

Abstract
Distinction between acute disseminated encephalomyelitis and acute multiple sclerosis is often clinically difficult. Perivenous demyelination is the pathological hallmark of acute disseminated encephalomyelitis, whereas confluent demyelination is the hallmark of acute multiple sclerosis. We investigated whether perivenous demyelination versus confluent demyelination distinguishes acute disseminated encephalomyelitis from multiple sclerosis. Patients with perivenous demyelination (n = 13; median age 43 years, range 5–67) on brain biopsy and/or autopsy, ascertained retrospectively, were compared with a cohort with confluent demyelination only (n = 91; 84% multiple sclerosis, 16% isolated syndrome at follow-up; median age 39 years, range 10–69). Clinical presentation, course and the International Paediatric Multiple Sclerosis Study Group clinical criteria for acute disseminated encephalomyelitis were assessed in both cohorts. Among the perivenous demyelination cohort, 10 patients had only perivenous demyelination and three also had confluent demyelination. All but one patient with perivenous demyelination only had a monophasic course, whereas two of three with both types had a relapsing course. The perivenous demyelination cohort was more likely than the confluent demyelination cohort to present with encephalopathy (P < 0.001), depressed level of consciousness (P < 0.001), headache (P < 0.001), meningismus (P = 0.04), cerebrospinal fluid pleocytosis (P = 0.04) or multifocal enhancing magnetic resonance imaging lesions (P < 0.001). A distinct pattern of cortical microglial activation and aggregation without associated cortical demyelination was found among six perivenous demyelination patients, all of whom had encephalopathy and four of whom had depressed level of consciousness. This pattern of cortical pathology was not observed in the confluent demyelination cohort, even in one patient with depressed level of consciousness. Clinical criteria were 80% sensitive and 91% specific for pathologically defined acute disseminated encephalomyelitis (perivenous demyelination), but misdiagnosed acute disseminated encephalomyelitis among 9% of patients with confluent demyelination and multiple sclerosis diagnosis at last follow-up. Perivenous demyelination is associated with meningoencephalopathic presentations and a monophasic course. Depressed level of consciousness is a more specific clinical criterion for pathologically confirmed acute disseminated encephalomyelitis than encephalopathy, which over-diagnosed acute disseminated encephalomyelitis among multiple sclerosis patients. A distinct pattern of cortical microglial activation without cortical demyelination may be the pathological correlate of depressed level of consciousness in acute disseminated encephalomyelitis. Although pathological evidence of perivenous demyelination may be superior to clinical criteria for diagnosing acute disseminated encephalomyelitis, the co-occurrence of perivenous and confluent demyelination in some individuals suggests pathogenic overlap between acute disseminated encephalomyelitis and multiple sclerosis and misclassification even with biopsy.
Keywords: multiple sclerosis, magnetic resonance imaging, neuropathology, immune-mediated demyelination, demyelinating disease
Introduction
A considerable proportion of children and adults with suspected acute disseminated encephalomyelitis (ADEM) ultimately are diagnosed with multiple sclerosis. Clinical criteria which distinguish monophasic ADEM from first presentations of relapsing multiple sclerosis are needed since patients with multiple sclerosis may benefit from early immunomodulatory or immunosuppressive therapy to prevent continuing inflammatory disease activity, whereas such therapies may not be warranted in those with monophasic ADEM. Consensus clinical diagnostic criteria for ADEM were recently proposed by the International Paediatric Multiple Sclerosis Study Group (Krupp et al., 2007). These clinical criteria diagnose ADEM in the setting of a first presentation of idiopathic inflammatory demyelinating disease in children that is acute or subacute, multifocal, polysymptomatic and marked by encephalopathy; encephalopathy was proposed as the factor best able to distinguish ADEM from a multifocal fulminant presentation of multiple sclerosis. The authors recommended prospective validation of these criteria in a multi-centre study of children with idiopathic inflammatory demyelinating diseases. Prior studies of clinically diagnosed ADEM were limited by the lack of standardized criteria and a diagnostic gold standard for ADEM (Dale et al., 2000; Hynson et al., 2001; Schwarz et al., 2001; Tenembaum et al., 2002; Murthy et al., 2002; Leake et al., 2004; Mikaeloff et al., 2004, 2006; Marchioni et al., 2005). Furthermore, previously published ADEM studies demonstrated considerable clinical overlap with first presentations of multiple sclerosis and have suggested ADEM may occur in a relapsing or recurrent form that is distinct from multiple sclerosis.
Pathological correlative data that might inform clinical distinction of ADEM from first presentations of acute multiple sclerosis are lacking. ADEM is a central nervous system idiopathic inflammatory demyelinating disease pathologically characterized by the presence of sleeves of perivenous demyelination that differ from the sharply demarcated confluent demyelinated plaques typical of multiple sclerosis (Turnbull and McIntosh, 1926; Van Bogaert, 1950; Lumsden, 1951; Uchimura and Shiraki, 1957; Greenfield and Norman, 1971; Oppenheimer, 1976). Most early pathological childhood and adult ADEM series were based on autopsy material from patients with fatal perivenous encephalomyelitis, with or without a prior history of infection or vaccination (Van Bogaert, 1950; Greenfield and Norman, 1971; Oppenheimer, 1976; Mizutani et al., 1977; Miller et al., 1993; Hafler and Hedley-Whyte, 1995; Wang et al., 1996; Shintaku and Matsumoto, 1998; Koch et al., 2005). The last large clinical pathological series of patients with post-infectious perivenous encephalomyelitis was published in 1975 (Hart and Earle, 1975). Since then, only case reports and small series of patients with clinically diagnosed ADEM have been described, accompanied by limited pathological information (de la Monte et al., 1986; Miller et al., 1993; Hasegawa et al., 1994; Kinoshita et al., 1994; Hafler and Hedley-Whyte, 1995; Khan et al., 1995; Horowitz et al., 1995; Dagher and Smirniotopoulos, 1996; Silver et al., 1997; Shintaku and Matsumoto, 1998; Olivero et al., 1999; Dale et al., 2000; Singh et al., 2000; Cohen et al., 2001; Tenembaum et al., 2002; Ravin and Hedley-Whyte, 2002; Lim et al., 2003; Leake et al., 2004; Tan et al., 2004; Mandrioli et al., 2004; Ohtake and Hirai, 2004; Koch et al., 2005; Malveira et al., 2005; Brinar and Poser, 2006). Few of these clinical pathological series document whether perivenous demyelination was present, nor distinguish between perivenous and confluent demyelination (Miller et al., 1993; Hafler and Hedley-Whyte, 1995; Silver et al., 1997; Shintaku and Matsumoto, 1998; Dale et al., 2000; Leake et al., 2004; Koch et al., 2005). An ADEM diagnosis in many of these reports is based on invalidated clinical criteria or the presence of clinical characteristics that are atypical for multiple sclerosis, rather than on pathology (Khan et al., 1995; Maranhao-Filho, 1996; Tsai and Hung, 1996; Dale et al., 2000; Unal et al., 2000; Hartel et al., 2002; Thomas and Hussain, 2004). No study has compared patients with perivenous and confluent demyelination.
We hypothesize that perivenous demyelination, the historical pathological hallmark of ADEM (Van Bogaert, 1950) reliably distinguishes monophasic ADEM from patients with confluent demyelination, the pathological hallmark of acute multiple sclerosis (Lumsden, 1951; Greenfield and Norman, 1971; Hart and Earle, 1975; Oppenheimer, 1976; Lucchinetti et al., 2005). To address this hypothesis we (i) describe the clinical, cerebrospinal spinal fluid and magnetic resonance imaging correlates of perivenous demyelination demonstrated on brain biopsy or autopsy; (ii) compare the clinical characteristics at presentation and follow-up of the perivenous demyelination cohort with an established multiple sclerosis cohort with biopsy proven confluent demyelination; and (iii) retrospectively apply clinical criteria for ADEM (Krupp et al., 2007) to both groups to assess their ability to distinguish ADEM from multiple sclerosis, as determined pathologically.
Materials and methods
Case ascertainment and data collection
This study was conducted under the Mayo Clinic Institutional Review Board approved protocol IRB# 2067-99. Cases of perivenous demyelination were retrospectively identified by searching the Mayo Clinic pathology database of patients who had a brain biopsy or autopsy at Mayo Clinic or had a brain tissue sample referred for neuropathological consultation between 1982 and 2006. Search terms were perivenous demyelination, ADEM, post-infectious encephalomyelitis and post-vaccination encephalomyelitis.
Perivenous demyelination cohort
The inclusion criteria for the perivenous demyelination cohort were (i) brain biopsy performed during diagnostic evaluation or autopsy; (ii) adequate tissue sampling to permit routine neuropathological assessment, including the following stains: haematoxylin and eosin, luxol fast blue or proteolipid protein immunostain for myelin, neurofilament protein immunostain or silver impregnation for axons; (iii) perivenous demyelination defined as limited circumferential perivenous inflammation and demyelination with relative axonal preservation; and (iv) availability of presenting clinical and follow-up information.
Patients were excluded when there was (i) pathological evidence of viral inclusions, bacterial or fungal infection, primary haemorrhage or infarction, neoplasm or other non-demyelinating disease; or (ii) retrospective clinical, laboratory or radiological evidence suggesting an alternative diagnosis.
A flow diagram summarizing case ascertainment is presented in Fig. 1. Of 26 possible cases, 13 were excluded because of no follow-up after brain biopsy (n = 6), insufficient tissue for pathological analysis (n = 2), no pathological evidence of demyelination (n = 2), confluent demyelination only (n = 2), and probable lymphoma (n = 1). Thirteen cases were included. A single brain biopsy was available for nine patients; brain biopsy followed by autopsy in 2; and autopsy alone in 2. The brain biopsies were performed either stereotactically (n = 9) or by open resection (n = 2).
Figure 1.

Case ascertainment.
Confluent demyelination cohort
Patients were identified from a cohort of 780 cases of biopsied confirmed central nervous system inflammatory demyelinating disease with detailed pathological, retrospective and prospective clinical and radiographic data and belonging to the Multiple Sclerosis Lesion Project (NMSS RG3184-B-3-02). Inclusion criteria were: (i) brain biopsy performed; (ii) confluent inflammatory demyelination consistent with multiple sclerosis, confirmed by a neuropathologist (BWS/JEP/CG); and (iii) follow-up neurological examination and brain magnetic resonance imaging. Cases of perivenous demyelination consistent with ADEM (Hart and Earle, 1975), as well as Devic’s neuromyelitis optica defined based on published criteria (Wingerchuk et al., 1999, 2006) were excluded from the confluent demyelination group. Ninety-one cases were included. Approximately 75% of the brain biopsies were performed stereotactically and 25% by open resection; no autopsies were included. Precise calculation of the type of biopsy was not possible because most were performed at a separate institution (87%) and most operative reports were unavailable.
Neuropathological assessment
Specimens were fixed in 10–15% neutral buffered formalin and paraffin embedded. Sections were stained with haematoxylin and eosin, luxol fast blue-periodic acid-Schiff and Bielschowsky silver impregnation. Immunohistochemistry was performed without modification using an avidin–biotin or an alkaline-phosphatase/anti-alkaline phosphatase technique as described earlier (Vass et al., 1986). The primary antibodies were specific for myelin proteins (proteolipid protein, polyclonal; Serotec, Oxford, USA), astrocytes [glial fibrillary acidic protein (Dako)], neurofilament protein (Dako), macrophages/microglial cells [CD68; KP-1, PGM-1 (Dako), KiM1P (Dr. Radzun, University of Göttingen, Germany)]. For immunohistochemistry, the primary antibodies were omitted in controls. All antibodies were incubated at 4°C overnight.
The pattern of demyelination was defined as perivenous when circumferentially restricted around vessels; coalescent when perivenous demyelination overlapped between adjacent vessels; and confluent when demyelination extended beyond a thin rim around the vessel. Stage of demyelinating activity was defined as active or inactive, based on the presence or absence of myelin degradation products within macrophages. Pathological data regarding number and distribution of demyelinating foci, type of demyelination (perivenous, coalescent, confluent), stage of demyelinating activity (active or inactive), composition of the inflammatory infiltrate (lymphocytes, granulocytes, macrophages) and the presence or absence of haemorrhage, necrosis, meningeal inflammation and cortical involvement were recorded. The cortex was further assessed for the presence and pattern of demyelination (perivenous, confluent or subpial) and inflammation, including microglial activation.
Clinical data collection
Clinical information was collected and evaluated blinded to pathological interpretation. Clinical and follow-up information was collected (NPY) on perivenous demyelination patients via retrospective chart review (n = 13) and telephone follow-up with patient (n = 6) or surviving family member (n = 2). Clinical data on the confluent demyelination cohort were obtained (CFL) by face-to-face clinical evaluation (n = 91). The following information was recorded: demographics, neurological symptoms prior to and at the time of brain biopsy or autopsy, treatment and response, symptoms of relapse, clinical course, medical co-morbidities, and laboratory data to evaluate for mimics and cerebrospinal fluid analysis. We evaluated for historical features associated with ADEM including encephalopathy, depressed level of consciousness, seizure, meningismus, prior infection or vaccination and headache. Encephalopathy was defined as a confusional state lasting >24 h (impaired attention, concentration, memory or other cognitive function) excluding isolated aphasia and post-ictal delirium improving <24 h. Depressed level of consciousness was defined by change in level of arousal requiring a painful stimulus to maintain wakefulness, mechanical ventilation or coma. Both the perivenous and confluent demyelination cohorts were analysed for whether they met individual ADEM clinical criteria according to the International Paediatric Multiple Sclerosis Study Group (Krupp et al., 2007) at initial presentation: (i) multifocal magnetic resonance imaging lesions (>1 cm); (ii) polysymptomatic; with (iii) encephalopathy.
Magnetic resonance imaging data were collected on the perivenous demyelination cohort based on retrospective review of images at initial presentation (n = 8) or written radiologist interpretation when images were not available (n = 4). Imaging data were unavailable for one patient. Neuroimaging data included the following: size, number, location of lesions; presence of cortical or deep grey matter T2 lesions; pattern of enhancement, whether the margins were well-defined or ill-defined, the combination of sole presence of well-defined lesions and ovoid lesions perpendicular to the corpus callosum (KIDMUS criteria) reported to be predictive of relapsing course in paediatric patients with first attacks of idiopathic inflammatory demyelination (Mikaeloff et al., 2004), and evolution of lesions on follow-up imaging. In the confluent demyelination cohort, magnetic resonance images obtained at the time of first attack (not time of brain biopsy) were reviewed for presence of multifocal T2 lesions, multifocal gadolinium enhancing lesions and KIDMUS criteria.
Statistical analysis
Presenting clinical, cerebrospinal fluid, neuroimaging and follow-up variables were compared between the perivenous and confluent demyelination cohorts. The ADEM clinical criteria were applied to all first attacks in both cohorts. The sensitivity and specificity of the ADEM clinical criteria were calculated using perivenous demyelination demonstrated on brain biopsy or autopsy as the diagnostic gold standard. Continuous variables are reported as medians and or ranges. Associations between nominal variables were analysed using Fisher’s exact test. Differences between continuous measures were analysed using Wilcoxon rank-sum tests. All tests were two sided.
Results
Neuropathological findings in perivenous demyelination cohort
The findings in individual cases are presented in Table 1. The median time from onset of symptoms to biopsy was 8 days (range 3 days to 22 months) and to autopsy was 23.5 days (range 6–50 days). The median number of tissue blocks analysed per biopsy was 3 (range 1–5), and for autopsies, 18 (range 8–24). White matter was available for analysis in all 13 included cases. The number of demyelinating lesions assessed per block ranged from <10 (n = 10), 10–50 (n = 6) and >50 (n = 3).
Table 1.
Pathological assessment of perivenous demyelination cohort
|
Pattern/location of demyelination |
Cortical |
Other |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Case | Sample | PVD | CD | WM | GM | Subpial | CMA | Haemorrhage | Necrosis | Meningitis |
| 1 | A | + | + | + | + | + | + | - | - | + |
| 2 | A | + | – | + | + | – | + | + | + | + |
| 3 | B/A | + | – | + | + | – | + | + | + | + |
| 4 | B/A | + | – | + | + | + | + | + | + | + |
| 5 | B | + | – | + | – | – | – | + | + | – |
| 6 | B | + | – | + | NA | NA | NA | – | – | NA |
| 7 | B | + | – | + | NA | NA | NA | – | – | NA |
| 8 | B | + | – | + | + | + | + | + | + | + |
| 9 | B | + | – | + | – | – | – | – | – | – |
| 10 | B | + | – | + | – | – | – | + | + | + |
| 11 | B | + | – | + | – | – | – | – | – | – |
| 12 | B | + | + | + | + | – | + | – | – | + |
| 13 | B | + | + | + | NA | NA | NA | – | – | NA |
A = autopsy; B = biopsy; PVD = perivenous demyelination; CD = confluent demyelination; WM = white matter; GM = grey matter; CMA = cortical migroglial activation; NA = adequate tissue not available.
White matter pathology
All 13 cases demonstrated perivenous demyelination (Fig. 2A), macrophage infiltration and mild to moderate perivenous lymphocytic predominant infiltrates with occasional plasma cells, eosinophils and polymorphonuclear cells. Multiple perivenous lesions coalescing into larger areas of demyelination were observed in five cases (Fig. 2B), with three cases demonstrating evidence of both perivenous and confluent demyelinated lesions (Fig. 2C). Active demyelination was noted in 12 of 13 cases, with all lesions from a given case demonstrating a uniform stage of demyelinating activity. Haemorrhage, ranging from remote as evidenced by the presence of haemosiderin laden macrophages or acute and gross haemorrhage, was observed in six cases.
Figure 2.

Representative brain biopsies illustrating the observed patterns of demyelination. (A) Perivenous sleeve of inflammation and demyelination (20×); (B) three coalescing perivenous lesions (60×); and (C) extensive region of confluent demyelination with areas of perivenous demyelination in the periplaque white matter (4×). Luxol-fast blue periodic acid-Schiff myelin stain (A–C).
Cortical pathology
Cerebral cortex was available for analysis in 10 of 13 perivenous demyelination cases (77%). Multifocal, patchy cortical pathology was observed in six cases, including all four autopsies. The spectrum of cortical lesions included perivenous demyelinating intracortical lesions (Fig. 3A), and subpial demyelination (n = 3) (Fig. 3B). A distinct pattern of microglial activation characterized by multifocal microglial aggregates, un-associated with cortical demyelination, was scattered throughout the sampled cortex in six cases (Fig. 3C), or was typically concentrated in cortical layer three, adjacent to large pyramidal neurons (n = 3) (Fig. 3D). The pattern of cortical microglial activation with aggregates dispersed throughout the cortex, often independent of any evidence of cortical demyelination, was not observed in the confluent demyelination cohort.
Figure 3.

Patterns of cortical pathology in perivenous demyelination cohort. (A) Perivenous intracortical demyelinated lesion (20×); (B) subpial demyelination (arrows) (4×); (C) multifocal aggregates of cortical migroglial activation (4×); (D) macrophage/microglial activation concentrated in cortical layer 3 (arrows) (4×). Immunocytochemistry for proteolipid protein (A/B) and KiM1P (C/D).
Presenting symptoms
The presenting symptoms among the perivenous demyelination cases are summarized in Table 2 and comparisons with the confluent demyelination only cohort are summarized in Table 3. There was no difference in median age at presentation, occurrence of preceding infection or vaccination, or the time from symptom onset to biopsy or autopsy between the perivenous and confluent demyelination cohorts. Patients with perivenous demyelination were more likely than those with confluent demyelination to present with encephalopathy (P < 0.004), depressed level of consciousness (P < 0.001), headache (P < 0.001), meningismus (P < 0.005), seizure (P < 0.05) or bilateral optic neuritis (P < 0.04). The distinct pattern of cortical microglial activation and aggregation observed in a subset of perivenous demyelination patients was present in 6 of 10 perivenous demyelination patients who presented with an encephalopathy, of whom four additionally had depressed level of consciousness. An additional perivenous demyelination case demonstrated a similar pattern of cortical pathology in a patient presenting with an isolated seizure and post-ictal depressed level of consciousness lasting <24 h, whereas a single perivenous demyelination case with depressed level of consciousness lacked this pattern of cortical pathology. Depressed level of consciousness was only observed in one of the 91 patients with confluent demyelination, occurring in the setting of confounding sepsis and benzodiazepine use.
Table 2.
Individual cases with perivenous demyelination
| Case | Age/Sex | Prior Attack | PI/V | Presenting symptoms | Time to maximal deficit (days) | Presenting MRI | IPMSSG criteria at presentation | CSF | Clinical course prior to biopsy/autopsy | Time to biopsy/ autopsy | Path | Treatment | Treatment response | Last EDSS | Clinical course | Onset to death or last follow up |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 25F | No | No | Headache Mild meningismus Encephalopathy DLC Quadriparesis Bilateral optic neuritis | 27 | Diffuse T2-weighted signal involving bilateral medulla, pons, cerebellum, midbrain, left temporal lobe; enhancement uncleara | Yes | WBC 10 PRO 50 GLU NA OCB 2 | NA | Autopsy day 27 | PVD CD +CMA | IVMP PLEX | None | 10 | Monophasic- fatal | 27 days |
| 2 | 67F | No | No | Encephalopathy DLC Quadriparesis Seizure | 20 | Multifocal bilateral large T2-weighted subcortical and cortical lesions without enhancement, mild mass effect | Yes | WBC 46 PRO 63 GLU 82 OCB 0 | NA | Autopsy day 20 | PVD +CMA | IVMP PLEX | Partial; died of cardiac arrest while neurologically improving | 10 | Monophasic- fatal | 20 days |
| 3 | 30F | No | No | Headache Encephalopathy (by 2 weeks) Ataxia Vomiting DLC (by 6 weeks) | 50 | MRI 6 weeks after onset: multiple bilateral small foci of increased periventricular, corpus callosum, brainstem T2-weighted signal minimal enhancement; clear deep grey matter involvement by 9 weeks with progression of lesions still with minimal enhancement | Yes | WBC 8 PRO 248 GLU 70 OCB 0 | Biopsy at 5 weeks followed by progression during treatment with IVMP and PLEX | Biopsy day 39 Autopsy day 50 | PVD +CMA | IVMP PLEX | None | 10 | Monophasic- fatal | 50 days |
| 4 | 37F | No | No | Headache Encephalopathy DLC Gait impairment Urinary incontinence | 2 | NA | Yes | NA | NA | Biopsy day 3 Autopsy day 6 | PVD +CMA | NA | None | 10 | Monophasic- fatal | 6 days |
| 5 | 17F | No | No | Headaches, severe Hemiparesis, left Arm weakness, right Bilateral visual impairment Abulia Encephalopathy DLC Required mechanical ventilation | 3.5 | Extensive areas of increased T2-weighted signal involving temporoparietal regions bilaterally, but also right occipital and left subfrontal areas; mild enhancement moderate mass effectb | Yes | WBC 540 PRO 130 GLU 61 OCB NT | NA | 8 days | PVD -CMA | IVMP Oral glucocortico steroid | Partial | 3.5 | Monophasic-survived | 13.6 years |
| 6 | 62F | No | URI | Headache Encephalopathy DLC Hemiparesis | 3 days | Day 3: numerous large (>2 cm) ill-defined, bilateral fluffy-appearing lesions with mass effect and oedema; faint rim of enhancement in subcortical white matter. Day 24 (after glucocorticosteroids): the enhancement resolved; residual T2 abnormality | Yes | NT | NA | 7 days | PVD Cortex NA | IV DEX | Partial | 1 | Monophasic- survived | 3 years |
| 7 | 43M | No | No | Partial seizures Epilepsy partialis continua Leg weakness, numbness | 2 weeks | Normal Day 9: normal Month 21: Focal parasagittal and paracentral white matter lesion with faint enhancement | No | WBC 4 PRO 31 GLU 61 OCB 0 | Partial seizures resolved with anti-epileptic drugs; no other treatment; no further attacks or progression; follow up MRI suggested glioma at 21 months | 22 months | PVD Cortex NA | None | NA; residual sensory loss after biopsy | 3 | Monophasic- survived | 16.7 years |
| 8 | 40M | No | No | Isolated seizure | NA | Incomplete ring enhancing left frontal mass with mass effect and oedema | No | NT | NA | 7 days | PVD +CMA | Oral glucocortico steroid | Complete | 0 | Monophasic- survived | 4.9 years |
| 9 | 53F | No | URI | Headache Encephalopathy Gerstmann syndrome Hemiparesis Gait ataxia | 2 | Day 2 MRI: Large ill-defined left hemispheric T2-weighted lesion with mild mass effect and shift; no enhancement; involved insula and thalamus; right hemisphere normal | Yes | NT | NA | 6 days | PVD –CMA | IV DEX | Complete | 0 | Monophasic- survived | 15.6 years |
| 10 | 61F | No | PV | Headache Encephalopathy DLC Nausea Meningismus, mild Hemiparesis Required mechanical ventilation | 3 | Large frontotemporal mass with oedema, midline shift and crossing the anterior corpus callosum with faint enhancement | Yes | NT | NA | 3 days | PVD –CMA | IVMP | Partial | 5 | Monophasic- survived | 2.5 years |
| 11 | 45F | No | URI | Encephalopathy Ataxia Neurogenic bladder Pseudobulbar affect | 10 weeks | Multifocal bilateral increased T2 in white matter, confluent; partial enhancementb | Yes | WBC 4 PRO 30 GLU 89 OCB 0 | Subacute stereotypical exacerbation | 10 weeks | PVD –CMA | IVMP | Complete initial response | 10 | Relapsing: recurrence at 6 and 8 months improved with IVMP and oral glucocorticosteroid, then severe recurrence at 9 months progressing to death despite IVMP and PLEX | 10 months |
| 12 | 5M | No | URI | Headache Vomiting Irritability Meningismus Ataxia Neck and leg pain Urinary retention Paraparesis | 4 weeks | CT scan: normal at presentation MRI 5 months later: multifocal bilateral T2 midbrain, pons, cervical cord, largest right PO and corpus callosum with atrophy (no gadolinium given) | Yes | WBC 40 PRO 18 GLU 51 OCB 0 | Initial syndrome spontaneously improved, then worsened with bilateral optic neuritis one month later improved with oral glucocorticosteroids, then relapse again prior to biopsy. | 7 months | PVD CD +CMA | IVMP | Near complete initial | 10 | Relapsing glucocorticosteroid responsive syndrome (at least 7); EDSS 2.5 16 months after onset; eventually unable to wean glucocorticosteroids; patient died secondary to status epilepticus 7 years after onset | 7 years |
| 13 | 68F | Yesb | No | Diplopia Dysarthria Gait ataxia Face numbness | 6 weeks | Patchy and punctuate solitary brain stem mass | No | WBC 2 PRO 49 GLU 59 OCB 0 | Possibly related 1–2 week spell of diplopia 30 years priorb | 7 weeks | PVD CD Cortex NA | IVMP | Partial | 7 | Relapsing stable at last follow up MRI lesion smaller, but leading edge of enhancement without new lesions | 1.5 years |
NT = not tested; NA = not available; PVD = perivenous demyelination; CD = Confluent demyelination; PC = perivenous demyelination with coalescence; CMA = cortical microglial activation; LOC = level of consciousness; DLC = depressed level of consciousness;WBC = white blood cell count; PRO = protein; EDSS = Expanded Disability Status Scale; GLU = glucose; OCB = oligoclonal bands; PLEX = plasma exchange; IV = intravenous; IVMP = intravenous methylprednisolone; CSF = cerebral spinal fluid; IPMSSG = International Paediatric Multiple Sclerosis Study Group ADEM criteria; DEX = dexamethasone.
a Description according to report; images unable to be obtained for review.
b Two episodes of binocular horizontal diplopia lasting 1–2 weeks, 30 years prior to index attack. The patient was not evaluated at the time.
Table 3.
Comparison of clinical and MRI characteristics in perivenous versus confluent demyelination cohorts
| Clinical and MRI characteristics | PVD (n = 13) | CD (n = 91) | Pa |
|---|---|---|---|
| Median age, years (range) | 44 (5-68) | 39 (8–69) | 0.24 |
| Female:male | 10:3 | 47:44 | 0.14 |
| Preceding infection/vaccination, n (%) | 4 (31) | 14 (15) | 0.09 |
| Multifocal T2 MRI lesions, n (%)b | 10 (77) | 54 (63) | 0.18 |
| Multifocal enhancing MRI lesions, n (%) | 4 of 10 (40%) | 25 (27) | <0.001 |
| Polysymptomatic, n (%)b | 11 (85) | 67 (74) | 0.51 |
| Encephalopathy, n (%)b | 10 (77) | 23 (25) | <0.001 |
| Depressed level of consciousness, n (%) | 8 (62) | 1 (1)c | <0.001 |
| Bilateral optic neuritis, n (%) | 2 (15%) | 1 (1) | 0.04 |
| Headache, n (%) | 8 (62) | 8 (9) | <0.001 |
| Meningismus, n (%) | 2 (15) | 1 (1) | 0.04 |
| Seizure, n (%) | 3 (23) | 5 (6) | 0.06 |
| Cerebellar, n (%) | 3 (23) | 31 (34) | 0.54 |
| Brainstem, n (%) | 2 (15) | 25 (27) | 0.51 |
| Cognitive, n (%)d | 10 (77) | 35 (38) | 0.01 |
| Motor, n (%) | 7 (54) | 47 (52) | 1.00 |
| Sensory syndrome, n (%) | 4 (31) | 36 (40) | 0.76 |
| KIDMUS MRI criteria, n (%)e | 0 (0) | 15 (17) | 0.35 |
| Absence of any KIDMUS MRI criteria, n (%) | 7 (64) | 23 (26) | 0.01 |
| Median days symptom onset to biopsy/autopsy (range) | 27 (3 days to 1.9 years) | 45 (4 days to 27.6 years) | 0.13 |
| Cerebrospinal fluid | |||
| White blood cells/mm3, median (range) | 19.0 (1.0–540.0) | 3.0 (0.0–1250.0) | 0.04 |
| Protein, median mg/dl (range) | 40.9 (10.0–215.0) | 40.5 (15.0–175.0) | 0.92 |
| Elevated immunoglobulin G synthesis rate, n (%) | 1 (14) | 17 (40) | 0.66 |
| Oligoclonal bands, n (%) | 1 of 7 (14) | 12 of 54 (22) | 1.0 |
| Follow up | |||
| Duration, median (range) | 9.6 (1.4–16.5) | 2.9 (0.1–18.8) | 0.25 |
| EDSS at presentation, median (range) | 9 (0–9.5) | 3 (0–9) | 0.003 |
| EDSS last follow up (living patients), median (range) | 3.5 (0–7) | 2.5 (0–8) | 0.53 |
PVD = perivenous demyelination; CD = confluent demyelination; EDSS = Expanded Disability Status Scale.
a The Wilcoxon rank sum test was used to compare continuous measures and Fishers exact test for discrete measures.
b Three major International Paediatric Multiple Sclerosis Study Group ADEM criteria (IPMSSG) criteria for patients presenting with first attacks of idiopathic inflammatory demyelination.
c Confounded by benzodiazepine and urosepsis.
d Cognitive includes depressed level of consciousness.
e Sole presence of well defined lesions AND ovoid lesions perpendicular to the long axis of corpus callosum.
Assessment of ADEM clinical criteria
Patients with perivenous demyelination were more likely to satisfy all ADEM clinical criteria at presentation than patients with confluent demyelination (77% versus 9%; P < 0.001). The ADEM clinical criteria were 80% (95% CI 0.44–0.96) sensitive and 91% (95% CI 0.83–0.96) specific (Fig. 2) for a pathological diagnosis of ADEM (perivenous demyelination alone; excluding those with both perivenous demyelination and confluent demyelination) and 77% sensitive (95% CI 0.46–0.94) and 91% specific (95% CI 0.83–0.96) for perivenous demyelination with or without coexistent confluent demyelination (Table 4). Both patients with perivenous demyelination alone, not meeting the clinical criteria, presented without encephalopathy apart from brief post-ictal delirium and had focal brain magnetic resonance imaging lesions.
Table 4.
Sensitivity and specificity of ADEM clinical criteria in perivenous demyelination
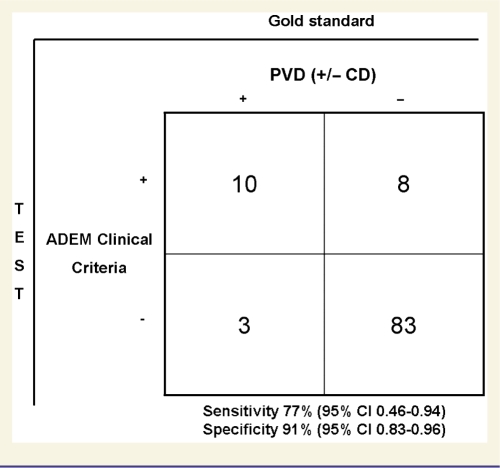 |
PVD = perivenous demyelination; CD = confluent demyelination.
When applied to the confluent demyelination cohort (n = 91), individual components of the ADEM clinical criteria at initial presentation were frequently satisfied: multifocal brain magnetic resonance imaging lesions (61%), polysymptomatic presentation (75%) and encephalopathy (22%). At the time of last follow up in the confluent demyelination cohort [median 2.9 years (0.1–18.8)], eight of eight adult patients with confluent demyelination initially meeting all ADEM clinical criteria at presentation eventually fulfilled McDonald criteria for multiple sclerosis; including one patient with a single attack (2.7 years follow up), multiple brain magnetic resonance imaging lesions and oligoclonal bands. Five patients in the confluent demyelination cohort had initial attacks that did not fulfil ADEM clinical criteria, which were followed by subsequent attacks with multifocal lesions and polysymptomatic presentation including encephalopathy that would have fulfilled ADEM clinical criteria had they been first attacks.
Cerebrospinal fluid
Cerebrospinal fluid was assessed in 8 of 13 (62%) patients in the perivenous demyelination cohort and 62 of 91 (68%) in the confluent demyelination cohort with results summarized in Table 3. The perivenous demyelination group had a higher median cerebrospinal fluid white blood cell count than the confluent demyelination group (median 19; range 1–540 mm3; interquartile range 3–43; P = 0.04). There was no difference in total protein or abnormal immunoglobulin G synthesis rate. Although the presence of supernumerary oligoclonal bands was not different between groups, a difference could not be excluded because of small sample sizes. The only patient having oligoclonal bands in the perivenous demyelination cohort also had confluent demyelination.
Presenting MRI correlates of perivenous demyelination
Brain magnetic resonance imaging abnormalities found in the perivenous demyelination cohort are summarized in Table 5 and representative examples illustrated in Fig. 4.
Table 5.
magnetic resonance imaging correlates of perivenous demyelination
| T2/fluid attenuated inversion recovery signal abnormalities |
Gadolinium enhancement |
Total number of MRI/time of last follow up MRI | Follow-up | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Case | Multiple | Bilateral | Single | ST | BS | C | DGM | WM | GM | >2 cm | Ill-defined | Well-defined | Present | Description | Summary of follow up MRI | |
| 1* | U | + | U | + | + | + | U | + | U | + | + | – | U | U | 1 | NA |
| 2 | + | + | – | + | – | – | – | + | + | + | + | – | No | NA | 1 | NA |
| 3 | + | + | – | + | + | – | – | + | – | – | + | + | No | NA | 1/9 weeks | Progressive increase in diffuse non-enhancing perivenous lesions, with DGM involvement |
| 4* | U | U | U | U | U | U | U | U | U | U | U | U | U | U | NA | NA |
| 5* | + | + | – | + | – | – | – | + | + | + | + | – | Yes | ‘Mild’, pattern unclear | 1/11 years | Residual ill-defined T2 signal without enhancement (11 years later), no new lesions |
| 6 | + | + | – | + | – | – | – | + | + | + | + | – | Yes | Faint rim | 6/1 year | Residual non-enhancing ill-defined T2 signal, no new lesions |
| 7 | – | – | + | + | – | – | – | + | + | + | + | – | Yes | Faint, punctate | 5/5 years | Biopsy tract, otherwise no change |
| 8 | – | – | + | + | – | – | – | + | + | + | – | + | Yes | Open ring | 5/4 years | Biopsy cavity, no new lesions |
| 9 | + | – | – | + | – | – | + | + | + | + | + | – | No | NA | 2/1 year | Residual T2 signal, no new lesions |
| 10 | + | + | – | + | – | – | – | + | + | + | + | – | Yes | Faint rim, faint central punctuate | 2/1 year | Residual T2 without enhancement, no new lesions |
| 11* | + | + | – | + | – | – | U | + | + | + | + | – | Yes | ‘Partial’ pattern unclear | 1/9 months | Increase in diffuse ST, DGM confluent T2 signal with patchy/punctuate enhancement. |
| 12* | + | + | – | + | + | U | U | + | U | + | + | – | U | Not performed | Not performed | |
| 13 | + ** | – | + | – | + | – | – | + | – | + | – | + | Yes | Partial enhancement of margin/leading edge with punctate central enhancement | 1/18 months | Brainstem T2-weighted lesion improved but persistent leading edge of enhancement, interval development of periventricular T2-weighted signal abnormalities, some perpendicular to corpus callosum, 18 months later. |
NA = not applicable; U = unknown; ST = supratentorial; C = cerebellar; DGM = deep grey matter; WM = white matter; GM = grey matter.
a Presenting MRI images were not available.
b Non-specific T2 abnormalities in the subcortical white matter.
Figure 4.

Magnetic resonance imaging correlates of perivenous demyelination. (A) Coronal fluid attenuated inversion recovery (FLAIR): multifocal large ill-defined supratentorial and brainstem lesions without enhancement; (B) axial T2: large ill-defined non-enhancing uni-hemispheric lesion; (C) T1 with gadolinium: single large open ring enhancing mass with surrounding oedema; (D) axial FLAIR: large brainstem lesion with (H) punctate central and peripheral rim of enhancement; (E and F) sagittal FLAIR: numerous multifocal bilateral non-enhancing T2 lesions in subcortical white matter, basal ganglion, cerebellum; (G) coronal T1 with gadolinium: numerous bilateral enhancing subcortical white matter lesions; (I) T1 with gadolinium: faint rim of enhancement of large ill-defined lesion with mass effect; (J) coronal FLAIR; residual confluent signal change crossing the corpus callosum; (K) axial FLAIR: residual signal change oriented perpendicular to corpus callosum becoming confluent; (L) new non-enhancing periventricular white matter lesions in patient with both perivenous demyelination and confluent demyelination.
T2 abnormalities
Multifocal, bilateral (Fig. 4A) or unilateral (Fig. 4B), supratentorial T2 lesions involving the white matter (n = 10) predominated, but single supratentorial (n = 1; Fig. 4C) or brainstem (n = 1; Fig. 4D) lesions were observed. Lesions extended into the juxtacortical white matter in eight cases. Deep grey matter involvement was uncommon (n = 1; Fig. 4F). Two cases had multiple uni-hemispheric lesions (i.e. the contralateral hemisphere appeared normal). One patient had a normal brain magnetic resonance imaging at presentation but at 21 months, a repeat study revealed a single ill-defined non-enhancing supratentorial lesion. The T2 signal abnormalities were usually ill-defined (n = 7) and >1 cm (n = 8). However, well-defined, discrete lesions were observed in two patients, one of whom had a single well-defined ‘open ring’ (Masdeu et al., 2000) enhancing supratentorial mass lesion (4 cm) with surrounding vasogenic oedema (Case 8, Fig. 4C) and another had a brainstem lesion (Case 13, Fig. 4D) with a mixture of enhancement at the edge of demyelination and punctate enhancement centrally. Case 3 presented with innumerable small (<1 cm), ill-defined, non-enhancing lesions perpendicular to the corpus callosum (Fig. 4E) that increased in number over the subsequent weeks (Fig. 4F).
Gadolinium enhancement
Gadolinium enhancement was present in six of nine cases (33%) with perivenous demyelination. The presence of enhancement was not associated with the interval between symptom onset to imaging. The patterns of enhancement were faint and punctate (n = 5) (Fig. 4G and H) and ring/arcs (n = 3) (Fig. 4C and I) with an overlap of both patterns observed in two patients. Although there was no difference in the frequency of multifocal T2 magnetic resonance imaging lesions between cohorts (77% perivenous demyelination, 63% confluent demyelination), patients with perivenous demyelination more frequently had multifocal enhancing lesions at first presentation of demyelinating disease compared with the confluent demyelination cohort (P < 0.001). However, the perivenous and confluent demyelination cohorts were equally likely to present with multiple lesions with and without enhancement. Enhancement of all lesions was not found in any patient with perivenous demyelination, contrary to common expectation for a monophasic demyelinating syndrome.
Follow-up magnetic resonance imaging
No monophasic-surviving patient developed additional lesions on brain magnetic resonance imaging (median interval to repeat imaging 5 years; range 1–11; median number of studies 5, range 1–6). The initial T2 lesions decreased in size but did not completely resolve (Fig. 4J and K) in all monophasic-surviving patients. A follow-up study was performed in 2 of 3 relapsing perivenous demyelination patients and both developed new non-enhancing T2 lesions in the periventricular white matter (Case 13, Fig. 4L), as well as subcortical white matter and deep grey matter (Case 11). Follow-up data were unavailable for one relapsing patient (Case 12) and three patients with a monophasic fatal course. Multifocal lesions were found in 86% of patients in the confluent demyelination cohort.
Treatment in perivenous demyelination cohort
Initial treatments included 1 g intravenous methylprednisolone for 3–5 days (n = 8), intravenous dexamethasone (n = 2), oral prednisone (n = 1) and no treatment (n = 2). The response to initial treatment was complete (no residual deficit; n = 2), partial (improved but residual deficit; n = 7) and no response (all to 5 days of intravenous methylprednisolone; n = 3). The three patients who did not improve after 5 days of 1 g intravenous methylprednisolone were treated with plasma exchange; two died from the neurological illness (Cases 1 and 3) and one who was neurologically improving during plasma exchange died of cardiac arrest (Case 2). Case 11 had a complete initial response to intravenous methylprednisolone maintained on oral prednisone, then partially responded to intravenous methylprednisolone at relapses occurring at 6 and 8 months, but did not respond to intravenous methylprednisolone followed by plasma exchange at a nine month relapse and died 10 months after onset. Case 4 died 6 days after onset before any treatment. Case 7 spontaneously improved without treatment and had a mild residual non-disabling sensory deficit.
Clinical course
Perivenous demyelination cohort
The clinical course is summarized in Table 6. The median duration of clinical follow-up among the entire perivenous demyelination cohort (onset to last contact among surviving patients) was 9.6 years (range 1.4–16.5). The median Expanded Disability Status Scale score among surviving patients at last follow-up was 3.5 (range 0–7). The clinical course was monophasic in 10 patients (77%; six non-fatal and four fatal outcomes) and relapsing in three (23%; two eventually fatal). Among the three patients with a relapsing clinical course, there was pathological evidence for overlap of perivenous and confluent demyelination in two; both cases developed new T2 lesions on follow-up brain magnetic resonance imaging and fulfilled criteria for multiple sclerosis. One patient was a 5-year-old male (Case 12) who presented with a syndrome initially meeting ADEM clinical criteria. The third patient (Case 1) presented with a fulminant attack meeting ADEM clinical criteria followed by death 20 days after the onset; at autopsy, perivenous and confluent demyelinating lesions were disseminated throughout the brain stem.
Table 6.
Clinical course
| PVD (n = 10) | PVD + CD (n = 3) | CD (n = 91) | |
|---|---|---|---|
| Median time to last follow up or death, years (range) | 3 (6 days to 16.5 years) | 1.5 (27 days to 7 years) | 2.9 (0.1–18.8) |
| IPMSSG criteria ADEM presentation | 8 (80%) | 2 | 8 (9%) |
| IPMSSG criteria for monophasic ADEM last follow up | 7 (70%) | 1 (fatal) | 0 |
| Criteria multiple sclerosis McDonald/probable poser | 0 | 1/1 | 69/7 |
| IPMSSG criteria recurrent ADEM | 1 (10%) | 0 | 0 |
| Monophasic, focal, without encephalopathy | 2 (20%) | 0 | 9 (10%) |
PVD = perivenous demyelination; CD = confluent demyelination; IPMSSG = International Paediatric Multiple Sclerosis Study Group clinical ADEM criteria.
Patients with perivenous demyelination alone were more likely to have a monophasic clinical course compared with patients with only confluent demyelination (90% versus 33%, P < 0.001). All six perivenous demyelination monophasic patients who survived had a short interval between symptom onset to maximal deficit (2–3.5 days); two presented with first seizure; six with one or more symptoms of meningoencephalitis (headache, seizure, encephalopathy), four with depressed level of consciousness, and all six had large (>2 cm), ill-defined T2 brain magnetic resonance imaging lesions that were either unifocal or mainly involved one hemisphere. Four fatal cases with perivenous demyelination alone had extensive multifocal bilateral hemispheric involvement on magnetic resonance imaging (Cases 2–4 and 11).
Confluent demyelination cohort
The median duration of clinical follow-up of the confluent demyelination cohort (onset to last contact) was 2.9 (0.1–18.8) years. The median Expanded Disability Status Scale score at last follow-up was 2.5 (range 0–8). Thirty of 91 patients in the confluent demyelination cohort had a single clinical attack, only two of which met ADEM clinical criteria (both eventually met McDonald Criteria for multiple sclerosis). At last follow-up, 76 of 91 individuals fulfilled multiple sclerosis criteria [McDonald (n = 69); probable Poser (n = 7)], and 15 patients (16%) had an isolated inflammatory demyelinating syndrome (median follow-up, 7.1 years). Clinical course and diagnosis at time of last follow-up for the 76 patients with multiple sclerosis were as follows: 54 (73%) relapsing remitting multiple sclerosis, 9 (12%) secondary progressive multiple sclerosis, 1 (1%) primary progressive multiple sclerosis, 7 (9%) monophasic (all probable or laboratory supported multiple sclerosis), 3 (4%) progressive-relapsing multiple sclerosis and unknown for two (3%) patients.
Discussion
This study is the first to test the hypothesis that perivenous demyelination may be a pathological gold standard for ADEM by defining and comparing cohorts based on pathological criteria for ADEM (perivenous demyelination) and multiple sclerosis (confluent demyelination). It is the largest clinical–radiological–pathological correlative study of perivenous demyelination since 1975 (Hart and Earle, 1975) and the first to assess the ADEM clinical criteria proposed by the International Paediatric Multiple Sclerosis Study Group using a hypothetical pathological gold standard of perivenous demyelination.
Spectrum of pathology associated with perivenous demyelination
Similar to prior autopsy series of patients with fatal post-infectious encephalomyelitis (Hart and Earle, 1975), we observed a characteristic spectrum of pathological abnormalities including perivenous inflammation and demyelination (all by definition), cortical microglial pathology (n = 6 of 10), microscopic or gross haemorrhage (n = 6 of 13) and a mild lymphocytic meningeal infiltration (n = 7 of 10). The presence of haemorrhage was not restricted to fatal cases, although the findings resembled those described in acute haemorrhagic leukoencephalitis (Hurst Disease) which is considered to be a severe lethal variant of ADEM.
Unique cortical pathology may be the pathological correlate of depressed level of consciousness in ADEM
We observed a multifocal pattern of cortical microglial activation in non-demyelinated cortex within the perivenous demyelination cohort (n = 6 of 10), but not in the confluent demyelination cohort. KIM1P macrophage staining was essential for detecting cortical microglial activation that would not have been detected using routine histological stains. In three cases, we observed a unique specificity of microglial activation concentrated around pyramidal neurons in the third cortical layer, of uncertain significance. Absence of cortical abnormalities in some cases with perivenous demyelination and those with confluent demyelination may result from sample available from biopsies targeting white matter lesions, sampling bias or absent pathology. Viral inclusions were absent in all cases. The pattern of cortical microglial we observed may be a non-specific reaction to the white matter process with secondary retrograde neuronal injury (Kutzelnigg et al., 2005). However, our finding of a unique pattern of microglial activation independent of cortical demyelination characterized by scattered microglial aggregates involving non-demyelinated cortex occurred in a subset of cases with perivenous demyelination, and was not observed among the confluent demyelination cohort. Interestingly, this pattern was over-represented among perivenous demyelination cases with a history of depressed level of consciousness. We therefore hypothesize that this diffuse pattern of microglial activation in perivenous demyelination patients may represent the pathological substrate of depressed level of consciousness. Whether cortical microglial activation reflects a truly unique pathogenic response operating in ADEM, possibly against a neurotropic virus or other infectious trigger, is speculative but merits further investigation.
Rare co-occurrence of perivenous and confluent demyelination
Both perivenous and confluent demyelination were found in three cases. Although there was no statistically significant difference in time of symptom onset to biopsy between the perivenous and confluent demyelination cohorts, the sample sizes are small and therefore we cannot exclude the possibility that perivenous demyelination is an early stage of a subset of lesions which will eventually become confluent. These cases are consistent with the rare ‘transitional’ cases demonstrating both ADEM (perivenous demyelination) and multiple sclerosis (confluent demyelination) pathology at autopsy associated with both a monophasic and relapsing course (Van Bogaert, 1950; Uchimura and Shiraki, 1957; Seitelberger et al., 1958; Oppenheimer, 1976; Mizutani et al., 1977; Prineas et al., 2002). Although pathological sampling error may be a limitation of case ascertainment in this and other studies, the three patients with both perivenous demyelination and confluent demyelination in this series were derived from a pool of ∼780 cases of biopsy or autopsy proven demyelinating disease examined at our institution as part of the Multiple Sclerosis Lesion Project. Thus co-occurrence of perivenous and confluent demyelination in a pathological sample is an uncommon phenomenon, an observation consistent with prior reports (Greenfield and Norman, 1971; Oppenheimer, 1976). Therefore, sampling error is unlikely to be a major limitation of assessing patterns of inflammatory demyelination. This contention is further supported by two cases in the series in which a brain biopsy was followed by an autopsy, which revealed the same pattern of demyelination (Cases 3 and 4). Whether co-occurrence of perivenous and confluent demyelination in rare cases indicates pathogenetic overlap between ADEM and multiple sclerosis or the existence of relapsing or recurrent ADEM is still uncertain (Van Bogaert, 1950; Hartung and Grossman, 2001; Garg, 2003; Dale and Branson, 2005). The rarity of overlap cases suggests that some patients are coincidentally found to have simultaneous evidence of two separate diseases (Oppenheimer, 1976; Prineas et al., 2002).
Clinical course is associated with pattern of demyelination
Unlike prior clinical-pathological series and case reports that described fatal cases (Turnbull and McIntosh, 1926; Van Bogaert, 1950; Hart and Earle, 1975), our series includes patients with perivenous demyelination alone who survived the initial attack. These patients are potentially informative with respect to the risk of relapse in ADEM. Long-term follow-up increases our confidence that this presumed monophasic illness does not ultimately prove to be relapsing. A monophasic course was observed in six of seven patients with perivenous demyelination alone who survived the initial attack (mean follow up 9.3 years; range 2.5–16.7). One patient with perivenous demyelination alone had a relapsing and ultimately fatal course over 10 months; no autopsy was performed therefore whether the perivenous demyelination pattern persisted or became associated with confluent demyelination is unknown.
Two of three patients with both perivenous demyelination and confluent demyelination ultimately developed a relapsing course and met criteria for multiple sclerosis. In addition to experiencing clinical relapses, both patients developed new areas of T2 signal on follow-up magnetic resonance imaging. The third patient with perivenous and confluent demyelination died 20 days after the onset of symptoms; thus, it is unknown whether a relapsing course would have followed had the patient survived. These cases, along with the high frequency of relapsing cases in the confluent demyelination cohort, suggest that the finding of confluent demyelination, even with perivenous demyelination, may be predictive of relapse and a subsequent diagnosis of multiple sclerosis.
Multifocal magnetic resonance imaging alone does not reliably distinguish monophasic ADEM from multiple sclerosis
Similar to the findings in clinically defined series of ADEM, 9 of 13 (69%) patients in this study presented with large, ill-defined T2 lesions that involved the white matter and extended to grey matter. Gadolinium enhancement, when present, was variable and occurred in several patterns, but no patient with ADEM presented with enhancement of all lesions. Although multifocal magnetic resonance imaging lesions have been emphasized in the ADEM clinical criteria and in prior clinical studies of ADEM, this series and others (Lucchinetti et al., 2008) report multifocal findings in the majority of patients at first presentations of multiple sclerosis as well. Thus, the presence of multifocal magnetic resonance imaging lesions alone is not a reliable criteria distinguishing ADEM from multiple sclerosis at presentation. Furthermore, we describe cases of ADEM (defined by perivenous demyelination) with single focal and unilateral lesions as well, suggesting there is a spectrum of overlapping magnetic resonance imaging imaging patterns in both ADEM and multiple sclerosis.
Magnetic resonance imaging KIDMUS criteria are specific but not sensitive in confluent demyelination
No patient with perivenous demyelination in this series presented with the magnetic resonance imaging KIDMUS criteria (‘sole presence of well-defined lesions and lesions perpendicular to the corpus callosum’) (Mikaeloff et al., 2004). The proportion of patients meeting KIDMUS criteria in the confluent demyelination cohort (16%) was similar to the defining study (Mikaeloff et al., 2004) and a subsequent study (Callen et al., 2008), which found that the combination of findings was 100% specific but insensitive (20–29%) predictor of future relapse and final diagnosis of multiple sclerosis in paediatric patients with first presentations of demyelinating disease. This study suggests that the KIDMUS criteria may also be useful for predicting future relapse and diagnosis of multiple sclerosis in adult patients, which is further supported by the association with confluent demyelination pathologically. Because of small sample sizes, this study does not exclude the possibility that patients with perivenous demyelination may present with KIDMUS criteria.
Clinical criteria over-diagnose ADEM in a pathologically defined multiple sclerosis cohort
Eight of ten individuals with perivenous demyelination alone met the ADEM clinical criteria. Seven patients had a monophasic clinical course and one patient developed a relapsing course meeting criteria for recurrent/multiphasic ADEM (Case 11). Among the perivenous demyelination cohort, only two patients were paediatric (aged 5 and 17 years). These findings suggest that the ADEM clinical criteria, although developed for paediatric ADEM, may also be appropriate for diagnosing ADEM in adults. However, we caution that the current ADEM clinical criteria may erroneously classify patients as ADEM who have pathology and clinical course most consistent with relapsing multiple sclerosis (9% in this study) because of the liberal use of encephalopathy as a criterion. The ADEM clinical criteria recognize that, rarely, ADEM may present with a focal lesion as we have found. Interestingly, the patients with focal lesions and perivenous demyelination in our series did not present with encephalopathy apart from a single individual who experienced a brief post-ictal delirium. Therefore the requirement of encephalopathy may not always capture patients with focal presentations of ADEM. In this group of patients, distinguishing a first presentation of multiple sclerosis from monophasic ADEM may be particularly challenging. The presence of encephalopathy broadly defined may overdiagnose ADEM in patients with more severe presentations of multiple sclerosis who may benefit from early immunomodulatory treatment. Depressed level of consciousness is a more specific criterion for ADEM that reliably distinguished patients with ADEM from multiple sclerosis in this study in which only one patient with multiple sclerosis presented with depressed level of consciousness in the setting of concomitant urosepsis and benzodiazepine treatment. If otherwise unexplained depressed level of consciousness rather than encephalopathy was a prerequisite for an ADEM clinical diagnosis, then fewer patients would have satisfied ADEM clinical criteria (n = 6; four fatal) in our series, and no patient with multiple sclerosis would have fulfilled these criteria. Relying on more restrictive criteria for ADEM would have increased specificity to 100% at the expense of reducing sensitivity to 46% when perivenous demyelination is used as the standard. However, the harm of failing to diagnose monophasic ADEM is probably less than missing a diagnosis of aggressive multiple sclerosis at an early point before the diagnosis can be confirmed based on standard criteria. Depressed level of consciousness is also likely to be more specific than using other criteria such as ‘atypical clinical symptoms of multiple sclerosis’ (including consciousness alteration, hypersomnia, seizures, encephalopathy, aphasia, severe motor deficit and bilateral optic neuritis) recently proposed based on a retrospective analysis of 60 adults presenting with fulminant demyelinating disease (de Seze et al., 2007).
Perivenous demyelination may be a pathological gold standard for ADEM
The clinical history, neuroimaging, clinical course and distinctive neuropathology in the patients with perivenous demyelination alone suggest they represent a clinicopathological entity distinct from multiple sclerosis and that brain biopsy may be useful in selected cases with diagnostic confusion. Although biopsy-confirmed perivenous demyelination may help distinguish ADEM from multiple sclerosis, such an invasive procedure is rarely considered or justified unless alternative aetiologies (especially neoplasm) cannot be excluded otherwise. Prospective studies applying proposed ADEM clinical criteria to children and adults combined with long-term follow-up and correlation with patterns of demyelination demonstrated on rare biopsies and autopsy may determine whether pathology is the appropriate standard for diagnosis of ADEM. Pathological confirmation of the clinical diagnosis of ADEM, even in rare cases, may help to refine and strengthen ADEM clinical criteria which are needed in clinical practice.
Funding
National Multiple Sclerosis Society (NMSS RG3184-B-3-02 and RG3185-B-3 to C.F.L.); and the National Institutes of Health (NS049577 to C.F.L.).
Acknowledgements
The authors thank Patricia Ziemer for technical assistance and Linda Linbo for assistance with study coordination.
Glossary
Abbreviations
- ADEM
acute disseminated encephalomyelitis
References
- Brinar VV, Poser CM. The spectrum of disseminated encephalomyelitis. Clin Neurol Neurosurg. 2006;108:295–310. doi: 10.1016/j.clineuro.2005.11.017. [DOI] [PubMed] [Google Scholar]
- Callen DJ, Shroff MM, Branson HM, Li DK, Lotze T, Stephens D, et al. Role of MRI in the differentiation of ADEM from MS in children. Neurology. 2008;72:968–73. doi: 10.1212/01.wnl.0000338630.20412.45. [DOI] [PubMed] [Google Scholar]
- Cohen O, Steiner-Birmanns B, Biran I, Abramsky O, Honigman S, Steiner I. Recurrence of acute disseminated encephalomyelitis at the previously affected brain site. Arch Neurol. 2001;58:797–801. doi: 10.1001/archneur.58.5.797. [DOI] [PubMed] [Google Scholar]
- Dagher AP, Smirniotopoulos J. Tumefactive demyelinating lesions. Neuroradiology. 1996;38:560–5. doi: 10.1007/BF00626098. [DOI] [PubMed] [Google Scholar]
- Dale RC, Branson JA. Acute disseminated encephalomyelitis or multiple sclerosis: can the initial presentation help in establishing a correct diagnosis? Arch Dis Child. 2005;90:636–9. doi: 10.1136/adc.2004.062935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dale RC, de Sousa C, Chong WK, Cox TC, Harding B, Neville BG. Acute disseminated encephalomyelitis, multiphasic disseminated encephalomyelitis and multiple sclerosis in children. Brain. 2000;123 (Pt 12):2407–22. doi: 10.1093/brain/123.12.2407. [DOI] [PubMed] [Google Scholar]
- de la Monte SM, Ropper AH, Dickersin GR, Harris NL, Ferry JA, Richardson EP., Jr Relapsing central and peripheral demyelinating diseases. Unusual pathologic features. Arch Neurol. 1986;43:626–9. doi: 10.1001/archneur.1986.00520060084027. [DOI] [PubMed] [Google Scholar]
- de Seze J, Debouverie M, Zephir H, Lebrun C, Blanc F, Bourg V, et al. Acute fulminant demyelinating disease: a descriptive study of 60 patients. Arch Neurol. 2007;64:1426–32. doi: 10.1001/archneur.64.10.1426. [DOI] [PubMed] [Google Scholar]
- Garg RK. Acute disseminated encephalomyelitis. Postgrad Med J. 2003;79:11–7. doi: 10.1136/pmj.79.927.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenfield JG, Norman RM. Demyelinating diseases. In: Blackwood W, McMenemey WH, Meyer M, Norman RM, Russell DS, editors. Greenfield's; neuropathology. London: Arnold; 1971. [Google Scholar]
- Hafler DA, Hedley-Whyte ET. Case records of the Massachusetts General Hospital. Weekly clinicopathological exercises. Case 37-1995. a 6-year-old boy with a rash, meningismus, and diplegia. N Engl J Med. 1995;333:1485–93. doi: 10.1056/NEJM199511303332208. [DOI] [PubMed] [Google Scholar]
- Hart MN, Earle KM. Haemorrhagic and perivenous encephalitis: a clinical-pathological review of 38 cases. J Neurol Neurosurg Psychiatry. 1975;38:585–91. doi: 10.1136/jnnp.38.6.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartel C, Schilling S, Gottschalk S, Sperner J. Multiphasic disseminated encephalomyelitis associated with streptococcal infection. Eur J Paediatr Neurol. 2002;6:327–9. doi: 10.1016/s1090-3798(02)90621-5. [DOI] [PubMed] [Google Scholar]
- Hartung HP, Grossman RI. ADEM: distinct disease or part of the MS spectrum? Neurology. 2001;56:1257–60. doi: 10.1212/wnl.56.10.1257. [DOI] [PubMed] [Google Scholar]
- Hasegawa H, Bitoh S, Koshino K, Obashi J, Iwaisako K, Fukushima Y. [Acute relapsing disseminated encephalomyelitis (ARDEM) mimicking a temporal lobe tumor] No Shinkei Geka. 1994;22:185–8. [PubMed] [Google Scholar]
- Horowitz MB, Comey C, Hirsch W, Marion D, Griffith B, Martinez J. Acute disseminated encephalomyelitis (ADEM) or ADEM-like inflammatory changes in a heart-lung transplant recipient: a case report. Neuroradiology. 1995;37:434–7. doi: 10.1007/BF00600082. [DOI] [PubMed] [Google Scholar]
- Hynson JL, Kornberg AJ, Coleman LT, Shield L, Harvey AS, Kean MJ. Clinical and neuroradiologic features of acute disseminated encephalomyelitis in children. Neurology. 2001;56:1308–12. doi: 10.1212/wnl.56.10.1308. [DOI] [PubMed] [Google Scholar]
- Khan S, Yaqub BA, Poser CM, al Deeb SM, Bohlega S. Multiphasic disseminated encephalomyelitis presenting as alternating hemiplegia. J Neurol Neurosurg Psychiatry. 1995;58:467–70. doi: 10.1136/jnnp.58.4.467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinoshita A, Kaseda S, Yagi K, Oda M, Tanabe H. [A case of acute disseminated encephalomyelitis with pathologically-proven acute demyelinating lesion in the peripheral nervous system] Rinsho Shinkeigaku. 1994;34:892–7. [PubMed] [Google Scholar]
- Koch M, den Dunnen W, Sie OG, De Keyser J. A fatal demyelinating illness in a young woman 10 weeks post partum. Lancet Neurol. 2005;4:129–34. doi: 10.1016/S1474-4422(05)00994-4. [DOI] [PubMed] [Google Scholar]
- Krupp LB, Banwell B, Tenembaum S for the International Pediatric MS Study Group. Consensus definitions proposed for pediatric multiple sclerosis and related disorders. Neurology. 2007;68:S7–12. doi: 10.1212/01.wnl.0000259422.44235.a8. [DOI] [PubMed] [Google Scholar]
- Kutzelnigg A, Lucchinetti CF, Stadelmann C, Bruck W, Rauschka H, Bergmann M, et al. Cortical demyelination and diffuse white matter injury in multiple sclerosis. Brain. 2005;128:2705–12. doi: 10.1093/brain/awh641. [DOI] [PubMed] [Google Scholar]
- Leake JA, Albani S, Kao AS, Senac MO, Billman GF, Nespeca MP, et al. Acute disseminated encephalomyelitis in childhood: epidemiologic, clinical and laboratory features. Pediatr Infect Dis J. 2004;23:756–64. doi: 10.1097/01.inf.0000133048.75452.dd. [DOI] [PubMed] [Google Scholar]
- Lim KE, Hsu YY, Hsu WC, Chan CY. Multiple complete ring-shaped enhanced MRI lesions in acute disseminated encephalomyelitis. Clin Imaging. 2003;27:281–4. doi: 10.1016/s0899-7071(02)00552-1. [DOI] [PubMed] [Google Scholar]
- Lucchinetti CF, Gavrilova RH, Metz I, Parisi JE, Scheithauer BW, Weigand S, et al. Clinical and radiographic spectrum of pathologically confirmed tumefactive multiple sclerosis. Brain. 2008;131:1759–75. doi: 10.1093/brain/awn098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucchinetti CF, Parisi J, Bruck W. The pathology of multiple sclerosis. Neurol Clin. 2005;23:77–105, vi. doi: 10.1016/j.ncl.2004.09.002. [DOI] [PubMed] [Google Scholar]
- Lumsden CE. Fundamental problems in the pathology of multiple sclerosis and allied demyelinating diseases. Br Med J. 1951;4714:1035–43. doi: 10.1136/bmj.1.4714.1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malveira GL, Deus-Silva L, Ferreira CM, Pirani C, Jr, Franca MC, Jr, Zanardi VA, et al. Alternating hemisphere tumefactive demyelinating disorder. Eur J Neurol. 2005;12:737–8. doi: 10.1111/j.1468-1331.2005.01046.x. [DOI] [PubMed] [Google Scholar]
- Mandrioli J, Ficarra G, Callari G, Sola P, Merelli E. Monofocal acute large demyelinating lesion mimicking brain glioma. Neurol Sci. 2004;25(Suppl 4):S386–8. doi: 10.1007/s10072-004-0349-6. [DOI] [PubMed] [Google Scholar]
- Maranhao-Filho PA. Multiple sclerosis or multiphasic disseminated encephalomyelitis? A new question about an old problem. Case report. Arq Neuropsiquiatr. 1996;54:505–9. doi: 10.1590/s0004-282x1996000300024. [DOI] [PubMed] [Google Scholar]
- Marchioni E, Ravaglia S, Piccolo G, Furione M, Zardini E, Franciotta D, et al. Postinfectious inflammatory disorders: subgroups based on prospective follow-up. Neurology. 2005;65:1057–65. doi: 10.1212/01.wnl.0000179302.93960.ad. [DOI] [PubMed] [Google Scholar]
- Masdeu JC, Quinto C, Olivera C, Tenner M, Leslie D, Visintainer P. Open-ring imaging sign: highly specific for atypical brain demyelination. Neurology. 2000;54:1427–33. doi: 10.1212/wnl.54.7.1427. [DOI] [PubMed] [Google Scholar]
- Mikaeloff Y, Adamsbaum C, Husson B, Vallee L, Ponsot G, Confavreux C, et al. MRI prognostic factors for relapse after acute CNS inflammatory demyelination in childhood. Brain. 2004;127:1942–7. doi: 10.1093/brain/awh218. Epub 2004 Aug 2. [DOI] [PubMed] [Google Scholar]
- Mikaeloff Y, Caridade G, Husson B, Suissa S, Tardieu M. Acute disseminated encephalomyelitis cohort study: Prognostic factors for relapse. Eur J Paediatr Neurol. 2006 doi: 10.1016/j.ejpn.2006.11.007. [DOI] [PubMed] [Google Scholar]
- Miller DH, Scaravilli F, Thomas DC, Harvey P, Hirsch NP. Acute disseminated encephalomyelitis presenting as a solitary brainstem mass. J Neurol Neurosurg Psychiatry. 1993;56:920–2. doi: 10.1136/jnnp.56.8.920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizutani T, Oda M, Tsuganezawa M, Abe H, Ohshio C, Suzuki K, et al. A case of demyelinating encephalomyelitis with some resemblance to collagen disease. J Neurol. 1977;217:43–52. doi: 10.1007/BF00316315. [DOI] [PubMed] [Google Scholar]
- Murthy SN, Faden HS, Cohen ME, Bakshi R. Acute disseminated encephalomyelitis in children. Pediatrics. 2002;110:e21. doi: 10.1542/peds.110.2.e21. [DOI] [PubMed] [Google Scholar]
- Ohtake T, Hirai S. Recurrence of acute disseminated encephalomyelitis after a 12-year symptom-free interval. Intern Med. 2004;43:746–9. doi: 10.2169/internalmedicine.43.746. [DOI] [PubMed] [Google Scholar]
- Olivero WC, Deshmukh P, Gujrati M. Bilateral enhancing thalamic lesions in a 10 year old boy: case report. J Neurol Neurosurg Psychiatry. 1999;66:633–5. doi: 10.1136/jnnp.66.5.633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oppenheimer DR. Demyelinating Diseases. In: Blackwood W, Corsellis JAN, editors. Greenfield's; Neuropathology. London: Edward Arnold Ltd; 1976. pp. 470–99. [Google Scholar]
- Prineas JW, McDonald WI, Franklin RJM. Demyelinating diseases. In: Graham D, Lantos P, editors. Greenfield's; neuropathology. Vol. 2. New York, NY: Oxford University Press; 2002. [Google Scholar]
- Ravin P, Hedley-Whyte ET. Case records of the Massachusetts General Hospital. Weekly clinicopathological exercises. Case 34-2002. A 55-year-old man with cognitive and sensorimotor findings and intracranial lesions. N Engl J Med. 2002;347:1433–40. doi: 10.1056/NEJMcpc020024. [DOI] [PubMed] [Google Scholar]
- Schwarz S, Mohr A, Knauth M, Wildemann B, Storch-Hagenlocher B. Acute disseminated encephalomyelitis: a follow-up study of 40 adult patients. Neurology. 2001;56:1313–8. doi: 10.1212/wnl.56.10.1313. [DOI] [PubMed] [Google Scholar]
- Seitelberger F, Jellinger K, Tschabitscher H. [Genesis of acute demyelinating encephalitis.] Wien Klin Wochenschr. 1958;70:453–9. [PubMed] [Google Scholar]
- Shintaku M, Matsumoto R. Disseminated perivenous necrotizing encephalomyelitis in systemic lupus erythematosus: report of an autopsy case. Acta Neuropathol. 1998;95:313–7. doi: 10.1007/s004010050804. [DOI] [PubMed] [Google Scholar]
- Silver B, McAvoy K, Mikesell S, Smith TW. Fulminating encephalopathy with perivenular demyelination and vacuolar myelopathy as the initial presentation of human immunodeficiency virus infection. Arch Neurol. 1997;54:647–50. doi: 10.1001/archneur.1997.00550170115023. [DOI] [PubMed] [Google Scholar]
- Singh S, Prabhakar S, Korah IP, Warade SS, Alexander M. Acute disseminated encephalomyelitis and multiple sclerosis: magnetic resonance imaging differentiation. Australas Radiol. 2000;44:404–11. doi: 10.1046/j.1440-1673.2000.00845.x. [DOI] [PubMed] [Google Scholar]
- Tan HM, Chan LL, Chuah KL, Goh NS, Tang KK. Monophasic, solitary tumefactive demyelinating lesion: neuroimaging features and neuropathological diagnosis. Br J Radiol. 2004;77:153–6. doi: 10.1259/bjr/26682607. [DOI] [PubMed] [Google Scholar]
- Tenembaum S, Chamoles N, Fejerman N. Acute disseminated encephalomyelitis: a long-term follow-up study of 84 pediatric patients. Neurology. 2002;59:1224–31. doi: 10.1212/wnl.59.8.1224. [DOI] [PubMed] [Google Scholar]
- Thomas GS, Hussain IH. Acute disseminated encephalomyelitis: a report of six cases. Med J Malaysia. 2004;59:342–51. [PubMed] [Google Scholar]
- Tsai ML, Hung KL. Multiphasic disseminated encephalomyelitis mimicking multiple sclerosis. Brain Dev. 1996;18:412–4. doi: 10.1016/0387-7604(96)00038-1. [DOI] [PubMed] [Google Scholar]
- Turnbull HM, McIntosh J. Encephalomyelitis following vaccination. Br J Exp Pahol. 1926;7:181–222. [Google Scholar]
- Uchimura I, Shiraki H. A contribution to the classification and the pathogenesis of demyelinating encephalomyelitis; with special reference to the central nervous system lesions caused by preventive inoculation against rabies. J Neuropathol Exp Neurol. 1957;16:139–203. doi: 10.1097/00005072-195704000-00001. discussion, 203–8. [DOI] [PubMed] [Google Scholar]
- Unal I, Cigdem Dogulu F, Saygi S, Firat M, Ruacan S, Karabudak R. Multiphasic disseminated encephalomyelitis: a 4-year follow-up. Eur Neurol. 2000;43:55–8. doi: 10.1159/000008131. [DOI] [PubMed] [Google Scholar]
- Van Bogaert L. Post-infectious encephalomyelitis and multiple sclerosis; the significance of perivenous encephalomyelitis. J Neuropathol Exp Neurol. 1950;9:219–49. doi: 10.1097/00005072-195007000-00001. [DOI] [PubMed] [Google Scholar]
- Vass K, Lassmann H, Wekerle H, Wisniewski HM. The distribution of Ia antigen in the lesions of rat acute experimental allergic encephalomyelitis. Acta Neuropathol. 1986;70:149–60. doi: 10.1007/BF00691433. [DOI] [PubMed] [Google Scholar]
- Wang PN, Fuh JL, Liu HC, Wang SJ. Acute disseminated encephalomyelitis in middle-aged or elderly patients. Eur Neurol. 1996;36:219–23. doi: 10.1159/000117253. [DOI] [PubMed] [Google Scholar]
- Wingerchuk DM, Hogancamp WF, O'B;rien PC, Weinshenker BG. The clinical course of neuromyelitis optica (Devic's; syndrome) Neurology. 1999;53:1107–14. doi: 10.1212/wnl.53.5.1107. [DOI] [PubMed] [Google Scholar]
- Wingerchuk DM, Lennon VA, Pittock SJ, Lucchinetti CF, Weinshenker BG. Revised diagnostic criteria for neuromyelitis optica. Neurology. 2006;66:1485–9. doi: 10.1212/01.wnl.0000216139.44259.74. [DOI] [PubMed] [Google Scholar]