

Abstract
Fragile X premutation carriers are at risk for developing a late-onset, progressive neurodegenerative disorder termed fragile X-associated tremor/ataxia syndrome (FXTAS). A growing body of evidence suggests the characteristic excess CGG repeat containing FMR1 mRNA observed in premutation carriers is pathogenic and leads to clinical features of FXTAS. The current model suggests premutation mRNA transcripts can induce the formation of intranuclear inclusions by the sequestration of RNA-binding proteins and other proteins. The sequestered proteins are prevented from performing their normal functions, which is thought to lead to the neuropathology-observed FXTAS. This paper discusses the existing evidence that microsatellite expansions at the level of RNA play a role in the disease pathogenesis of FXTAS and some of the approaches that may uncover downstream effects of expanded riboCGG expression.
Keywords: FXTAS, intranuclear inclusion, neurodegeneration, premutation allele, riboCGG-mediated toxicity, RNA-binding protein
The X-linked FMR1 gene has a polymorphic CGG repeat located in its 5′ untranslated region (UTR). The alleles of the FMR1 gene can be grouped into three distinct classes depending on the size of the repeat. Among individuals in the general population, repeat length ranges between five and 54 triplets, with the majority of X chromosomes carrying either 29 or 30 repeats. A second allele class is known as the full mutation, wherein upon expansion of the CGG repeat tract to more than 200 triplets, the repeat sequence itself and an adjacent CpG island become hypermethylated and associated histones are modified. As a result, FMR1 transcription is silenced [1–7]. Thus, fragile X syndrome (FXS), the most commonly inherited form of mental retardation, results from the loss of function of FMR1. The mechanism for CGG repeat expansion is still under investigation, however, evidence suggests the expansion to the full mutation occurs in female gametes [8,9]. Male and female carriers of FXS represent the third allele class, known as the premutation allele. Premutation carriers have an intermediate number of CGG triplets between 55 and 200 repeats, which show no unusual methylation. Although both males and females can transmit premutation alleles to their offspring, only alleles obtained via maternal transmission expand to a full mutation and cause FXS in a single generation. Younger premutation carriers have normal cognitive abilities and are not affected with FXS. However, reports of tremor and cognitive decline in grandfathers of FXS families with known probands sparked further study of older males carrying premutation alleles in families where FXS was present. Case studies of male premutation carriers led to the description of a new late-onset neurodegenerative disorder termed fragile X-associated tremor/ataxia syndrome (FXTAS) [10–12].
The most common clinical features of FXTAS include a progressive action tremor with ataxia. More advanced or severe cases may show a progressive decline in cognition that ranges from executive and memory deficits to dementia [13]. FXTAS patients may also exhibit psychological symptoms such as depression, increased anxiety and mood instability [14,15]. Approximately 30% of patients complain of muscle weakness, numbness and pain in the lower extremities [16], suggesting that the disorder is not confined to the CNS. A useful differential diagnostic criterion for FXTAS is increased T2 intensities of the middle cerebellar peduncle (MCP) and adjacent white matter observed by MRI [17]. These criteria distinguish the disorder from other late-onset movement disorders and dementias. FXTAS patients also demonstrate mild-to-moderate global brain atrophy, which is most common in the frontal and parietal regions, as well as in the pons and cerebellum. In addition, degeneration of the cerebellum including Purkinje cell loss, Bergman gliosis, spongiosis of the deep cerebellar white matter and swollen axons have been observed in nearly all case studies of autopsy brains of symptomatic premutation carriers [18,19].
A study of penetrance revealed that more than a third of all carriers aged 50 years and older will show symptoms of FXTAS and the penetrance of this disorder exceeds 50% for men over 70 years of age [20]. The prevalence of premutation alleles is approximately one in 800 for males and one in 250 for females in the general population [21,22]; however, it is estimated that approximately one in 3000 men of more than 50 years of age in the general population will show symptoms of FXTAS [23]. These estimates do not consider the influence of size on penetrance, which predicts a correlation between the age of onset of symptoms to the size of the repeat. Recent studies have correlated the age of onset of clinical symptoms with the length of expanded repeats, demonstrating that larger repeats represent an increased risk factor for the development of FXTAS [23,24]. The degree of brain atrophy [24] and severity of the tremor and ataxia [24], as well as the risk of developing cognitive impairments [25], have also been correlated with CGG repeat length [24].
Although few, there are cases of FXTAS clinical features in female premutation carriers [26–29]. The lower frequency of FXTAS in females is thought to be due to the partial protection offered by random X-inactivation, leading to approximately half of cells expressing only the normal allele in most female carriers. Recent studies have been aimed at defining differences between male and female FXTAS, as well as female penetrance. One such study, led by Adams et al., demonstrated milder whole brain and cerebellar volumetric loss in FXTAS females than in FXTAS males; however, FXTAS females exhibit significantly reduced brain volume compared with unaffected premutation carrier females or control females without the premutation [30]. Similar patterns of brain atrophy and white matter disease were observed in both males and females affected by FXTAS. Unlike FXTAS males, FXTAS females do not show significant correlation between reduced cerebellar volume and both increased FXTAS severity and increased CGG repeat length [30]. A study of FXTAS penetrance in 85 women with premutation alleles predicted that approximately 16.5% of female carriers over 50 years of age will exhibit signs of FXTAS [31].
Female premutation carriers are at an increased risk for developing other medical and neurological problems. It has been established that between 16 and 24% of female premutation carriers will exhibit fragile X-associated primary ovarian insufficiency (FXPOI), which results in the cessation of menstruation before the age of 40 years [32,33]. Although the basis for FXPOI is not understood, the penetrance and age of onset appear to correlate with CGG repeat length [34]; however, a later study suggests a nonlinear relationship between age of menopause and premutation size, in which female carriers with premutations in the midsize range are at a greater risk for FXPOI, while larger repeat tracts are associated with lower risk [35]. Aside from FXTAS and FXPOI, increased prevalence of thyroid disease, fibromyalgia and hypertension have been observed in female premutation carriers [31,36].
Neuropathology: intranuclear inclusions
The neuropathological hallmark and post-mortem criterion for definitive FXTAS is the presence of eosinophilic, ubiquitin-positive intranuclear inclusions. FXTAS inclusions are negative for tau and α-synuclein, which rule out FXTAS as a tauopathy or synucleinopathy [37–39]. The inclusions are located in broad distributions throughout the brain – in neurons, astrocytes and the spinal column. FXTAS inclusions share the ubiquitin-positive hallmark with several other inclusion disorders, such as the polyglutamine disorders; however, FXTAS inclusions do not stain with antibodies that recognize polyglutamine. It is important to note that, unlike the polyglutamine disorders, there is no known structurally abnormal protein with FXTAS. The presence of ubiquitin and heat-shock proteins (HSP) in the FXTAS inclusions strongly suggests a defect in the proteasome degradation or protein-folding pathways; however, the link between these processes and RNA-mediated neurodegeneration is unknown.
Iwahashi et al. purified human FXTAS frontal cortex inclusions using fluorescence-activated flow-based methods [40]. Both mass spectroscopic analysis of purified inclusions and immunohistochemical analysis of isolated nuclei and tissue sections uncovered 19 proteins that constitute the protein complement of the inclusions (Table 1). The protein components of FXTAS inclusions fell into eight major functional categories, including: histone family; intermediate filament; microtubule; myelin-associated proteins; RNA-binding proteins (RBPs); stress-related proteins; chaperones and ubiquitin–proteasome-related proteins. Several proteins previously identified as components of FXTAS inclusions, such as ubiquitin, HSP70 and 20S subunit of the proteasome, were identified and purified by immunofluorescent staining of isolated nuclei of FXTAS frontal cortex. In addition, proteins such as tau and α-synuclein, which were not found in immunohistochemical analysis of FXTAS brains, were also not present in isolated nuclei of FXTAS frontal cortex in this study.
Table 1. Protein components of CGG-induced inclusions identified by various methods.
| Model | Candidate protein | Category | Ref. |
|---|---|---|---|
| Humans, mice and flies | Ubiquitin | Ubiquitin–proteasome system | [66,81,84,102,103] |
| Humans | 11S regulator to 20S proteasome | Ubiquitin–proteasome system | [40] |
| Mice and flies | 20S proteasome | Ubiquitin–proteasome system | [40,66] |
| Mice | Homolog of yeast gene Rad23A | DNA repair and ubiquitin–proteasome system | [104] |
| Mice | Homolog of yeast gene Rad23B | DNA repair and ubiquitin–proteasome system | [102,104] |
| Humans | H2B histone family | Histone family | [40] |
| Humans | Similar to H2A histone family, member Z | Histone family | [40] |
| Humans | HIST I H4D protein | Histone family | [40] |
| Humans | H2A histone family A (L) | Histone family | [40] |
| Humans | H2A histone family, member Q (O) | Histone family | [40] |
| Humans | Neurofilament 3 | Intermediate filament | [40] |
| Humans and mice | Lamin A/C | Intermediate filament | [40,78,84] |
| Humans | Vimentin | Intermediate filament | [40] |
| Humans | Internexin neuronal intermediate filament | Intermediate filament | [40] |
| Humans | NEFL protein | Intermediate filament | [40] |
| Humans | Glial fibrillary acidic protein (GFAP) | Intermediate filament | [40] |
| Humans | β5-tubulin | Microtubule | [40] |
| Humans | Tubulin, α6 | Microtubule | [40] |
| Humans | Tubulin, alpha and ubiquitous | Microtubule | [40] |
| Humans | Myelin/oligodendrocyte glycoprotein β3 | Myelin-associated protein | [40] |
| Humans | 2′,3′-cyclic nucleotide 3′-phosphodiesterase (CNPase) | Myelin-associated protein | [40] |
| Humans | Myelin basic protein | Myelin-associated protein | [40] |
| Humans | Muscleblind-like 1 | RNA-binding protein | [40] |
| Humans | Heterogenous nuclear ribonucleoprotein A2/B1 | RNA-binding protein | [40] |
| Humans and flies | Purα | RNA-binding protein | [92] |
| Humans | αβ-crystallin | Stress-related protein | [40,78] |
| Humans and flies | Hsp70 | Chaperone | [40,66] |
| Humans | Hsp27 | Chaperone | [40] |
| Mice | Hsp40 | Chaperone | [81,102] |
Molecular correlates of FXTAS
Before the discovery of FXTAS, premutation carriers were generally regarded as clinically unaffected [41–44]. No alterations in FMR1 mRNA or FMRP levels could be detected using semiquantitative reverse transcription-PCR and immunohistochemistry [45–48]. Despite these findings, reports of learning disabilities [43,49,50] and psychiatric dysfunction [11,16,51–56] in both male and female premutation carriers raised suspicions that carriers may exhibit molecular abnormalities due to the premutation expansion in the FMR1 mRNA [57]. The clinical observations warranted a more quantitative investigation of FMR1 mRNA and FMRP levels in carriers. Tassone et al. used quantitative fluorescence PCR to demonstrate that relative FMR1 mRNA levels in premutation carrier blood leukocyte samples were up to tenfold higher than that of control individuals [58]. In addition, immunocytochemical staining of premutation lymphocytes revealed a reduction in FMRP-positive staining, indicating a reduction in FMRP production. Other studies also revealed a reduction in FMRP levels in blood leukocytes of some premutation carriers with cognitive deficits [59]. Kenneson et al. developed a slot-blot-based assay to measure FMRP levels in cell lysate and demonstrated significantly diminished FMRP levels in carriers that negatively correlated with repeat number; however, despite reduced FMRP levels, the carriers overexpressed FMR1 mRNA, which resulted in a positive correlation between repeat number and FMR1 message levels [60].
Several possible mechanisms for FMR1 message overexpression have been put forward. One feedback mechanism suggests that the cell attempts to compensate for reduced levels of FMRP by increasing the amount of available FMR1 transcript [61]. Alternatively, expansion of CGG repeats may change the distances between potential factor-binding sites, leading to a more open promoter that could favor increased access of transcription factors. Another suggested mechanism is that increased stability of the premutation FMR1 transcript may be the cause of the message overexpression. After measuring primary transcription and stability of FMR1 mRNA, Tassone et al. determined that the mechanism of elevated FMR1 transcript levels results from increased transcription at the FMR1 locus and not from increased stability [62]. Reduced translational efficiency of premutation alleles has been shown to result from a reduced association of the premutation mRNA with polysomes [63]. This decline in translation efficiency parallels increasing CGG repeat length. Other studies have considered the effect of strong secondary or higher order structures to impede ribosome entry or scanning of the 5′ UTR of FMR1 mRNA [64]. In addition, changes in FMRP may result from altered cellular localization of the transcripts carrying expanded repeats. It is possible that large fractions of the expanded transcripts are sequestered in the nuclear inclusions, and are therefore not accessible to the ribosomes; however, as of yet there is no evidence for this hypothesis.
RNA-mediated neurodegeneration model for FXTAS
A threshold in triplet repeat mutation length in FMR1 defines two molecularly distinct diseases. The presence of elevated FMR1 mRNA carrying repeats in premutation carriers along with the absence of FXTAS in older adults with FXS led to the proposal of RNA-mediated toxicity mechanism that describes the contribution of premutation RNA to FXTAS pathogeneisis [11,65,66]. Several clues regarding the molecular mechanisms involved in FXTAS come from the RNA gain-of-function model for myotonic dystrophy (DM), a typically adult-onset, dominant, muscle disorder caused by an abnormal expansion of a CUG triplet repeat present in the 3′ untranslated region of a protein kinase gene, DMPK [67]. In DM1 cells, mRNA transcripts with expanded CUG repeats are sequestered in nuclear RNA foci [68] along with decreased DMPK protein levels [69]. Expanded CUG repeats in DMPK mRNA can form hairpins that are thought to block the export of the RNA from the nucleus [70]. An RBP, MBNL1, can colocalize with the DM RNA foci [71]. Ultimately, disruption of MBNL1 leads to downstream misregulated splicing of MBNL1 targets that result in DM pathology [72]. The RNA toxicity model has been demonstrated in a DM mouse model expressed a transgene with expanded CTG repeats in the DMPK gene that resulted in myotonia [73,74]. A similar mechanism leading to FXTAS pathology has been recently tested (Figure 1). The proposed mechanism for RNA pathogenesis for FXTAS states that CGG-repeat-containing FMR1 transcripts recruit RNA binding and other proteins that accumulate as intranuclear inclusions. The sequestration of these proteins is thought to prevent them from performing their normal functions that would lead to downstream alterations in cellular function. The observation of expanded FMR1 RNA transcripts in the FXTAS inclusions of a 70-year-old male who died with FXTAS helped to further support for the RNA toxicity hypothesis [75].
Figure 1. RNA toxicity model in a premutation cell.
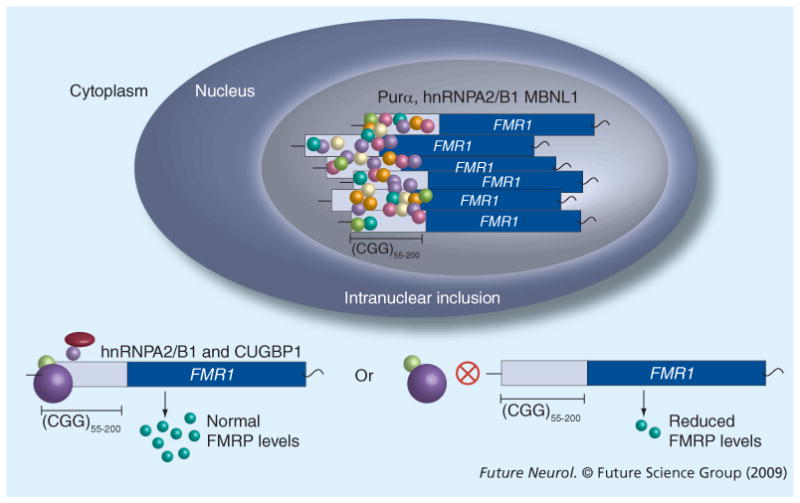
Via unknown mechanisms, increased transcription of premutation alleles of the FMR1 gene leads to elevated levels of FMR1 mRNA. The excess RNA is thought to exert its toxic effect on the cell via sequestration of RNA-binding proteins, ubiquitin, proteasome components and other unknown proteins, which would lead to abnormal regulation of cellular processes normally regulated by the sequestered proteins. Sequestration of proteins into intranuclear inclusions may lead to downstream neurodegeneration. In addition, sequestration of the toxic RNA into intranuclear inclusions could be a mechanism that the cell uses for survival. Premutation RNA exported to the cytoplasm may be translated normally. Reduced FMRP levels may result from, first, interactions of RNA-binding proteins in the cytoplasm with the expanded RNA, which could block ribosome entry; second, complex RNA structures due to expanded CGG repeats; third, reduced translation owing to nuclear sequestration of RNA-binding proteins required for translation; and/or last, reduced translation due to sequestration of premutation RNA in the inclusion.
The major parallels between FXTAS and DM are the induction of intranuclear inclusions and nuclear foci as a result of expanded repeat RNA. If these aberrant structures are responsible for pathology, what are the proteins that might be altered in abundance by them? Attempts to define proteins in the inclusions in FXTAS have revealed a number of candidates (Table 1). Among these, is a mammalian ortholog of the Drosophila protein muscleblind, MBNL1. Interestingly, MBNL1 is present in both FXTAS intranuclear inclusions [40] and DM nuclear foci suggesting at least a potentially parallel mechanism for FXTAS pathogenesis. A major caveat to the parallel with DM is demonstrated by considering different clinical features of FXTAS and DM. DM is a multisystemic disease that can present at any age from birth to old age. The major pathologies observed in DM are muscle wasting and myotonia, which are not observed in FXTAS. Although DM1 neuronal nuclear foci have been observed, it is uncertain whether the foci directly contribute to the neuronal dysfunction [76]. In DM muscle, there is evidence to support the direct involvement of RNA foci in disease pathogenesis through mechanisms that involve sequestration of MBNL1 and other proteins resulting in misregulation of genes that undergo alternative splicing [72,77]. MBNL1 is an RBP that functions as regulator of alternative splicing. In the case of DM1, human brain samples exhibited splicing misregulation with reversion to fetal isoforms of NMDAR1, APP and Tau genes [76]. The altered splicing that results from misregulation of MBNL1 may lead to the mental retardation and other cognitive defects observed in some DM patients. Changes in alternative splicing have not been described for FXTAS; however, the cellular and murine models of FXTAS provide adequate tools for investigation of CGG-mediated splicing alterations.
Evidence for an RNA-mediated neurodegeneration mechanism for FXTAS pathology
Cellular models
Garcia-Arocena et al. demonstrated that expanded CGG repeats expressed in untranslated mRNA are toxic to cells and can induce inclusion formation [78]. In this study, human neural cells transfected with green fluorescent protein (GFP) reporters possessing FMR1 5′UTR with 88 CGG repeats, not only exhibited cytotoxicity and inclusions but also changes in nuclear morphology and nuclear lamin structure. Handa et al. performed microarray analysis on RNA from cells transfected with plasmids expressing transcribed but untranslated premutation length CGG repeats upstream of FMR1 [79]. The microarray studies revealed upregulation of genes required for apoptosis, which were validated by quantitative-PCR, demonstrating that premutation CGG repeat RNA is toxic to cells.
Animal models
Fly models
Jin et al. developed a series of transgenic Drosophila to test the role of CGG in RNA as a toxic agent [66]. The authors made use of the Gal4-upstream activation sequence system to drive expression of expanded, untranslated riboCGG repeats upstream of the coding sequence for the EGFP reporter gene and demonstrated that ribo-CGG was sufficient to cause degeneration of neurons in the fly eye [66]. This model provided the first in vivo evidence for RNA toxicity in an animal model. The riboCGG induced neurodegeneration was neuron specific, dose sensitive and directly dependent on CGG repeat length. A repeat length of 90 CGGs was sufficient to cause neurodegeneration of photoreceptors. In addition, the flies developed inclusions that were positive for ubiquitin, Hsp70 chaperone and the proteasome. The finding that Hsp70 could suppress the riboCGG-mediated neurodegeneration of the fly eye [66] made this model particularly useful for the discovery of other genetic modifiers (see section ‘Genetic and chemical modifiers of CGG-mediated phenotypes in FXTAS models’).
Murine models
Knock-in mouse models of FXTAS: Oostra & Usdin mice
The Oostra group at Erasmus University, Rotterdam, The Netherlands, created a knock-in mouse model that replaced the eight endogenous murine CGG repeats with an expanded repeat of human origin (∼100 repeats) [80]. This model exhibited up to 3.5-fold more Fmr1 mRNA in the brain compared with controls. The mice developed intranuclear ubiquitin-positive inclusions in several neuronal cell types [81], but not in Purkinje neurons of the cerebellum. An increase in both the number and size of the inclusions was correlated with the age of the animals. As in human post-mortem samples, the inclusions in this model were positive for Hsp40, Rad23B and the 20S catalytic core complex of the proteasome. These mice demonstrated cognitive decline, neuromotor and behavioral disturbances that were assessed by a battery of tests [82]. Possibly the most significant piece of evidence to support the RNA-mediated toxicity model for FXTAS was that these mice displayed no alterations in FMRP levels, as assessed by western blot, despite elevation in Fmr1 mRNA levels [81]. Animals derived from the original knock-in with CGG repeat expansions greater than 200 triplets have now been developed after many generations of selecting offspring with longer repeats. Repeats of this length would result in FXS in humans. Interestingly, these mice do not develop molecular or phenotypic characteristics of FXS, and no methylation is observed at the Fmr1 locus. Elevated Fmr1 mRNA levels were only observed in the lower range for premutation alleles and Fmrp levels were reduced in the upper premutation range in these animals [83]. Loss of the intranuclear inclusions was observed in animals with greater than 200 triplets [83], which raises the question of the relevance of inclusions to disease pathology.
A second knock-in mouse model that has small serial ligated, short stable CGG•CGG repeat tracts knocked into exon 1 of the endogenous murine Fmr1 gene was created by the Usdin group at the NIH [84]. This model exhibited genetic and pathophysiological changes that were similar to human FXTAS but not observed in the previous mouse model. For example, the Usdin knock-in repeats were found in rare cases to expand to a full mutation-sized repeat without evidence of methylation in a single generation. In addition, this model has Purkinje neuron pathology in the form of abnormal calbindin staining, swollen axonal torpedos and Purkinje neuron cell loss. A correlation between the repeat number and the Fmr1 mRNA levels was also observed.
Transgenic mouse model of FXTAS
Purkinje neuron-specific trangenic model of FXTAS
Both knock-in mouse models suggest a direct involvement of riboCGG in neuropathology; however, it remained unclear which part of the expanded rCGG transcript was necessary or whether FMRP plays a role. Our group developed transgenic mouse models to explore the molecular requirement for CGG-mediated neurodegeneration observed in FXTAS. The mice expressed 90 CGG repeats in the 5′ untranslated region (UTR) of either the Fmr1 gene or reporter gene EGFP under the regulation of the Purkinje neuron-specific promoter, L7/Pcp2 [85]. As controls L7Fmr1 and L7EGFP lines were also created without expanded repeats. We found that mice with CGG-containing transcripts outside the context of Fmr1 are sufficient to cause Purkinje neuron neurodegeneration and behavioral anomalies in our models. The presence of large and frequent Purkinje neuronal inclusions is unique to our models. This model has provided the first evidence in a mammalian system that rCGG outside the context of Fmr1 was sufficient to cause neurodegeneration, as only CGG-containing repeat mice exhibited significant Purkinje neuron cell loss compared with wild-type or L7Fmr1 mice. In additon, detailed examination of Purkinje neurons in transgenics revealed axonal swellings. Ubiquitin-positive inclusion formation was observed in mice expressing L7CGG90Fmr1 or L7CGG90EGFP transgenes suggesting that inclusion formation is not directly correlated to Fmr1 levels but rather, expression of expanded CGG repeats. Purkinje neuronal inclusions in our rCGG-containing transgenics also stained positive for antibodies against Rad23B, Hsp40 and the 20S core subunit of the proteasome, as assessed by immunohistochemical analysis. Our transgenics also exhibited a decline in motor-learning abilities as assessed by the accelerating rotarod. L7CGG90Fmr1 mice developed an age-dependent decline in rotarod walking since mice at 40 weeks performed worse than mice at 20 weeks, while L7Fmr1 transgenics showed no age effect-related decline in rotarod performance. Each of the animal models has different neurological phenotypes, which strongly suggests differences in CGG repeat length, RNA expression and proteins levels may play an essential role in FXTAS pathology (Table 2).
Table 2. Phenotypic comparisons between mouse models of fragile X-associated tremor/ataxia syndrome.
| Mouse model | Brain Fmr1 mRNA levels | Brain FMRP levels | Ubiquitin-positive inclusions | Purkinje neuronal phenotype | Onset of neuromotor behavioral symptoms (weeks) | Onset of inclusions (weeks) | Ref. |
|---|---|---|---|---|---|---|---|
| CGG98 knock-in | 2–3.5-fold increase relative to wild-type | Normal | + | N/R | N/R | 30 | [81] |
| Expanded CGG100–150 | 2–3.5-fold increase relative to wild-type | Normal | + | N/R | 52 | N/R | [105] |
| Expanded CGG151–200 | Normal | Decreased | + | N/R | N/R | N/R | [105] |
| Expanded CGG>200 | Normal | Decreased | Few/absent | N/R | N/R | N/R | [105] |
| Usdin CGG knock-in | Increased | Decreased (in some brain regions) | + | Swollen axons, cell death and abnormal calbindin staining | N/R | N/R | [84] |
| L7CGG90 transgenics | N/A | N/A | + (only in Purkinje neurons) | Inclusions, swollen axons and cell death | 20 | 8 | [102] |
Positive;
N/A: Not applicable;
N/R: Not reported.
Yeast artificial chromosome transgenics
Our group also developed yeast artificial chromosome (YAC) transgenic mice carrying a premutation-sized CGG repeat within the entire human FMR1 gene in order to study instability of the FXS repeat [86]. While they are not as well characterized as the models previously described, the YAC transgenics develop significant pathology but lack detectable intranuclear inclusions. In particular, Purkinje neurons show accelerated degeneration and neuropathology in the form of axonal torpedoes as animals age (Figure 2). These animals overexpress the human RNA and protein; whether differences result from human versus mouse overexpression is a factor remains unknown. Interestingly, SCA1 transgenic mice expressing ataxin-1 with 82 glutamine repeats develop ataxia and Purkinje cell degeneration [87,88] in the absence of nuclear inclusions [89]. These studies suggest that inclusion formation may not be required for disease pathogenesis. However, conditional SCA1 transgenics [90], as well as a conditional mouse model of Huntington's disease (HD) expressing 94 CAG repeats upstream of the lacZ promoter under the regulation of a Tet inducible promoter [91] can recover from polyglutamine-induced inclusions. It would be of interest to determine whether similar inducible models of FXTAS pathogenesis show the ability to recover.
Figure 2. Expanded CGG repeats in a yeast artificial chromosome transgenic model using human FMR1 lead to neuropathology with very few nuclear inclusions.
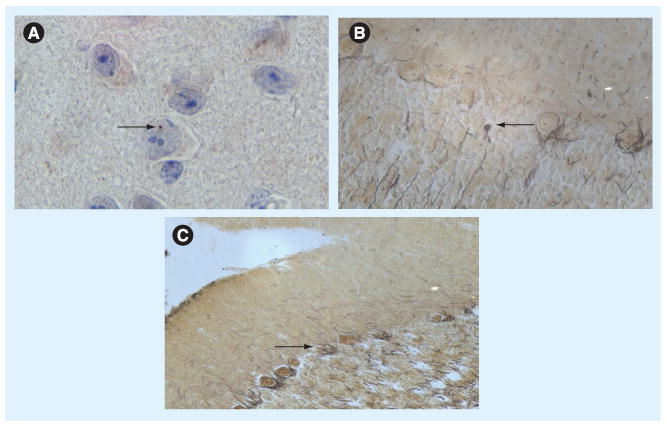
(A) Infrequent ubiquitin-stained inclusions seen in hippocampus, CA3, mammilary nucleus and hypothalamus. (B & C) Cerebellar pathology including axonal torpedos (B, swellings) and empty baskets (C, Purkinje neuron loss).
Genetic & chemical modifiers of CGG-mediated phenotypes in FXTAS models
Our group and others made use of the transgenic fly model by performing biased genetic screens that uncovered RBPs capable of modifying the neurodegenerative eye phenotype in the fly (Table 3). For example, overexpression of RBP, Purα, suppresses the riboCGG-mediated neurodegeneration in the Drosophila eye [92]. Jin et al. found Purα in both the riboCGG-induced fly inclusions as well as human FXTAS sup–mid temporal cortex inclusions [92]. However, Iwashashi et al. did not detect Purα in human FXTAS inclusions purified from the cerebral cortex [40]. In addition, Purα-positive inclusions have not been observed in either the Oostra knock-in [Willemsen R, Erasmus University, Pers. Comm.] nor our Purkinje neuron-specific transgenics. The reasons behind the differences in inclusion protein content from human to human or from human to mouse and fly are unclear; however, it is not difficult to imagine that inclusions from one brain region to the next will not mirror exactly in protein content, as gene expression varies from region to region; the same can be said for species to species gene expression differences. In addition, variations in technique to assess inclusion protein compositions may yield different results.
Table 3. Genes that modify the RNA-mediated neurodegeneration caused by the fragile-X premutation riboCGG repeat in Drosophila melanogaster.
| Gene | Protein present in fly inclusions | Ref. |
|---|---|---|
| Hsp70 | Yes | [66] |
| Purα | Yes | [92] |
| Cugbp1 | No | [96] |
| hnRNPA2/B1 | No | [96] |
| riboGCC repeats | N/A | [101] |
N/A: not applicable.
Although there is conflicting data regarding the presence of Purα in inclusions, the protein may still play a role in FXTAS pathology as Purα directly interacts with riboCGG repeats in vitro and in vivo [92]. Interestingly, Purα-null mice develop many of the hallmark features of human FXTAS. Similar to humans with FXTAS, Purα-null mice exhibit loss of neurons in the cerebellar granular layer as well as the Purkinje cell layer and develop tremor upon motion [93]. Even Purα-heterozygous mice develop neurological features, suggesting that haploinsufficiency is problematic, at least in the brain. Purα has been implicated in dendritic RNA transport and translation [94,95]. The RNA-toxicity model of FXTAS would suggest that sequestration of Purα into inclusions or even alterations in the Purα protein levels may lead to alterations in RNA transport or translation of Purα targets.
In another biased screen for RBPs that modify the transgenic fly neurodegenerative phenotype, Sofola et al. observed that overexpression of two additional RBPs, hnRNPA2/B1 and CUGBP1, could suppress the riboCGG-induced eye neurodegeneration and hnRNPA2/B1 could interact directly with the riboCGG repeats [96]. CUGBP1 has been implicated in the pathology of DM1. CUGBP1 steady-state levels are increased in DM1, which induces downstream misregulated alternative splicing events. Sofola et al. demonstrated CUGBP1 could interact with rCGG repeats via hnRNPA2/B1 in vitro [96].
Several studies indicate that riboCGGs can fold into hairpin and tetraplex structures that could reduce the efficiency of translation of premutation FMR1 mRNA. The reduced translational efficiency of premutation RNA results from its reduced association with polysomes [63]. Interestingly, hnRNPA2 can mediate the nonenzymatic destabilization of quadruplex forms of (CGG)n in both DNA and RNA [97]. Khateb et al. demonstrated, using an in vitro translation assay, that premutation repeats in the promoter region of a construct expressing the firefly reporter gene could reduce its translational efficiency [98]. In addtion, a similar effect was demonstrated in vivo; however, in the presence of overexpressed hnRNPA2, not only could the cell more efficiently translate the premutation RNA but also diminish the production of excess premutation RNA in vivo.
Ofer et al. recently demonstrated that TMPyP4, a quadruplex ribo-CGG destabilizing porphyrin, on its own and in cooperation with overexpressed hnRNPA2, can significantly increase the efficacy of translation of premutation RNA in vivo making it an attractive candidate for FXTAS therapeutics [99]. Although the mechanism that allows TMPyP4 to increase efficiency of premutation mRNA translation and diminish excess premutation RNA accumulation is unknown, it may be useful to decrease RNA toxicity in carriers affected by FXTAS. According to the RNA toxicity model, if hnRNPA2/B1 is sequestered to the cytoplasmic inclusions in flies, it may serve to enhance translation of premutation RNA, thereby maintaining the levels of FMRP in the cell. Since hnRNPA2/B1 can be found in both the nucleus and the cytoplasm, sequestration of hnRNPA2/B1 to human FXTAS nuclear inclusions may hamper its cytoplasmic functions and lead to reduced translation of FMR1 mRNAs.
All of these observations taken together have led to the hypothesis that the neurodegeneration and inclusion formation observed in FXTAS are the result of a RNA-mediated toxicity caused by the increased expression of expanded ribo-CGG containing mRNA. A significant correlation between CGG repeat length and the frequency of intranuclear inclusions, in both neurons and astrocytes of human premutation carriers, further supports these models. In addition, intranuclear inclusions can be induced in primary progenitor cells and established cell lines when premutation-sized CGG repeats fused to FMR1 are expressed in these cells [78]. Given the high prevalence of fragile X premutation carriers in the general population and their risk for developing clinical features of FXTAS, it is essential to understand the molecular mechanisms of FXTAS, and further analysis of the pathogenic effects of ribo-CGG in all of the models is essential for determining the mechanism of FXTAS.
Conclusion
Considerable data are now available implicating fragile X premutation mRNA in disease pathogenesis. While the cell, fly and mouse models have provided useful molecular and genetic tools that will aid in our understanding of FXTAS disease progression, the precise mechanisms involved remain obscure and need to be investigated further. Early events that lead to inclusion formation and neuronal death are not understood. The relevance of inclusion formation in neurodegeneration remains a mystery since FXTAS pathology is observed in animal models that express greater than 200 CGG repeats and YAC transgenics expressing 90 CGG repeat and human FMR1, but these rarely ever show inclusions. A reasonable suggestion for inclusion formation would be to sequester pathogenic RNA; however, the protective versus harmful effects of inclusion formation have been hotly debated in polyQ inclusions disorders [100]. The argument that inclusions have a role in neuronal dysfunction suggests that a key step in the pathogenesis of various polyQ disorders result from aberrant interactions of misfolded protein with cellular proteins. The interaction would inhibit the sequestered proteins from performing their normal activities and trigger a cascade of events that lead to neuronal dysfunction and subsequently, inclusion formation. The argument for a neuroprotective role for inclusions suggests they function to sequester misfolded proteins from the pool of correctly folded proteins.
It is still unknown how the premutation and other pathogenic RNAs recruit RBPs and other proteins into inclusions or whether these proteins play some role in the inclusions. The downstream effects of sequestration of Purα, Hsp70, hnRNPA2/B1, MBNL1 and other as yet unidentified proteins, are essential to our understanding of the consequences of their depletion in cells. It is reasonable to suggest that, owing to the nature of the sequestered proteins, alterations in proteasome degradation and changes in neuronal function may be to blame for the clinical features of FXTAS. The variability of Fmr1 transcript expression in knock-in mice with various repeat lengths may help us understand FXTAS penetrance and disease severity. Understanding how and why cells form inclusions in response to expanded RNA, as well as the nature of the inclusions will likely lead to new therapeutic approaches that will combat neurodegeneration and improve cognitive and motor performance. Therapy could take many approaches from gene delivery of genetic modifiers to prevent inclusion formation. One example is provided by a study aimed at the RNA interference knockdown of premutation CGG repeats in Drosophila that has already demonstrated that gene delivery can suppress ribo-CGG-mediated neurodegeneration [101].
Future perspective
The future of FXTAS should focus on genetic modifiers of neurodegeneration and behavioral anomalies. Additional studies that provide more insight into the roles of proteins such as Purα, hnRNPA2/B1 and MBNL1 in intranuclear inclusions are necessary to determine the alterations required for disease pathogenesis. In addition, the presence of these proteins in inclusions may provide useful information regarding the downstream effects of protein sequestration. Mouse models that employ a Tet ON/OFF or tissue-specific inducible system for the expression of premutation CGG repeats would prove invaluable for the study of the consequence of repeat expression on inclusion recovery, behavioral and cognitive rescue studies, cell morphology and function. The differences between inclusion content as well as an increased tolerance for expanded repeat length in mice must also be considered. As previously mentioned, Purα and MBNL1 have not been observed in murine inclusions, which may result in the use of different pathways for induction of inclusion formation between humans and mice. In addition, differences in phenotypes between CGG-repeat knock-in and Purkinje-specific CGG90 transgenic mice exist. The reasons for the variation in phenotypes between the animal models are unknown; however, differences such as the developmental timing of the expression of the CGG-containing transcript may account for the differences in phenotype. Both knock-in models of FXTAS use the endogenous mouse Fmr1 regulatory sequences, while our transgenics use a robust Purkinje neuron-specific promoter active after birth, which may also account for the early inclusion phenotype observed in our transgenics. The differences in onset or severity of the pathogenic and behavioral phenotypes observed in our models may also be due to variations in the level of transgene expression due to positional effects. Better understanding of parallel pathways between FXTAS and other noncoding dominant repeat disorders may provide a useful insight into FXTAS pathogenesis.
The Drosophila model of FXTAS aids our understanding of the effect of varying levels of CGG-transcript expression and phenotype severity as we have observed a correlation between increased levels of CGG-transcript expression and RNA-mediated toxicity in the flies [Sofola O, Baylor College of Medicine, Uunpublished Data]. The fly has helped to uncover modifiers of the neurodegenerative phenotype, and points to potential pathways involved in the disorder. The fly offers important advantages to the study of RNA-mediated CGG repeat toxicity, such as the ability to perform rapid genetic screens for modifiers and to assess behavioral anomalies quickly. These flies can be used to investigate drug and gene therapies for FXTAS. Although the fly model of FXTAS may help us uncover additional modifiers of the neurodegenerative phenotype, it is important to consider that the Drosophila inclusions were almost always cytoplasmic, which is unlike those observed in patients or in mouse models. Therefore, the composition of the inclusions and pathways involved in neurodegeneration may differ significantly.
Studies that identify mechanisms for overexpression of CGG-containing FMR1 mRNA, as well as downstream molecular mechanisms for expanded CGG-repeat FMR1 mRNA to trigger downstream events leading to pathology, are required to understand of FXTAS disease progression. A focus on defining the cell death mechanisms and markers of CNS toxicity will help us understand the events that precede inclusion formation. Identification of genetic and environmental risk and protective factors associated with the penetrance of FXTAS is required to develop treatments for the disorder. FXTAS is a relatively new disorder; therefore, there is clearly a need for additional animal models to increase our knowledge of disease pathology, molecular mechanisms and drug treatment. One thing is clear, expanded CGG-repeat containing mRNAs play an integral role in FXTAS pathology.
Executive summary.
Background
Premutation alleles of FMR1 are linked to fragile X-associated tremor/ataxia syndrome (FXTAS) and fragile X-associated primary ovarian insufficiency (FXPOI).
Approximately a third of premutation allele carriers will exhibit clinical features of FXTAS.
The radiological criterion for definitive FXTAS is increased T2 intensities observed by MRI.
This paper presents a number of observations/conclusions that provide evidence for a pathogenic RNA gain-of-function mechanism for FXTAS.
Neuropathology: intranuclear inclusions
The presence of ubiquitin-positive eosinophilic intranuclear inclusions throughout the brain and spinal column are a major neuropathological hallmark of FXTAS.
FXTAS is not associated with mutations or repeat expansion in FMRP.
Molecular correlates of FXTAS
FXTAS patients display no signs of fragile X syndrome and vice versa.
Premutation carriers have elevated levels of expanded CGG-containing FMR1 mRNA.
Premutation carriers have normal to slightly reduced FMRP levels.
Evidence for an RNA-mediated neurodegeneration mechanism for FXTAS pathology
Premutation length riboCGGs as RNA are toxic to cells.
A fly model of FXTAS provided the first in vivo evidence of an RNA-toxicity mechanism for FXTAS.
Two premutation CGG knock-in mouse models exhibit ubiquitin-positive intranuclear inclusion formation and correlations between repeat length and Fmr1 mRNA levels.
CGG90Fmr1 and CGG90EGFP Purkinje neuron-specific transgenic mice exhibit ubiquitin-positive intranuclear inclusions.
FXTAS-like neuropathology observed in yeast artificial chromosome transgenics suggest neuropathology is independent of inclusion formation.
Genetic & chemical modifiers of CGG-mediated phenotypes in FXTAS models
The transgenic fly model of FXTAS and cellular models are useful tools to uncover genetic and chemical modifiers.
Future perspective
Modifiers of CGG-induced neurodegeneration may provide essential clues for downstream events leading to FXTAS pathology.
Understanding the role of FXTAS inclusion formation and elucidating inclusion protein composition may help us understand the molecular mechanism of FXTAS.
Footnotes
Financial & competing interests disclosure: The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Contributor Information
Jocelyn N Galloway, Email: jg148007@bcm.tmc.edu, Baylor College of Medicine, Interdepartmental Program in Cell & Molecular Biology, One Baylor Plaza, Room 904E, Houston, TX 77030, USA, Tel.: +1 713 798 7898, Fax.: +1 713 798 1116.
David L Nelson, Email: nelson@bcm.tmc.edu, Baylor College of Medicine, Department of Molecular & Human Genetics, One Baylor Plaza, Room 902EA, Houston, TX 77030, USA, Tel.: +1 713 798 4787, Fax.: +1 713 798 1116.
Bibliography
Papers of special note have been highlighted as:
• of interest
- 1.El-Osta A. FMR1 silencing and the signals to chromatin: a unified model of transcriptional regulation. Biochem Biophys Res Commun. 2002;295:575–581. doi: 10.1016/s0006-291x(02)00682-4. [DOI] [PubMed] [Google Scholar]
- 2.Fu YH, Kuhl DP, Pizzuti A, et al. Variation of the CGG repeat at the fragile X site results in genetic instability: resolution of the Sherman paradox. Cell. 1991;67:1047–1058. doi: 10.1016/0092-8674(91)90283-5. [DOI] [PubMed] [Google Scholar]
- 3.Gheldof N, Tabuchi TM, Dekker J. The active FMR1 promoter is associated with a large domain of altered chromatin conformation with embedded local histone modifications. Proc Natl Acad Sci USA. 2006;103:12463–12468. doi: 10.1073/pnas.0605343103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hansen RS, Gartler SM, Scott CR, et al. Methylation analysis of CGG sites in the CpG island of the human FMR1 gene. Hum Mol Genet. 1992;1:571–578. doi: 10.1093/hmg/1.8.571. [DOI] [PubMed] [Google Scholar]
- 5.Hornstra IK, Nelson DL, Warren ST, et al. High resolution methylation analysis of the FMR1 gene trinucleotide repeat region in fragile X syndrome. Hum Mol Genet. 1993;2:1659–1665. doi: 10.1093/hmg/2.10.1659. [DOI] [PubMed] [Google Scholar]
- 6.Oberle I, Rousseau F, Heitz D, et al. Instability of a 550-base pair DNA segment and abnormal methylation in fragile X syndrome. Science. 1991;252:1097–1102. doi: 10.1126/science.252.5009.1097. [DOI] [PubMed] [Google Scholar]
- 7.Sutcliffe JS, Nelson DL, Zhang F, et al. DNA methylation represses FMR-1 transcription in fragile X syndrome. Hum Mol Genet. 1992;1:397–400. doi: 10.1093/hmg/1.6.397. [DOI] [PubMed] [Google Scholar]
- 8.Malter HE, Iber JC, Willemsen R, et al. Characterization of the full fragile X syndrome mutation in fetal gametes. Nat Genet. 1997;15:165–169. doi: 10.1038/ng0297-165. [DOI] [PubMed] [Google Scholar]
- 9.Moutou C, Vincent MC, Biancalana V, et al. Transition from premutation to full mutation in fragile X syndrome is likely to be prezygotic. Hum Mol Genet. 1997;6:971–979. doi: 10.1093/hmg/6.7.971. [DOI] [PubMed] [Google Scholar]
- 10.Hagerman PJ, Greco CM, Hagerman RJ. A cerebellar tremor/ataxia syndrome among fragile X premutation carriers. Cytogenet Genome Res. 2003;100:206–212. doi: 10.1159/000072856. [DOI] [PubMed] [Google Scholar]
- 11.Hagerman RJ, Leehey M, Heinrichs W, et al. Intention tremor, parkinsonism, and generalized brain atrophy in male carriers of fragile X. Neurology. 2001;57:127–130. doi: 10.1212/wnl.57.1.127. [DOI] [PubMed] [Google Scholar]
- 12.Leehey MA, Munhoz RP, Lang AE, et al. The fragile X premutation presenting as essential tremor. Arch Neurol. 2003;60:117–121. doi: 10.1001/archneur.60.1.117. [DOI] [PubMed] [Google Scholar]
- 13.Grigsby J, Brega AG, Jacquemont S, et al. Impairment in the cognitive functioning of men with fragile X-associated tremor/ataxia syndrome (FXTAS) J Neurol Sci. 2006;248:227–233. doi: 10.1016/j.jns.2006.05.016. [DOI] [PubMed] [Google Scholar]
- 14.Bacalman S, Farzin F, Bourgeois JA, et al. Psychiatric phenotype of the fragile X-associated tremor/ataxia syndrome (FXTAS) in males: newly described fronto–subcortical dementia. J Clin Psychiatry. 2006;67:87–94. doi: 10.4088/jcp.v67n0112. [DOI] [PubMed] [Google Scholar]
- 15.Hessl D, Tassone F, Loesch DZ, et al. Abnormal elevation of FMR1 mRNA is associated with psychological symptoms in individuals with the fragile X premutation. Am J Med Genet B Neuropsychiatr Genet. 2005;139B:115–121. doi: 10.1002/ajmg.b.30241. [DOI] [PubMed] [Google Scholar]
- 16.Jacquemont S, Hagerman RJ, Leehey M, et al. Fragile X premutation tremor/ataxia syndrome: molecular, clinical, and neuroimaging correlates. Am J Hum Genet. 2003;72:869–878. doi: 10.1086/374321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brunberg JA, Jacquemont S, Hagerman RJ, et al. Fragile X premutation carriers: characteristic MR imaging findings of adult male patients with progressive cerebellar and cognitive dysfunction. AJNR Am J Neuroradiol. 2002;23:1757–1766. [PMC free article] [PubMed] [Google Scholar]
- 18.Greco CM, Berman RF, Martin RM, et al. Neuropathology of fragile X-associated tremor/ataxia syndrome (FXTAS) Brain. 2006;129:243–255. doi: 10.1093/brain/awh683. [DOI] [PubMed] [Google Scholar]
- 19.Greco CM, Hagerman RJ, Tassone F, et al. Neuronal intranuclear inclusions in a new cerebellar tremor/ataxia syndrome among fragile X carriers. Brain. 2002;125:1760–1771. doi: 10.1093/brain/awf184. [DOI] [PubMed] [Google Scholar]
- 20.Jacquemont S, Hagerman RJ, Leehey MA, et al. Penetrance of the fragile X-associated tremor/ataxia syndrome in a premutation carrier population. JAMA. 2004;291:460–469. doi: 10.1001/jama.291.4.460. [DOI] [PubMed] [Google Scholar]
- 21.Dombrowski C, Levesque S, Morel ML, et al. Premutation and intermediate-size FMR1 alleles in 10572 males from the general population: loss of an AGG interruption is a late event in the generation of fragile X syndrome alleles. Hum Mol Genet. 2002;11:371–378. doi: 10.1093/hmg/11.4.371. [DOI] [PubMed] [Google Scholar]
- 22.Rousseau F, Rouillard P, Morel ML, et al. Prevalence of carriers of premutation-size alleles of the FMRI gene – and implications for the population genetics of the fragile X syndrome. Am J Hum Genet. 1995;57:1006–1018. [PMC free article] [PubMed] [Google Scholar]
- 23.Jacquemont S, Leehey MA, Hagerman RJ, et al. Size bias of fragile X premutation alleles in late-onset movement disorders. J Med Genet. 2006;43:804–809. doi: 10.1136/jmg.2006.042374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Leehey MA, Berry-Kravis E, Goetz CG, et al. FMR1 CGG repeat length predicts motor dysfunction in premutation carriers. Neurology. 2008;70:1397–1402. doi: 10.1212/01.wnl.0000281692.98200.f5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sevin M, Kutalik Z, Bergmann S, et al. The penetrance of marked cognitive impairment in older male carriers of the FMR1 gene premutation. J Med Genet. 2009 doi: 10.1136/jmg.2008.065953. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 26.Berry-Kravis E, Potanos K, Weinberg D, et al. Fragile X-associated tremor/ataxia syndrome in sisters related to X-inactivation. Ann Neurol. 2005;57:144–147. doi: 10.1002/ana.20360. [DOI] [PubMed] [Google Scholar]
- 27.Hagerman RJ, Leavitt BR, Farzin F, et al. Fragile-X-associated tremor/ataxia syndrome (FXTAS) in females with the FMR1 premutation. Am J Hum Genet. 2004;74:1051–1056. doi: 10.1086/420700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jacquemont S, Orrico A, Galli L, et al. Spastic paraparesis, cerebellar ataxia, and intention tremor: a severe variant of FXTAS? J Med Genet. 2005;42:E14. doi: 10.1136/jmg.2004.024190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zuhlke C, Budnik A, Gehlken U, et al. FMR1 premutation as a rare cause of late onset ataxia – evidence for FXTAS in female carriers. J Neurol. 2004;251:1418–1419. doi: 10.1007/s00415-004-0558-1. [DOI] [PubMed] [Google Scholar]
- 30.Adams JS, Adams PE, Nguyen D, et al. Volumetric brain changes in females with fragile X-associated tremor/ataxia syndrome (FXTAS) Neurology. 2007;69:851–859. doi: 10.1212/01.wnl.0000269781.10417.7b. [DOI] [PubMed] [Google Scholar]
- 31.Rodriguez-Revenga L, Madrigal I, Pagonabarraga J, et al. Penetrance of FMR1 premutation associated pathologies in fragile X syndrome families. Eur J Hum Genet. 2009 doi: 10.1038/ejhg.2009.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Allingham-Hawkins DJ, Babul-Hirji R, Chitayat D, et al. Fragile X premutation is a significant risk factor fo r premature ovarian failure: the International Collaborative POF in Fragile X study – preliminary data. Am J Med Genet. 1999;83:322–325. [PMC free article] [PubMed] [Google Scholar]
- 33.Murray A, Ennis S, MacSwiney F, et al. Reproductive and menstrual history of females with fragile X expansions. Eur J Hum Genet. 2000;8:247–252. doi: 10.1038/sj.ejhg.5200451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sullivan AK, Marcus M, Epstein MP, et al. Association of FMR1 repeat size with ovarian dysfunction. Hum Reprod. 2005;20:402–412. doi: 10.1093/humrep/deh635. [DOI] [PubMed] [Google Scholar]
- 35.Ennis S, Ward D, Murray A. Nonlinear association between CGG repeat number and age of menopause in FMR1 premutation carriers. Eur J Hum Genet. 2006;14:253–255. doi: 10.1038/sj.ejhg.5201510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Coffey SM, Cook K, Tartaglia N, et al. Expanded clinical phenotype of women with the FMR1 premutation. Am J Med Genet A. 2008;146A:1009–1016. doi: 10.1002/ajmg.a.32060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chung KK, Dawson VL, Dawson TM. The role of the ubiquitin-proteasomal pathway in Parkinson's disease and other neurodegenerative disorders. Trends Neurosci. 2001;24:S7–S14. doi: 10.1016/s0166-2236(00)01998-6. [DOI] [PubMed] [Google Scholar]
- 38.Lee VM, Goedert M, Trojanowski JQ. Neurodegenerative tauopathies. Annu Rev Neurosci. 2001;24:1121–1159. doi: 10.1146/annurev.neuro.24.1.1121. [DOI] [PubMed] [Google Scholar]
- 39.Taylor JP, Hardy J, Fischbeck KH. Toxic proteins in neurodegenerative disease. Science. 2002;296:1991–1995. doi: 10.1126/science.1067122. [DOI] [PubMed] [Google Scholar]
- 40.Iwahashi CK, Yasui DH, An HJ, et al. Protein composition of the intranuclear inclusions of FXTAS. Brain. 2006;12:9, 256–271. doi: 10.1093/brain/awh650. [DOI] [PubMed] [Google Scholar]; • Following purification of fragile X-associated tremor/ataxia syndrome (FXTAS) inclusions, mass spectroscopy was used to identify components of the human FXTAS inclusions. RNA-binding proteins of interest found in inclusions were hnRNPA2/B1 and MBNL1.
- 41.Mazzocco MM, Pennington BF, Hagerman RJ. The neurocognitive phenotype of female carriers of fragile X: additional evidence for specificity. J Dev Behav Pediatr. 1993;14:328–335. [PubMed] [Google Scholar]
- 42.Reiss AL, Freund L, Abrams MT, et al. Neurobehavioral effects of the fragile X premutation in adult women: a controlled study. Am J Hum Genet. 1993;52:884–894. [PMC free article] [PubMed] [Google Scholar]
- 43.Rousseau F, Heitz D, Tarleton J, et al. A multicenter study on genotype-phenotype correlations in the fragile X syndrome, using direct diagnosis with probe StB12.3: the first 2,253 cases. Am J Hum Genet. 1994;55:225–237. [PMC free article] [PubMed] [Google Scholar]
- 44.Taylor AK, Safanda JF, Fall MZ, et al. Molecular predictors of cognitive involvement in female carriers of fragile X syndrome. JAMA. 1994;271:507–514. [PubMed] [Google Scholar]
- 45.Devys D, Lutz Y, Rouyer N, et al. The FMR-1 protein is cytoplasmic, most abundant in neurons and appears normal in carriers of a fragile X premutation. Nat Genet. 1993;4:335–340. doi: 10.1038/ng0893-335. [DOI] [PubMed] [Google Scholar]
- 46.Feng Y, Lakkis L, Devys D, et al. Quantitative comparison of FMR1 gene expression in normal and premutation alleles. Am J Hum Genet. 1995;56:106–113. [PMC free article] [PubMed] [Google Scholar]
- 47.Pieretti M, Zhang FP, Fu YH, et al. Absence of expression of the FMR-1 gene in fragile X syndrome. Cell. 1991;66:817–822. doi: 10.1016/0092-8674(91)90125-i. [DOI] [PubMed] [Google Scholar]
- 48.Siomi H, Siomi MC, Nussbaum RL, et al. The protein product of the fragile X gene, FMR1, has characteristics of an RNA-binding protein. Cell. 1993;74:291–298. doi: 10.1016/0092-8674(93)90420-u. [DOI] [PubMed] [Google Scholar]
- 49.Loesch DZ, Hay DA, Sutherland GR, et al. Phenotypic variation in male-transmitted fragile X: genetic inferences. Am J Med Genet. 1987;27:401–417. doi: 10.1002/ajmg.1320270219. [DOI] [PubMed] [Google Scholar]
- 50.Snow K, Doud LK, Hagerman R, et al. Analysis of a CGG sequence at the FMR-1 locus in fragile X families and in the general population. Am J Hum Genet. 1993;53:1217–1228. [PMC free article] [PubMed] [Google Scholar]
- 51.Jacquemont S, Farzin F, Hall D, et al. Aging in individuals with the FMR1 mutation. Am J Ment Retard. 2004;109:154–164. doi: 10.1352/0895-8017(2004)109<154:AIIWTF>2.0.CO;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cronister A, Schreiner R, Wittenberger M, et al. Heterozygous fragile X female: historical, physical, cognitive, and cytogenetic features. Am J Med Genet. 1991;38:269–274. doi: 10.1002/ajmg.1320380221. [DOI] [PubMed] [Google Scholar]
- 53.Steyaert J, Borghgraef M, Gaulthier C, et al. Cognitive profile in adult, normal intelligent female fragile X carriers. Am J Med Genet. 1992;43:116–119. doi: 10.1002/ajmg.1320430117. [DOI] [PubMed] [Google Scholar]
- 54.Hull C, Hagerman RJ. A study of the physical, behavioral, and medical phenotype, including anthropometric measures, of females with fragile X syndrome. Am J Dis Child. 1993;147:1236–1241. doi: 10.1001/archpedi.1993.02160350110017. [DOI] [PubMed] [Google Scholar]
- 55.Dorn MB, Mazzocco MM, Hagerman RJ. Behavioral and psychiatric disorders in adult male carriers of fragile X. J Am Acad Child Adolesc Psychiatry. 1994;33:256–264. doi: 10.1097/00004583-199402000-00015. [DOI] [PubMed] [Google Scholar]
- 56.Sobesky WE, Pennington BF, Porter D, et al. Emotional and neurocognitive deficits in fragile X. Am J Med Genet. 1994;51:378–385. doi: 10.1002/ajmg.1320510416. [DOI] [PubMed] [Google Scholar]
- 57.Heitz D, Devys D, Imbert G, et al. Inheritance of the fragile X syndrome: size of the fragile X premutation is a major determinant of the transition to full mutation. J Med Genet. 1992;29:794–801. doi: 10.1136/jmg.29.11.794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tassone F, Hagerman RJ, Taylor AK, et al. Elevated levels of FMR1 mRNA in carrier males: a new mechanism of involvement in the fragile-X syndrome. Am J Hum Genet. 2000;66:6–15. doi: 10.1086/302720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tassone F, Hagerman RJ, Taylor AK, et al. Clinical involvement and protein expression in individuals with the FMR1 premutation. Am J Med Genet. 2000;91:144–152. doi: 10.1002/(sici)1096-8628(20000313)91:2<144::aid-ajmg14>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- 60.Kenneson A, Zhang F, Hagedorn CH, et al. Reduced FMRP and increased FMR1 transcription is proportionally associated with CGG repeat number in intermediate-length and premutation carriers. Hum Mol Genet. 2001;10:1449–1454. doi: 10.1093/hmg/10.14.1449. [DOI] [PubMed] [Google Scholar]
- 61.Tassone F, Hagerman PJ. Expression of the FMR1 gene. Cytogenet Genome Res. 2003;100:124–128. doi: 10.1159/000072846. [DOI] [PubMed] [Google Scholar]
- 62.Tassone F, Beilina A, Carosi C, et al. Elevated FMR1 mRNA in premutation carriers is due to increased transcription. RNA. 2007;13:555–562. doi: 10.1261/rna.280807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Primerano B, Tassone F, Hagerman RJ, et al. Reduced FMR1 mRNA translation efficiency in fragile X patients with premutations. RNA. 2002;8:1482–1488. [PMC free article] [PubMed] [Google Scholar]
- 64.Kozak M. Interpreting cDNA sequences: some insights from studies on translation. Mamm Genome. 1996;7:563–574. doi: 10.1007/s003359900171. [DOI] [PubMed] [Google Scholar]
- 65.Hagerman PJ, Hagerman RJ. The fragile-X premutation: a maturing perspective. Am J Hum Genet. 2004;74:805–816. doi: 10.1086/386296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Jin P, Zarnescu DC, Zhang F, et al. RNA-mediated neurodegeneration caused by the fragile X premutation rCGG repeats in Drosophila. Neuron. 2003;39:739–747. doi: 10.1016/s0896-6273(03)00533-6. [DOI] [PubMed] [Google Scholar]; • First in vivo evidence of ribo-CGG-induced neurodegeneration.
- 67.Ranum LP, Cooper TA. RNA-mediated neuromuscular disorders. Annu Rev Neurosci. 2006;29:259–277. doi: 10.1146/annurev.neuro.29.051605.113014. [DOI] [PubMed] [Google Scholar]
- 68.Davis BM, McCurrach ME, Taneja KL, et al. Expansion of a CUG trinucleotide repeat in the 3′ untranslated region of myotonic dystrophy protein kinase transcripts results in nuclear retention of transcripts. Proc Natl Acad Sci USA. 1997;94:7388–7393. doi: 10.1073/pnas.94.14.7388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Salvatori S, Fanin M, Trevisan CP, et al. Decreased expression of DMPK: correlation with CTG repeat expansion and fibre type composition in myotonic dystrophy type 1. Neurol Sci. 2005;26:235–242. doi: 10.1007/s10072-005-0466-x. [DOI] [PubMed] [Google Scholar]
- 70.Koch KS, Leffert HL. Giant hairpins formed by CUG repeats in myotonic dystrophy messenger RNAs might sterically block RNA export through nuclear pores. J Theor Biol. 1998;192:505–514. doi: 10.1006/jtbi.1998.0679. [DOI] [PubMed] [Google Scholar]
- 71.Fardaei M, Larkin K, Brook JD, et al. In vivo co-localisation of MBNL protein with DMPK expanded-repeat transcripts. Nucleic Acids Res. 2001;29:2766–2771. doi: 10.1093/nar/29.13.2766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kanadia RN, Johnstone KA, Mankodi A, et al. A muscleblind knockout model for myotonic dystrophy. Science. 2003;302:1978–1980. doi: 10.1126/science.1088583. [DOI] [PubMed] [Google Scholar]
- 73.Orengo JP, Chambon P, Metzger D, et al. Expanded CTG repeats within the DMPK 3′ UTR causes severe skeletal muscle wasting in an inducible mouse model for myotonic dystrophy. Proc Natl Acad Sci USA. 2008;105:2646–2651. doi: 10.1073/pnas.0708519105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Seznec H, Agbulut O, Sergeant N, et al. Mice transgenic for the human myotonic dystrophy region with expanded CTG repeats display muscular and brain abnormalities. Hum Mol Genet. 2001;10:2717–2726. doi: 10.1093/hmg/10.23.2717. [DOI] [PubMed] [Google Scholar]
- 75.Tassone F, Iwahashi C, Hagerman PJ. FMR1 RNA within the intranuclear inclusions of fragile X-associated tremor/ataxia syndrome (FXTAS) RNA Biol. 2004;1:103–105. doi: 10.4161/rna.1.2.1035. [DOI] [PubMed] [Google Scholar]; • First demonstration of FXTAS premutation mRNA in the ubiquitin positive intranuclear inclusions.
- 76.Jiang H, Mankodi A, Swanson MS, et al. Myotonic dystrophy type 1 is associated with nuclear foci of mutant RNA, sequestration of muscleblind proteins and deregulated alternative splicing in neurons. Hum Mol Genet. 2004;13:3079–3088. doi: 10.1093/hmg/ddh327. [DOI] [PubMed] [Google Scholar]
- 77.Mankodi A, Logigian E, Callahan L, et al. Myotonic dystrophy in transgenic mice expressing an expanded CUG repeat. Science. 2000;289:1769–1773. doi: 10.1126/science.289.5485.1769. [DOI] [PubMed] [Google Scholar]
- 78.Arocena DG, Iwahashi CK, Won N, et al. Induction of inclusion formation and disruption of lamin A/C structure by premutation CGG-repeat RNA in human cultured neural cells. Hum Mol Genet. 2005;14:3661–3671. doi: 10.1093/hmg/ddi394. [DOI] [PubMed] [Google Scholar]
- 79.Handa V, Goldwater D, Stiles D, et al. Long CGG-repeat tracts are toxic to human cells: implications for carriers of fragile X premutation alleles. FEBS Lett. 2005;579:2702–2708. doi: 10.1016/j.febslet.2005.04.004. [DOI] [PubMed] [Google Scholar]
- 80.Bontekoe CJ, Bakker CE, Nieuwenhuizen IM, et al. Instability of a (CGG)98 repeat in the Fmr1 promoter. Hum Mol Genet. 2001;10:1693–1699. doi: 10.1093/hmg/10.16.1693. [DOI] [PubMed] [Google Scholar]
- 81.Willemsen R, Hoogeveen-Westerveld M, Reis S, et al. The FMR1 CGG repeat mouse displays ubiquitin-positive intranuclear neuronal inclusions; implications for the cerebellar tremor/ataxia syndrome. Hum Mol Genet. 2003;12:949–959. doi: 10.1093/hmg/ddg114. [DOI] [PubMed] [Google Scholar]; • Demonstrates FXTAS pathology in knock-in mouse model with approximately 100 CGG repeats with elevated Fmr1 levels but no change in FMRP levels.
- 82.Van Dam D, Errijgers V, Kooy RF, et al. Cognitive decline, neuromotor and behavioural disturbances in a mouse model for fragile-X-associated tremor/ataxia syndrome (FXTAS) Behav Brain Res. 2005;162:233–239. doi: 10.1016/j.bbr.2005.03.007. [DOI] [PubMed] [Google Scholar]
- 83.Brouwer JR, Huizer K, Severijnen LA, et al. CGG-repeat length and neuropathological and molecular correlates in a mouse model for fragile X-associated tremor/ataxia syndrome. J Neurochem. 2008;107(6):1671–1682. doi: 10.1111/j.1471-4159.2008.05747.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Entezam A, Biacsi R, Orrison B, et al. Regional FMRP deficits and large repeat expansions into the full mutation range in a new fragile X premutation mouse model. Gene. 2007;395:125–134. doi: 10.1016/j.gene.2007.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]; • Expanded repeat mouse model exhibits proportional increase in repeat length and Fmr1 mRNA levels.
- 85.Hashem V, Galloway JN, Mori M, et al. Ectopic expression of CGG containing mRNA is neurotoxic in mammals. Hum Mol Genet. 2009;18:2443–2451. doi: 10.1093/hmg/ddp182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Peier AM, Nelson DL. Instability of a premutation-sized CGG repeat in FMR1 YAC transgenic mice. Genomics. 2002;80:423–432. doi: 10.1006/geno.2002.6849. [DOI] [PubMed] [Google Scholar]
- 87.Burright EN, Clark HB, Servadio A, et al. SCA1 transgenic mice: a model for neurodegeneration caused by an expanded CAG trinucleotide repeat. Cell. 1995;82:937–948. doi: 10.1016/0092-8674(95)90273-2. [DOI] [PubMed] [Google Scholar]
- 88.Clark HB, Burright EN, Yunis WS, et al. Purkinje cell expression of a mutant allele of SCA1 in transgenic mice leads to disparate effects on motor behaviors, followed by a progressive cerebellar dysfunction and histological alterations. J Neurosci. 1997;17:7385–7395. doi: 10.1523/JNEUROSCI.17-19-07385.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Klement IA, Skinner PJ, Kaytor MD, et al. Ataxin-1 nuclear localization and aggregation: role in polyglutamine-induced disease in SCA1 transgenic mice. Cell. 1998;95:41–53. doi: 10.1016/s0092-8674(00)81781-x. [DOI] [PubMed] [Google Scholar]
- 90.Zu T, Duvick LA, Kaytor MD, et al. Recovery from polyglutamine-induced neurodegeneration in conditional SCA1 transgenic mice. J Neurosci. 2004;24:8853–8861. doi: 10.1523/JNEUROSCI.2978-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Yamamoto A, Lucas JJ, Hen R. Reversal of neuropathology and motor dysfunction in a conditional model of Huntington's disease. Cell. 2000;101:57–66. doi: 10.1016/S0092-8674(00)80623-6. [DOI] [PubMed] [Google Scholar]
- 92.Jin P, Duan R, Qurashi A, et al. Purα binds to rCGG repeats and modulates repeat-mediated neurodegeneration in a Drosophila model of fragile X tremor/ataxia syndrome. Neuron. 2007;5:5, 556–564. doi: 10.1016/j.neuron.2007.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]; • Demonstrates that overexpression of Purα can suppress the riboCGG-induced neurodegeneration in flies and is also a component of fly and human inclusions.
- 93.Khalili K, Del Valle L, Muralidharan V, et al. Purα is essential for postnatal brain development and developmentally coupled cellular proliferation as revealed by genetic inactivation in the mouse. Mol Cell Biol. 2003;23:6857–6875. doi: 10.1128/MCB.23.19.6857-6875.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Johnson EM, Kinoshita Y, Weinreb DB, et al. Role of Purα in targeting mRNA to sites of translation in hippocampal neuronal dendrites. J Neurosci Res. 2006;83:929–943. doi: 10.1002/jnr.20806. [DOI] [PubMed] [Google Scholar]
- 95.Li LB, Yu Z, Teng X, et al. RNA toxicity is a component of ataxin-3 degeneration in Drosophila. Nature. 2008;453:1107–1111. doi: 10.1038/nature06909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Sofola OA, Jin P, Qin Y, et al. RNA-binding proteins hnRNP A2/B1 and CUGBP1 suppress fragile X CGG premutation repeat-induced neurodegeneration in a Drosophila model of FXTAS. Neuron. 2007;5:5, 565–571. doi: 10.1016/j.neuron.2007.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]; • Study demonstrates that interaction between hnRNPA2/B1 and riboCGG and also implicates CUGBP1 in FXTAS.
- 97.Khateb S, Weisman-Shomer P, Hershco I, et al. Destabilization of tetraplex structures of the fragile X repeat sequence (CGG)n is mediated by homolog-conserved domains in three members of the hnRNP family. Nucleic Acids Res. 2004;32:4145–4154. doi: 10.1093/nar/gkh745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Khateb S, Weisman-Shomer P, Hershco-Shani I, et al. The tetraplex (CGG)n destabilizing proteins hnRNP A2 and CBF-A enhance the in vivo translation of fragile X premutation mRNA. Nucleic Acids Res. 2007;35:5775–5788. doi: 10.1093/nar/gkm636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Ofer N, Weisman-Shomer P, Shklover J, et al. The quadruplex r(CGG)n destabilizing cationic porphyrin TMPyP4 cooperates with hnRNPs to increase the translation efficiency of fragile X premutation mRNA. Nucleic Acids Res. 2009;37:2712–2722. doi: 10.1093/nar/gkp130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Imarisio S, Carmichael J, Korolchuk V, et al. Huntington's disease: from pathology and genetics to potential therapies. Biochem J. 2008;412:191–209. doi: 10.1042/BJ20071619. [DOI] [PubMed] [Google Scholar]
- 101.Sofola OA, Jin P, Botas J, et al. Argonaute-2-dependent rescue of a Drosophila model of FXTAS by FRAXE premutation repeat. Hum Mol Genet. 2007;16:2326–2332. doi: 10.1093/hmg/ddm186. [DOI] [PubMed] [Google Scholar]
- 102.Hashem V, Galloway JN, Mori M, et al. Ectopic expression of CGG containing mRNA is neurotoxic in mammals. Hum Mol Genet. 2009;1:8, 2443–2451. doi: 10.1093/hmg/ddp182. [DOI] [PMC free article] [PubMed] [Google Scholar]; • First evidence of CGG repeats outside the context of Fmr1 to cause neurodegeneration in a mammalian system.
- 103.Louis E, Moskowitz C, Friez M, et al. Parkinsonism, dysautonomia, and intranuclear inclusions in a fragile X carrier: a clinical–pathological study. Mov Disord. 2006;21:420–425. doi: 10.1002/mds.20753. [DOI] [PubMed] [Google Scholar]
- 104.Bergink S, Severijnen LA, Wijgers N, et al. The DNA repair-ubiquitin-associated HR23 proteins are constituents of neuronal inclusions in specific neurodegenerative disorders without hampering DNA repair. Neurobiol Dis. 2006;23:708–716. doi: 10.1016/j.nbd.2006.06.005. [DOI] [PubMed] [Google Scholar]
- 105.Brouwer JR, Huizer K, Severijnen LA, et al. CGG-repeat length and neuropathological and molecular correlates in a mouse model for fragile X-associated tremor/ataxia syndrome. J Neurochem. 2008;10:7, 1671–1682. doi: 10.1111/j.1471-4159.2008.05747.x. [DOI] [PMC free article] [PubMed] [Google Scholar]; • Demonstrates the loss of intranuclear inclusion formation in expanded knock-in mice with greater than 200 CGG triplets.