

Abstract
Analysis of the complex I NDUFS8 gene from Leigh syndrome patients with isolated complex I deficiency revealed that one patient with late-onset disease and partial complex I defect was a compound heterozygote for two novel mutations in NDUFS8 gene. Western blot analysis revealed a deficiency in the NDUFS8 polypeptide, but also reductions in other nuclear subunits of complex I, suggesting that this subunit is essential for either the assembly or stability of complex I.
Leigh syndrome (LS) is one of the most common mitochondrial diseases with an early childhood onset of loss of motor and intellectual milestones, hypotonia, bilateral striatal necrosis, and early death. Deleterious mutations have been demonstrated in six of the nuclear DNA (nDNA) encoded complex I genes, most having presented with LS.1 So far, only one LS patient with a severe phenotype has been reported with deleterious mutations in the NDUFS8 gene.2
Here, we describe a LS patient with an isolated complex I defect who is a compound heterozygote for two new missense mutations in the NDUFS8 gene. However, unlike the previous report our patient has milder symptoms and a later onset.
Case report
A 9-year-old girl born to non-consanguineous parents after an uncomplicated full-term pregnancy appeared normal up to the age of 7, at which time she began intermittently walking on her toes. After 6 months she began to fall and her balance started to deteriorate. Dysarthric speech was observed at this time, although her cognitive skills remained normal. Cranial nerve examination revealed a marked horizontal and vertical nystagmus on lateral upgaze. Motor examination revealed normal strength and tone at rest, but involuntary movements of her hands and a mild degree of dystonic posture. The patient slowly worsened, falling more frequently when walking with increased dysarthria.
Urine organic acids and plasma amino acids were normal except for a mild increase in alanine. Blood and urine carnitines were in the normal range. Measurements of lactate and pyruvate concentrations in blood, urine, and CSF were also repeatedly normal. A head MRI showed abnormal bilateral, symmetric lucencies involving the posterior half of the putamen, lesions compatible with LS. Electromyography and nerve conduction studies at age 7 did not reveal any abnormalities. No known history of toxin exposure, infection, or trauma and no family history of LS were reported. Histochemistry and electron microscopy performed on the skeletal muscle were unremarkable. The muscle mitochondrial DNA (mtDNA) screening for rearrangement mutations by Southern blotting and common point mutations was negative.
Methods
Biochemistry
Mitochondria from both muscle and lymphoblastoid cell line were isolated and used for OXPHOS enzyme analyses.3
DNA analysis
Skeletal muscle and lymphoblast DNA were isolated using the Puregene DNA Isolation Kit (Gentra System, Minneapolis, MN). Oligonucleotides were designed from the NDUFS8 gene sequence (Genbank number AF038406). Primers to amplify and sequence exon 1 were 1F 5′-CACCCACGGCGAAAGCACTC-3′, 1R 5′-AAGGCCCTCCTGGAACTGTC-3′; exons 2 to 5, 2F 5′-GTGCGAGTAGAGGGCAAAGT-3′, 2R 5′-ACATCAAGCAGGGGCTAGG-3′; and exons 6 and 7, 6F 5′-GGCGACAGAGCGAGACTCTA-3′, 6R 5′-GTGGCGACCTAGTCCAGTTG-3′. Two additional primers were used for sequencing exons 6 and 7 (3F 5′-AACAGGCTCAGAGAAGAAC-3′, 4F 5′-GAGAGGTGTGGTGAGTGTA-3′). Cycle sequencing reactions were performed and separated on an ABI 377-XL DNA sequencer according to the manufacturer (Perkin-Elmer-Applied Biosystems).
Western blot analysis
Skeletal muscle and lymphoblast mitochondria were analyzed for complex I proteins by Western blot4 using antibodies provided by Prof. Lunardi (23, 49, and 51 kDa) and Prof. Capaldi (39 kDa). A monoclonal antibody directed against complex IV COI subunit was used according to the manufacturer (Molecular Probes, Eugene, OR). The anti-porin antibody (Calbiochem) used as a control was diluted 1:100,000.
Results
Respiratory chain enzyme and citrate synthase activities were assayed on mitochondria isolated from skeletal muscle and a lymphoblastoid cell line. Complex I was specifically reduced to 31% and 43% of control values after normalization by citrate synthase in muscle and lymphoblast cells. Complex IV activity was identical to controls (table).
Table.
Respiratory chain complex activities in the patient’s skeletal muscle and lymphoblastoid cell line
| Mean specific activity
|
Ratio
|
||||
|---|---|---|---|---|---|
| Complex | CI | CIV | CS | I/CS | IV/CS |
| Skeletal muscle | |||||
| Patient | 26 | 1,351 | 563 | 0.046 | 2.4 |
| Controls (n = 25), mean ± SD | 106 ± 46 | 1,583 ± 370 | 685 ± 157 | 0.15 ± 0.03 | 2.3 ± 0.01 |
| Lymphoblasts | |||||
| Patient | 20 | 1,276 | 612 | 0.032 | 2.1 |
| Controls (n = 8), mean ± SD | 50 ± 18 | 1,432 ± 343 | 669 ± 174 | 0.074 ± 0.06 | 2.1 ± 0.04 |
Complex I (CI) and IV (CIV) enzyme activities were normalized by citrate synthase (CS), a matrix enzyme. Patient activities were compared to control values for muscle and lymphoblast mitochondria. Specific enzymes activities as nmol of substrate/min/mg of mitochondrial protein.
To determine the molecular basis of the complex I defect in the proband and 22 other LS patients with isolated complex I defects, the seven exons of the NDUFS8 gene were analyzed. Only the proband of this study with late-onset LS was a compound heterozygote harboring two base substitutions in NDUFS8: a C254T which replaced a proline at position 85 with a leucine (P85L) and a G413A which replaced an arginine at position 138 with a histidine (R138H). Each parent was heterozygous for one of the two missense mutations. These mutations were absent in 100 DNA controls and were not found in any available EST databases.
The P85L amino acid substitution is upstream from the two cysteine motifs that are thought to coordinate the two [4Fe-4S] clusters, while the R138H amino acid substitution is located between these two iron sulfur clusters which have been shown to generate the complex I N2 clusters.5 Alignment of the homologous proteins to the human NDUFS8 showed that both mutant amino acid positions are extremely well conserved (figure 1). To investigate the molecular implications of the NDUFS8 mutations in our patient, we analyzed the protein levels of the 23 kDa NDUFS8 subunit as well as the 39 kDa membrane and the 49 and 51 kDa peripheral proteins of complex I by Western blot. The 23 kDa protein level was reduced in both lymphoblast and muscle mitochondria, but so were the levels of the 39, 49, and 51 kDa proteins of complex I (figure 2). The complex IV mtDNA COI subunit protein level used as a control was increased in patient muscle consistent with previous studies.6
Figure 1.

Sequence conservation of NDUFS8 between different species. Alignment of amino acid sequences from human to bacteria. Arrows indicate the mutations at the position P85L, strictly conserved in all species down through bacteria, and at the position R138H, conserved in all species except the bacterium E coli.
Figure 2.
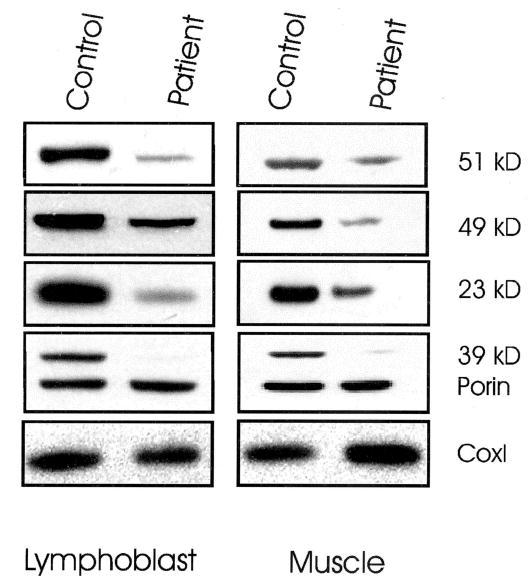
Immunodetection of nuclear-encoded subunits of complex I of mitochondrial proteins isolated from lymphoblast cell lines and skeletal muscle from the patient compound heterozygous for mutations in NDUFS8 gene and a normal age-matched control. Antibodies against the 23-, 39-, 49-, and 51-kDa subunits of complex I and against subunit I of complex IV were applied to Western blots of proteins from control and patient cell lines and skeletal muscle (10 μg protein). An antibody against porin was used as a control to normalize the amount of protein loaded. Patient shows decreased amounts of nuclear complex I subunits compared to the control. Complex IV COI subunit was increased in patient’s skeletal muscle compared to the control.
Discussion
LS usually presents during the first 1 or 2 years of life with a rapidly progressive degenerative condition associated with bilateral degeneration of the basal ganglia. However, in this article we report an unusual LS patient with a partial complex I enzyme defect and a late-onset presentation. This patient was a compound heterozygote for two new recessive mutations in the NDUFS8 subunit, P85L and R138H. Only one other LS patient has been reported with compound heterozygous mutations (P79L and R102H) in the NDUFS8 gene associated with a severe metabolic failure apparent on the first day of life and a cardiomyopathy. This patient had elevated lactate and pyruvate in blood and CSF and died at 11 weeks due to massive cardiorespiratory failure.2 The biochemical significance of the mutations was confirmed by their introduction into the Y lipolytica homologue of human NDUFS8.7
Thus, the phenotype of our NDUFS8 compound heterozygote patient was strikingly less severe than the previous case. It has been argued that mitochondrial diseases are more severe when caused by nDNA mutations than by mtDNA mutations. However, our patient had a milder phenotype than has been often observed in most LS cases associated with the mtDNA T8993G mutation in the ATP6 gene. The reduced pathogenicity of our patient’s complex I disease might be attributable to the nature of the two new missense mutations. While both mutations changed highly conserved amino acids, neither disrupted the cysteine motifs thought to be associated with the [4Fe-4S] clusters. Hence, electron transport through this subunit should be partially preserved. Moreover, our patient had reduced levels of all complex I subunits examined consistent with a proposed function for the NDUFS8 subunit in complex I assembly, specifically in connecting the membrane and peripheral domains of complex I.8 It is therefore possible that our patient had decreased respiratory complex I activity due to faulty assembly or increased instability (see figure 2). In contrast to the complex I protein, the complex IV COI subunit was elevated in the patient’s skeletal muscle. Perhaps this induction of mitochondrial gene expression is an attempt to compensate for the complex I defect.6
Assembly defects in complex I have been proposed for other patients based on steady state levels of nuclear complex I subunits determined using monoclonal antibodies.9 Our patient differed from the previous NDUFS8 compound heterozygote patient in having a drastic reduction in the 39-kDa subunit. It has been suggested that the 39-kDa subunit, like the NDUFS8 23-kDa subunit, is located at the boundary of the peripheral and the membrane domains.10 Both nuclear subunits may interact together and mutations in NDUFS8 in our patient could disrupt this interaction.
Two independent LS patients have now been described with different compound heterozygous NDUFS8 mutations. However, these patients differed markedly in the severity of their phenotype and in the stability of other complex I subunits. Hence, a more complete understanding of the structure and function of the complex I subunits will be required before accurate correlations can be made between a patient’s mutant genotype and clinical phenotype.
Footnotes
Supported by a J. Worley Brown Fellowship awarded to V.P. and NIH grant NS21324 awarded to D.C.W.
References
- 1.Triepels RH, Van Den Heuvel LP, Trijbels JM, Smeitink JA. Respiratory chain complex I deficiency. Am J Med Genet. 2001;106:37–45. doi: 10.1002/ajmg.1397. [DOI] [PubMed] [Google Scholar]
- 2.Loeffen J, Smeitink J, Triepels R, et al. The first nuclear-encoded complex I mutation in a patient with Leigh syndrome. Am J Hum Genet. 1998;63:1598–1608. doi: 10.1086/302154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Trounce IA, Kim YL, Jun AS, Wallace DC. Assessment of mitochondrial oxidative phosphorylation in patient muscle biopsies, lymphoblasts, and transmitochondrial cell lines. Methods Enzymol. 1996;264:484–509. doi: 10.1016/s0076-6879(96)64044-0. [DOI] [PubMed] [Google Scholar]
- 4.Procaccio V, Mousson B, Beugnot R, et al. Nuclear DNA origin of mitochondrial complex I deficiency in fatal infantile lactic acidosis evidenced by transnuclear complementation of cultured fibroblasts. J Clin Invest. 1999;104:83–92. doi: 10.1172/JCI6184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chevallet M, Dupuis A, Issartel JP, Lunardi J, van Belzen R, Albracht SP. Two EPR-detectable [4Fe-4S] clusters, N2a and N2b, are bound to the NuoI (TYKY) subunit of NADH:ubiquinone oxidoreductase (Complex I) from Rhodobacter capsulatus. Biochim Biophys Acta. 2003;1557:51–66. doi: 10.1016/s0005-2728(02)00398-5. [DOI] [PubMed] [Google Scholar]
- 6.Heddi A, Stepien G, Benke PJ, Wallace DC. Coordinate induction of energy gene expression in tissues of mitochondrial disease patients. J Biol Chem. 1999;274:22968–22976. doi: 10.1074/jbc.274.33.22968. [DOI] [PubMed] [Google Scholar]
- 7.Ahlers PM, Garofano A, Kerscher SJ, Brandt U. Application of the obligate aerobic yeast Yarrowia lipolytica as a eucaryotic model to analyse Leigh syndrome mutations in the complex I core subunits PSST and TYKY. Biochim Biophys Acta. 2000;1459:258–265. doi: 10.1016/s0005-2728(00)00160-2. [DOI] [PubMed] [Google Scholar]
- 8.Chevallet M, Dupuis A, Lunardi J, van Belzen R, Albracht SP, Issartel JP. The NuoI subunit of the Rhodobacter capsulatus respiratory Complex I (equivalent to the bovine TYKY subunit) is required for proper assembly of the membraneous and peripheral domains of the enzyme. Eur J Biochem. 1997;250:451–458. doi: 10.1111/j.1432-1033.1997.0451a.x. [DOI] [PubMed] [Google Scholar]
- 9.Triepels RH, Hanson BJ, van den Heuvel LP, et al. Human complex I defects can be resolved by monoclonal antibody analysis into distinct subunit assembly patterns. J Biol Chem. 2001;276:8892–8897. doi: 10.1074/jbc.M009903200. [DOI] [PubMed] [Google Scholar]
- 10.Antonicka H, Ogilvie I, Taivassalo T, et al. Identification and characterization of a common set of complex I assembly intermediates in mitochondria from patients with complex I deficiency. J Biol Chem. 2003;278:43081–43088. doi: 10.1074/jbc.M304998200. [DOI] [PubMed] [Google Scholar]