. Trends Mol. Med. 2002;8:232–236. doi: 10.1016/s1471-4914(02)02310-9. [DOI] [PubMed] [Google Scholar]
- 32.Levine B, Kroemer G. Autophagy in the Pathogenesis of Disease. Cell. 2008;132:27–42. doi: 10.1016/j.cell.2007.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fergestad T, et al. Neuropathology in Drosophila Mutants With Increased Seizure Susceptibility. Genetics. 2008;178:947–956. doi: 10.1534/genetics.107.082115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vance JE. Phosphatidylserine and phosphatidylethanolamine in mammalian cells: two metabolically related aminophospholipids. J. Lipid Res. 2008;49:1377–1387. doi: 10.1194/jlr.R700020-JLR200. [DOI] [PubMed] [Google Scholar]
- 35.Di Paolo G, De Camilli P. Phosphoinositides in cell regulation and membrane dynamics. Nature. 2006;443:651–657. doi: 10.1038/nature05185. [DOI] [PubMed] [Google Scholar]
- 36.Bosveld F, et al. De novo CoA biosynthesis is required to maintain DNA integrity during development of the Drosophila nervous system. Hum. Mol. Genet. 2008;17:2058–2069. doi: 10.1093/hmg/ddn105. [DOI] [PubMed] [Google Scholar]
- 37.Rubio-Gozalbo ME, Bakker JA, Waterham HR, Wanders RJ. Carnitine-acylcarnitine translocase deficiency, clinical, biochemical and genetic aspects. Molecular Aspects of Medicine. 2004;25:521–532. doi: 10.1016/j.mam.2004.06.007. [DOI] [PubMed] [Google Scholar]
- 38.Shobab LA, Hsiung G-YR, Feldman HH. Cholesterol in Alzheimer's Disease. Lancet Neurol. 2005;4:841–852. doi: 10.1016/S1474-4422(05)70248-9. [DOI] [PubMed] [Google Scholar]
- 39.Tschape JA, et al. The neurodegeneration mutant lochrig interferes with cholesterol homeostasis and Appl processing. EMBO J. 2002;21:6367–6376. doi: 10.1093/emboj/cdf636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Towler MC, Hardie DG. AMP-activated protein kinase in metabolic control and insulin signaling. Circ. Res. 2007;100:328–341. doi: 10.1161/01.RES.0000256090.42690.05. [DOI] [PubMed] [Google Scholar]
- 41.Breitling R. Greased hedgehogs: new links between hedgehog signaling and cholesterol metabolism. Bioessays. 2007;29:1085–1094. doi: 10.1002/bies.20663. [DOI] [PubMed] [Google Scholar]
- 42.Pfrieger FW. Cholesterol homeostasis and function in neurons of the central nervous system. Cell. Mol. Life Sci. 2003;60:1158–1171. doi: 10.1007/s00018-003-3018-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Freeman MR, Dobritsa A, Gaines P, Segraves WA, Carlson JR. The dare gene: steroid hormone production, olfactory behavior, and neural degeneration in Drosophila. Development. 1999;126:4591–4602. doi: 10.1242/dev.126.20.4591. [DOI] [PubMed] [Google Scholar]
- 44.Dobritsa AA. Ph.D. Thesis. Dept. of Molecular, Cellular, and Developmental Biology, Yale University; 2003. Molecular genetics of odor reception and development in Drosophila. [Google Scholar]
- 45.Griffin LD, Gong W, Verot L, Mellon SH. Niemann-Pick type C disease involves disrupted neurosteroidogenesis and responds to allopregnanolone. Nat. Med. 2004;10:704–711. doi: 10.1038/nm1073. [DOI] [PubMed] [Google Scholar]
- 46.Spasic MR, Callaerts P, Norga KK. Drosophila alicorn is a neuronal maintenance factor protecting against activity-induced retinal degeneration. J. Neurosci. 2008;28:6419–6429. doi: 10.1523/JNEUROSCI.1646-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tasken K, Aandahl EM. Localized Effects of cAMP Mediated by Distinct Routes of Protein Kinase A. Physiol. Rev. 2004;84:137–167. doi: 10.1152/physrev.00021.2003. [DOI] [PubMed] [Google Scholar]
- 48.Teng FY, Tang BL. Axonal regeneration in adult CNS neurons--signaling molecules and pathways. J. Neurochem. 2006;96:1501–1508. doi: 10.1111/j.1471-4159.2006.03663.x. [DOI] [PubMed] [Google Scholar]
- 49.Kim Y, et al. PINK1 controls mitochondrial localization of Parkin through direct phosphorylation. Biochem. Biophys. Res. Commun. 2008;377:975–980. doi: 10.1016/j.bbrc.2008.10.104. [DOI] [PubMed] [Google Scholar]
- 50.Narendra D, Tanaka A, Suen D-F, Youle RJ. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J. Cell Biol. 2008;183:795–803. doi: 10.1083/jcb.200809125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Moore DJ. Parkin: a multifaceted ubiquitin ligase. Biochem. Soc. Trans. 2006;34:749–753. doi: 10.1042/BST0340749. [DOI] [PubMed] [Google Scholar]
- 52.Mukhopadhyay D, Riezman H. Proteasome-Independent Functions of Ubiquitin in Endocytosis and Signaling. Science. 2007;315:201–205. doi: 10.1126/science.1127085. [DOI] [PubMed] [Google Scholar]
- 53.Plun-Favreau H, et al. The mitochondrial protease HtrA2 is regulated by Parkinson's disease-associated kinase PINK1. Nat. Cell Biol. 2007;9:1243–1252. doi: 10.1038/ncb1644. [DOI] [PubMed] [Google Scholar]
- 54.Vande Walle L, Lamkanfi M, Vandenabeele P. The mitochondrial serine protease HtrA2/Omi: an overview. Cell Death Differ. 2008;15:453–460. doi: 10.1038/sj.cdd.4402291. [DOI] [PubMed] [Google Scholar]
- 55.Challa M, et al. Drosophila Omi, a mitochondrial-localized IAP antagonist and proapoptotic serine protease. EMBO J. 2007;26:3144–3156. doi: 10.1038/sj.emboj.7601745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Igaki T, et al. Evolution of mitochondrial cell death pathway: Proapoptotic role of HtrA2/Omi in Drosophila. Biochem. Biophys. Res. Commun. 2007;356:993–997. doi: 10.1016/j.bbrc.2007.03.079. [DOI] [PubMed] [Google Scholar]
- 57.Umbach JA, et al. Presynaptic dysfunction in Drosophila csp mutants. Neuron. 1994;13:899–907. doi: 10.1016/0896-6273(94)90255-0. [DOI] [PubMed] [Google Scholar]
- 58.Zinsmaier KE, Eberle KK, Buchner E, Walter N, Benzer S. Paralysis and early death in cysteine string protein mutants of Drosophila. Science. 1994;263:977–980. doi: 10.1126/science.8310297. [DOI] [PubMed] [Google Scholar]
- 59.Fernandez-Chacon R, et al. The synaptic vesicle protein CSP alpha prevents presynaptic degeneration. Neuron. 2004;42:237–251. doi: 10.1016/s0896-6273(04)00190-4. [DOI] [PubMed] [Google Scholar]
- 60.Chandra S, Gallardo G, Fernandez-Chacon R, Schluter OM, Sudhof TC. a-Synuclein cooperates with CSPa in preventing neurodegeneration. Cell. 2005;123:383–396. doi: 10.1016/j.cell.2005.09.028. [DOI] [PubMed] [Google Scholar]
- 61.Lee VM, Trojanowski JQ. Mechanisms of Parkinson's disease linked to pathological alpha-synuclein: new targets for drug discovery. Neuron. 2006;52:33–38. doi: 10.1016/j.neuron.2006.09.026. [DOI] [PubMed] [Google Scholar]
- 62.Greenspan RJ, Finn JA, Jr, Hall JC. Acetylcholinesterase mutants in Drosophila and their effects on the structure and function of the central nervous system. J. Comp. Neurol. 1980;189:741–774. doi: 10.1002/cne.901890409. [DOI] [PubMed] [Google Scholar]
- 63.Hall JC, Alahiotis SN, Strumpf DA, White K. Behavioral and biochemical defects in temperature-sensitive acetylcholinesterase mutants of Drosophila melanogaster. Genetics. 1980;96:939–965. doi: 10.1093/genetics/96.4.939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rival T, et al. Decreasing glutamate buffering capacity triggers oxidative stress and neuropil degeneration in the Drosophila brain. Curr. Biol. 2004;14:599–605. doi: 10.1016/j.cub.2004.03.039. [DOI] [PubMed] [Google Scholar]
- 65.Palladino MJ, Bower JE, Kreber R, Ganetzky B. Neural dysfunction and neurodegeneration in Drosophila Na+/K+ ATPase alpha subunit mutants. J. Neurosci. 2003;23:1276–1286. doi: 10.1523/JNEUROSCI.23-04-01276.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sweeney ST, Davis GW. Unrestricted synaptic growth in spinster - a late endosomal protein implicated in TGF-beta-mediated synaptic growth regulation. Neuron. 2002;36:403–416. doi: 10.1016/s0896-6273(02)01014-0. [DOI] [PubMed] [Google Scholar]
- 67.Dermaut B, et al. Aberrant lysosomal carbohydrate storage accompanies endocytic defects and neurodegeneration in Drosophila benchwarmer. J. Cell Biol. 2005;170:127–139. doi: 10.1083/jcb.200412001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Celotto AM, et al. Mitochondrial Encephalomyopathy in Drosophila. J. Neurosci. 2006;26:810–820. doi: 10.1523/JNEUROSCI.4162-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kaukonen J, et al. Role of Adenine Nucleotide Translocator 1 in mtDNA Maintenance. Science. 2000;289:782–785. doi: 10.1126/science.289.5480.782. [DOI] [PubMed] [Google Scholar]
- 70.Wallace DC. A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicine. Annu. Rev. Genet. 2005;39:359–407. doi: 10.1146/annurev.genet.39.110304.095751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Pridgeon JW, Olzmann JA, Chin L-S, Li L. PINK1 Protects against Oxidative Stress by Phosphorylating Mitochondrial Chaperone TRAP1. PLoS Biol. 2007;5:e172. doi: 10.1371/journal.pbio.0050172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Mattson MP. Apoptosis in neurodegenerative disorders. Nat. Rev. Mol. Cell Biol. 2000;1:120–129. doi: 10.1038/35040009. [DOI] [PubMed] [Google Scholar]
- 73.Gandhi S, et al. PINK1-Associated Parkinson's Disease Is Caused by Neuronal Vulnerability to Calcium-Induced Cell Death. Mol. Cell. 2009;33:627–638. doi: 10.1016/j.molcel.2009.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Freeman MR, Doherty J. Glial cell biology in Drosophila and vertebrates. Trends Neurosci. 2006;29:82–90. doi: 10.1016/j.tins.2005.12.002. [DOI] [PubMed] [Google Scholar]
- 75.Xiong WC, Montell C. Defective glia induce neuronal apoptosis in the repo visual system of Drosophila. Neuron. 1995;14:581–590. doi: 10.1016/0896-6273(95)90314-3. [DOI] [PubMed] [Google Scholar]
- 76.Buchanan RL, Benzer S. Defective glia in the Drosophila brain degeneration mutant drop-dead. Neuron. 1993;10:839–850. doi: 10.1016/0896-6273(93)90200-b. [DOI] [PubMed] [Google Scholar]
- 77.Phillips SE, Woodruff EA, 3rd, Liang P, Patten M, Broadie K. Neuronal loss of Drosophila NPC1a causes cholesterol aggregation and age-progressive neurodegeneration. J. Neurosci. 2008;28:6569–6582. doi: 10.1523/JNEUROSCI.5529-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Palladino MJ, Keegan LP, O'Connell MA, Reenan RA. A-to-I pre-mRNA editing in Drosophila is primarily involved in adult nervous system function and integrity. Cell. 2000;102:437–449. doi: 10.1016/s0092-8674(00)00049-0. [DOI] [PubMed] [Google Scholar]
- 79.McGuire SE, Mao Z, Davis RL. Spatiotemporal Gene Expression Targeting with the TARGET and Gene-Switch Systems in Drosophila. Science STKE. 2004;2004:p16. doi: 10.1126/stke.2202004pl6. [DOI] [PubMed] [Google Scholar]
- 80.Pfeiffer BD, et al. Tools for neuroanatomy and neurogenetics in Drosophila. Proc. Natl. Acad. Sci. U.S.A. 2008;105:9715–9720. doi: 10.1073/pnas.0803697105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lee T, Luo L. Mosaic analysis with a repressible cell marker for studies of gene function in neuronal morphogenesis. Neuron. 1999;22:451–461. doi: 10.1016/s0896-6273(00)80701-1. [DOI] [PubMed] [Google Scholar]
- 82.Wang T, Montell C. Phototransduction and retinal degeneration in Drosophila. Pflugers Arch. - Eur. J. Physiol. 2007;454:821–847. doi: 10.1007/s00424-007-0251-1. [DOI] [PubMed] [Google Scholar]
- 83.Stowers RS, Schwarz TL. A genetic method for generating Drosophila eyes composed exclusively of mitotic clones of a single genotype. Genetics. 1999;152:1631–1639. doi: 10.1093/genetics/152.4.1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Franceschini N. In: Information Processing in the Visual Systems of Arthropods. Wehner R, editor. Berlin: Springer-Verlag; 1972. pp. 75–82. [Google Scholar]
- 85.Ahmed Y, Hayashi S, Levine A, Wieschaus E. Regulation of armadillo by a Drosophila APC inhibits neuronal apoptosis during retinal development. Cell. 1998;93:1171–1182. doi: 10.1016/s0092-8674(00)81461-0. [DOI] [PubMed] [Google Scholar]
- 86.Zhai RG, et al. Drosophila NMNAT maintains neural integrity independent of its NAD synthesis activity. PLoS Biol. 2006;4:e416. doi: 10.1371/journal.pbio.0040416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.McQuibban GA, Lee JR, Zheng L, Juusola M, Freeman M. Normal mitochondrial dynamics requires rhomboid-7 and affects Drosophila lifespan and neuronal function. Curr. Biol. 2006;16:982–989. doi: 10.1016/j.cub.2006.03.062. [DOI] [PubMed] [Google Scholar]
- 88.Mast JD, Tomalty KM, Vogel H, Clandinin TR. Reactive oxygen species act remotely to cause synapse loss in a Drosophila model of developmental mitochondrial encephalopathy. Development. 2008;135:2669–2679. doi: 10.1242/dev.020644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Phillips JP, et al. Subunit-destabilizing mutations in Drosophila copper/zinc superoxide dismutase: neuropathology and a model of dimer dysequilibrium. Proc. Natl. Acad. Sci. U.S.A. 1995;92:8574–8578. doi: 10.1073/pnas.92.19.8574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Rimkus SA, et al. Mutations in String/CDC25 inhibit cell cycle re-entry and neurodegeneration in a Drosophila model of Ataxia telangiectasia. Genes Dev. 2008;22:1205–1220. doi: 10.1101/gad.1639608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Muraro NI, Moffat KG. Down-regulation of torp4a, encoding the Drosophila homologue of torsinA, results in increased neuronal degeneration. J. Neurobiol. 2006;66:1338–1353. doi: 10.1002/neu.20313. [DOI] [PubMed] [Google Scholar]
- 92.Matthies HJG, Broadie K. Techniques to dissect cellular and subcellular function in the Drosophila nervous system. Methods Cell Biol. 2003;71:195–265. doi: 10.1016/s0091-679x(03)01011-2. [DOI] [PubMed] [Google Scholar]
- 93.Magyar JP, et al. Degeneration of neural cells in the central nervous system of mice deficient in the gene for the adhesion molecule on Glia, the beta 2 subunit of murine Na,K-ATPase. J. Cell Biol. 1994;127:835–845. doi: 10.1083/jcb.127.3.835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Watase K, et al. Motor discoordination and increased susceptibility to cerebellar injury in GLAST mutant mice. Eur. J. Neurosci. 1998;10:976–988. doi: 10.1046/j.1460-9568.1998.00108.x. [DOI] [PubMed] [Google Scholar]
- 95.Aoyama K, et al. Neuronal glutathione deficiency and age-dependent neurodegeneration in the EAAC1 deficient mouse. Nat. Neurosci. 2006;9:119–126. doi: 10.1038/nn1609. [DOI] [PubMed] [Google Scholar]
- 96.Pavlidis P, Ramaswami M, Tanouye MA. The Drosophila easily shocked gene: a mutation in a phospholipids synthetic pathway causes seizure, neuronal failure, and paralysis. Cell. 1994;79:23–33. doi: 10.1016/0092-8674(94)90397-2. [DOI] [PubMed] [Google Scholar]
- 97.Bettencourt da Cruz A, et al. Disruption of the MAP1B-related protein FUTSCH leads to changes in the neuronal cytoskeleton, axonal transport defects, and progressive neurodegeneration in Drosophila. Mol. Biol. Cell. 2005;16:2433–2442. doi: 10.1091/mbc.E04-11-1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Liu W, et al. Mutations in cytochrome c oxidase subunit VIa cause neurodegeneration and motor dysfunction in Drosophila. Genetics. 2007;176:937–946. doi: 10.1534/genetics.107.071688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Greene JC, Whitworth AJ, Andrews LA, Parker TJ, Pallanck LJ. Genetic and genomic studies of Drosophila parkin mutants implicate oxidative stress and innate immune responses in pathogenesis. Hum. Mol. Genet. 2005;14:799–811. doi: 10.1093/hmg/ddi074. [DOI] [PubMed] [Google Scholar]
- 100.Greene JC, et al. Mitochondrial pathology and apoptotic muscle degeneration in Drosophila parkin mutants. Proc. Natl. Acad. Sci. U.S.A. 2003;100:4078–4083. doi: 10.1073/pnas.0737556100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Tschape JA, Bettencourt da Cruz A, Kretzschmar D. In: Advances in Neurodegeneration. Horowski R, et al., editors. Wien: Springer-Verlag; 2003. pp. 51–62. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.