

Abstract
Like all herpesviruses, Kaposi's sarcoma associated herpesvirus (KSHV) can produce either latent or lytic infection. The latent v-FLIP gene is a strong activator of NF-κB, and in primary effusion lymphoma (PEL) cells, blockade of NF-κB activation is associated with enhanced lytic gene expression, while over-expression of p65 impairs expression of reporter genes driven by lytic promoters. This has led to the suggestion that NF-κB activation may promote latency by suppressing lytic reactivation. Here we examine in detail the effects of NFκB activation on KSHV replication in several cell types. In accord with earlier work, we find that inhibition of NFκB signaling in PEL cells is associated with enhanced lytic reactivation of KSHV. Similarly, in de novo KSHV infection of primary endothelial cells, inhibition of NF-κB signaling leads to an increase in lytic gene expression and enhanced virion production. By contrast, KSHV-infected human foreskin fibroblasts (HFF) show no increase in spontaneous lytic reactivation when NFκB is inhibited. Moreover, if NFκB activation is always inhibitory to lytic gene expression, one might expect its activation to be suppressed during the lytic cycle. However, we find that NFκB signaling is strongly and consistently activated in lytically infected cells of all lineages. Together these data indicate that (i) the relationship of NFκB activation to latency and lytic reactivation is not uniform, but is dependent on the cellular context; and (ii) even though NF-κB activation is inhibitory to lytic gene expression in some contexts, such inhibition is at least partially bypassed or overridden during lytic growth.
Introduction
Kaposi's sarcoma-associated herpesvirus (KSHV, also called human herpesvirus 8) is the etiologic agent of Kaposi's sarcoma (KS), an inflammatory and proliferative lesion affecting microvascular endothelium. KSHV also targets B lymphocytes, and is linked to two rare lymphoproliferative syndromes, multicentric Castleman's disease (MCD) and primary effusion lymphoma (PEL) (Arvanitakis et al., 1996; Moore and Chang, 1998; Soulier et al., 1995). Like all herpesviruses, KSHV can execute two different genetic programs, known as latency and lytic growth. In latency, viral gene expression is strongly restricted, with only a handful of the virus' nearly 100 genes being expressed. The latent viral genome is maintained as a circular nuclear plasmid, and no viral progeny are produced. Latency is usually the default pathway following experimental infection in cultured cells (Vieira et al 2001;Bechtel et al., 2003). However, if latently infected cells are exposed to certain stimuli (such as: phorbol esters or HDAC inhibitors) latency can be disrupted and lytic replication triggered. The lytic program proceeds via a temporally regulated cascade of gene expression, in which lytic cycle-specific genes are activated in at least 3 classes: immediate-early (IE), delayed-early (DE) and late (L). Viral DNA replication follows DE gene expression, and progeny genomes are encapsidated into virions and released.
KS tumors are primarily latently infected, but a small number of cells in the lesion are lytically infected (Staskus et al., 1997), and both latency and lytic replication have been postulated to play important roles in KS pathogenesis (Ganem, 2006). A single viral gene, RTA (Replication and Transcription Activator) controls this genetic switch; forced expression of RTA in latency triggers lytic reactivation, and mutational inactivation of RTA blocks lytic reactivation (Sun et al 1998; Lukac et al 1999). However, little is known of the physiologic stimuli that activate RTA expression to trigger lytic growth, and still less is known about how latency, once established, is stably maintained (that is to say, how the expression of RTA and other lytic genes is prevented).
Several years ago, Brown et al (2003) demonstrated that treatment of PEL cells with Bay 11-7082, a known pharmacological inhibitor of NFκB activation, triggered enhanced spontaneous lytic reactivation. Because KSHV latency is associated with expression of the v-FLIP gene, which tonically activates NFκB by binding to NEMO and activating IKK (Field et al., 2003; Liu et al., 2002), latently infected cells have elevated NFκB activity. A model has accordingly been proposed that NFκB activation opposes lytic reactivation; in support of this notion, Brown et al (2003) reported that overexpression of p65 inhibits expression of luciferase reporter genes driven by several KSHV lytic promoters. Here we have examined the role of NFκB in lytic reactivation in several cell types other than PEL cells (which have been selected for stable latency in vivo and display a strong dependence on v-FLIP-mediated NFκB activation (Guasparri, Keller, and Cesarman, 2004)). We find that the dependence of latency on NFκB activation is variable and dependent upon the cellular context – primary endothelial cells behave similarly to PEL cells, but equally permissive human fibroblasts show no enhancement of lytic reactivation when the NFκB activation pathway is inhibited. Moreover, NFκB is strongly activated during lytic replication in all cell types, indicating that if there is a block to lytic gene expression mediated by p65 it must be at least partially bypassed or overcome in the context of the lytic program.
Materials and Methods
Cells and KSHV infection
Human umbilical vein endothelial cells (HUVECs) were purchased from Clonetics and cultured in EGM-2 media supplemented with the endothelial supplement pack (Clonetics). Human foreskin fibroblasts (HFFs) were purchased from ATCC and cultured in DMEM supplemented with 10% fetal bovine serum, penicillin, streptomycin and l-glutamine. BCBL-1 cells were carried in RPMI 1640 supplemented with 10% fetal bovine serum, penicillin, streptomycin, glutamine and β-mercaptoethanol. KSHV was concentrated from supernatants of induced BCBL-1 cells as previously described (Bechtel et al., 2003; Lagunoff et al., 2002). KSHV infections were done in media containing 2μg/ml polybrene and incubated with cells for six hours, after which cells were rinsed and media was added back. In the case of lytic infection, Adeno-RTA (1×109 particles/ml) was incubated in media plus 0.5ug/ml polylysine (Sigma) for 2 hours before being applied to cells after the removal of the KSHV containing media.
Retrovirus production and infection
Retroviruses were produced using the amphotropic Phoenix packaging cell line transfected with the Moloney Murine Leukemia Virus based vector pMXpie (a kind gift from Lewis Lanier). Phoenix cells were transfected using FuGENE 6 (Roche) according to the manufacturer's specifications. 36 hours after transfection supernatants were collected and concentrated at 5,000rpm for 16hrs. Concentrated retroviruses were resuspended in EGM-2 media with 6μg/ml polybrene and filtered through a 0.2μm filter. Concentrated retrovirus was diluted in EGM-2 media with 6μg/ml polybrene and applied to cells. These cultures were spun at 2,000rpm for 1.5 hours after which virus-containing media was removed and regular culture media was added back. In cases were selection was employed, media containing the selective agent was added to cells at the stated concentration 24 hours after transduction.
Immunofluorescence and Western Blotting
Immunofluorescence assays were performed as previously described using anti-ORF59 (ABI) and an anti-mouse rhodamine (Santa Cruz Biotechnology) secondary antibody was used at 1:300.
Lysates for western blots were obtained by rinsing cells twice with ice cold PBS, scraping adherent cells from the plate, spinning cells at 1000rpm for 5min and resuspending pellets in 2-3x the pellet volume of RIPA lysis buffer. Lysates were quantified using Biorad Protein Assay. 20ug of protein were run on 4-15% Tris-HCl gels and protein was transferred to PVDF at 100mV for 1hr at 4°C. Blots were blocked for 1hr at room temperature in 5% milk in TBS plus 0.1% Tween. Primary antibodies were incubated overnight at 4°C at 1:1000 anti-PARP (Cell Signaling Technology), 1:5000 anti-β-actin (A1978, Sigma); secondary antibodies, anti-mouse-HRP and anti-rabbit-HRP (Santa Cruz) were incubated at 1:5000 for 1hr at room temperature. Signal was detected using ECL Detection Reagent (Amersham).
Bay 11-7082 treatment of BCBL-1 cells
Bay 11-7082 was purchased from Calbiochem, diluted in DMSO and used at the concentrations stated. For reactivation assays, BCBL-1 cells were resuspended in media containing Bay 11-7082, incubated for 30 minutes, then spun down and resuspended in fresh media. RNA and protein lysates were harvested at 48hrs post Bay 11-7082 treatment.
KSHV virion DNA isolation
Media was removed from virus-producing cells, filtered through a 0.45uM filter and stored at 4°C. 20u/ml DNase was added to filtered supernatants and incubated at 37°C for 1hr. Supernatants were then chilled on ice and spun at 15,000 rpm for 2 hours at 4°C. Media was then removed and pelleted virus was resuspended in 600ul lysis buffer (20 mM Tris-HCl pH 8, 10 mM EDTA, 100 mM NaCl, 0.5% SDS) and incubated at room temperature for 10 minutes. 0.7mg/ml Proteinase K and linearized plasmid encoding an unrelated malaria gene were diluted in 100ul lysis buffer and added to the 600ul resuspended virions. The 700ul total volume was incubated at 55°C for 2 hours; this was then extracted once with 700ul phenol/chloroform/isoamyl. 600ul of the aqueous phase was removed and to it was added 3ul glycogen, 100ul 3M Sodium Acetate and 700ul isopropanol; this was then spun at 15,000 rpm for 15 minutes at 4°C. The resulting pellet was rinsed once with 70% ethanol, spun and air dried before being resuspended in 85ul of water. 2ul of this was used in the subsequent Taqman and Sybergreen assays for PAN promoter and spiked-in malaria gene as a normalizing factor.
Taqman and Sybergreen Real-time PCR assays
For all Taqman assays, 200ng of RNA was reverse transcribed using SuperScript III and oligo d(T) primer (Invitrogen) according to manufacturer's instructions. 2ul of this reaction was used in all subsequent real-time PCR Taqman assays. Primer and probe sequences used for lytic gene expression ORF50 is previously described (Bechtel, Grundhoff, and Ganem, 2005). Other primers used were ORF 57 Forward: TGGACATTATGAAGGGCATCC, Reverse: CGGGTTCGGACAATTGCT; gB Forward: TCGCCGCACCAATACCATA, Reverse: CCTGCGATCTACGTCGGG; PAN promoter Forward: GCCAGCTTGAGTCAGTTTAGCA, Reverse: CGAGCACAAAATCCATAGGTG; Malaria DNA Forward: AGGACCCGATCAACAACAT, Reverse: AAGCTGAACAAGAACGCGAT. Taqman reactions (all but malaria DNA) were performed using Taqman Universal Master Mix (Applied Biosystems) and SyberGreen reactions (malaria DNA) were performed using SybergreenER mastermix (Invitrogen) as per the manufacturer's specifications.
NF-κB EMSA
Nuclear enriched lysates were made from cells by incubation in hypotonic buffer (20mM HEPES pH7.8, 5mM KCl, 1.5mM MgCl2, 1mM DTT and protease inhibitors), followed by pelleting and disruption of nuclei by incubation in high salt buffer (0.4M KCl, 50mM HEPES pH7.9, 0.1% NP40, 0.5μM EDTA, 10% glycerol and protease inhibitors). 5μg of lysate was incubated with 32P-labled oligonucleotide encoding the NF-κB consensus sequence (Santa Cruz Biotechnology), dI:dC DNA (Sigma) in binding buffer (20μM HEPES pH7.9, 50mM KCl, 10% glycerol, 1mM EDTA, 1mM MgCl2, 1mM DTT). Reactions were incubated at room temperature for 30 minutes without the labled probe and an additional 30 minutes after addition of the probe. Complexes were resolved in a 1x TBE, 4% acrylamide gel.
NFκB luciferase assay
293T cells were transfected using Fugene 6 (Roche) as per the manufacturer's specifications with increasing amounts for the following constructs: pNFκB-luc (BD Biosciences), pCDNA3.1-RTA, pCD8 (Mylteni Biotech). 36 hours post transfection cells were passed over anti-CD8 MACS columns (Mylteni Biotech) to enrich for cells expressing CD8 and RTA. The separated cells were plated and allowed to recover overnight. The next morning cells were either mock infected or infected with KSHV in triplicate. 48 hours post infection luciferase levels were assayed as per the manufacturer's specifications (Promega).
Results
Inhibition of NFκB in PEL cells leads to increased lytic gene expression and apoptosis
Although Brown et al. (2003) reported that NFκB inhibition upregulated lytic gene expression, other investigators have not observed lytic derepression in this setting (Keller, Schattner, and Cesarman, 2000). We therefore re-examined this issue. In accord with previous work (Brown et al., 2003), we found that inhibition of NFκB signaling in KSHV positive PEL cells leads to increased expression of an array of lytic genes. BCBL-1 cells, a line persistently infected with KSHV, were treated with a range of doses (1, 2, 4, 6μM) of the compound Bay 11-7082 (in DMSO), an irreversible inhibitor of IκBα phosphorylation, or a corresponding amount of DMSO. Total RNA was isolated from the cells and expression of several lytic genes was assayed by Taqman real time RT-PCR. As shown in Fig 1A, cells treated with Bay showed a marked increase in expression of an immediate early lytic gene (ORF 50), a delayed early gene (ORF 57) and a late lytic gene (gB). This increase was seen at all assayed doses and followed a dose-dependent trend.
Figure 1.
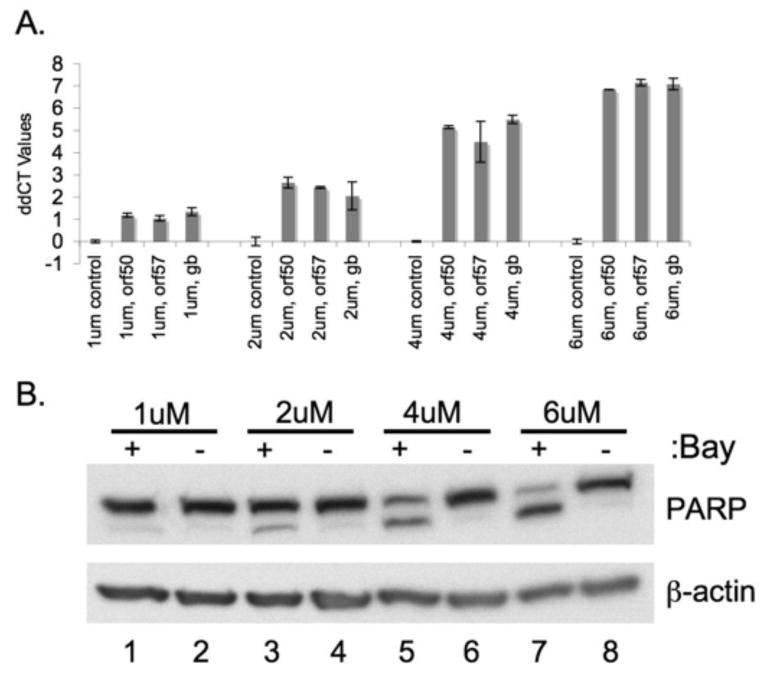
Inhibition of NFκB in BCBL-1 cells leads to increased lytic gene expression as well as increased apoptosis.
BCBL-1 cells were treated with the indicated amounts of Bay 11-7082 or vehicle (DMSO) for 30min at 37°C. Cells were resuspended in fresh media and total RNA and total protein were harvested at 48 hours post treatment. Taqman real time PCR was performed on cDNA made from total RNA. ddCT values (log based 2 indicator of fold change) for lytic genes Orf 50, Orf57 and gB are shown in panel (A). All values were normalized to GAPDH levels, and are expressed for each gene in BCBL-1 cells treated with Bay 1170-82 relative to those treated with the same amount of DMSO. Panel (B) shows a dose dependent increase in apoptosis as measured by PARP cleavage with increasing doses of the Bay 11-7082 compound. 20ug of total protein was blotted for PARP protein and β actin as a loading control.
NFκB inhibition in PEL cells has been reported to be associated with increased apoptosis of the PEL cells (Keller et al., 2006; Keller, Schattner, and Cesarman, 2000). Accordingly, we examined apoptosis levels by immunoblotting for PARP, a downstream cleavage target of caspases 3 and 7 in the apoptotic cascade (Fig 1B). As expected, increased cleavage of PARP was seen in BCBL-1 cells treated with Bay 11-7082 (lanes 1,3,5,7) but not the corresponding vehicle, DMSO (lanes 2,4,6,8). Because apoptosis was assayed in the mass culture rather than at the single cell level, we do not know if the cell population experiencing lytic reactivation was the same or different from that undergoing apoptosis.
De Novo KSHV infection of endothelial cells in the context of NFκB inhibition leads to increased cytotoxicity, lytic reactivation and apoptosis
Previous experiments investigating the link between the NFκB signaling pathway and the KSHV latent-lytic switch have been done almost exclusively in PEL lines (Brown et al., 2003; Guasparri, Keller, and Cesarman, 2004; Keller et al., 2006; Keller, Schattner, and Cesarman, 2000; Sgarbanti et al., 2004). But PEL cells are very far removed from the initial latent infection, having been selected in vivo for stable episome maintenance despite rapid growth – a selection that we know requires epigenetic changes that are likely not present in KS-derived endothelial (spindle) cells or most latently infected cells established in culture (Grundhoff and Ganem, 2004). Therefore, PEL cells may not be fully representative of all cells in which latent KSHV infection can be observed. Accordingly, we have examined several other cell types in which de novo KSHV infection can produce latency, and which are permissive for lytic reactivation. These include primary human umbilical vein endothelial cells (HUVEC) and secondary human foreskin fibroblasts (HFF).
To examine the role of NFκB in HUVEC cells, we generated a HUVEC culture stably expressing a degradation-resistant mutant of IκBα. This version of IκBα (IκB super-repressor, IκBSR) contains two Ser/Ala mutations at positions 32 and 36, rendering it resistant to phosphorylation by the IκBα kinase (IKK) and therefore refractory to subsequent degradation by the proteasome. This results in a stabilization of IκBα-NFκB complexes in the cytoplasm of the cell, inhibiting the ability of the NFκB transcription factor to translocate into the nucleus. As previously described (Grossmann et al., 2006), HUVECs were transduced with retrovirus encoding IκBSR and selected for a short time with puromycin. These cells were assayed for inhibition of NFκB signaling by treatment with TNFα (10ng/ml) for 2hrs, isolation of nuclear extracts, followed by electromobility shift assay (EMSA) to determine NFκB DNA binding activity. Figure 2A shows complete inhibition of inducible NFκB DNA binding in IκBSR HUVECs upon treatment with TNFα, in contrast to those expressing the empty vector, which display strong NF-κB induction.
Figure 2.
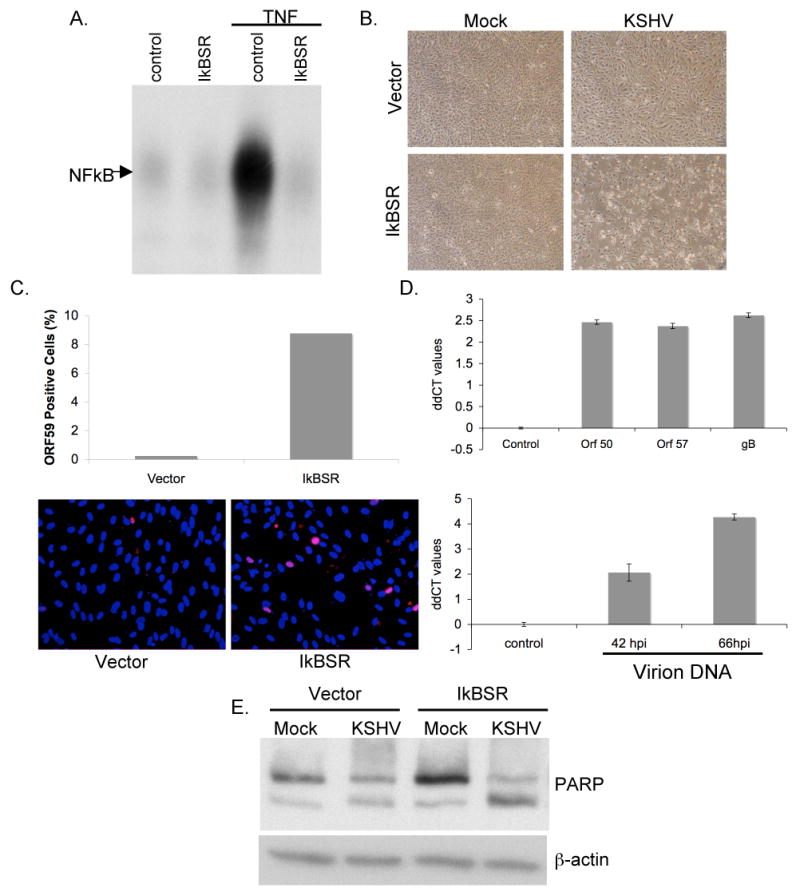
Inhibition of NFκB in HUVEC cells leads to increased cellular toxicity, increased lytic gene expression, virion production and apoptosis upon de novo infection with KSHV.
(A) HUVECs were transduced with retroviruses encoding a degradation-resistant mutant of IκBα (IκBSR). NFκB DNA binding activity in these cells was blocked upon treatment with 10ng/ml TNFα for 2hrs, as compared to those expressing vector alone.
(B) HUVECs expressing IκBSR show increased cellular toxicity upon de novo latent KSHV infection. HUVECs expressing either vector or IκBSR were infected with KSHV for 6hrs, pictures were taken at 66 hours post infection.
(C) KSHV infected IκBSR HUVECs show increased staining for ORF 59 (red) as compared to HUVECs expressing vector alone. Cells were fixed and stained at 66 hours post infection.
(D) Taqman real time PCR ddCT values (log base 2) for ORF 50, ORF 57 and gB (upper panel) and PAN promoter values (lower panel) show increased lytic gene expression and enhanced virion production in IκBSR expressing HUVECs. All values were normalized to GAPDH or spike-in DNA levels, and are expressed for each gene in IκBSR HUVECs relative to vector HUVECs. (Please see Materials and Methods sections for details on virion DNA isolation)
(E) IκBSR HUVECs show increased apoptosis upon infection with KSHV. Total protein was isolated from all cells (floating and adherent to the dish) from vector and IκBSR HUVECs at 66 hours post infection. 20ug of total protein was blotted for PARP protein and β actin as a loading control.
Next, IκBSR and control HUVECs were infected with KSHV at levels that were previously determined to give 100% LANA positive cells and low (< 1%) spontaneous lytic reactivation; infected and mock-infected cells were then followed for 3 days. Figure 2B shows representative pictures of the mock- or KSHV- infected HUVEC monolayers at 66 hours post infection. Upon KSHV infection, IκBSR expressing cells showed reproducible increases in cytotoxicity as compared to control cells. In some cases, this toxicity was visible at time points as early as 18 hours post infection (data not shown), but was most pronounced at the 66 hour time point.
To determine whether the observed increases in cell toxicity correlated with increased spontaneous lytic reactivation of the virus, IκBSR and control HUVECs previously infected with KSHV were fixed and stained for the delayed early protein ORF 59 (Fig 2C). The lower panel of Fig 2C shows representative fluorescence images of cells stained for ORF 59 expression (Red) and DAPI (Blue) at 66 hours post infection (hpi). As is evident in the picture, and is quantified in the histogram in the upper panel, IκBSR HUVECs showed a significant increase in ORF 59 positive cells. In order to better quantify the increase in lytic gene expression in IκBSR HUVECs, total RNA was harvested from cells at 66hpi. Real time quantitative RT-PCR was performed to determine the relative abundance of mRNAs that spanned the multiple phases of temporal regulation of the lytic cycle. As is shown in the upper panel of Fig 2D, all three lytic genes assayed, (ORF50, ORF57 and gB) showed increased expression in IκBSR HUVECs over control cells. Values are normalized to levels of the endogenous GAPDH gene for each sample, and are displayed as ddCT values (log base 2) showing the relative increase in expression of the genes in IκBSR HUVECs over control cells.
To determine whether the observed increase in lytic gene expression in the IκBSR HUVECs corresponded to an increased production of KSHV virions, supernatants from infected cells were collected at 42 and 66hpi. Supernatants were cleared of dead cells and cellular debris and the remaining virions were concentrated by centrifugation. Exogenous DNA was removed by DNase digestion; following inactivation of the nuclease, virions were lysed and virion DNA was extracted as described in the Materials and Methods section. Real time quantitative Taqman PCR was performed on the isolated virion DNA, and the abundance of viral genomes was quantified by measuring the relative levels of the PAN promoter sequence (Fig 2D, lower panel), normalized to a spiked-in control. IκBSR HUVECs showed increased production and release of virions into the supernatant at both the 42 and 66 hpi time points, reaching a maximum of 8-fold. In conjunction with the observed increases in staining for lytic protein and lytic gene expression, the increased production of KSHV virions supports the view that inhibition of the NFκB pathway in primary human endothelial cells leads to increased spontaneous lytic reactivation of KSHV upon de novo infection.
Although much of the cytotoxicity observed in the infected IkB-SR HUVECS could be attributed to cell necrosis from lytic infection, we also asked whether there was an increase in apoptosis in this setting. Lysates taken at 66 hpi were blotted with an anti-PARP antibody to assess the extent of PARP cleavage in the individual cultures (Fig 2E). Increased PARP cleavage was observed in the KSHV infected IκBSR HUVECs (lane 4) as compared to the infected control cells (lane 2), suggesting enhanced levels of apoptosis in these cells. This is not entirely surprising, as many lytic herpesviral gene products can trigger apoptosis. Additionally, since NF-κB is known to upregulate an antiapoptotic cascade in endothelial cells (Stehlik et al., 1998), inhibition of this pathway in infected cells might be expected to trigger enhanced apoptosis.
KSHV-infected human fibroblasts do not display enhanced lytic replication or cytotoxicity in the presence of NFκB inhibition
To determine whether the cell death associated with KSHV infection in the context of NFκB inhibition is common to all cell types, human foreskin fibroblasts (HFF) expressing the IκBSR were constructed. HFFs have been previously shown to be fully permissive for both latent and lytic KSHV infection (Bechtel et al., 2003; Vieira and O'Hearn, 2004). As expected, IκBSR HFFs display strong inhibition of NFκB signaling in response to TNFα (Fig 3A), relative to control HFFs transduced with an empty retroviral vector. These cells were then infected with KSHV under conditions promoting latent infection of nearly all cells. Despite robust latent infection, as evaluated by staining for LANA (green, Fig 3C), no increase in cell death (Fig 3B) or ORF 59 staining (red, Fig 3C) was detected in IκBSR HFFs as compared to control cells. Likewise, no increase in lytic mRNA expression or virion production was observed in these cells (Fig 3D). (In fact, for unclear reasons the levels of lytic transcripts modestly decreased in the presence of IkB-SR). Thus, neither lytic reactivation nor cell injury is an ineluctable consequence of KSHV infection in the absence of NFκB activation. Clearly, the relationship between NFκB signaling and spontaneous lytic reactivation is dependent on the cellular context.
Figure 3.
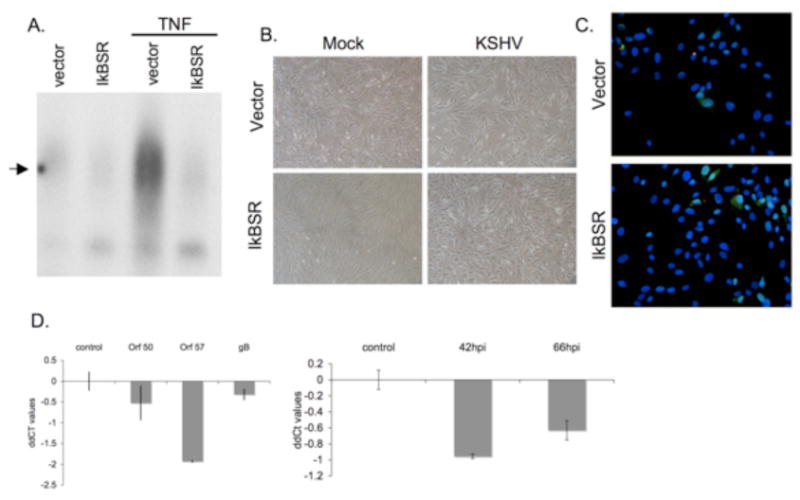
Inhibition of NFkB in human foreskin fibroblasts (HFF) does not lead to increased cellular toxicity, increased lytic gene expression and virion production upon de novo infection with KSHV.
(A) HFFs were transduced with retrovirus encoding a degradation-resistant mutant of IκBα (IκBSR). NFκB DNA binding activity in these cells was blocked upon treatment with 10ng/ml TNFα for 2hrs, as compared to those expressing vector alone.
(B) HFFs expressing IκBSR show no increase in cellular toxicity upon de novo latent KSHV infection. HFFs expressing either vector or IκBSR were infected with KSHV for 6hrs, pictures were taken at 66 hours post infection.
(C) No increase in ORF 59 staining was seen in IκBSR HFFs as compared to vector despite extensive latent KSHV infection as determined by staining for LANA (green). Cells were fixed and stained at 66 hours post infection.
(D) Taqman real time PCR ddCT values for ORF 50, ORF 57 and gB (left panel) and PAN promoter values (right panel) show no increase in lytic gene expression and or virion production in IκBSR expressing HFFs. All values were normalized to GAPDH or spike-in DNA levels, and are expressed for each gene in IκBSR HFFs relative to vector HFFs.
NFκB signaling is activated in lytically infected cells of all lineages
Data showing that inhibition of NFκB induces lytic gene expression in HUVEC and PEL cells suggests that NFκB activation is inhibitory to lytic gene expression. Consistent with this, Brown et al (2003) reported that overexpression of recombinant p65 can extinguish gene expression from cotransfected reporter genes driven by lytic KSHV promoters. These data suggest that NFκB activation may be incompatible with lytic gene expression and lead to the prediction that the lytic cycle might actively suppress NFκB activation. To test this prediction, we examined the state of NFκB activity at several time points during the progression of lytic KSHV infection. Contrary to the predictions of this model, we found robust NFκB activation in several cell types including HUVECs, HFFs and 293T. The NFκB activation state of HUVECs and HFFs was examined during both latent and lytic KSHV infections. Cells were infected with either KSHV alone or KSHV followed by an adenovirus encoding the lytic transactivator RTA (AdRTA). At 48 hpi nuclear extracts were prepared from the cells and NFκB DNA binding was assessed by EMSA (Fig 4A). Increased NFκB DNA binding could be seen in HUVECs upon latent infection (upper panel, lane 3) and was further enhanced upon reactivation with AdRTA (upper panel, lane 4). HFFs showed similarly increased NFκB DNA binding upon lytic KSHV infection (Fig 4A, lower panel). Moreover, this enhanced NFκB was functionally active, as judged by its ability to upregulate expression of an NFκB-dependent luciferase reporter. 293T cells were cotransfected with plasmids encoding CD8, RTA, and an NFκB-luciferase reporter. 36 hours post transfection, cells expressing CD8 (and therefore RTA) were enriched for by binding to magnetic beads bearing anti-CD8. The next day, these cells were either infected with KSHV or mock infected; at 48 hours post infection, luciferase levels were assayed (Fig 3B). KSHV-infected, RTA-expressing cells showed enhanced luciferase readings, indicating increased NFκB-dependent transcriptional activation under these conditions. (We note that the extent of upregulation of the reporter observed here is surely an underestimate of the degree of NFκB activation, owing to the global turnover of host mRNAs during lytic KSHV replication (Glaunsinger and Ganem, 2004)).
Figure 4.
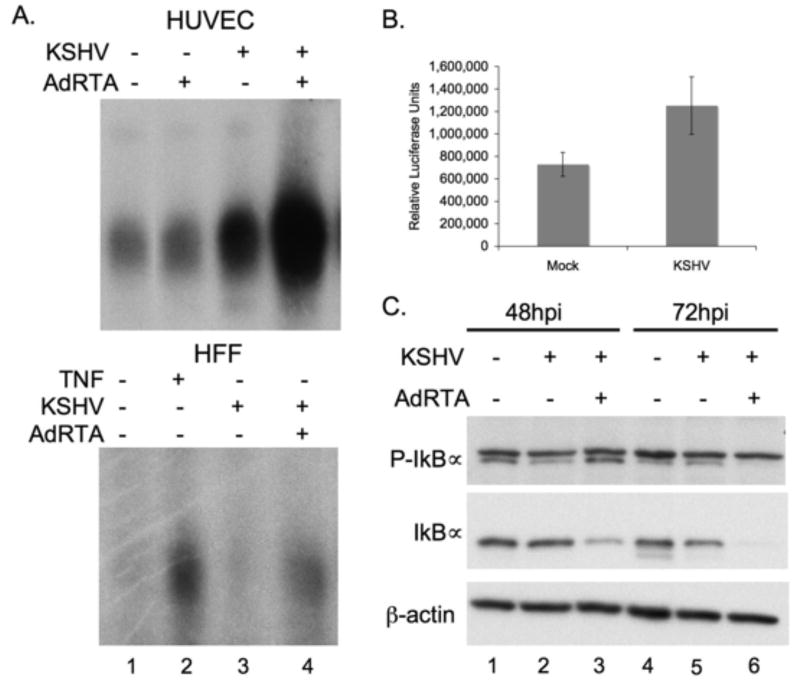
NFκB signaling is activated in lytically infected cells.
(A) Increased NFκB DNA binding activity is observed in lytically infected HUVECs (upper panel) and HFFs (lower panel), as compared to both uninfected, and latently infected cells. Nuclear fractions were harvested at 48 hours post infection, 5ug of protein was used in each binding reaction.
(B) Increased NFκB transcription is shown in 293T cells cotransfected with RTA and NFκB-luciferase reporter at 48 hours post infection. Both Mock and KSHV cells were transfected with CD8, RTA and luciferase plasmids for 36 hrs before infection, and enriched for CD8, and therefore RTA positive cells, using anti-CD8 coupled beads over magnetic columns.
(C) Lytically infected HUVECs show increased phosphorylation of IκBα (top blot, lower band) and corresponding decrease in levels of total IκBα (middle blot) demonstrating active NFkB signal transduction.
As another indication of the activation state of the NFκB pathway during lytic growth, we assayed for phosphorylation of IκBα by the IKK complex in lytically infected HUVECs. Cells were infected with either KSHV alone or KSHV plus Ad-RTA; extracts were prepared at 48 and 72 hpi and assayed by immunoblotting for phospho-IκBα and total levels of IκBα. Figure 4C shows increased phosphorylation of IκBα in lytically infected cells by 48hpi (top panel, lane 3, bottom band) and a corresponding decrease in steady state levels of total IκBα protein in these cells (middle panel, lane 3). By 72 hpi the intracellular pool of IkBα is virtually totally depleted, most likely as a result of proteasome action, and possibly exacerbated by KSHV-mediated mRNA turnover, which is very pronounced late in infection (Glaunsinger and Ganem, 2004). These data affirm that the NFκB pathway is upregulated during lytic replication, and that this induction is occurring at or above the level of IKK activation.
Discussion
Previous studies have described complicated interactions between the NFκB signaling pathway and KSHV infection in the context of PEL cell lines (Brown et al., 2003; Guasparri, Keller, and Cesarman, 2004; Keller et al., 2006; Keller, Schattner, and Cesarman, 2000; Sgarbanti et al., 2004). These studies have shown increased lytic gene induction upon chemical inhibition of NFκB (Brown et al., 2003), enhanced apoptosis upon chemical inhibition of NFκB or knockdown of vFLIP, a potent NFκB inducer (Guasparri, Keller, and Cesarman, 2004; Keller et al., 2006), and even production of defective KSHV particles upon genetic inhibition of NFκB in TPA-induced PEL cells (Sgarbanti et al., 2004). Additionally, inhibition of RTA-mediated transactivation of lytic gene promoters was shown in the context of overexpression of the p65 subunit of NFκB (Brown et al., 2003). Taken together, these accounts have led some to propose a model of suppression of lytic gene expression and function by NFκB activation in PEL cells.
In our hands, inhibition of NFκB in BCBL-1 cells by treatment with Bay 11-7082 lead to increases in both lytic gene expression, in agreement with the work of Brown et al (2003), and enhanced apoptosis, in accord with the observations of Guasparri et al (2004). Results in HUVEC cells were largely congruent with these PEL observations; however, KSHV-infected human fibroblasts behaved completely differently – they displayed neither enhanced cytotoxicity nor increased spontaneous lytic gene reactivation. These results lead us to conclude that the relationship between NFκB and spontaneous reactivation of KSHV is complex, non-uniform and dependent on the cellular context. Cell specific differences in KSHV biology are not without precedent. For example, while many cell types can be latently infected in vitro, only a small subset of these can efficiently support full lytic replication following treatment with chemical inducers (Bechtel et al., 2003). Additionally, we and others have reported modifications in cellular morphology upon latent KSHV infection that occur only within the context of human primary endothelium (Ciufo et al., 2001; Grossmann et al., 2006; Grundhoff and Ganem, 2004; Pan, Zhou, and Gao, 2004; Tang et al., 2003); differences in KSHV gene expression upon de novo infection and in stable latency have also been reported to differ between cell type (Krishnan et al., 2004; Rivas et al., 2001). Given these differences, which indicate intricate and complex interactions of the KSHV genome with the machinery of host gene expression, it is perhaps not surprising that the relationship of lytic induction to NFκB activation is not invariant in different cellular environments.
Finally, despite the earlier finding (Brown et al., 2003) that p65-overexpression can impair reporter genes driven by KSHV lytic promoters (outside of the contect of viral infection), we find that lytic replication is associated with increased NFκB activity in endothelial, epithelial and fibroblast cell lines upon lytic infection. Increased NFκB activity has also been observed in PEL cell lines during lytic reactivation (Sgarbanti et al., 2004), though those studies were complicated by the use of TPA as an inducer (and the fact that isogenic KSHV-negative PEL cells are not available as controls). Therefore, NFκB activation and lytic gene expression cannot be strictly incompatible under all circumstances. Whatever inhibitory influence p65 activation may have on lytic promoters, it clearly can be at least partially bypassed or overcome during full lytic infection. (That this bypass is not complete is indicated by our recent observation that when TPA is used to induce lytic reactivation in infected HUVECs, titers of released virions are 2-3 fold higher in the presence of IkB-SR expression; C.G., unpublished data). Exactly how this bypass is accomplished will require further investigation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Arvanitakis L, Mesri EA, Nador RG, Said JW, Asch AS, Knowles DM, Cesarman E. Establishment and characterization of a primary effusion (body cavity-based) lymphoma cell line (BC-3) harboring kaposi's sarcoma-associated herpesvirus (KSHV/HHV-8) in the absence of Epstein-Barr virus. Blood. 1996;88(7):2648–54. [PubMed] [Google Scholar]
- Bechtel J, Grundhoff A, Ganem D. RNAs in the virion of Kaposi's sarcoma-associated herpesvirus. J Virol. 2005;79(16):10138–46. doi: 10.1128/JVI.79.16.10138-10146.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bechtel JT, Liang Y, Hvidding J, Ganem D. Host range of Kaposi's sarcoma-associated herpesvirus in cultured cells. J Virol. 2003;77(11):6474–81. doi: 10.1128/JVI.77.11.6474-6481.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown HJ, Song MJ, Deng H, Wu TT, Cheng G, Sun R. NF-kappaB inhibits gammaherpesvirus lytic replication. J Virol. 2003;77(15):8532–40. doi: 10.1128/JVI.77.15.8532-8540.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciufo DM, Cannon JS, Poole LJ, Wu FY, Murray P, Ambinder RF, Hayward GS. Spindle cell conversion by Kaposi's sarcoma-associated herpesvirus: formation of colonies and plaques with mixed lytic and latent gene expression in infected primary dermal microvascular endothelial cell cultures. J Virol. 2001;75(12):5614–26. doi: 10.1128/JVI.75.12.5614-5626.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Field N, Low W, Daniels M, Howell S, Daviet L, Boshoff C, Collins M. KSHV vFLIP binds to IKK-gamma to activate IKK. J Cell Sci. 2003;116(Pt 18):3721–8. doi: 10.1242/jcs.00691. [DOI] [PubMed] [Google Scholar]
- Ganem D. KSHV infection and the pathogenesis of Kaposi's sarcoma. Ann Rev Pathology. 2006;1:273–296. doi: 10.1146/annurev.pathol.1.110304.100133. [DOI] [PubMed] [Google Scholar]
- Glaunsinger B, Ganem D. Lytic KSHV infection inhibits host gene expression by accelerating global mRNA turnover. Mol Cell. 2004;13(5):713–23. doi: 10.1016/s1097-2765(04)00091-7. [DOI] [PubMed] [Google Scholar]
- Grossmann C, Podgrabinska S, Skobe M, Ganem D. Activation of NF-kappaB by the latent vFLIP gene of Kaposi's sarcoma-associated herpesvirus is required for the spindle shape of virus-infected endothelial cells and contributes to their proinflammatory phenotype. J Virol. 2006;80(14):7179–85. doi: 10.1128/JVI.01603-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grundhoff A, Ganem D. Inefficient establishment of KSHV latency suggests an additional role for continued lytic replication in Kaposi sarcoma pathogenesis. J Clin Invest. 2004;113(1):124–36. doi: 10.1172/JCI200417803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guasparri I, Keller SA, Cesarman E. KSHV vFLIP is essential for the survival of infected lymphoma cells. J Exp Med. 2004;199(7):993–1003. doi: 10.1084/jem.20031467. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Keller SA, Hernandez-Hopkins D, Vider J, Ponomarev V, Hyjek E, Schattner EJ, Cesarman E. NF-kappaB is essential for the progression of KSHV- and EBV-infected lymphomas in vivo. Blood. 2006;107(8):3295–302. doi: 10.1182/blood-2005-07-2730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller SA, Schattner EJ, Cesarman E. Inhibition of NF-kappaB induces apoptosis of KSHV-infected primary effusion lymphoma cells. Blood. 2000;96(7):2537–42. [PubMed] [Google Scholar]
- Krishnan HH, Naranatt PP, Smith MS, Zeng L, Bloomer C, Chandran B. Concurrent expression of latent and a limited number of lytic genes with immune modulation and antiapoptotic function by Kaposi's sarcoma-associated herpesvirus early during infection of primary endothelial and fibroblast cells and subsequent decline of lytic gene expression. J Virol. 2004;78(7):3601–20. doi: 10.1128/JVI.78.7.3601-3620.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagunoff M, Bechtel J, Venetsanakos E, Roy AM, Abbey N, Herndier B, McMahon M, Ganem D. De novo infection and serial transmission of Kaposi's sarcoma-associated herpesvirus in cultured endothelial cells. J Virol. 2002;76(5):2440–8. doi: 10.1128/jvi.76.5.2440-2448.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Eby MT, Rathore N, Sinha SK, Kumar A, Chaudhary PM. The human herpes virus 8-encoded viral FLICE inhibitory protein physically associates with and persistently activates the Ikappa B kinase complex. J Biol Chem. 2002;277(16):13745–51. doi: 10.1074/jbc.M110480200. [DOI] [PubMed] [Google Scholar]
- Lukac DM, Kirshner JR, Ganem D. Transcriptional activation by the product of open reading frame 50 of Kaposi's sarcoma-associated herpesvirus is required for lytic viral reactivation in B cells. J Virol. 1999;73:9348–9361. doi: 10.1128/jvi.73.11.9348-9361.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore PS, Chang Y. Kaposi's sarcoma (KS), KS-associated herpesvirus, and the criteria for causality in the age of molecular biology. Am J Epidemiol. 1998;147(3):217–21. doi: 10.1093/oxfordjournals.aje.a009440. [DOI] [PubMed] [Google Scholar]
- Pan H, Zhou F, Gao SJ. Kaposi's sarcoma-associated herpesvirus induction of chromosome instability in primary human endothelial cells. Cancer Res. 2004;64(12):4064–8. doi: 10.1158/0008-5472.CAN-04-0657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivas C, Thlick AE, Parravicini C, Moore PS, Chang Y. Kaposi's sarcoma-associated herpesvirus LANA2 is a B-cell-specific latent viral protein that inhibits p53. J Virol. 2001;75(1):429–38. doi: 10.1128/JVI.75.1.429-438.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sgarbanti M, Arguello M, tenOever BR, Battistini A, Lin R, Hiscott J. A requirement for NF-kappaB induction in the production of replication-competent HHV-8 virions. Oncogene. 2004;23(34):5770–80. doi: 10.1038/sj.onc.1207707. [DOI] [PubMed] [Google Scholar]
- Soulier J, Grollet L, Oksenhendler E, Cacoub P, Cazals-Hatem D, Babinet P, d'Agay MF, Clauvel JP, Raphael M, Degos L, et al. Kaposi's sarcoma-associated herpesvirus-like DNA sequences in multicentric Castleman's disease. Blood. 1995;86(4):1276–80. [PubMed] [Google Scholar]
- Staskus KA, Zhong W, Gebhard K, Herndier B, Wang H, Renne R, Beneke J, Pudney J, Anderson DJ, Ganem D, Haase AT. Kaposi's sarcoma-associated herpesvirus gene expression in endothelial (spindle) tumor cells. J Virol. 1997;71(1):715–9. doi: 10.1128/jvi.71.1.715-719.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stehlik C, de Martin R, Kumabashiri I, Schmid JA, Binder BR, Lipp J. Nuclear factor (NF)-kappaB-regulated X-chromosome-linked iap gene expression protects endothelial cells from tumor necrosis factor alpha-induced apoptosis. J Exp Med. 1998;188(1):211–6. doi: 10.1084/jem.188.1.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun R, Lin SF, Gradoville L, Yuan Y, Zhu F, Miller G. A viral gene that activates lytic cycle expression of Kaposi's sarcoma-associated herpesvirus. Proc Natl Acad Sci U S A. 1998;95(18):10866–71. doi: 10.1073/pnas.95.18.10866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang J, Gordon GM, Muller MG, Dahiya M, Foreman KE. Kaposi's sarcoma-associated herpesvirus latency-associated nuclear antigen induces expression of the helix-loop-helix protein Id-1 in human endothelial cells. J Virol. 2003;77(10):5975–84. doi: 10.1128/JVI.77.10.5975-5984.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vieira J, O'Hearn P, Kimball L, Chandran B, Corey L. Activation of Kaposi's sarcoma-associated herpesvirus (human herpesvirus 8) lytic replication by human cytomegalovirus. J Virol. 2001;75:1378–1386. doi: 10.1128/JVI.75.3.1378-1386.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vieira J, O'Hearn PM. Use of the red fluorescent protein as a marker of Kaposi's sarcoma-associated herpesvirus lytic gene expression. Virology. 2004;325(2):225–40. doi: 10.1016/j.virol.2004.03.049. [DOI] [PubMed] [Google Scholar]