

Abstract
Background
Tropomyosin (TM), an essential actin-binding protein, is central to the control of calcium-regulated striated muscle contraction. Although TPM1α (also called α-TM) is the predominant TM isoform in human hearts, the precise TM isoform composition remains unclear.
Methods and Results
In this study, we quantified for the first time the levels of striated muscle TM isoforms in human heart, including a novel isoform called TPM1κ. By developing a TPM1κ specific antibody, we found that the TPM1κ protein is expressed and incorporated into organized myofibrils in hearts and its level is increased in human dilated cardiomyopathy and heart failure. To investigate the role of TPM1κ in sarcomeric function, we generated transgenic mice overexpressing cardiac specific TPM1κ. Incorporation of increased levels of TPM1κ protein in myofilaments leads to dilated cardiomyopathy. Physiological alterations include decreased fractional shortening, systolic and diastolic dysfunction, and decreased myofilament calcium sensitivity with no change in maximum developed tension. Additional biophysical studies demonstrate less structural stability and weaker actin-binding affinity of TPM1κ as compared to TPM1α.
Conclusion
This functional analysis of TPM1κ provides a possible mechanism for the consequences of the TM isoform switch observed in dilated cardiomyopathy and heart failure patients.
Keywords: Tropomyosin, Dilated Cardiomyopathy, Heart failure, Transgenic mice, Contractile function, Myofilament calcium sensitivity
The heart adapts to different challenges presented by an array of mechanical, hormonal and nutritional signals in the process of maintaining its circulatory function. Isoform switching of sarcomeric proteins is one mode the heart employs to adapt to those challenges along with alterations in the relative abundance and phosphorylation status of contractile and regulatory proteins.1 These changes in isoform expression and phosphorylation status also play an essential role during cardiac development and in response to cardiac hypertrophy and heart failure (HF). Although sarcomeric protein isoforms are subject to developmental regulation, cardiomyopathy and HF primarily elicit changes in thick filament protein isoforms.2 The only thin filament protein to change isoform expression in the failing human heart is troponin T.3, 4 Furthermore, altered phosphorylation of troponin I, myosin binding protein C, and other sarcomeric proteins have dramatic effects on cardiac function in the failing human myocardium.5
To understand the specific role of another thin filament protein, tropomyosin (TM), in the normal and pathologic heart, it is essential to define the TM isoform expression profile. Tropomyosins comprise a family of actin-binding proteins encoded by four different genes (TPM1, TPM2, TPM3 and TPM4). Each gene uses alternative splicing, alternative promoters, and differential processing to encode multiple striated muscle, smooth muscle, and cytoskeletal transcripts. For example, the TPM1 gene utilizes 15 exons to encode at least nine distinct isoforms.6,7 There are three primary striated muscle TM isoforms, α-TM, β-TM and γ-TM, which are products of the TPM1, TPM2 and TPM3 genes, respectively. These isoforms are highly homologous, yet exhibit unique physiological properties.8,9 Recently, a novel striated muscle isoform of the TPM1 gene was identified in human cardiac tissue.10 This isoform contains an exon pattern similar to striated muscle TPM1α mRNA (also called α-TM) except for the substitution of the smooth muscle specific exon 2a for 2b. This TM isoform is designated as TPM1-kappa (TPM1κ).10 Our quantification results of human cardiac RNA reveals that ∼50% of the striated muscle TPM1 mRNA is TPM1κ, with the remainder being TPM1α. Although TPM1κ mRNA has been identified, TPM1κ protein expression in human hearts had not been confirmed. To address this, we developed a TPM1κ specific antibody and quantified protein levels in the hearts of normal and cardiomyopathy patients. We now report for the first time the TM protein composition in adult human hearts and determined that TPM1κ protein is expressed and incorporated into cardiac myofibrils. Interestingly, TPM1κ protein levels are differentially regulated such that cardiomyopathy patients with end-stage HF exhibit increased expression.
TMs are central to the control of calcium regulated thin filament function and striated muscle contraction. To investigate the role of TPM1κ in sarcomeric function, we generated transgenic (Tg) mice overexpressing TPM1κ in the hearts. Results show that incorporation of TPM1κ protein in myofilaments leads to dilated cardiomyopathy (DCM). Physiological changes include decreased fractional shortening of the left ventricle, systolic and diastolic dysfunction, decreased myofilament calcium sensitivity, and weaker actin-binding affinity. In conclusion, our Tg mouse studies and in vitro biophysical results demonstrate significant functional and structural differences between TPM1κ and TPM1α which provide a possible mechanism for the consequences of the TM isoform switch that is observed in DCM and HF patients.
Methods
An expanded Methods section is in the Data Supplement.
Study subjects and human samples
This study was performed in accordance with the Declaration of Helsinki as adopted and promulgated by the US National Institutes of Health as well as rules and regulations of the University of Cincinnati's Institutional Ethics Committee. The study group consisted of hearts excised from patients undergoing cardiac transplantation at the University of Cincinnati, and human cardiac protein samples from previously published work.11 The clinical data of the HF patients is presented in the Supplement Table 1. Three healthy hearts procured from brain dead patients/organ donors with no history of cardiac disease served as controls. Normal human RNAs from adult and fetal cardiac and skeletal muscle, uterus and lung were procured from commercial sources (Stratagene).
Generation of TPM1κ transgenic mice
Transgenic mice (FVB/N background) were generated using a cDNA encoding human TPM1κ cloned into the cardiac-specific α-MHC expression vector.12 Animal experiments were approved by the University of Cincinnati's Institutional Animal Care and Use Committee.
Cardiac function
Cardiac performance of the Tg mice was assessed by physiological studies including echocardiography, isolated anterograde perfused heart model, and skinned fiber preparations which are described in the Data Supplement.
Quantitative RT-PCR analysis, bacterial recombinant protein expression, circular dichroism measurements, actin-binding assay and structure modeling analysis
Details regarding the methods used are presented in the Data Supplement.
Statistical analysis
All values, unless otherwise mentioned, are presented as mean ± SD. Protein data were analyzed using the Wilcoxon rank sum test. The isoproterenol response data and NEM-S1 data were analyzed using the Kruskal Wallis test with post hoc analysis using the Wilcoxon rank sum test after adjusting the level of significance. In addition, the NEM-S1 data was examined using a repeated measure analysis of variance with Bonferonni post hoc analysis with a significance of P<0.05.
The authors had full access to and take full responsibility for the integrity of the data. All authors have read and agree to the manuscript as written.
Results
Expression Profile of TPM1α and TPM1κ in Human Hearts
Although the expression of TPM1κ mRNA was identified in the human heart,10 the relative levels of TPM1κ and TPM1α transcripts are unknown. To quantify TM transcript levels in human hearts, we conducted quantitative RT-PCR using striated muscle TM isoform specific primers. Results show the TPM1α and TPM1κ isoforms are expressed in equal amounts (∼50% each) in both fetal and adult hearts (Figure 1A). Interestingly, both isoform levels increase in adult hearts by 3.1 fold when compared to fetal hearts and normalized to GAPDH expression. Further analysis shows TPM1κ is expressed only in human cardiac muscle and not in skeletal muscle, uterus, or lung (data not shown). Additional quantitative RT-PCR results show that β-TM is expressed at equivalent levels in fetal and adult human hearts, but that γ-TM is expressed at a ∼30 fold increase in adult versus fetal myocardium (data not shown).
Figure 1.
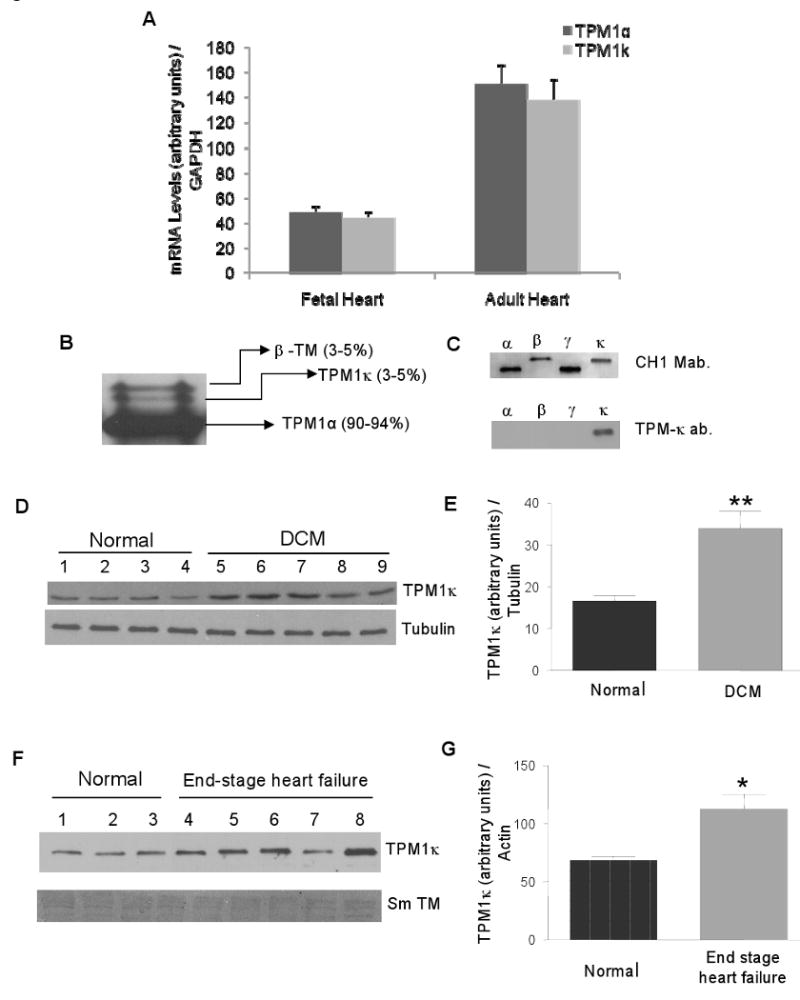
TPM1κ expression in human hearts. A, Real-time RT-PCR quantification of the TPM1α and TPM1κ mRNAs in normal fetal and adult hearts. B, TM protein profile in the normal adult human heart analyzed by Western blotting using striated muscle TM-specific antibody. C, Migration of TM isoforms and TM antibody specificity. D and E, TPM1κ expression and quantification in cardiac total protein lysate from DCM patients using anti-TPM1κ and anti-tubulin antibodies. ** P < 0.01, normal vs. DCM. F and G, TPM1κ protein expression and quantification in cardiac myofibrillar preparations from normal and heart failure patients using anti-TPM1κ and anti-smooth muscle tropomyosin (Sm TM) antibodies. * P < 0.02, normal vs. end-stage HF patients.
To determine TM protein composition in human hearts, we conducted Western blot analyses using a striated muscle specific TM antibody. Results reveal the ratio of TM isoforms in the adult human heart: TPM1α (90-94%), TPM1κ (3-5%) and β-TM (3-5%) (Figure 1B, C). Additional experiments show no expression of γ-TM protein in the adult human heart (data not shown). Interestingly, although TPM1κ and TPM1α mRNAs are expressed in equivalent amounts, 90-94% of the total translated TM is the TPM1α isoform with the remaining being TPM1κ and β-TM.
To distinguish the TPM1κ protein from other TM isoforms, we developed an isoform specific antibody with an epitope that resides in exon 2a sequence which is encoded within TPM1κ but not in TPM1α protein. We used this antibody to quantify TPM1κ protein levels in hearts from several normal, dilated cardiomyopathy (DCM) and HF patients. The TPM1κ protein levels from human left ventricular free walls were normalized to tubulin or actin and quantified (Figure 1D through G). Results show that TPM1κ is expressed in normal patients, and these levels are increased in DCM and HF patients. Both control and patient samples show minimal reactivity to a smooth muscle TM antibody, thereby verifying the specificity of the TPM1κ antibody (Figure 1F). These results demonstrate the differential expression of TPM1κ protein in DCM and HF patients. We also observe a tendency of downregulation of β-TM levels in those patients (data not shown), but due to the unavailability of a β-TM specific antibody, we are unable to quantify the exact level of change. Furthermore, the levels of TPM1α protein are so high in the heart that subtle changes in TM levels are not easily quantified. It should be noted that the relative migratory positions of β-TM and TPM1κ proteins can vary dependent upon gel composition (Supplement Figure 1).
TPM1κ Tg Mouse Hearts Autoregulate TM Isoform Expression
To elucidate relations between sarcomeric TM isoforms and contractile behavior in striated muscle, we developed a Tg mouse model to overexpress the TPM1κ isoform in the heart. The transgene construct was generated by ligating the human TPM1κ cDNA (containing the entire amino acid coding region and the 3′untranslated region) into the murine α-MHC cardiac-specific expression vector. There is 100% amino acid conservation between mouse and human TPM1α as well as exon2a sequence. Three independent transgenic mouse lines (lines 72, 77 and 80) were established that express varied levels of TPM1κ protein in the heart (90%, 32%, and 51%, respectively, of TPM1κ/total TM normalized to actin) (Figure 2A and B). The expression of TPM1κ is coupled with a concomitant downregulation of endogenous TPM1α, with the total TM levels remaining unchanged. Transgenic lines 72 and 80 were used for further functional analyses. Interestingly, unlike human hearts, wild type mouse hearts do not express TPM1κ. We confirmed this in various mouse strains including FVB/N, Black Swiss, C57-BL/6 and BALB/c (data not shown). Immunohistological analysis of the Tg hearts using the anti-TPM1κ antibody reveal incorporation of the TPM1κ protein into organized myofibrillar bundles while it is absent in non-transgenic (Ntg) mouse hearts (Figure 2C). The epitope of TPM1κ against which the antibody was raised is also found in smooth muscle TM and due to antibody cross-reactivity, there is a positive reaction in vascular smooth muscle regions of Ntg hearts; this is more prominent in mouse lung sections (Figure 2C).
Figure 2.
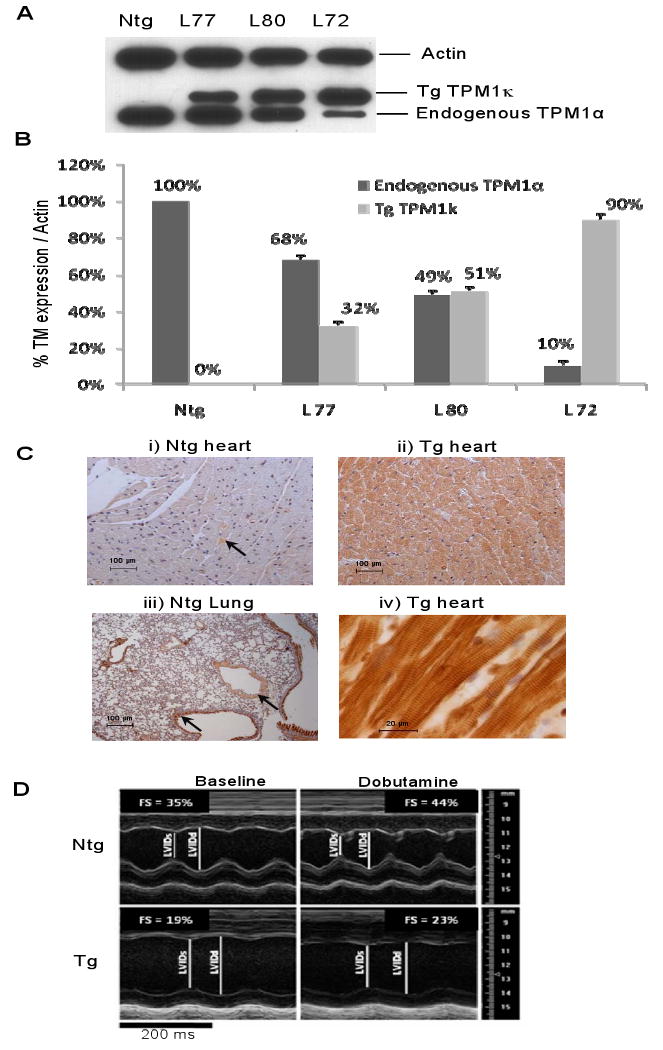
Characterization of the TPM1κ Tg mice. A and B, Western blot analysis of myofilament preparations from TPM1κ Tg mouse hearts using striated muscle TM and actin antibodies. The TM level found in Ntg hearts was set to 100%, and the signal intensity of the TM isoforms in Tg mice were quantified and normalized to striated muscle α-actin. C, Immunohistochemistry of ventricular sections of Ntg and TPM1κ Tg hearts using anti-TPM1κ antibody. Arrows indicate the regions of expression for vascular smooth muscle TM in the Ntg tissue sections. Panel (iv) shows the incorporation of TPM1κ protein into sarcomeres as reflected by the striated staining pattern. D, Representative AM-mode echocardiographic images of adult left ventricular (LV) response to dobutamine. Short axis view AM-Mode recording of the LV from Ntg and Tg mice, before and after infusion of dobutamine. LVIDs = end systolic LV internal dimension; LVIDd = end-diastolic LV internal dimension; FS = fractional shortening.
TPM1κ Mice Exhibit Altered Cardiac Morphology and Function
None of the founder TPM1κ mice or their progeny demonstrates any differences in percentage heart weight-to-body weight ratio or reduced viability when compared with Ntg controls. Histological analysis of hearts from 3, 6 and 12 month-old Tg mice reveal no detectable changes in gross chamber morphology, interstitial fibrosis, or myocyte disarray when compared with age and sex matched Ntg littermates (data not shown). However, echocardiographic analyses show the TPM1κ mice have increased end-systolic and end-diastolic left ventricular dimensions (Figure 2D; Supplement Table 2).
To obtain a better understanding of the physiological significance of TPM1κ protein in the regulation of cardiac contractility, we implemented echocardiography, the work-performing heart model, and skinned fiber preparations. In vivo physiologic assessment of cardiac function in young (2 mo) and adult (5 mo) mice using Doppler echocardiographic analyses demonstrated a progressive decrease in fractional shortening and ejection fraction (Supplement Table 1). Stroke volume and cardiac output were maintained at the expense of an increased end-diastolic volume. To test whether impairment of cardiac function could be reversed by stimulation of β-adrenergic receptors, we subjected the hearts to dobutamine, a positive inotropic agent (Figure 2D). Results show the Tg hearts responded appropriately to dobutamine, but maintained their inherent physiological impairment (Supplement Table 3).
To assess cardiac function ex vivo, we implemented the work-performing heart model in the absence of hormonal stimulation and under controlled loading conditions. Studies show that overexpression of wild type TPM1α in Tg mouse hearts does not lead to alterations in morphology or physiological performance of the heart.13-15 As summarized in Supplement Table 4, the TPM1κ Tg hearts show decreased baseline contraction (+dP/dt) and relaxation (-dP/dt). The magnitude of impairment was greater for line 72 versus line 80, consistent with higher levels of transgene expression. Heart rates were unaltered in either line compared to Ntg. An increased time to peak pressure (TPP) in both lines and half-time to relaxation (RT½) in line 72 were observed, consistent with the systolic and diastolic dysfunction.
Since there is systolic and diastolic dysfunction, we determined how hearts from the TPM1κ mice would respond to acute functional stress induced by stimulation of the β-adrenergic cardio-stimulatory pathway. The reduced contractile and lusitropic performance by TPM1κ Tg hearts was assessed during stimulation with isoproterenol, a β-adrenergic agonist that augments muscle contraction and relaxation by increasing the rate of Ca2+ cycling. Although Tg hearts show an isoproterenol dose response, they are unable to reach control levels of contraction or relaxation (Figure 3). Thus, in isolated heart preparations, the cumulative dose response to isoproterenol of both contractile (+dP/dt, TPP) and relaxation (-dP/dt, RT½) parameters is similar in the Tg and Ntg hearts even though functional differences remain. These results show that although TPM1κ mice demonstrate significantly impaired indicators of contractility at basal loading conditions, this magnitude of hemodynamic impairment is not sufficient to provoke compensatory chronic activation of sympathetic drive and desensitization of β-adrenergic signal transduction mechanisms.
Figure 3.
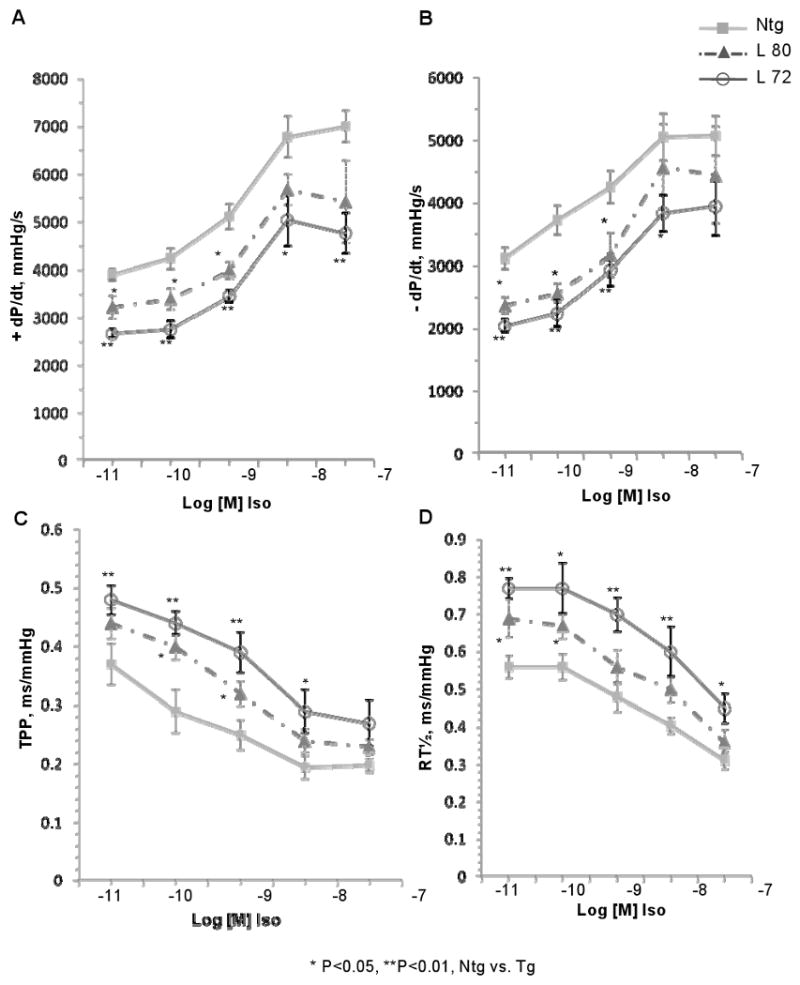
Isoproterenol dose response curves in Ntg and TPM1κ Tg mouse hearts at 5 months of age. A through D, Hearts from Ntg and Tg lines 72 and 80 were subject to isolated heart analyses with increasing concentrations of isoproterenol (10-11 to 10-7 mol/L).
TPM1κ Mice Exhibit a Decreased Myofilament Ca2+ Sensitivity
We compared the tension-pCa relationship of myofilaments from Ntg and Tg hearts (line 72). Results (Figure 4A) show a significant decrease in the Ca2+ sensitivity of Tg myofilaments (Ntg: pCa50 = 5.91 ± 0.012; Tg: pCa50 = 5.76 ± 0.006; P < 0.05) with no change in the maximum developed tension (Ntg: Fmax = 29.59 ± 0.76 mN/mm2; Tg: Fmax = 30.84 ± 0.93 mN/mm2). Compared to Ntg, the Hill coefficient of Tg myofilaments was significantly increased (Ntg: nH = 4.22 ± 0.09; Tg: nH = 5.84 ± 0.21; P < 0.05).
Figure 4.
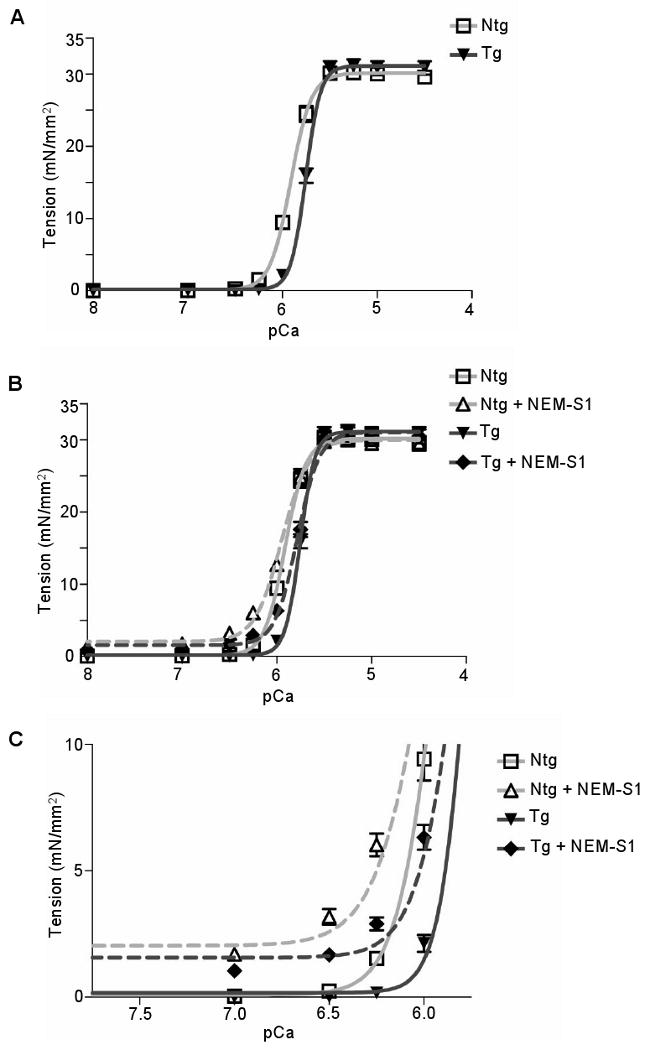
Tension-pCa relationship of myofiber preparations from Ntg and TPM1κ Tg hearts. A, Tension-pCa relationship with a sarcomeric length set at 2.0 μm. B, Tension-pCa relationship before and after treatment with NEM-S1. C, Expanded scale of tension generated at low Ca2+ concentrations which enhances recruitment of crossbridges by NEM-S1.
To assess cooperative activation of the myofilaments, we employed NEM-S1 to mimic the effect of strong cross-bridge binding. NEM-S1 is known to probe cooperative processes and to enhance endogenous cross-bridge cycling.16 In the presence of NEM-S1, no change in maximum developed tension was observed between the two groups (Ntg: Fmax = 29.35 ± 0.75 mN/mm2; Tg: Fmax = 30.45 ± 0.97 mN/mm2) (Figure 4B). Also, Tg myofilaments are less sensitive to Ca2+ than Ntg (Ntg + NEM-S1: pCa50 = 5.95 ± 0.013; Tg + NEM-S1: pCa50 = 5.78 ± 0.009; P < 0.05). At low Ca2+ values, NEM-S1 induces activation of the thin filament to a greater extent in Ntg versus Tg myofilaments (Ntg + NEM-S1: pCa 6.5 = 3.18 ± 1.3; Tg + NEM-S1: 1.68 ± 0.81; P < 0.0001) (Figure 4C). Although decreased in both cases, the Hill coefficient remained significantly different (P < 0.05) [Ntg + NEM-S1 (nH = 3.17 ± 0.06) versus Tg + NEM-S1 (nH = 3.98 ± 0.17)].
Altered Thermal Stability and Actin-Binding of TPM1κ
To investigate whether the physiological alterations in TPM1κ Tg hearts are due to an altered protein structure, we used circular dichroism to study the thermal stability of recombinant TPM1κ protein. Results show a decreased TPM1κ homodimer stability when compared to TPM1α at temperatures above 37°C which may influence actin binding and myofilament Ca2+ sensitivity (Figure 5A).
Figure 5.
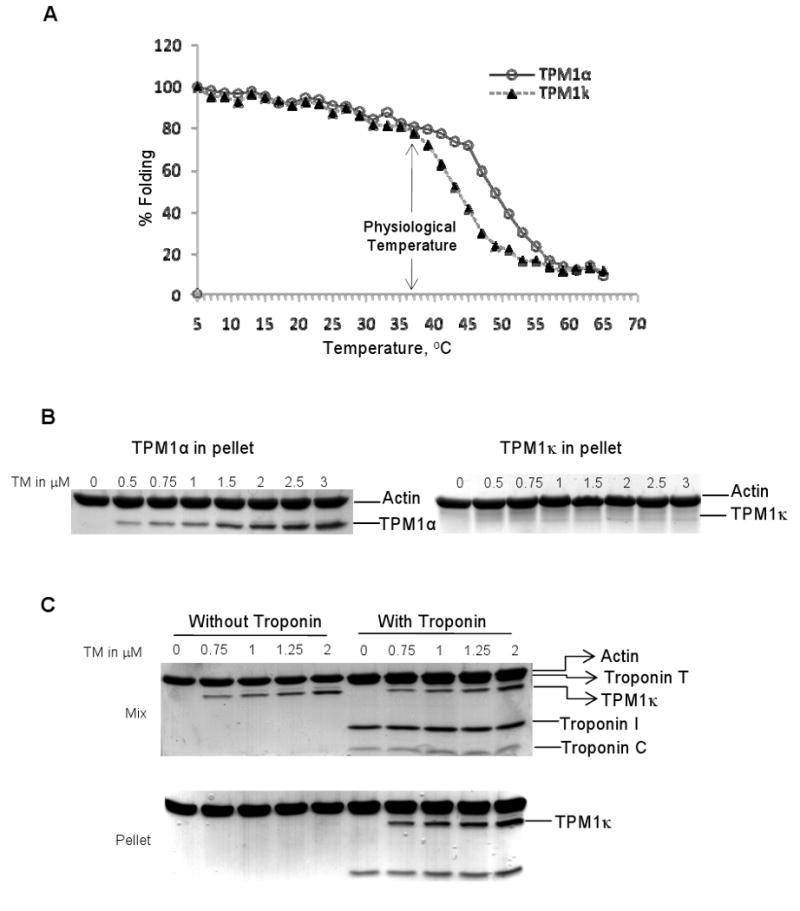
Biochemical analyses of TPM1κ. A, Thermal denaturation profiles of recombinant TPM1α and TPM1κ proteins monitored by circular dichroism measurements. The curves show the percentage folding vs. temperature and was calculated based on the ellipticity at a wavelength of 222 nm. B, Cosedimentation assay of TPM1α and TPM1κ binding to cardiac actin. C, Cosedimentation assay of TPM1κ binding to actin with and without human cardiac troponin. The mixture and the pellet fractions were analyzed by SDS-PAGE and Coomassie blue staining.
We conducted a series of cosedimentation assays to assess TM's ability to bind actin. Results show binding of TPM1κ protein to actin was barely detectable with a much weaker affinity than TPM1α (Figure 5B). This result is in agreement with previous observations using unacetylated recombinant TMs.17 Addition of human cardiac troponin dramatically increased the actin-binding affinity of TPM1κ protein (Figure 5C), which agrees with previous observations that troponin increases the affinity of TM for actin.18 The dependence of TPM1κ on troponin for binding to actin and its increased expression in failing hearts suggests that the TM isoform switch is commensurate with the isoform switch of troponin T that occurs in HF.
Discussion
In this study, we report for the first time that the normal adult human heart expresses TPM1α, TPM1κ and β-TM proteins and that the TPM1κ level is differentially regulated in cardiomyopathy patients. Previous results demonstrate there are functional differences among various striated muscle TM isoforms19,20 and our results provide insight into the structure-function relations of TPM1κ and its influence on cardiac function. Cardiac specific overexpression of β-TM leads to a phenotype with diastolic dysfunction and increased myofilament calcium sensitivity.15, 20 Overexpression of γ-TM leads to a hyperdynamic effect on systolic and diastolic function, but decreased myofilament calcium sensitivity and without any morphological abnormalities in the hearts.21 Interestingly, TPM1κ mouse hearts elicit functional properties that are a combination of β-TM and γ-TM isoforms: systolic and diastolic impairment coupled with a decrease in myofilament calcium sensitivity. This physiological profile is similar to the α-TM E54K mouse model that causes DCM;14 the TPM1κ Tg hearts also exhibit a dilated cardiac chamber phenotype.
The relative amounts of the different TM isoforms that are expressed in a muscle cell are characteristic of the species, muscle fiber type, stage of development, and other factors.22 Exons 2a and 2b of the TPM1 gene are spliced in a mutually exclusive manner with exon 2b being the default exon in the mRNA of most cell types; exon 2a had previously been restricted to smooth muscle TM.6,7 That TPM1κ, a striated muscle TM that includes exon 2a, is incorporated into the sarcomere is a novel finding. The significance of this TM region (amino acids 41-80) is illustrated by the occurrence of various cardiomyopathies in humans with mutations found in exon 2b leading to familial hypertrophic cardiomyopathy (FHC) (Glu62Gln; Ala63Val; Lys70Thr) and DCM (Glu40Lys and Glu54Lys).9,14 Furthermore, both DCM mutations and a FHC mutation (Glu62Gln) found in exon 2b have conserved amino acids in the same positions in exon 2a. Hence, the potential of the TPM1κ isoform replacing the TPM1α isoform containing mutant exon 2b in such cardiomyopathic conditions may be critical.
An intriguing question is why high levels of TPM1κ mRNA are present in adult human hearts when the protein levels are low. Similar discordant mRNA and protein levels were previously found with α-myosin heavy chain expression in human ventricles.23, 24 Possible explanations for this mismatch between mRNA and protein levels may be associated with translational regulation or the involvement of microRNAs.25
Our results demonstrate that the TPM1κ protein affects tension developed at sub-maximal Ca2+ concentrations, the level at which normal heart function occurs, without altering maximum tension. The decreased binding affinity of TPM1κ to actin in the absence of troponin may contribute to the decrease in myofilament Ca2+ sensitivity. Substitution of exon 2a for 2b, which happens with TPM1κ, may alter the propagation of force through the myofilament. Interestingly, with DCM TM mutations that occur in exon 2b – the same region of the TM protein, myofilaments also exhibit decreased sensitivity to Ca2+.14, 26 In fact, TPM1κ expression is increased in human patients with DCM and HF. Although the properties of the DCM α-TM E54K and TPM1κ isoforms are similar in decreasing myofilament Ca2+ sensitivity, the novel TM isoform does not affect maximum tension (Figure 4A), which contrasts with the E54K mutation.14 This difference in tension development could be due to structural effects of the E54K mutation/TPM1κ isoform. Previous studies demonstrate that TM mutations can result in structural alterations that affect TM stability27 and binding to actin17, 28 which can influence myofilament function. With TPM1κ, saturation of troponin C with high Ca2+ concentrations is able to fully activate the myofilaments.
Our evidence points to a mechanism in which thin filaments regulated by TPM1κ demonstrate reduced activation by both Ca2+ and strong crossbridges in the form of NEM-S1. The steeper dependence of force on Ca2+ in the Tg fibers indicates a greater reliance on crossbridge dependent activation, a process of higher cooperativity in the face of little change in Ca2+ binding, a relatively non-cooperative process. Evidence that crossbridge dependent activation is blunted in the Tg fibers comes from our data of thin filament activation at low Ca2+ (Figure 4C). The data show that induction of force at or near relaxing levels of free Ca2+ was greater in Ntg than in Tg fibers. This effect is likely due to lower stability and weaker actin-binding affinity of TPM1κ as compared to TPM1α.
TM is an alpha helical, coiled-coil dimer characterized by a heptapeptide repeat motif (a-b-c-d-e-f-g) in which residues in the hydrophobic core positions a and d are the primary determinants of folding and stability.29 The difference between TPM1κ and TPM1α is a stretch of 40 amino acids encoded by exon 2a/b, which contains a number of substituted residues (26 of 40 amino acid differences). Alanine residues in TPM1α promote flexibility in the helical structure and enhance binding to actin.30 Replacement of these interface alanines with other residues would be expected to decrease actin affinity due to localized helix stabilization and loss of flexibility. Structural modeling of the TPM1κ isoform reveals several regions where substituted amino acids are likely to decrease the overall stability of the TPM1 coiled-coil dimer, as well as a few potentially stabilizing substitutions (Figure 6 and Supplement Table 5). The destabilizing substitutions include several residues located in the hydrophobic central region of the coiled-coil (the a and d positions, including L43I, L46K, T53S, L57R, and Y60V) that lose well-packed hydrophobic interactions in the TPM1κ isoform (Supplement Table 5). In addition, there are a few destabilizing substitutions in the e and g positions that typically form polar or salt bridge interactions; these include K49L, A63E, Q68E, and K70S. The few substitutions predicted to stabilize the dimer (Q47E, S61L, and K77A) all occur in the e or g positions. Substitutions on the outer surface of the TPM1 dimer (positions b, c, and f) are not involved in the dimer interface but could have direct effects on the interaction with actin or troponin. There are also a few differences in predicted stability between the TPM1κ/TPM1κ (least probable in vivo dimer) and the TPM1κ/TPM1α isoforms (most probable in vivo dimer). For example, the Q47E substitution is predicted to stabilize the homodimer κ/κ isoform through the addition of a salt bridge with L46K from the opposing helix, but this salt bridge would not form in the heterodimer κ/α isoform. Thus, the variation in amino acid sequence between exons 2a and 2b may contribute to differences in thermal denaturation and actin binding of TPM1κ compared to TPM1α.
Figure 6.
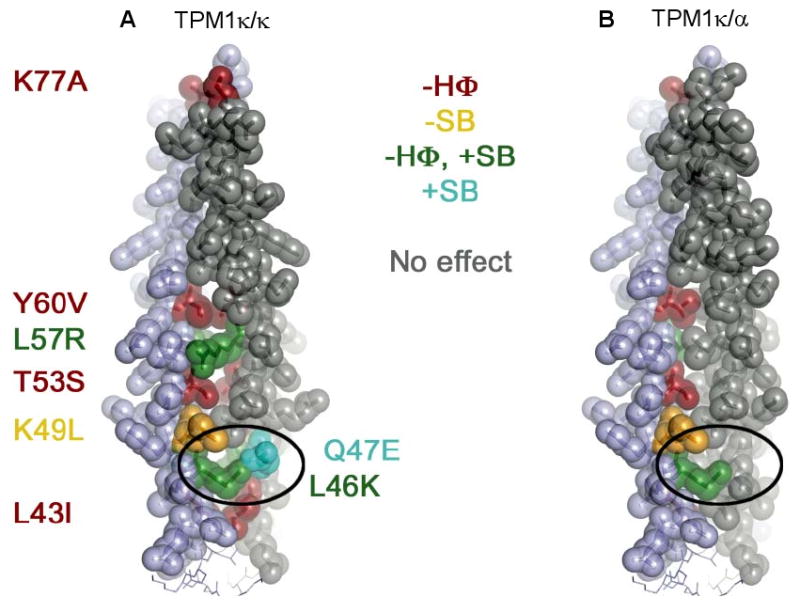
Structural models of TPM1κ/κ and κ/α isoforms. A and B, Substitutions in the κ isoform which are predicted to alter the stability of the TPM1 coiled coil are highlighted in color. Red and yellow indicate amino acid substitutions that are likely to destabilize the coiled coil, whereas green indicates a mixed effect and cyan indicates putative stabilizing changes. The oval highlights residues that are predicted to form a salt bridge in a κ/κ coiled coil but would be destabilizing in the κ/α isoform. Legend abbreviations are as follows: -HΦ, loss of hydrophobic contact; -SB, loss of salt bridge; +SB, potential gain of a salt bridge.
What is the biological significance for expressing the TPM1κ isoform? In terminally-differentiated cells, such as cardiomyocytes, there is a need for adaptation to changing environments. Increasing protein isoform diversity through processes, such as alterative splicing, meets this need. The TPM1κ isoform, one of 10 distinct products of the TPM1 gene, provides the opportunity to modulate sarcomeric performance during changing conditions, such as exercise, stress, or cardiac disease. Several studies reported that during end-stage HF, the contractile apparatus is characterized by an increased Ca2+ sensitivity.5, 31, 32 That TPM1κ compensates by decreasing calcium sensitivity without affecting maximum tension is logical for patients exhibiting chronic DCM. Although the increased level of TPM1κ is low when compared to the total TM, the relative percentage of TPM1κ increase is significant. Also, there is a possibility of a regional distribution of TPM1κ expression within the heart to match the functional demands of a specific region. Similar regional preference of β-MHC isoform expression versus α-MHC was previously demonstrated to improve cardiac function.33 The fact that human patients with chronic DCM and HF symptoms exhibit a cardiac pathology and physiology similar to TPM1κ Tg mice is striking considering the small, but significant, increase in TPM1κ protein levels in these patients. We speculate that during normal cardiac function, the low levels of TPM1κ (which confers decreased myofilament Ca2+ sensitivity) are offset by the low levels of β-TM (which confers increased Ca2+ sensitivity). Recent studies found that normalization of myofilament calcium sensitivity can rescue mice exhibiting cardiac hypertrophy.34, 35 In DCM and HF patients, there is decreased cardiac performance which correlates with increased TPM1κ levels; whether this increased expression is the cause or a consequence of the DCM/HF phenotype remains to be determined.
Supplementary Material
Acknowledgments
We thank Jon Neumann for production of the transgenic mice, Ms. Maureen Bender for her daily care of the animals, and Drs. R. Shukla and A. Dwivedi of the University of Cincinnati Center for Biostatistical Services. We also thank Ms. Emily Schulz and Dr. Vikram Prasad for editorial assistance. The human cardiac troponin was graciously provided by Dr. Brandon Biesiadecki.
Sources of Funding: This work was supported in part by NHLBI HL081680 awarded to DFW, HL79032 to BMW, and HL22231 and HL062426 to RJS.
Footnotes
Clinical Perspective: Understanding the function of tropomyosin (TM) is important from a clinical perspective because mutations in the protein can result in familial hypertrophic cardiomyopathy and dilated cardiomyopathy. Four TM genes have been identified, each of which undergoes differential promoter usage and alternative splicing to generate multiple isoforms. In the heart, TM is an essential sarcomeric protein that controls calcium-regulated muscle contraction. Although much is known about TM, little is known about its expression in human hearts. In this study, we determine and quantify the expression levels of striated muscle TM isoforms in the human heart, including a novel TM isoform called TPM1κ. Our data show that TPM1κ is incorporated into cardiac myofilaments, and its level increases by 100% during human dilated cardiomyopathy and heart failure. We also develop a mouse model to understand the physiological function of TPM1κ. Since there is 100% amino acid sequence identity between human and mouse TM, these studies have direct relevance to TM function. Our mouse model shows that increased expression of TPM1κ results in increased end-systolic and end-diastolic left ventricular dimensions, similar to patients with dilated cardiomyopathy. Physiological assessment of the hearts from our mouse model show decreased fractional shortening, systolic and diastolic dysfunction, and decreased myofilament calcium sensitivity which is similar to patients with heart disease. These studies provide for a possible mechanism for the clinical features that are observed in dilated cardiomyopathy and heart failure patients.
Disclosures: None
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Sudarsan Rajan, Department of Molecular Genetics, Biochemistry and Microbiology, University of Cincinnati Medical Center, Cincinnati, OH 45267 USA.
Ganapathy Jagatheesan, Department of Molecular Genetics, Biochemistry and Microbiology, University of Cincinnati Medical Center, Cincinnati, OH 45267 USA.
Chehade N. Karam, Department of Physiology and Biophysics, University of Illinois at Chicago, IL.
Marco L. Alves, Department of Physiology and Biophysics, University of Illinois at Chicago, IL.
Ilona Bodi, Institute of Molecular Pharmacology & Biophysics, University of Cincinnati Medical Center, Cincinnati, OH 45267 USA.
Arnold Schwartz, Institute of Molecular Pharmacology & Biophysics, University of Cincinnati Medical Center, Cincinnati, OH 45267 USA.
Christian F. Bulcao, Department of Surgery, Section of Cardiothoracic Surgery, University of Cincinnati Medical Center, Cincinnati, OH 45267 USA.
Karen M. D'Souza, Department of Surgery, Section of Cardiothoracic Surgery, University of Cincinnati Medical Center, Cincinnati, OH 45267 USA.
Shahab A. Akhter, Department of Surgery, Section of Cardiothoracic Surgery, University of Cincinnati Medical Center, Cincinnati, OH 45267 USA.
Greg P. Boivin, Department of Pathology and Laboratory Medicine, University of Cincinnati Medical Center, Cincinnati, OH 45267 USA.
Dipak K. Dube, Department of Medicine, SUNY Upstate Medical University, Syracuse, NY.
Natalia Petrashevskaya, Department of Medicine, University of Maryland Medical Center, Baltimore.
Andrew B. Herr, Department of Molecular Genetics, Biochemistry and Microbiology, University of Cincinnati Medical Center, Cincinnati, OH 45267 USA.
Roger Hullin, Department of Cardiology, CHUV, University of Lausanne, Lausanne, Switzerland.
Stephen B. Liggett, Department of Medicine, University of Maryland Medical Center, Baltimore.
Beata M. Wolska, Department of Physiology and Biophysics, University of Illinois at Chicago, IL.
R. John Solaro, Department of Physiology and Biophysics, University of Illinois at Chicago, IL.
David F. Wieczorek, Department of Molecular Genetics, Biochemistry and Microbiology, University of Cincinnati Medical Center, Cincinnati, OH 45267 USA.
References
- 1.Schiaffino S, Reggiani C. Molecular diversity of myofibrillar proteins: gene regulation and functional significance. Physiol Rev. 1996;76:371–423. doi: 10.1152/physrev.1996.76.2.371. [DOI] [PubMed] [Google Scholar]
- 2.Palmer BM. Thick filament proteins and performance in human heart failure. Heart Fail Rev. 2005;10:187–197. doi: 10.1007/s10741-005-5249-1. [DOI] [PubMed] [Google Scholar]
- 3.Marston SB, Redwood CS. Modulation of thin filament activation by breakdown or isoform switching of thin filament proteins: physiological and pathological implications. Circ Res. 2003;93:1170–1178. doi: 10.1161/01.RES.0000105088.06696.17. [DOI] [PubMed] [Google Scholar]
- 4.Purcell IF, Bing W, Marston SB. Functional analysis of human cardiac troponin by the in vitro motility assay: comparison of adult, foetal and failing hearts. Cardiovasc Res. 1999;43:884–891. doi: 10.1016/s0008-6363(99)00123-6. [DOI] [PubMed] [Google Scholar]
- 5.van der Velden J, Papp Z, Zaremba R, Boontje NM, de Jong JW, Owen VJ, Burton PB, Goldmann P, Jaquet K, Stienen GJ. Increased Ca2+-sensitivity of the contractile apparatus in end-stage human heart failure results from altered phosphorylation of contractile proteins. Cardiovasc Res. 2003;57:37–47. doi: 10.1016/s0008-6363(02)00606-5. [DOI] [PubMed] [Google Scholar]
- 6.Lees-Miller JP, Helfman DM. The molecular basis for tropomyosin isoform diversity. Bioessays. 1991;13:429–437. doi: 10.1002/bies.950130902. [DOI] [PubMed] [Google Scholar]
- 7.Wieczorek DF, Smith CW, Nadal-Ginard B. The rat alpha-tropomyosin gene generates a minimum of six different mRNAs coding for striated, smooth, and nonmuscle isoforms by alternative splicing. Mol Cell Biol. 1988;8:679–694. doi: 10.1128/mcb.8.2.679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pieples K, Wieczorek DF. Tropomyosin 3 increases striated muscle isoform diversity. Biochemistry. 2000;39:8291–8297. doi: 10.1021/bi000047x. [DOI] [PubMed] [Google Scholar]
- 9.Wieczorek DF, Jagatheesan G, Rajan S. The role of tropomyosin in heart disease. Adv Exp Med Biol. 2008;644:132–142. doi: 10.1007/978-0-387-85766-4_11. [DOI] [PubMed] [Google Scholar]
- 10.Denz CR, Narshi A, Zajdel RW, Dube DK. Expression of a novel cardiac-specific tropomyosin isoform in humans. Biochem Biophys Res Commun. 2004;320:1291–1297. doi: 10.1016/j.bbrc.2004.06.084. [DOI] [PubMed] [Google Scholar]
- 11.Hullin R, Matthes J, von Vietinghoff S, Bodi I, Rubio M, D'Souza K, Friedrich Khan I, Rottlander D, Hoppe UC, Mohacsi P, Schmitteckert E, Gilsbach R, Bunemann M, Hein L, Schwartz A, Herzig S. Increased expression of the auxiliary beta2-subunit of ventricular L-type Ca2+ channels leads to single-channel activity characteristic of heart failure. PLoS ONE. 2007;2:e292. doi: 10.1371/journal.pone.0000292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Subramaniam A, Jones WK, Gulick J, Wert S, Neumann J, Robbins J. Tissue-specific regulation of the alpha-myosin heavy chain gene promoter in transgenic mice. J Biol Chem. 1991;266:24613–24620. [PubMed] [Google Scholar]
- 13.Prabhakar R, Boivin GP, Grupp IL, Hoit B, Arteaga G, Solaro JR, Wieczorek DF. A familial hypertrophic cardiomyopathy alpha-tropomyosin mutation causes severe cardiac hypertrophy and death in mice. J Mol Cell Cardiol. 2001;33:1815–1828. doi: 10.1006/jmcc.2001.1445. [DOI] [PubMed] [Google Scholar]
- 14.Rajan S, Ahmed RP, Jagatheesan G, Petrashevskaya N, Boivin GP, Urboniene D, Arteaga GM, Wolska BM, Solaro RJ, Liggett SB, Wieczorek DF. Dilated cardiomyopathy mutant tropomyosin mice develop cardiac dysfunction with significantly decreased fractional shortening and myofilament calcium sensitivity. Circ Res. 2007;101:205–214. doi: 10.1161/CIRCRESAHA.107.148379. [DOI] [PubMed] [Google Scholar]
- 15.Wolska BM, Keller RS, Evans CC, Palmiter KA, Phillips RM, Muthuchamy M, Oehlenschlager J, Wieczorek DF, de Tombe PP, Solaro RJ. Correlation between myofilament response to Ca2+ and altered dynamics of contraction and relaxation in transgenic cardiac cells that express beta-tropomyosin. Circ Res. 1999;84:745–751. doi: 10.1161/01.res.84.7.745. [DOI] [PubMed] [Google Scholar]
- 16.Swartz DR, Moss RL. Influence of a strong-binding myosin analogue on calcium-sensitive mechanical properties of skinned skeletal muscle fibers. J Biol Chem. 1992;267:20497–20506. [PubMed] [Google Scholar]
- 17.Cho YJ, Hitchcock-DeGregori SE. Relationship between alternatively spliced exons and functional domains in tropomyosin. Proc Natl Acad Sci U S A. 1991;88:10153–10157. doi: 10.1073/pnas.88.22.10153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Heald RW, Hitchcock-DeGregori SE. The structure of the amino terminus of tropomyosin is critical for binding to actin in the absence and presence of troponin. J Biol Chem. 1988;263:5254–5259. [PubMed] [Google Scholar]
- 19.Jagatheesan G, Rajan S, Petrashevskaya N, Schwartz A, Boivin G, Vahebi S, DeTombe P, Solaro RJ, Labitzke E, Hilliard G, Wieczorek DF. Functional importance of the carboxyl-terminal region of striated muscle tropomyosin. J Biol Chem. 2003;278:23204–23211. doi: 10.1074/jbc.M303073200. [DOI] [PubMed] [Google Scholar]
- 20.Muthuchamy M, Grupp IL, Grupp G, O'Toole BA, Kier AB, Boivin GP, Neumann J, Wieczorek DF. Molecular and physiological effects of overexpressing striated muscle beta-tropomyosin in the adult murine heart. J Biol Chem. 1995;270:30593–30603. doi: 10.1074/jbc.270.51.30593. [DOI] [PubMed] [Google Scholar]
- 21.Pieples K, Arteaga G, Solaro RJ, Grupp I, Lorenz JN, Boivin GP, Jagatheesan G, Labitzke E, DeTombe PP, Konhilas JP, Irving TC, Wieczorek DF. Tropomyosin 3 expression leads to hypercontractility and attenuates myofilament length-dependent Ca(2+) activation. Am J Physiol Heart Circ Physiol. 2002;283:H1344–1353. doi: 10.1152/ajpheart.00351.2002. [DOI] [PubMed] [Google Scholar]
- 22.Heeley DH, Dhoot GK, Perry SV. Factors determining the subunit composition of tropomyosin in mammalian skeletal muscle. Biochem J. 1985;226:461–468. doi: 10.1042/bj2260461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Coumans JV, Yeoh T, Seeto RK, Keogh A, Brennan K, Gunning P, Hardeman E, dos Remedios CG. Variations in the relative mRNA levels of actins and myosin heavy chains do not produce corresponding differences in their proteins in the adult human heart. J Mol Cell Cardiol. 1997;29:895–905. doi: 10.1006/jmcc.1996.0317. [DOI] [PubMed] [Google Scholar]
- 24.Miyata S, Minobe W, Bristow MR, Leinwand LA. Myosin heavy chain isoform expression in the failing and nonfailing human heart. Circ Res. 2000;86:386–390. doi: 10.1161/01.res.86.4.386. [DOI] [PubMed] [Google Scholar]
- 25.Zhu S, Si ML, Wu H, Mo YY. MicroRNA-21 targets the tumor suppressor gene tropomyosin 1 (TPM1) J Biol Chem. 2007;282:14328–14336. doi: 10.1074/jbc.M611393200. [DOI] [PubMed] [Google Scholar]
- 26.Chang AN, Harada K, Ackerman MJ, Potter JD. Functional consequences of hypertrophic and dilated cardiomyopathy-causing mutations in alpha-tropomyosin. J Biol Chem. 2005;280:34343–34349. doi: 10.1074/jbc.M505014200. [DOI] [PubMed] [Google Scholar]
- 27.Heller MJ, Nili M, Homsher E, Tobacman LS. Cardiomyopathic tropomyosin mutations that increase thin filament Ca2+ sensitivity and tropomyosin N-domain flexibility. J Biol Chem. 2003;278:41742–41748. doi: 10.1074/jbc.M303408200. [DOI] [PubMed] [Google Scholar]
- 28.Golitsina N, An Y, Greenfield NJ, Thierfelder L, Iizuka K, Seidman JG, Seidman CE, Lehrer SS, Hitchcock-DeGregori SE. Effects of two familial hypertrophic cardiomyopathy-causing mutations on alpha-tropomyosin structure and function. Biochemistry. 1997;36:4637–4642. doi: 10.1021/bi962970y. [DOI] [PubMed] [Google Scholar]
- 29.Tripet B, Wagschal K, Lavigne P, Mant CT, Hodges RS. Effects of side-chain characteristics on stability and oligomerization state of a de novo-designed model coiled-coil: 20 amino acid substitutions in position “d”. J Mol Biol. 2000;300:377–402. doi: 10.1006/jmbi.2000.3866. [DOI] [PubMed] [Google Scholar]
- 30.Brown JH, Kim KH, Jun G, Greenfield NJ, Dominguez R, Volkmann N, Hitchcock-DeGregori SE, Cohen C. Deciphering the design of the tropomyosin molecule. Proc Natl Acad Sci U S A. 2001;98:8496–8501. doi: 10.1073/pnas.131219198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hajjar RJ, Schwinger RH, Schmidt U, Kim CS, Lebeche D, Doye AA, Gwathmey JK. Myofilament calcium regulation in human myocardium. Circulation. 2000;101:1679–1685. doi: 10.1161/01.cir.101.14.1679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wolff MR, Buck SH, Stoker SW, Greaser ML, Mentzer RM. Myofibrillar calcium sensitivity of isometric tension is increased in human dilated cardiomyopathies: role of altered beta-adrenergically mediated protein phosphorylation. J Clin Invest. 1996;98:167–176. doi: 10.1172/JCI118762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Krenz M, Sadayappan S, Osinska HE, Henry JA, Beck S, Warshaw DM, Robbins J. Distribution and structure-function relationship of myosin heavy chain isoforms in the adult mouse heart. J Biol Chem. 2007;282:24057–24064. doi: 10.1074/jbc.M704574200. [DOI] [PubMed] [Google Scholar]
- 34.Coutu P, Bennett CN, Favre EG, Day SM, Metzger JM. Parvalbumin corrects slowed relaxation in adult cardiac myocytes expressing hypertrophic cardiomyopathy-linked alpha-tropomyosin mutations. Circ Res. 2004;94:1235–1241. doi: 10.1161/01.RES.0000126923.46786.FD. [DOI] [PubMed] [Google Scholar]
- 35.Jagatheesan G, Rajan S, Petrashevskaya N, Schwartz A, Boivin G, Arteaga GM, Solaro RJ, Liggett SB, Wieczorek DF. Rescue of tropomyosin-induced familial hypertrophic cardiomyopathy mice by transgenesis. Am J Physiol Heart Circ Physiol. 2007;293:H949–958. doi: 10.1152/ajpheart.01341.2006. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.