

Abstract
We describe a systematic study of the scope and relationship between ligand structure and activity for a highly efficient and selective class of catalysts for the amination of heteroaryl and aryl chlorides, bromides and iodides containing sterically hindered chelating alkylphosphines. In the presence of this catalyst, aryl and heteroaryl chlorides, bromides and iodides react with many primary amines in high yields with part-per-million quantities of palladium precursor and ligand. Many reactions of primary amines with both heteroaryl and aryl chlorides, bromides and iodides occur to completion with 0.0005-0.05 mol % catalysts. A comparison of the reactivity of this catalyst for coupling of primary amines at these loadings is made with catalysts generated from hindered monophosphines and carbenes, and these data illustrate the benefits of chelation. Thus, these complexes constitute a fourth-generation catalyst for the amination of aryl halides, whose activity complements catalysts based on monophosphines and carbenes.
Introduction
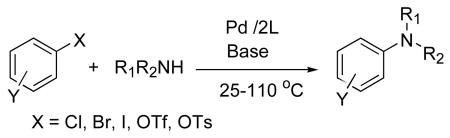
The palladium-catalyzed cross coupling of amines with aryl halides has become a principal method for the formation of C-N bonds at aromatic systems (Eq. 1).1-5 In the past 10 years, extensive effort from different groups have led to the discovery of a variety of catalysts that are capable of coupling a wide scope of amines with aryl halides under mild reaction conditions. However, previously reported catalyst loadings for this transformation were often higher than are needed to make the synthesis of pharmaceutical intermediates on large scale or bulk fine chemicals (> 0.1 mol %) economical and to make the removal of palladium from the final product simple to conduct. These factors have limited the use of the coupling of amines to produce less expensive intermediates and commodity building blocks.6,7
 |
(1) |
Three general classes of reagents react slowly and require high loading of catalyst, even with most of the most recently developed systems:8 (1) primary alkylamines,9-23 which form substantial amounts of product from diarylation in the absence of an ortho substituent on the haloarene reagent or an excess of the primary amines; (2) heteroaryl halides,11,12,15,17,19,24-32 which are important for medicinal chemistry applications, but have reacted more slowly, with narrower scope, and with higher catalyst loadings than aryl halides; and (3) aryl iodides, which react more slowly and provide lower yields than aryl bromides in couplings with amine nucleophiles,15,17,33-45 despite the higher reactivity of aryl iodides in most other palladium-catalyzed cross-coupling processes.46-49
Replacement of phosphine ligands in the catalyst by primary amine nucleophiles or by heteroaryl substrates through a basic heterocyclic nitrogen is one possible side reaction that limits the lifetime of the catalyst. Ammonia,50 pyridine50 and benzylamine51 have been shown to displace the phosphine from the arylpalladium(II) halide complex {Pd[P(o-tolyl)3](Ar)(Br)}2 (Ar = p-tol, p-tBuC6H4).50 More recently, benzylamine and pyridine have been shown do displace the phosphine from Pd[P(t-Bu)3](Ar)(Br) (Ar=o-tol, Ph) to form [Pd(pyridine)2(o-tol)Br] and [Pd(PhCH2NH2)2(Ph)Br] and free ligand.52 Apparently the high basicity of the alkylphosphine does not sufficiently stabilize the Pd(II)-P bond toward the less hindered nitrogen donors. These amine and pyridine complexes do not catalyze the reactions of amines with aryl chlorides. Therefore, a more reactive catalyst for the coupling of primary amines or heteroaryl halides might contain a bisphosphine ligand that 1) binds the metal strongly enough to prevent the ligand replacement and 2) has the properties to promote oxidative addition and reductive elimination.
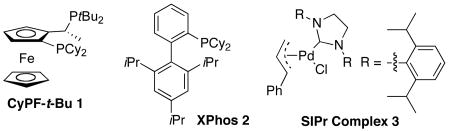
The Josiphos ligand with one di-tert-butylphopshino and one dicyclohexylphosphino group (CyPF-t-Bu)53 is now commercially available, and the ligand is stable to air and moisture (vide infra). The conformation of the backbone of this ligand is more rigid than that of many other bisphosphines because the orientation of the methyl group and the ferrocenyl group directs the phosphorus electron pairs toward the metal center. Thus, we considered that the combination of the severe steric hindrance, conformational preferences of the backbone and strong electron donation of this Josiphos ligand could create a catalyst for the amination of heteroaryl halides that would be reactive, but also long lived.36,54
In a preliminary communication,52 we showed that the complex generated from this ligand coupled primary nitrogen nucleophiles with heteroaryl and aryl chlorides with catalyst loadings of only 10 to 100 ppm. Here, we report a broad range of studies of C-N couplings catalyzed by a combination of Pd(OAc)2 and CyPF-t-Bu. We show that the coupling occurs in high yield and with high turnover numbers with alkylamines and some arylamines. Comparisons of the activity of this catalyst to those of catalysts containing monophosphines and carbenes show the value of the Pd(OAc)2 and CyPF-t-Bu system for the reactions of primary amines, and studies of derivatives of this ligand illustrate the importance of the conformational rigidity to obtain high activity.
Results and Discussion
A. Identification of Conditions for Coupling of Chloropyridines with ppm-Levels of Palladium
To begin to assess the activity of catalysts generated from a palladium precursor and hindered Josiphos ligands, we focused on the challenging reaction of the unactivated heteroaryl chloride 3-chloropyridine with an unhindered primary amine. This reaction was studied in the presence of palladium catalysts containing equimolar amounts of the Josphos ligand CyPF-t-Bu and a palladium precursor in a variety of solvents, with different bases at various temperatures. The yields of the desired N-octyl-3-aminopyridine were evaluated after 10 h by GC analysis.
Reactions catalyzed by complexes derived from Pd(dba)2 and PdCl2(PhCN)2 occurred over a similar time and in similar yields. However, reactions catalyzed by the complex generated from Pd(OAc)2 were much faster than those generated from Pd(dba)2. The reaction catalyzed by the combination of Pd(OAc)2 and CyPF-t-Bu was even complete after 1 h with only 0.05 mol% catalyst. We attribute this difference in reactivity to the slow dissociation of dba from complexes of the type (chelate)Pd(dba).55 Reactions in the presence of NaO-t-Bu occurred in high yield, while reactions of this heteroaryl chloride with weaker bases, such as Cs2CO3, K2CO3, and K3PO4, led to no products or low conversions. (For reactions of more activated aryl halides with the weaker bases see section I.2.) Reactions conducted with NaO-t-Bu as base in DME (dimethoxyethane) were faster than those in toluene, THF, and 1,4-dioxane, but the coupling reaction did occur in these relatively non-polar, aprotic solvents, and useful procedures for coupling reactions in toluene were developed (vide infra). Reactions in other more polar solvents, such as DMF and DMSO, formed only trace amounts of the desired products.
Studies to determine the efficiency of the catalyst showed that the reactions of 3-chloropyridine with octylamine in DME occurred to completion after 24 h at 90 °C in 93% isolated yield with only 50 ppm of catalyst. These data correspond to a turnover number of 18,600. Reactions conducted with 10 ppm of Pd(OAc)2 and ligand occurred to partial (65%) conversion at 100 °C after 48 h. Reactions in toluene were slower but did occur to completion after 24 h at 90 °C with 0.05 mol% of catalyst and to 80% conversion with only 0.01 mol% catalyst.
It was important to mix the catalyst components before they contacted the reagents or base. Although an effect of premixing the catalyst has been noted previously,56 it is particularly important in the current work because the concentrations of catalyst and ligand are so low. At such low concentrations, the rate of complexation of the ligand to palladium is expected to be slow. Without performing the complex, Pd(OAc)2 would be expected to decompose in the presence of amine and base to form palladium black. Thus, the reactions were conducted by first mixing 1.0 M solutions of Pd(OAc)2 and CyPF-t-Bu without any added reagents or base. This solution was then diluted, and a portion of the solution was added to the haloarene and base in DME prior to addition of amine.
B. Scope of the Amination of Heteroaryl and Aryl Chlorides, Bromides and Iodides with Primary Alkylamines
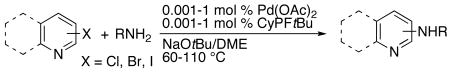
Reactions of heteroaryl chlorides, bromides and iodides with primary alkylamines are summarized in Table 1. The reaction conditions optimized for the amination of aryl chlorides were also effective for the amination of aryl bromides and iodides. Generally, the rates for amination of heteroaryl bromides were slightly faster than those for amination of heteroaryl chlorides, and equal or slightly lower loadings of catalyst could be used. In contrast, the rates for amination of heteroaryl iodides were slower than those for amination of heteroaryl bromides and more catalyst was needed, but reactions of aryl iodides occurred with much greater efficiency than with previous catalysts33,36,38,41 and with sufficient scope and efficiency to create a useful process.
Table 1.
Coupling of Heteroaryl Halides with Primary Alkylamines Catalyzed by Pd(OAc)2 and CyPF-t-Bu (1:1).a
 | ||||||
|---|---|---|---|---|---|---|
| Entry | Ar | X | R | Cat. (%) |
Conditions | Yield (%)b |
| 1 | 2-Py | Cl | Octyl | 0.001 | 100 °C, 48 h | 86 |
| 2 | Br | Octyl | 0.0005 | 110 °C, 12 h | 84 | |
| 3 | I | Octyl | 0.005 | 110 °C, 12 h | 96 | |
| 4 | Cl | Bn | 0.001 | 100 °C, 10 h | 85 | |
| 5 | Br | Bn | 0.0005 | 110 °C, 12 h | 83 | |
| 6 | Cl | Cyclohexyl | 0.01 | 70 °C, 12 h | 98 | |
| 7 | Br | Cyclohexyl | 0.005 | 60 °C, 12 h | 96 | |
| 8 | I | sec-Bu | 0.05 | 110 °C, 12 h | 98 | |
| 9d | Cl | 1-Methylbenzylf | 0.05 | 100 °C, 24 h | 99 | |
| 10 | 3-Me-2-Py | Cl | Octyl | 0.005 | 90 °C, 24 h | 96 |
| 11 | 3-Py | Cl | Octyl | 0.005 | 90 °C, 24 h | 93 |
| 12c | Cl | Octyl | 0.005 | 100 °C, 24 h | 99 | |
| 13 | Br | Octyl | 0.005 | 100 °C, 36 h | 99 | |
| 14 | Cl | Bn | 0.01 | 100 °C, 24 h | 95 | |
| 15 | Br | Bn | 0.005 | 100 °C, 48 h | 99 | |
| 16 | Br | secBu | 0.005 | 100 °C, 48 h | 93 | |
| 17 | I | isoBu | 0.05 | 100 °C, 12 h | 96 | |
| 18 | Cl | Cyclohexyl | 0.01 | 100 °C, 48 h | 79 | |
| 19 | I | Cyclohexyl | 0.05 | 100 °C, 12 h | 78 | |
| 20 | Cl | tert-Bu | 1.0 | 70 °C, 16 h | 67 | |
| 21d | Cl | 1-Methylbenzylg | 0.05 | 100 °C, 24 h | 91 | |
| 22 | 4-Py | Cl | Octyl | 0.01 | 90 °C, 24 h | 83 |
| 23 | Br | Octyl | 0.005 | 100 °C, 3 6h | 93 | |
| 24 | I | Octyl | 0.05 | 100 °C, 48 h | 80 | |
| 25 | Cl | Phenylcarbonyl | 1.0 | 70 °C, 10 h | 75 | |
| 26 | 2-Pyrazinyl | Cl | Octyl | 0.005 | 100 °C, 16 h | 82 |
| 27 | I | Octyl | 0.05 | 100 °C, 36 h | 83 | |
| 28e | 1,3-Pyrimidyl-5- | Br | Cyclohexyl | 1.0 | 100 °C, 48 h | 80 |
| 29 | 3-Quinolinyl | Cl | Cyclohexyl | 0.01 | 100 °C, 48 h | 60 |
| 30 | 1-isoQuinolinyl | Cl | Octyl | 0.005 | 90 °C, 15 h | 91 |
| 31 | 4-isoQuinolinyl | Br | Octyl | 0.005 | 100 °C, 36 h | 93 |
Reactions conducted with a 1:1 ratio of metal to ligand, 1 mmol aryl halide, 1.2 equiv amine, and 1.4 equiv NaOtBu in 1 mL DME.
Isolated yield;
Reaction performed without using a drybox.
from phenethylamine that is stated to be 99% ee.
Using K3PO4 as the base.
96% ee.
95% ee.
Pyridyl chlorides, bromides and iodides bearing the halogen in the 2, 3, or 4-position underwent reaction with a variety of primary alkylamines in high yield with loadings of catalyst between 5 and 500 ppm (Table 1, entries 1-19, 21-24 and 26-31). For example, the reaction of octylamine with 2-bromopyridine occurred with only 5 ppm of the catalyst at 60 °C for 48 h to afford 98% of the product (Table 1, entry 2). These results correspond to a turnover number of 196,000, which is the highest turnover number observed for any palladium-catalyzed amination of an aryl halide. The average turnover frequency is roughly 4000/h. The analogous process with chloride and iodide leaving groups occurred in high yield (entries 1 and 3). Other comparative data in this table consistently show that the yield and turnover numbers are as high or higher for reactions of aryl bromides as for aryl chlorides, and that high yields are obtained for reactions of aryl iodides with only 5-10 times more catalyst. Not only reactions of pyridyl halides, but reactions of quinolinyl, isoquinolinyl and pyrazyl chlorides, bromides and iodides occurred in high yield with catalyst loadings (Table 1, entries 26-27 and 29-31) that are nearly two to three orders of magnitude lower than those reported for the reaction of pyridyl halides with any primary amine.12 As one comparison, the coupling of benzylamine with 2-chloropyridine occurred in 85% yield with only 10 ppm of the catalyst, corresponding to a turnover number of 85,000 (Table 1, entry 4), whereas the same reaction conducted with 2-(di-t-butylphosphino)biphenyl occurred with 196 turnovers.12
The steric hindrance of the catalyst did not prohibit reactions of more sterically hindered primary amines. tert-Butylamine reacted in good yield with 3-chloropyridine (Table 1, entry 20), although 1.0 mol % of palladium was needed. The reactions of less hindered α-branched primary amines, such as cyclohexylamine and sec-butylamine, occurred in high yield with only 0.005-0.05 mol % palladium (Table 1, entry 6-9, 16, 18-19, 21, 29). This loading is more than an order of magnitude lower than that of previous reactions of α-branched primary amines with the corresponding haloarenes.
Enantiomerically enriched amines containing stereogenic centers α to the nitrogen have been shown to undergo racemerization in competition with cross-coupling when Pd(0) and monodendate ligands, such as P(o-tolyl)3 or PPh3, were used as the catalyst.57,58 This racemerization is proposed to result from reversible β-hydrogen elimination, which should occur more readily from complexes containing monophosphine ligands than from those containing bisphosphine ligands.59,60 Consistent with this hypothesis, the coupling of 2- or 3-chloropyridine with enantioenriched (99% ee) α-phenethylamine gave the heteroarylamine products in high yield with > 95% ee in the presence of only 0.05 mol % of the catalyst (Table 1, entries 9 and 21).
In addition to the remarkable reactivity and stability of this catalyst, it is highly chemoselective for monoarylation of primary amines. Although noted above Table 1, it is important to emphasize that no diarylation products were observed in any case by GC/MS.
Finally, this coupling is not particularly sensitive to oxygen. CyPF-t-Bu is air stable, both as a solid and in solution. Samples left in air as solids or as solutions in toluene or DME for at least 24 h were unchanged, as determined by 1H NMR and 31P NMR spectroscopy. Thus, reactions conducted with Pd(OAc)2 and CyPF-t-Bu can be assembled easily outside of a drybox. Identical catalytic activity was observed for reactions performed in a dry box or under inert atmosphere using common Schlenk techniques. For example, the reaction of 3-chloropyridine with octylamine assembled inside a drybox with 0.005 mol % of the catalyst occurred in 95% yield, and the same reaction conducted outside of a drybox using Schlenk techniques occurred in 99% yield.
Considering that these heteroaryl systems are electron-poor, the coupling reactions might occur, at least in part, by uncatalyzed processes. Thus, reactions with and without catalyst were conducted in parallel.61 Only reactions of quinolines and isoquinolines generated any measurable quantity of products. Conversions were below 40%, even at 100 °C for 48 h, in these cases. Reactions of 2- and 4-chloropyridines occur more readily in polar solvents or under high pressure, but are typically conducted at temperatures closer to 150 °C.62 Nucleophilic substitution of 3-choropyridines are often 104-105 slower than those of 2- and 4-chloropyridines,63-65 and we obtained less than 10% product from the uncatalyzed reaction of 3-chloropyridine with octylamine in DME and less than 20% from the uncatalyzed reaction in DMSO after 24 h at 110 °C.
C. Comparison to the Amination of Heteroaryl Chlorides Catalyzed by Complexes of CyPF-t-Bu to Reactions Catalyzed by Complexes of Xphos And SiPr
Palladium catalysts based on dialkylphosphinobiaryl ligands developed by Buchwald and coworkers are highly active for the coupling of amines with aryl halides.12,13,32 Recently, this group compared the two catalyst systems for the reaction of hexylamine with 4-chlorophenol,32 in part, to assess the importance of chelation on the amination of heteroaryl systems. This reaction conducted with the catalyzed based on Xphos occurred with faster rates and in higher yield than the one we reported using CyPF-t-Bu as ligand. However, this example required the highest catalyst loading of any of the reactions we published using CyPF-t-Bu, and this work made no comparisons of catalysts lifetimes.
Our interest in the use of sterically hindered chelating ligands for the C-N coupling has focused on identifying catalysts that are reactive, but also long-lived.52 To address this issue more specifically, we have now compared the yields and turnover numbers for reactions of primary amines with 3-chloropyridine using low loadings of several different catalysts. We previously emphasized that palladium catalysts generated from N-heterocyclic carbene ligands form N,N-dialkylaminopyridines from secondary amines and halopyridines with high turnover numbers.11 Thus, we also tested reactions of primary amines with 3-chloropyridine using the allylpalladium halide adduct of this ligand (NHC)Pd(R-allyl)Cl, which Nolan recently reported to be highly efficient for some combinations of aryl halides and amines.31,66
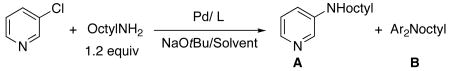
Table 2 provides a comparison of the activity and selectivity of catalysts containing CyPF-t-Bu 1, the most recently developed biaryl phosphine XPhos 2, and the catalyst containing the SiPr carbene 3 for the reaction of 3-chloropyridine with ocylamine. The data in this table correspond to reactions under the reported optimal conditions for each of the catalyst systems.12,66 Reaction of 3-chloropyridine with 1.2 equiv of octylamine in the presence of 0.005 mol % of Pd(OAc)2/CyPF-t-Bu afforded a 92% yield of the N-octyl aminopyridine with no side product from diarylation that could be detected by GC/MS or 1H NMR spectroscopy. In contrast, the reaction conducted with an order of magnitude more catalyst from X-phos ligand 2 (0.05 mol %) led to less than 40% conversion. This reaction with 100-200 times more catalyst (0.1-0.5 mol %) occurred to full conversion, but the selectivity for monoarylation vs. diarylation was only 2.4:1-2.6:1. Consistent with the published work,12 reactions in DME were much slower than those in toluene. Reactions conducted with Pd(dba)2, instead of Pd(OAc)2, occurred with similar selectivity (3.0:1-4.8:1).
Table 2.
Comparison of the Activity of CyPF-t-Bu, XPhos, and SIPr for the Reactions of a Heteroaryl Chloride with a Primary Alkylamine.a
 | |||||||
|---|---|---|---|---|---|---|---|
| Entry | Pd/L | Loading | Solvent | T[°C]b | t [ h] | Conversionc (%) |
A/B |
| 1 | Pd(OAc)2/1 | 0.005 | DME | 90 | 24 | 100(92) | 100:0 |
| 2 | Pd(OAc)2/2 | 0.05 | Toluene | 90 | 24 | 40 | 5.4:1 |
| 3 | Pd(OAc)2/2 | 0.1 | Toluene | 90 | 24 | 85(80) | 2.4:1 |
| 4 | Pd(OAc)2/2 | 0.1 | DME | 90 | 24 | 14 | - |
| 5 | Pd(dba)2/2 | 0.1 | Toluene | 90 | 24 | 47 | 4.8/1 |
| 6 | Pd(OAc)2/2 | 0.5 | Toluene | 90 | 12 | 88(81) | 2.6:1 |
| 7 | Pd(OAc)2/2 | 0.5 | DME | 90 | 24 | 74 | 4.1:1 |
| 8 | Pd(dba)2/2 | 0.5 | Toluene | 90 | 12 | 83 | 3.0/1 |
| 9 | 3 | 0.005 | DME | 110 | 24 | <10 | - |
| 10 | 3 | 0.05 | DME | 110 | 48 | 58 | >25:1 |
| 11 | 3 | 0.5 | DME | 110 | 12 | 100(71) | 7.6:1 |
 | |||||||
The experiments were conducted with a 1:1 ratio of Pd/CyFP-t-Bu or 1:2 ratio of Pd/XPhos, 1 mmol of 3-chloropyridine and 1.2 equiv 1-octylamine, and 1.4 equiv base in 1.0 mL solvent.
Bath temperature.
Determined by 1H NMR analysis of the crude product. Isolated yields are indicated in parentheses.
Likewise, reactions of 3-chloropyridine with 1.2 equiv of octylamine in the presence of 0.005 mol % (NHC)Pd(R-allyl)Cl (3) occurred to less than 50% conversion. The same reaction in the presence of a higher loading of (NHC)Pd(R-allyl)Cl occurred to full conversion, but with moderate selectivity (7.6:1) for formation of monoarylation vs. diarylation products. The published reactions were conducted with KOtBu as base; in our hands, these reactions were faster when conducted with NaOtBu as base, and the data from these faster reactions are reported in Table 2.
These results demonstrate that catalysts generated from CyPF-t-Bu ligand 1 are particularly effective for the coupling of heteroaryl chlorides with primary alkylamines. In combination with the previous data comparing different catalysts for the reaction of 4-chlorophenol,32 our data also illustrate that different catalysts possess different beneficial properties, and the optimal catalyst often depends on the synthetic context.
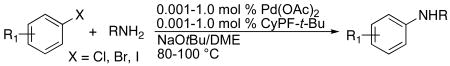
D. Scope of the Amination of Aryl Chlorides, Bromides and Iodides with Primary Alkylamines
Reactions of aryl chlorides, bromides and iodides with primary alkylamines occurred in high yield without changing the reaction conditions from that of entry 11 in Table 1. The reactions of a series of aryl halides with several primary alkylamines in the presence of NaO-t-Bu base and the catalyst generated from Pd(OAc)2 and CyPF-t-Bu 1 are summarized in Table 3. In general, high yields of coupled products were formed from the amination of electron-deficient and electron-neutral aryl halides when using only 10-500 ppm of catalyst. Higher catalyst loadings (0.1-1.0 mol %) were required to obtain high yields of products from the amination of electron-rich aryl halides, but these reactions also occurred in high yields.
Table 3.
Coupling of Aryl Halides with Primary Alkylamines Catalyzed by Pd(OAc)2 and CyPF-t-Bu (1:1).a
 | ||||||
|---|---|---|---|---|---|---|
| Entry | R1 | X | R | Cat (%) |
Conditions | Yield (%)b |
| 1 | H | Cl | Cyclohexyl | 0.05 | 100 °C, 36 h | 99 |
| 2 | Br | Cyclohexyl | 0.01 | 100 °C, 24 h | 96 | |
| 3 | Cl | Bn | 0.005 | 100 °C, 48 h | 99 | |
| 4 | Br | Bn | 0.001 | 100 °C, 36 h | 97 | |
| 5 | 2-Me | Cl | Octyl | 0.01 | 100 °C, 48 h | 98 |
| 6 | Br | Octyl | 0.005 | 100 °C, 36 h | 99 | |
| 7 | I | Octyl | 0.05 | 100 °C, 8 h | 96 | |
| 8 | 2-MeO | Br | Cyclohexyl | 0.05 | 100 °C, 24 h | 94 |
| 9 | 4-Me | Cl | isoBu | 0.1 | 100 °C, 48 h | 83 |
| 10c | Br | isoBu | 0.05 | 100 °C, 24 h | 90 | |
| 11 | I | isoBu | 0.05 | 100 °C, 18 h | 75 | |
| 12d | I | isoBu | 0.02 | 100 °C, 36 h | 81 | |
| 13 | Br | secBu | 0.05 | 100 °C, 24 h | 87 | |
| 14 | I | secBu | 0.5 | 100 °C, 24 h | 82 | |
| 15 | 4-MeO | Cl | Octyl | 0.1 | 100 °C, 48 h | 92 |
| 16 | I | Octyl | 1.0 | 100 °C, 36 h | 67 | |
| 17 | 4-Cyano | Cl | isoBu | 0.005 | 80 °C, 24 h | 99 |
| 18 | Br | Cyclohexyl | 0.005 | 80 °C, 48 h | 92 | |
| 19 | 3-MeO | Cl | Bn | 0.005 | 80 °C, 48 h | 98 |
| 20 | I | Bn | 0.005 | 100 °C, 48 h | 99 | |
| 21 | Br | Cyclohexyl | 0.01 | 100 °C, 36 h | 99 | |
| 22 | I | Cyclohexyl | 0.05 | 100 °C,18 h | 94 | |
| 23 | Naphathyl | I | Octyl | 0.05 | 100 °C, 8 h | 97 |
| 24 | 2-isoPr | Br | isoBu | 0.05 | 100 °C, 48 h | 95 |
| 25 | 2,6-Di-Me | Cl | Octyl | 0.1 | 100 °C, 36 h | 97 |
| 26 | Br | Octyl | 0.05 | 100 °C, 48 h | 98 | |
| 27 | Br | secBu | 0.5 | 100 °C, 24 h | 97 | |
Reactions conducted with a 1:1 ratio of metal to ligand 1 mmol ArX (X = Cl, Br, I), 1.2 equiv amine and 1.4 equiv NaOtBu in 1 mL DME.
Isolated yields.
3.0 equivalent of octylamine used.
Reaction with 5.5 g of 4-iodotoluene (25.0 mmol).
Reactions of linear primary amines were fast and occurred in high yield with 0.005-0.1 mol % catalyst (Table 3, entries 3-7, 9-12, 19-20, 23-24). For example, reaction of benzylamine with bromobenzene and chlorobenzene occurred to completion in 97% and 99% isolated yield with only 10 and 50 ppm of catalyst (Table 3, entries 4 and 3), corresponding to turnover numbers of 97,000 and 19,800. Reactions of several other haloarenes occurred in similarly high yield with a similar amount of catalyst. The maximum turnover numbers for the reactions of primary alkylamines with aryl chlorides exceeded those achieved previously for reactions of this type by nearly two orders of magnitude.12 They also exceeded the maximum turnover numbers for the reactions of primary alkyl amines with aryl bromides by nearly an order of magnitude.56
High yields were also obtained for reactions of aryl iodides with linear primary amines in the presence of just 5-10 times more catalyst than needed for reactions of aryl bromides and chlorides. For instance, the reaction of octylamine with 2-bromotoluene and 2-iodotoluene occurred to completion with 0.005 mol % and 0.05 mol% of catalyst and formed the coupled product in 99% and 96% yield, respectively (Table 3, entries 6 and 7). Reactions of several other iodoarenes occurred in similarly high yield with similar amounts of catalyst. These reactions exceeded the maximum turnover numbers for the reactions of primary alkyl amines with aryl iodides by more than an order of magnitude.35
Hindered, ortho-substituted aryl halides also reacted with primary alkylamines with low catalyst loadings (Table 3, entries 5-8, 24). For example, the reaction of 1-bromo-2-iso-propylbenzene with iso-butylamine formed the coupled product in 95% yield in DME after 48 h at 100 °C in the presence of 0.05 mol % catalyst, and the reaction of 2,6-disubstituted aryl bromides and chlorides occurred with octylamine with only 0.05-0.1 mol % catalyst. However, reactions of 2,6-disubstituted aryl bromides with α-branched primary alkylamines required a higher 0.5 mol % catalyst (Table 3, entry 27) to occur in high yield. The most hindered bromoarenes occurred with lower loadings in the presence of catalysts generated from monodentate ligands. Nolan reported that the reaction of benzylamine with 2,6-dimethyl-1-bromobenzene at 80 °C using just 0.001 mol % of a carbene-ligated allyl-Pd complex occurred to form 88% yield of coupled product.66 Reactions of less hindered bromoarenes and of aliphatic amines with palladium-carbene ctalysts are either unreported66 or require loadings near 1 mol %.16
Reactions of α-branched primary amines were somewhat slower and required more catalyst than those of linear primary amines, but the required loadings remained lower than those used previously. For example, the reaction of cyclohexylamine with chlorobenzene occurred in high yield with 0.05 mol % palladium (TON = 1980) (Table 3, entry 2), and this loading of catalyst is more than an order of magnitude lower than that of previously reported reactions of α-branched primary amines with chloroarenes.12 Reactions of the acyclic, α-branched sec-butylamine occurred in high yield with 0.01-1.0 mol % of the catalyst.
Like the coupling of primary amines with heteroaryl halides, the coupling or primary amines with electron-poor or neutral aryl halides occurred without the formation of diarylamines in amounts detectable by GC/MS. Products from diarylation were observed by GC/MS only for the reaction of octylamine with chloroanisole and iodoanisole.12 However, the diarylation side- product was reduced to 5% if excess of octylamine (3.0 equiv) was used.
To develop a better understanding of the reactivity of the aryl chlorides, bromides and iodides, several competition experiments were conducted that would reveal the origin of the differences in relative rates. In the first experiment, equimolar amounts of 4-bromotoluene and 4-chlorotoluene were allowed to react with 1.0 equiv of octylamine in the presence of 0.01 mol % of the catalyst. As shown in Scheme 1, more than 95% conversion of the bromide was observed after 10 h at 110 °C, while less than 5% conversion of the chloride occurred. In a separate experiment, the reaction of 1-bromo-4-chlorobenzene with octylamine at 110 °C for 24 h occurred to full conversion to afford N-octyl-4-chloroaniline in 95% yield. No N-octyl-4-bromoaniline was detected by GC/MS. Thus, the catalyst generated from Pd(OAc)2 and CyPF-t-Bu is highly reactive toward chloroarenes, but it maintains the conventional high selectivity for reaction of a bromoarene over a chloroarene.
Scheme 1.

Comparison of the Reactivity of Aryl Chlorides vs. Aryl Bromides and Aryl Bromides vs. Aryl Iodides in the Presence of Pd(OAc)2 and CyPF-t-Bu (1:1).
Reagents and conditions: a Isolated Yield. i. 0.01 mol % Pd(OAc)2, 0.01 mol % CyPF-t-Bu, 1.4 equiv. NaOtBu, DME, 100 °C, 10 h; ii. 0.005 mol % Pd(OAc)2, 0.005 mol % CyPF-t-Bu, 1.4 equiv. NaOtBu, DME, 100 °C, 20 h; iii. 0.05 mol % Pd(OAc)2, 0.05 mol % CyPF-t-Bu, 1.4 equiv. NaOtBu, DME, 100 °C, 20 h; 0.5 mol % Pd(OAc)2, 0.5 mol % CyPF-t-Bu, 1.4 equiv. NaOtBu, DME, 100 °C, 20 h.
Similar experiments showed that the aryl iodide functionality was more reactive than the aryl bromide. Although this greater reactivity of an iodoarene is typically observed for palladium-catalyzed cross-coupling, the greater reactivity of aryl bromides than aryl iodides toward primary amines in separate vessels with the current catalyst made it unclear whether this order of reactivity would be observed when the two halides were present in the same system. Competition studies reveal an inhibitory effect of the iodide.
In one experiment, equimolar amounts of 4-iodotoluene and 4-bromotoluene were allowed to react with 1.0 equiv of octylamine in the presence of 0.05 mol % of the catalyst. Although bromotoluene alone reacted to full conversion with octylamine in much less than 20 h in the presence of 0.05% catalyst, little bromotoluene reacted in the system containing iodotoluene. As shown in Scheme 1, 66% conversion of the iodide was observed after 20 h at 100 °C, while less than 6% conversion of the bromide was observed. Likewise, the reaction of 1-bromo-4-iodobenzene with octylamine at 110 °C for 20 h with the same amount of catalyst occurred to 57% conversion after 20 h at 100 °C, and N-octyl-4-bromoaniline was the exclusive product. Thus, the catalyst was fully selective for reactions of aryl iodides over aryl bromides, but the presence of the aryl iodide inhibits the reaction of the aryl bromide. Although further data are needed to reveal the origin of this inhibitory effect, we presume that the alkali metal iodide product, not the iodoarene itself, retards the rate of the reaction. Consistent with this hypothesis, the addition of NaI to the reaction of bromotoluene in DME retarded the reaction.
E. Comparison to the Amination of Aryl Halides Catalyzed by Complexes of CyPF-t-Bu to Reactions Catalyzed by Complexes of Xphos And SiPr
A comparison of the reactivity of catalysts generated from CyPF-t-Bu, X-phos, and SiPr for the coupling of octylamine with a prototypical chlorarene is provided in Table 4. These studies again showed the unusually long lifetimes and high monoarylation selectivity of the Pd-CyPF-t-Bu system. Reactions of 4-chlorotoluene with 1.2 equiv of octylamine in the presence of 0.01 mol % of Pd(OAc)2/CyPF-t-Bu afforded 95% yields of the N-octyltoluidine without detectable amounts of side products from diarylation. In contrast, the same reaction conducted with 0.01 mol% of the catalyst generated from X-phos ligand 2 occurred to less than 25% conversion. This reaction conducted with higher loadings of Pd(OAc)2 and X-phos (0.5 mol %) occurred to full conversion, but formed only a 2.7:1 ratio of monoarylation to diarylation product. Reactions conducted with this ligand and Pd(dba)2 instead of Pd(OAc)2 as palladium precursor occurred with similar selectivity (2.3:1). Reactions of 4-chlorotoluene with 1.2 equiv of octylamine in the presence of 0.005 mol % (NHC)Pd(R-allyl)Cl 3 occurred to less than 50% conversion. The same reaction in the presence of a 0.5 mol % of (NHC)Pd(R-allyl)Cl occurred to full conversion, but formed a 4.3:1 ratio of products from monoarylation and diarylation.
Table 4.
Comparison of the Activity of CyPF-t-Bu, XPhos, and SiPr for the Reactions of 4-Chlorotoluene with a Primary Alkylamine.a
 | |||||||
|---|---|---|---|---|---|---|---|
| Entry | Ligand | Loading | Solvent | T[°C]b | t [ h] | Conversionc (%) |
A:B |
| 1 | Pd(OAc)2/1 | 0.01 | DME | 100 | 48 | 100(95) | 100:0 |
| 2 | Pd(OAc)2/2 | 0.05 | Toluene | 100 | 48 | 13 | 18:1 |
| 3 | Pd(dba)2/2 | 0.1 | Toluene | 100 | 48 | 25 | 10:1 |
| 4 | Pd(OAc)2/2 | 0.1 | Toluene | 100 | 48 | 16 | 39:1 |
| 5 | Pd(OAc)2/2 | 0.5 | Toluene | 100 | 12 | 96(91) | 2.7:1 |
| 6 | Pd(dba)2/2 | 0.5 | Toluene | 100 | 12 | 100(93) | 2.3:1 |
| 7 | 3 | 0.005 | Toluene | 110 | 48 | 40 | >50:1 |
| 8 | 3 | 0.05 | Toluene | 110 | 48 | 76 | 15.3:1 |
| 9 | 3 | 0.5 | Toluene | 110 | 12 | 100 | 4.3:1 |
 | |||||||
The experiments were conducted with a 1:1 ratio of Pd/CyFP-t-Bu or 1:2 ratio of Pd/XPhos, 1 mmol of 4-chlorotoluene and 1.2 equiv 1-octylamine, and 1.4 equiv base in 1.0 mL solvent.
Bath temperature.
Determined by 1H NMR analysis of the crude product. Isolated yields are indicated in parentheses.
F. Catalytic Amination of Heteroaryl and Aryl Chlorides, Bromides and Iodides with Primary Amines in Toluene
In some settings, toluene would be preferred over DME as solvent. The reactions of a series of heteroaryl and aryl halides with several primary amines in toluene in the presence of Pd(OAc)2 and CyPF-t-Bu are summarized in Table 5. These reactions occurred in similarly high yields as the reactions in DME. These reactions were somewhat slower than those in DME, and the yields in Table 5 were obtained with two to ten times more catalyst than those in DME. Nevertheless, these loadings are one to two orders of magnitude lower than those reported for the corresponding reaction of an aryl halide with any primary amine.12,56 Although speculative at this stage, we have considered that the ability of DME to ligate an alkali metal might make the alkoxide base stronger and increase the rate of formation of the amido intermediate. Alternatively, the properties of the solvent and base could affect the relative concentration of active and dormant complexes in the system.
Table 5.
Coupling of Heteroaryl and Aryl Halides with Primary Amines Catalyzed by Pd(OAc)2 and CyPF-t-Bu (1:1) in Toluene.a
 | |||||||
|---|---|---|---|---|---|---|---|
| Entry | Ar | X | R | Cat (%) |
Yield (%)b |
||
| 1 | 2-Py | Cl | Octyl | 0.01 | 98 | ||
| 2 | 3-Py | Cl | Octyl | 0.05 | 98 | ||
| 3 | 3-Py | Cl | 4-Tolylamino | 0.1 | 66 | ||
| 4 | Ph | Cl | Octyl | 0.05 | 99 | ||
| 5 | 4-Tol | Cl | isoBu | 0.02 | 99 | ||
| 6 | 2-Py | Br | Octyl | 0.01 | 92 | ||
| 7 | 3-Py | Br | Bn | 0.01 | 99 | ||
| 8 | Ph | Br | Bn | 0.005 | 99 | ||
| 9 | 3-Py | I | isoBu | 0.2 | 99 | ||
| 10 | 2-Tol | I | Cyclohexyl | 0.05 | 93 | ||
Reactions conducted with a 1:1 ratio of metal to ligand, 1 mmol ArX (X = Cl, Br, I), 1.2 equiv amine and 1.4 equiv NaOtBu in 1 mL Toluene.
isolated yield.
G. Scope of the Amination of Heteroaryl and Aryl Chorides, Bromides and Iodides with Primary Arylamines
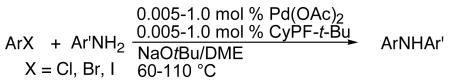
The reactions of primary arylamines with aryl halides are simpler to catalyze than those of primary alkylamines because β-hydrogen elimination does not compete with reductive elimination, and many palladium complexes catalyze the couplings of aryl halides with arylamines. However, catalyst loadings are often quite high, and diarylation of the aniline can be a competing process. To address issues of catalyst lifetime and selectivity, we have studied the reactions of heteroaryl and aryl chlorides, bromides and iodides with primary aromatic amines catalyzed by the combination of Pd(OAc)2 and CyPF-t-Bu.
The scope of the reactions of primary aromatic amines with heteroaryl and aryl halides are summarized in Table 6. Although these reactions were slower than the reactions of primary alkylamines, high yields of reactions of heteroaryl and aryl halides with electron rich primary aryl amines were obtained using only 0.005-0.1 mol % catalyst (Table 6, entries 1-5, 7-8, 11-13, 17-22 and 24). These catalyst loadings are one or two orders of magnitude lower than those of prior catalyst systems for reactions of aryl chlorides or bromides with unhindered primary aryl amines.12 Nolan reported one example of the extremely hindered 2,6-di-iso-propylaniline with the hindered chloroarene 2,6-dimethyl-1-chlorobenzene in the presence of 0.01 ml% of catalyst to give 93% isolated yield.66
Table 6.
Coupling of Heteroaryl and Aryl Halides with Primary Arylamines Catalyzed by Pd(OAc)2 and CyPF-t-Bu (1:1).a
 | ||||||
|---|---|---|---|---|---|---|
| Entry | Ar | X | Ar′ | Cat (%) |
Conditions | Yield (%)b |
| 1 | 2-Py | Cl | 4-Tol | 0.1 | 100 °C, 24 h | 86 |
| 2 | Br | 4-Tol | 0.05 | 60 °C, 48 h | 95 | |
| 3 | Cl | 1-Naphathyl | 0.05 | 100 °C, 24 h | 98 | |
| 4 | 3-Py | Cl | 4-Tol | 0.05 | 100 °C, 24 h | 93 |
| 5 | Cl | 2-Anisyl | 0.05 | 100 °C, 24 h | 99 | |
| 6 | Cl | 2-Py | 0.5 | 100 °C, 24 h | 97 | |
| 7 | Br | 4-Anisyl | 0.05 | 100 °C, 48 h | 80 | |
| 8 | I | 4-Anisyl | 0.05 | 100 °C, 24 h | 97 | |
| 9 | Br | 2-Pyrimidyl | 1.0 | 110 °C, 20 h | 96 | |
| 10 | Br | 2-Prazinyl | 1.0 | 110 °C, 20 h | 92 | |
| 11c | 1,3-Pyrimidyl-5- | Br | 4-Tol | 1.0 | 100 °C, 48 h | 52 |
| 12 | 4-isoQuinolinyl | Br | 4-Tol | 0.05 | 100 °C, 38 h | 84 |
| 13 | Ph | Cl | 4-Tol | 0.05 | 100 °C, 24 h | 99 |
| 14 | Br | 4-Tol | 0.005 | 110 °C, 20 h | 95 | |
| 15 | Br | 2-Py | 1.0 | 100 °C, 30 h | 81 | |
| 16 | Br | 2-benzothiazolyl | 1.0 | 100 °C, 36 h | 62 | |
| 17 | 4-Tol | Cl | 4-Anisyl | 0.05 | 100 °C, 24 h | 99 |
| 18 | Br | 4-Anisyl | 0.05 | 100 °C, 48 h | 97 | |
| 19 | 3-Anisyl | Cl | 4-Anisyl | 0.05 | 100 °C, 24 h | 99 |
| 20 | Br | 4-Anisyl | 0.05 | 100 °C, 48 h | 94 | |
| 21 | 2-Tol | Br | 4-Tol | 0.05 | 100 °C, 48 h | 96 |
| 22 | I | 4-Tol | 0.05 | 100 °C, 24 h | 99 | |
| 23 | 2,5-DiMephenyl | Br | 4-Tol | 0.5 | 100 °C, 48 h | 89 |
| 24 | 2-Cyclohexylphenyl | Br | 4-Tol | 0.05 | 100 °C, 48 h | 89 |
Reactions conducted with a 1:1 ratio of metal to ligand, 1 mmol ArX (X = Cl, Br, I), 1.2 equiv amine and 1.4 equiv NaOtBu in 1 mL DME.
isolated yield.
Using K3PO4 as the base.
Reactions of electron-deficient primary heteroaryl amines also occurred in the presence of the combination of Pd(OAc)2 and CyPF-t-Bu, but were slower and required higher catalyst loadings than the reactions of electron-rich primary arylamines (Table 6, entries 6, 9-10 and 15-16).26 Nevertheless, 2-aminopyridine, 2-aminopyrimidine and 2-aminobenzothiazole (Table 6, entries 6, 9, 10, 15 and 16) coupled with chlorobenzene and or bromobenzene to afford the desired products in 62-97% yields in the presence of 0.5-1.0 mol % catalyst.
H. Catalytic Amination of Heteroaryl and Aryl Chlorides and Bromides with Secondary Amines
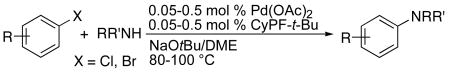
The combination of Pd(OAc)2 and the Josiphos ligand CyPF-t-Bu is highly selective for monoarylation of primary amines. Thus, the reactions of secondary amines are likely to be slower. Consistent with this logic, only a limited number of substrate combinations reacted in high yield at low catalyst loading. These data are as summarized in Table 7.
Table 7.
Coupling of Aryl Chlorides and Bromides with Secondary Amines Catalyzed by Pd(OAc)2 and CyPF-t-Bu (1:1).a
 | |||||
|---|---|---|---|---|---|
| Entry | Ar | Amine | Cat (%) |
Conditions | Yield (%)b |
| 1 | 2-Py | N-Methyl-aniline | 1.0 | 70 °C, 3 h | 57 |
| 2 | 3-Py | Morpholine | 1.0 | 90 °C, 12 h | 69 |
| 3 | 4-Py | Di-4-anisidine | 1.0 | 70 °C, 12 h | 50 |
| 4 | 1-isoquinolinyl | N-Me-aniline | 1.0 | 70 °C, 10 h | 84 |
| 5 | Ph | N-Me-aniline | 0.05 | 100 °C, 48 h | 56 |
| 6 | Dibutylamine | 0.5 | 100 °C, 24 h | 69 | |
| 7 | Diphenylamine | 0.5 | 100 °C, 20 h | 86 | |
| 8 | 4-Tol | Morpholine | 0.1 | 100 °C, 48 h | 86 |
| 9 | 3-Anisyl | Morpholine | 0.05 | 100 °C, 48 h | 82 |
Reactions conducted with a 1:1 ratio of metal to ligand, 1 mmol ArX (X = Cl, Br), 1.2 equiv amine and 1.4 equiv NaOtBu in 1 mL DME.
isolated yield.
Reactions of aryl chlorides with secondary amines were slow, and substantial amounts of hydrodehalogenation product were observed. However, reactions of heteroaryl chlorides with cyclic and acyclic amines occurred in moderate to good yields in the presence of 1.0 mol % of the catalyst. For example, the amination of 3-chloropyridine with morpholine in DME gave the coupled product in 69% yield. Reactions of aryl bromides with secondary amines also occurred in higher yield than reactions of aryl chlorides. For instance, morpholine reacted with 4-bromotoluene and 3-bromoanisole in the presence of 0.1 and 0.05 mol % catalyst to give the desired products in 86% and 82% yield, respectively (Table 7, entries 8 and 9). The acyclic secondary amines dibutylamine and diphenylamine required 0.5 mol % catalyst to form the coupled product in acceptable yield (Table 7, entries 6 and 7). We presume that the slower rate of reactions of acyclic secondary amines results from slower formation of the amido complex from the more hindered secondary amines.
Thus, coupling of secondary amines are currently best conducted with third-generation catalysts containing sterically bulky, electron-rich monodentate alkylphosphines or carbenes.11,31,66,67 Yet, if one wished to use a single catalyst for coupling of primary amines and secondary amines, the catalyst containing the Josiphos ligand would be competent for coupling of many examples of both classes of amines.
I. Scope of the Amination of Functionalized Heteroaryl and Aryl Chlorides, Bromides and Iodides with Primary Alkylamines
1. Catalytic Amination of Functionalized Heteroaryl Chlorides with Primary Alkylamines
Despite advances in the scope of palladium-catalyzed amination reactions, reactions of amines with functionalized heteroaryl chlorides remains challenging.32,68-71 The strong NaOtBu base, more than the palladium catalyst, limits the tolerance of the process to ancillary functionality. Several approaches have been followed to address functional group compatability.72-75 By one approach, reactions between secondary amines and chloroarenes containing protic functionality have been shown to occur when using lithium bis(trimethylsilyl)amide (LiN(SiMe3)2) as base.74,75
Here we show that the combination of CyPF-t-Bu as ligand and LiN(SiMe3)2 as based leads to the coupling of a variety of primary amines with heteroaryl chlorides containing protic functionality. These reactions generally required higher catalyst loadings (0.5 mol %) than reactions of chloroarenes lacking protic functionality, but they generally occurred in good to excellent yields. Reactions of these substrates conducted with the weaker bases K3PO4 or Cs2CO3, instead of LiN(SiMe3)2, occurred to low conversion, and reactions conducted with NaOtBu gave only trace amount of the desired coupled product.
The results in Table 8 summarize the scope of the couplings of heteroaryl chlorides containing protic functionality. Each reaction occurred to full conversion and formed the coupled product in high yield with little side product. In some cases, the isolated yields were lower than those for reactions of less functionalized products because of the loss of some of these highly polar products during purification by column chromatography. For example, the reactions of N-(5-chloro-2-pyridinyl)acetamide with octylamine and sec-butyl amine in the presence of 0.5 mol % of palladium and ligand afforded 65% and 96% isolated yields of the desired products, respectively. Likewise, heteroaryl chlorides containing functional groups, such as a free alcohol and phenol, reacted in acceptable isolated yields with 0.5 mol % catalyst.
Table 8.
Coupling of Functionalized Heteroaryl Chlorides with Primary Amines Catalyzed by Pd(OAc)2 and CyPF-t-Bu (1:1).a
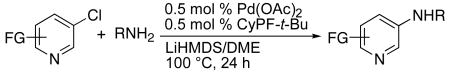 | ||||
|---|---|---|---|---|
| Entry | Halide | R | Cat. (%) |
Yield (%)b |
| 1 | 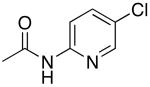 |
Bn | 0.5 | 73 |
| 2 | secBu | 0.5 | 96 | |
| 3 | 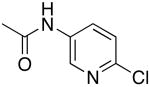 |
Bn | 0.5 | 59 |
| 4 | isoBu | 0.5 | 78 | |
| 5 | 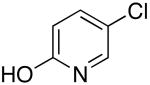 |
Octyl | 0.5 | 53 |
| 6 | 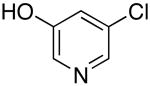 |
Cyclohexyl | 0.5 | 67 |
| 7 | 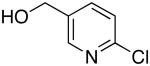 |
Octyl | 0.5 | 79 |
| 8 | Bn | 0.5 | 55 | |
Reactions conducted with a 1:1 ratio of metal to ligand 1 mmol ArCl, 1.2 equiv amine and 1.4 equiv LiN(SiMe3)2 in 1 mL DME.
Isolated yields.
2. Catalytic Amination of Functionalized Aryl Chlorides, Bromides and Iodides with Primary Alkylamines
The reactions of primary amines with haloarenes containing the same types of functional groups occurred in high yield in the presence of LiN(SiMe3)2 as base (Table 9). Aryl chlorides, bromides and iodides containing functional groups, such as a free alcohol, phenol, carboxylic acid, primary amide or secondary amide reacted in high yields with only 0.05-0.5 mol % catalyst loading. Reactions of aryl chlorides containing an enolizable ketone also gave the desired products in good yield in the presence of LiN(SiMe3)2, while reactions of aryl bromides and iodides gave more complicated mixtures, even when 2.0 mol % of the catalyst was used. These reactions were better conducted with K3PO4 as base (vide infra). The reaction of 4′-chloro-acetophenone with primary amines occurred in higher yield under somewhat more dilute conditions. The side product formed from α–arylation was minimized, though not fully eliminated, when the concentration of the reaction solution was decreased from 1 M to 0.2 M (Table 9, entry 7).
Table 9.
Coupling of Functionalized Aryl Halides with Primary Amines Catalyzed by Pd(OAc)2 and CyPF-t-Bu (1:1).a
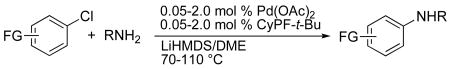 | ||||||
|---|---|---|---|---|---|---|
| Entry | -FG | X | R | Cat. (%) |
Conditions | Yieldb (%) |
| 1 | -4-COOH | Cl | Octyl | 0.05 | 100 °C, 20 h | 81 |
| 2 | Br | Octyl | 0.05 | 100 °C, 20 h | 72 | |
| 3 | Br | secBu | 0.05 | 100 °C, 20 h | 92 | |
| 4 | I | secBu | 0.05 | 100 °C, 48 h | 78 | |
| 5 | -4-CONH2 | Cl | isoBu | 0.5 | 100 °C, 20 h | 67 |
| 6 | Br | isoBu | 0.05 | 100 °C, 20 h | 85 | |
| 7c | -4-COCH3 | Cl | Octyl | 0.5 | 70 °C, 20 h | 91 |
| 8d | Cl | Octyl | 0.5 | 110 °C, 24 h | 74 | |
| 9 | Cl | Octyl | 0.05 | 100 °C, 16 h | 61 | |
| 10d | I | Octyl | 1.0 | 110 °C, 24 h | 78 | |
| 11c | Cl | Cyclohexyl | 0.5 | 70 °C, 20 h | 87 | |
| 12d | Br | Cyclohexyl | 1.0 | 110 °C, 24 h | 82 | |
| 13 | -4-NHCOCH3 | Br | Octyl | 0.05 | 100 °C, 20 h | 98 |
| 14 | I | Octyl | 0.5 | 110 °C, 20 h | 67 | |
| 15 | -3-NHCOCH3 | Cl | Cyclohexyl | 0.05 | 100 °C, 24 h | 74 |
| 16 | -4-CH2OH | Br | isoBu | 0.05 | 100 °C, 48 h | 72 |
| 17 | I | isoBu | 0.05 | 100 °C, 20 h | 47 | |
| 18 | -4-CH(OH)CH3 | Cl | secBu | 0.5 | 100 °C, 18 h | 83 |
| 19 | -4-CH2CH2OH | Cl | Octyl | 0.5 | 100 °C, 20 h | 89 |
| 20 | -3-OH | Cl | Bn | 0.5 | 100 °C, 20 h | 84 |
| 21 | Br | Bn | 0.5 | 100 °C, 20 h | 85 | |
| 22 | Br | Cyclohexyl | 0.5 | 100 °C, 36 h | 81 | |
| 23 | I | Cyclohexyl | 1.0 | 100 °C, 20 h | 70 | |
| 24 | -4-OH | Cl | Octyl | 2.0 | 100 °C, 18 h | 72 |
| 25d | -4-CO2Me | Cl | isoBu | 1.0 | 110 °C, 24 h | 94 |
| 26d | -4-CO2Et | I | secBu | 2.0 | 110 °C, 48 h | 77 |
| 27d | -3-CO2Me | Cl | isoBu | 1.0 | 110 °C, 24 h | 91 |
| 28d | Br | isoBu | 1.0 | 110 °C, 24 h | 97 | |
| 29d | -2-CO2Me | Cl | Bn | 2.0 | 110 °C, 24 h | 82 |
| 30d | -4-NO2 | I | Octyl | 1.0 | 110 °C, 24 h | 79 |
Reactions conducted with a 1:1 ratio of metal to ligand, 1.0 mmol ArX (X = Cl, Br, I), 1.2 equiv amine, and 2.4 equiv LiN(SiMe3)2 in 1 mL DME.
Isolated yields.
Reactions run at 0.2 M.
Using K3PO4 as the base.
As mentioned earlier in this paper, reactions catalyzed by complexes of the hindered Josiphos ligand conducted with weak bases13,14,56,76,77 occur more slowly than reactions catalyzed by complexes of monodentate ligands conducted with weak bases. Nevertheless, reactions of haloarenes containing electron-withdrawing groups, such as enolizable ketone, carboalkoxy, acyl or nitro groups, occurred fast enough to give the coupled product in high yield when K3PO4 was used as base. Reactions of aryl chlorides, bromides and iodides bearing an enolizable keto group in the para-position or a carbomethoxy group in the meta or para position occurred to completion and gave good to excellent yields of the coupled product (Table 9, entries 8, 10 and 25-28). Reactions of purely aliphatic amines with the chloroarene that contained a carboalkoxy group ortho to the chloride were slower. However, acceptable yields were obtained for the reaction of methyl-2-chlorobenzoate with benzylamine using 2.0 mol % catalyst at 110 °C (Table 9, entry 29).
Previous reactions of primary alkylamines with aryl chlorides, bromides, and iodides possessing potentially reactive functional groups were limited to those of electron-poor aryl chlorides and bromides containing an ester or nitro functionality.13,76 No examples of reactions of primary alkylamines with chloro-, bromo- or iodo-arenes containing the functional groups of Table 9, other than an ester or nitro group, have been published previously using catalysts other than that containing the Josiphos CyPF-t-Bu ligand.
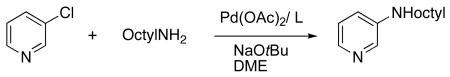
J. Comparison of the Reactivity of Catalysts Generated from Analogs of CyPF-t-Bu
To identify the structural elements of CyPF-t-Bu that give rise to high activity and stability, we evaluated the catalytic activity of complexes containing several analogs of this ligand. These analogs (Table 10) include derivatives of CyPF-t-Bu that lack the methyl group of the backbone or bear a more bulky iso-propyl group, contain less hindered alkyl groups on the phosphorus, are partially oxidized, or have the positions of the two phosphino groups interchanged. In addition, we prepared ligands that contain backbones mimicking the structures of the ferrocenyl bisphosphines.
Table 10.
Comparison of the Activity of Josiphos CyPF-t-Bu Analogs for the Coupling of Heteroaryl Chloride with Primary Alkylamine.
 | |||||
|---|---|---|---|---|---|
| Entry | Ligand | Loading | Temp. | Time (h) | Yield |
| 1 | CyPF-t-Bu 1 | 0.005 | 90 °C | 24 | 93 |
| 2 | PPF-t-Bu 4 | 1.0 | 90 °C | 24 | 67 |
| 3 | MePF-t-Bu 5 | 0.005 | 90 °C | 24 | < 5% |
| 4 | EtPF-t-Bu 6 | 0.005 | 90 °C | 24 | < 5% |
| 5 | CyPF-Cy 7 | 1.0 | 90 °C | 24 | 46 |
| 6 | CyPF-Ph 8 | 1.0 | 90 °C | 24 | 48 |
| 7 | tBuPF-Cy 9 | 0.005 | 90 °C | 24 | 62 |
| 8 | 10 | 0.005 | 90 °C | 24 | 50 |
| 9 | 11 | 0.001 | 100 °C | 48 | 16 |
| 10 | 11 | 0.005 | 90 °C | 24 | 93 |
| 11 | 12 | 0.005 | 90 °C | 24 | < 5% |
| 12 | 13 | 0.005 | 90 °C | 24 | < 5% |
| 13 | 14 | 0.005 | 90 °C | 24 | < 5% |
| 14 | 15 | 0.005 | 90 °C | 24 | < 5% |
| 15 | 16 | 0.005 | 90 °C | 24 | < 5% |
| 16 | 17 | 0.005 | 90 °C | 24 | < 5% |
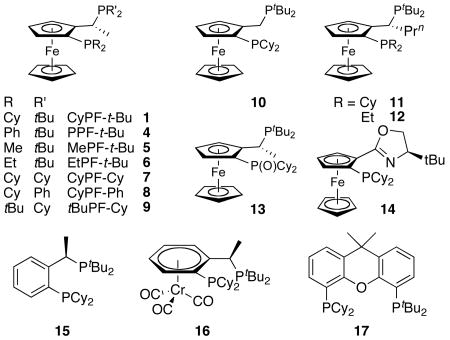 | |||||
To determine whether the true catalyst in this system contains both phosphino groups bound to palladium, ligand 13, in which one phosphino group has been oxidized, and ligand 14, in which one phosphino group was replaced by an oxazoline, were studied. The reactivity of catalysts generated from these ligands for the coupling of 3-chloropyridine with octylamine were compared to that of catalysts generated from CyPF-t-Bu. The reaction conducted with CyPF-t-Bu occurred in 93% isolated yield after 24 h at 90 °C in the presence of 0.005 mol % of Pd(OAc)2/CyPF-t-Bu. In contrast, this reaction conducted with 13 or 14 as ligand formed only small amounts of the coupled product under similar reaction conditions. This experiment suggests that palladium complexes of the bis(phosphine)ligand are the catalytically active species in the reaction and that both of the phosphino groups in CyPF-t-Bu are important for the high catalytic activity in the amination process.
Ligands 5-7 containing less hindered alkylphosphino groups formed much less active catalysts than did CyPF-t-Bu. The catalysts generated from these ligands formed the coupled product in <5% to 48% yields (Table 10, entries 3-5). The catalysts generated from ligands 4 or 8 containing diphenylphosphino groups were more active than those containing dimethylphosphino or diethylphosphino groups, but formed the coupled product in only 67 and 48% yields, even with 1.0 mol % loading (Table 10, entries 2, 6). Moreover, ligand 9, in which the two phosphino groups of the most reactive ligand 1 were transposed, led to a less active catalyst (Table 10, entry 8). We do not have a firm explanation for the lower reactivity of transposed ligand 9 vs. ligand 1,
Ligand 10 lacking the backbone methyl group formed a catalyst that coupled 3-chloropyridine with octylamine. However, the reaction of these substrates in the presence of the catalyst containing ligand 10 occurred at least five times slower than the reaction catalyzed by the complex containing CyPF-t-Bu (Table 10, entry 8). The catalyst generated from ligand 11 containing a slightly larger propyl group reacted with rates and lifetimes (Table 10, entries 9-10) that are similar to those for reactions conducted with catalysts generated from ligand 1.
Even ligands 15-16 containing backbones related to the 1,2-disubstituted ferrocenyl group generated much less reactive catalysts at 0.005 mol % loadings. Ligand 15 should generate a complex with a slightly smaller bite angle than that generated from ligand 1. However, the backbone would be expected to be substantially less rigid without the combination of the anti arrangement of the ferrocenyl unit and backbone methyl group in the coordinated ligand. The bite angle in the complex generated from ligand 16 would also be slightly smaller than that generated from PPF-t-Bu because the complex of 16 would contain a six-membered ring fused to the chelate ring. The backbone of this ligand could be more rigid than that in 15 because of the relative stereochemistry of the chromium carbonyl unit and the backbone methyl group. However, the carbonyl groups bound to chromium can suffer nucleophilic attack by the combination of amine and base, and this attack could open a pathway to catalyst decomposition.
The complex of ligand 17 containing a Xanthane backbone in place of the ferrocenyl-1-ethyl group would contain a slightly larger bite angle than that of CyPF-t-Bu, and the value of 4,6-diphenylphosphino-10,10-dimethylxantane (Xantphos) as ligand for cross-coupling has been attributed to its large bite angle. However, the reaction of 3-chloropyridine with octylamine catalyzed by Pd(OAc)2 and 17 occurred in much lower conversion than it did when catalyzed by Pd(OAc)2 and CyPF-t-Bu.
From these results, we conclude that the following structural features that are specific to CyPF-t-Bu lead to the high efficiency of the Pd-CyPF-t-Bu catalyst. 1) a rigid backbone resulting from the relative stereochemistry of the backbone methyl group and ferrocenyl unit, which causes the ligand to bind tightly to palladium and prevents the displacement of the Pd(II) complex of this ligand by primary amines and basic heterocycles; 2) strong electron donation of the two alkylphosphino groups, which promotes the oxidative addition of less reactive haloarenes; 3) steric bulk of the phosphino groups, which discourages diarylation of the primary amine substrates, facilitates dissociation of ligand from (chelate)2Pd(0) to form the reactive intermediate (chelate)Pd(0), and increases the rate of reductive elimination from the arylpalladium amido complexes vs. those containing less hindered chelating alkylphosphines; and 4) stability of the ferrocene unit and hindered backbone methane hydrogen to the reagents in the system.
Conclusions
In summary, we have shown that palladium complexes generated from the Josiphos ligand CyPF-t-Bu are general, highly efficient catalysts for the coupling of heteroaryl and aryl chlorides and bromides with primary nitrogen nucleophiles. Reactions catalyzed by the complexes generated in situ from Pd(OAc)2 and ligand 1 occur with turnover numbers that are typically one or two orders of magnitude higher than those of related couplings by previous catalysts. The process exhibits a broad scope and a high tolerance for functional groups, including cyano, keto, free carboxylate, amido, carboalkoxy, aromatic and aliphatic hydroxyl and amino groups.
The efficiency of palladium catalysts containing this ligand for the amination process is believed to result from an unusual combination of 1) a rigid backbone which causes the ligand to bind to palladium tightly enough to prevent displacement of the ligand by primary amines and basic heterocycles; 2) strong electron donation, which promotes the oxidative addition of less reactive haloarenes; and 3) steric bulk that disfavors diarylation, facilitates the generation of the (chelate)Pd(0) intermediate, and promotes reductive elimination from the arylpalladium amido complexes.
The strengths of the catalyst reported here complement those of previously reported catalysts that contain hindered monodentate ligands. The catalyst in the current work is less reactive toward secondary amines than primary amines, while the catalysts for aminations of chloroarenes reported previously are less reactive toward primary amines than secondary amines.
In future work, we will define more precisely the mechanism of the coupling process with this catalyst, and will follow these design principles with the goal of developing readily accessible ligands for the reactions of secondary amines and nitrogen heterocycles with low loading of catalyst.
Supplementary Material
Acknowledgments
We thank the NIH (GM-55382) for support of this work, Johnson-Matthey for PdCl2, and Solvias for the Josiphos ligands.
Footnotes
Supporting Information Available: All experimental procedures and spectroscopic data of new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Hartwig JF. In: Modern Arene Chemistry. Austruc C, editor. Wiley-VCH; Weinheim: 2002. p. 107. [Google Scholar]
- 2.Hartwig JF. Handbook of Organopalladium Chemistry for Organic Synthesis. 2002;1:1051. [Google Scholar]
- 3.Muci AR, Buchwald SL. Top Curr Chem. 2002;219:131. [Google Scholar]
- 4.Belfield AJ, Brown GR, Foubister AJ, Ratcliffe PD. Tetrahedron. 1999;55:13285. [Google Scholar]
- 5.Yang BH, Buchwald SL. J Organomet Chem. 1999;576:125. [Google Scholar]
- 6.Buchwald SL, Mauger C, Mignani G, Scholz U. Adv Synth Catal. 2006;348:23. [Google Scholar]
- 7.King AO, N Y. In: Organometallics in process chemistry. Larsen RD, editor. Springer; Berlin: 2004. p. 205. [Google Scholar]
- 8.Christmann U, Vilar R. Angew Chem Int Ed. 2005;44:366. doi: 10.1002/anie.200461189. [DOI] [PubMed] [Google Scholar]
- 9.Littke AF, Fu GC. Angew Chem Int Ed. 2002;41:4176. doi: 10.1002/1521-3773(20021115)41:22<4176::AID-ANIE4176>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 10.Stambuli JP, Kuwano R, Hartwig JF. Angew Chem Int Ed. 2002;41:4746. doi: 10.1002/anie.200290036. [DOI] [PubMed] [Google Scholar]
- 11.Stauffer SR, Lee S, Stambuli JP, Hauck SI, Hartwig JF. Org Lett. 2000;2:1423. doi: 10.1021/ol005751k. [DOI] [PubMed] [Google Scholar]
- 12.Wolfe JP, Tomori H, Sadighi JP, Yin J, Buchwald SL. J Org Chem. 2000;65:1158. doi: 10.1021/jo991699y. [DOI] [PubMed] [Google Scholar]
- 13.Huang X, Anderson KW, Zim D, Jiang L, Klapars A, Buchwald SL. J Am Chem Soc. 2003;125:6653. doi: 10.1021/ja035483w. [DOI] [PubMed] [Google Scholar]
- 14.Kataoka N, Shelby Q, Stambuli JP, Hartwig JF. J Org Chem. 2002;67:5553. doi: 10.1021/jo025732j. [DOI] [PubMed] [Google Scholar]
- 15.Urgaonkar S, Xu JH, Verkade JG. J Org Chem. 2003;68:8416. doi: 10.1021/jo034994y. [DOI] [PubMed] [Google Scholar]
- 16.Viciu MS, Kissling RM, Stevens ED, Nolan SP. Org Lett. 2002;4:2229. doi: 10.1021/ol0260831. [DOI] [PubMed] [Google Scholar]
- 17.Grasa GA, Viciu MS, Huang J, Nolan SP. J Org Chem. 2001;66:7729. doi: 10.1021/jo010613+. [DOI] [PubMed] [Google Scholar]
- 18.Li GY. Angew Chem Int Ed. 2001;40:1513. doi: 10.1002/1521-3773(20010417)40:8<1513::aid-anie1513>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- 19.Rataboul F, Zapf A, Jackstell R, Harkal S, Riermeier T, Monsees A, Dingerdissen U, Beller M. Chem Eur J. 2004;10:2983. doi: 10.1002/chem.200306026. [DOI] [PubMed] [Google Scholar]
- 20.Dai Q, Gao WZ, Liu D, Kapes LM, Zhang XM. J Org Chem. 2006;71:3928. doi: 10.1021/jo060321e. [DOI] [PubMed] [Google Scholar]
- 21.Xie XM, Zhang TY, Zhang ZG. J Org Chem. 2006;71:6522. doi: 10.1021/jo060945k. [DOI] [PubMed] [Google Scholar]
- 22.Ackermann L, Spatz JH, Gschrei CJ, Born R, Althammer A. Angew Chem Int Ed. 2006;45:7626. doi: 10.1002/anie.200602222. [DOI] [PubMed] [Google Scholar]
- 23.Singer RA, Dore M, Sieser JE, Berliner MA. Tetrahedron Lett. 2006;47:3727. [Google Scholar]
- 24.Wagaw S, Buchwald SL. J Org Chem. 1996;61:7240. doi: 10.1021/jo9612739. [DOI] [PubMed] [Google Scholar]
- 25.Hooper MW, Utsunomiya M, Hartwig JF. J Org Chem. 2003;68:2861. doi: 10.1021/jo0266339. [DOI] [PubMed] [Google Scholar]
- 26.Yin J, Zhao MM, Huffman MA, McNamara JM. Org Lett. 2002;4:3481. doi: 10.1021/ol0265923. [DOI] [PubMed] [Google Scholar]
- 27.Jonckers THM, Maes BUW, Lemiere GLF, Dommisse R. Tetrahedron. 2001;57:7027. [Google Scholar]
- 28.Maes BUW, Loones KTJ, Lemiere GLF, Dommisse RA. Synlett. 2003:1822. [Google Scholar]
- 29.Burton G, Cao P, Li G, Rivero R. Org Lett. 2003;5:4373. doi: 10.1021/ol035655u. [DOI] [PubMed] [Google Scholar]
- 30.Marion N, Ecarnot EC, Navarro O, Amoroso D, Bell A, Nolan SP. J Org Chem. 2006;71:3816. doi: 10.1021/jo060190h. [DOI] [PubMed] [Google Scholar]
- 31.Navarro O, Marion N, Mei J, Nolan SP. Chem Eur J. 2006;12:5142. doi: 10.1002/chem.200600283. [DOI] [PubMed] [Google Scholar]
- 32.Anderson KW, Tundel RE, Ikawa T, Altman RA, Buchwald SL. Angew Chem Int Ed. 2006;45:6523. doi: 10.1002/anie.200601612. [DOI] [PubMed] [Google Scholar]
- 33.Wolfe JP, Buchwald SL. J Org Chem. 1996;61:1133. [Google Scholar]
- 34.Driver MS, Hartwig JF. J Am Chem Soc. 1996;118:7217. [Google Scholar]
- 35.Wolfe JP, Buchwald SL. J Org Chem. 1997;62:6066. [Google Scholar]
- 36.Hamann BC, Hartwig JF. J Am Chem Soc. 1998;120:7369. [Google Scholar]
- 37.Huang J, Grasa G, Nolan SP. Org Lett. 1999;1:1307. [Google Scholar]
- 38.Ali MH, Buchwald SL. J Org Chem. 2001;66:2560. doi: 10.1021/jo0008486. [DOI] [PubMed] [Google Scholar]
- 39.Arterburn JB, Pannala M, Gonzalez AM. Tetrahedron Lett. 2001;42:1475. [Google Scholar]
- 40.Maes BUW, Loones KTJ, Jonckers THM, Lemiere GLF, Dommisse RA, Haemers A. Synlett. 2002:1995. [Google Scholar]
- 41.Parrot I, Ritter G, Wermuth CG, Hibert M. Synlett. 2002:1123. [Google Scholar]
- 42.Urgaonkar S, Nagarajan M, Verkade JG. J Org Chem. 2003;68:452. doi: 10.1021/jo0205309. [DOI] [PubMed] [Google Scholar]
- 43.Enguehard C, Allouchi H, Gueiffier A, Buchwald SL. J Org Chem. 2003;68:4367. doi: 10.1021/jo0341463. [DOI] [PubMed] [Google Scholar]
- 44.Meyers C, Maes BUW, Loones KFJ, Bal G, Lemiere GLF, Dommisse RA. J Org Chem. 2004;69:6010. doi: 10.1021/jo049774e. [DOI] [PubMed] [Google Scholar]
- 45.Gerristma D, Timothy B, McNulty J, Capretta A. Tetrahedron Lett. 2004;45:8319. [Google Scholar]
- 46.Farina V. Adv Synth Catal. 2004;346:1553. [Google Scholar]
- 47.Beletskaya IP, Cheprakov AV. J Organometallic Chem. 2004;689:4055. [Google Scholar]
- 48.Diederich F, Stang PJ. Metal-Catalyzed Cross-Coupling Reactions. Wiley-VCH; New York: 1998. [Google Scholar]
- 49.Tsuji J. Transition Metal Reagents and Catalysis: Innovations in Organic synthesis. Wiley; Chichester: 2000. [Google Scholar]
- 50.Paul F, Patt J, Hartwig JF. Organometallics. 1995;14:3030. [Google Scholar]
- 51.Widenhoefer RA, Buchwald SL. Organometallics. 1996;15:3534. [Google Scholar]
- 52.Shen Q, Shekhar S, Stambuli JP, Hartwig JF. Angew Chem Int Ed. 2005;44:1371. doi: 10.1002/anie.200462629. [DOI] [PubMed] [Google Scholar]
- 53.(R)-(-)-1-[(S)-2-(dicyclohexylphosphino)ferrocenyl]ethyl-di-tert-butylphosphine [158923-11-6], Strem catalog number 26-0975.
- 54.Roy AH, Hartwig JF. J Am Chem Soc. 2003;125:8704. doi: 10.1021/ja035835z. [DOI] [PubMed] [Google Scholar]
- 55.Amatore C, Jutand A, Khalil F, M'Barki MA, Mottier L. Organometallics. 1993;12:3168. [Google Scholar]
- 56.Wolfe JP, Buchwald SL. J Org Chem. 2000;65:1144. doi: 10.1021/jo9916986. [DOI] [PubMed] [Google Scholar]
- 57.Optically active amines. Wagaw S, Rennels RA, Buchwald SL. J Am Chem Soc. 1997;119:8451. [Google Scholar]
- 58.Catellani M, Catucci C, Celentano G, Ferraccioli R. Synlett. 2001;6:803. [Google Scholar]
- 59.Hartwig JF, Richards S, Baranano D, Paul F. J Am Chem Soc. 1996;118:3626. [Google Scholar]
- 60.Hartwig JF. J Am Chem Soc. 1996;118:7010. [Google Scholar]
- 61.Control experiments without catalyst were conducted under reaction conditions that were identical to those conducted with catalyst. A 10 μL aliquot was withdrawn from the reaction vial and analyzed for conversion by GC.
- 62.Giam CS. In: Pyridine and its derivatives. Abramovitch RA, editor. Vol. 3. John Wiley & Sons; New York: 1974. p. 41. [Google Scholar]
- 63.Hashimoto S, Otani S, Okamoto T, Matsumoto K. Heterocycles. 1988;27:319. [Google Scholar]
- 64.Miller J. Aromatic Nucleophilic Substitution. Vol. 8 Elsevier; Amsterdam: 1968. [Google Scholar]
- 65.Vinter-Pasquier K, Jamart-Gregoire B, Caubere P. Heterocycles. 1997;45:2113. [Google Scholar]
- 66.Marion N, Navarro O, Mei J, Stevens ED, Scott NM, Nolan SP. J Am Chem Soc. 2006;128:4101. doi: 10.1021/ja057704z. [DOI] [PubMed] [Google Scholar]
- 67.Nishiyama M, Yamamoto T, Koie Y. Tetrahedron Lett. 1998;39:617. [Google Scholar]
- 68.Scott JP. Synlett. 2006:2083. [Google Scholar]
- 69.Queiroz MJRP, Begouin A, Ferreira ICFR, Kirsch G, Calhelha RC, Barbosa S, Estevinho LM. Eur J Org Chem. 2004:3679. [Google Scholar]
- 70.Luker TJ, Beaton HG, Whiting M, Mete A, Cheshire DR. Tetrahedron Lett. 2000;41:7731. [Google Scholar]
- 71.Castellote I, Vaquero JJ, Alvarez-builla J. Tetrahedron Lett. 2004;45:769. [Google Scholar]
- 72.Wolfe JP, Buchwald SL. Tetrahedron Lett. 1997;38:6359. [Google Scholar]
- 73.Lee DY, Hartwig JF. Org Lett. 2005;7 doi: 10.1021/ol050141b. in press. [DOI] [PubMed] [Google Scholar]
- 74.Harris MC, Huang X, Buchwald SL. Org Lett. 2002;4:2885. doi: 10.1021/ol0262688. [DOI] [PubMed] [Google Scholar]
- 75.Urgaonkar S, Verkade JG. Adv Synth Cat. 2004;346:611. [Google Scholar]
- 76.Old DW, Wolfe JP, Buchwald SL. J Am Chem Soc. 1998;120:9722. [Google Scholar]
- 77.Hartwig JF, Kawatsura M, Hauck SI, Shaughnessy KH, Alcazar-Roman LM. J Org Chem. 1999;64:5575. doi: 10.1021/jo990408i. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.