

Abstract
Chemoprevention of prostate cancer by second-generation selenium compounds in reference to selenomethionine holds strong promise to deal with the disease at the root. Here we used the transgenic adenocarcinoma mouse prostate (TRAMP) model to establish the efficacy of methylseleninic acid (MSeA) and methylselenocysteine (MSeC) against prostate carcinogenesis and to characterize potential mechanisms. Eight-week-old male TRAMP mice (C57B/6 background) were given a daily oral dose of water, MSeA or MSeC at 3 mg Se/kg body weight and were euthanized at either 18 or 26 weeks of age. By 18 weeks of age, the genito-urinary (GU) tract and dorsal-lateral prostate (DLP) weights for the MSeA and MSeC-treated groups were lower than for the control (p<0.01). At 26 weeks, 4 out of 10 control mice had GU weight over 2 g, only 1 out of 10 in each of the Se groups did. The efficacy was accompanied by delayed lesion progression, increased apoptosis and decreased proliferation without appreciable changes of T-antigen expression in the DLP of Se-treated mice and decreased serum IGF-1 when compared to control mice. In another experiment, giving MSeA to TRAMP mice from 10-weeks or 16-weeks of age increased their survival to 50 weeks of age, and delayed the death due to synaptophysin-positive neuroendocrine carcinomas and synaptophysin-negative prostate lesions and seminal vesicle hypertrophy. Wild-type mice receiving MSeA from 10 weeks did not exhibit decreased body weight, GU weight or increased serum ALT than the control mice. Therefore, these selenium compounds may effectively inhibit this model of PCa carcinogenesis.
Keywords: methylselenol precursor compounds, prostate cancer, transgenic mice, synaptophysin
Introduction
One in six American men will be diagnosed in their lifetime with prostate cancer (PCa), which is the second leading cause of male cancer death in United States (1). Treatment options by hormone ablation therapy, radiation and surgery for advanced PCa do not offer cure but delay the inevitable recurrence of the lethal hormone-refractory disease (2). Available chemotherapeutic drugs including docetaxel offer little or no survival benefit to patients with such late stage disease. Chemoprevention of prostate carcinogenesis is considered a necessary and practical approach to deal with the problem at the root (3, 4).
Previous studies have suggested that selenium (Se) may modify the risk of and prevent PCa (5, 6). A clinical trial led by the late Dr. Larry Clark demonstrated that Se supplementation in the form of selenized yeast significantly decreased PCa incidence in skin-cancer patients compared to those receiving placebo in a randomized trial (7, 8). Prompted by these encouraging results, the Selenium and Vitamin E Cancer Prevention Trial (SELECT) was testing the PCa preventive efficacy of L-selenomethionine (SeMet), the major species of Se present in selenized yeast, and/or vitamin E (all rac-alpha-tocopheryl acetate) supplementation in North American men(9). The study was a randomized, placebo-controlled trial of 35 533 men from 427 participating sites in the United States, Canada, and Puerto Rico randomly assigned to 4 groups (Se, vitamin E, Se + vitamin E, and placebo) in a double-blind fashion between August 22, 2001, and June 24, 2004. Baseline eligibility included age 50 years or older (African American men) or 55 years or older (all other men), a serum prostate-specific antigen level of 4 ng/mL or less, and a digital rectal examination not suspicious for prostate cancer. Oral Se (200 microgram/d from L-selenomethionine) and matched vitamin E placebo, vitamin E (400 IU/d of all rac-alpha-tocopheryl acetate) and matched Se placebo, Se + vitamin E, or placebo + placebo for a planned follow-up of minimum of 7 years and a maximum of 12 years. The National Cancer Institute stopped the trial in late October 2008, several years ahead of the scheduled completion date due to a possible increase of diabetes risk in SeMet-supplemented subjects and a rise in prostate cancer risk by the vitamin E-supplemented subjects(10). Specifically, as of October 23, 2008(10), median overall follow-up was 5.46 years (range, 4.17-7.33 years). Hazard ratios (99% confidence intervals [CIs]) for prostate cancer were 1.13 (99% CI, 0.95-1.35; n = 473) for vitamin E, 1.04 (99% CI, 0.87-1.24; n = 432) for Se, and 1.05 (99% CI, 0.88-1.25; n = 437) for Se + vitamin E vs 1.00 (n = 416) for placebo. There were no significant differences (all P>.15) in any other prespecified cancer end points. There were statistically nonsignificant increased risks of prostate cancer in the vitamin E group (P =.06) and type 2 diabetes mellitus in the Se group (relative risk, 1.07; 99% CI, 0.94-1.22; P =.16) but not in the Se + vitamin E group.
In hindsight, experiments with preclinical PCa models conducted prior to (11) and since SELECT was initiated including our study with xenograft models (12, 13) did not support any in vivo anti-cancer activity of SeMet. Because of the metabolic and biological differences that have been well documented between SeMet and other Se forms (13), the failure of SeMet in the SELECT should not be equated to all Se forms as “ineffective” for PCa prevention. Whereas the SELECT results convincingly ruled out SeMet for PCa prevention, we argue that the quest for effective Se agents takes on even greater significance and urgency because there is no other clinically-proven effective PCa preventive agent other than the controversial 5-alpha reductase II inhibitor Finasteride that blocks the intra-prostatic generation of the active androgen dihydrotestesterone but with significant side effects and questionable survival benefit (14-16).
Cell culture and preclinical animal studies have suggested that the overall efficacy of Se supplementation depends on the specific form and dosage administered as well as the stage of the disease process (17, 18). Methylselenol has been considered an in vivo active anti-cancer metabolite (17, 18). Studies conducted by us and others have shown that the putative methylselenol precursor compound methylseleninic acid (MSeA) can inhibit in vitro growth of hormone-refractory advanced human prostate carcinoma DU145 (19) (20) and PC-3 cells (21) and androgen-dependent human prostate carcinoma LNCaP cells (22-24) by cell cycle arrest and induction of apoptosis via caspase activation as well as inhibition of androgen signaling in the latter. We have recently shown that orally administered MSeA and another methylselenol precursor methylselenocysteine (MSeC) inhibit the in vivo growth of DU145 and PC-3 xenograft in nude mice and SeMet was ineffective (13). These studies led us to hypothesize that these “second-generation” Se compounds (in reference to SeMet) may prevent or delay prostate primary carcinogenesis, leading to a survival benefit. To our knowledge, such Se forms have not been rigorously tested in any animal prostate primary carcinogenesis model.
The transgenic adenocarcinoma of mouse prostate (TRAMP) model, originally developed by Dr. N Greenberg (25), to some extent mimics, the natural history and progression of human PCa (26). The probasin promoter-driven SV-40 T-antigen expression in the dorsolateral prostate (DLP) glands abrogates p53 and Rb tumor suppressor gene functions to spontaneously propel the progression of PCa from prostatic intraepithelial neoplasia (PIN) to more advanced stages of lesions with morphological features of adenocarcinomas and poorly differentiated (PD) carcinomas (26). In this model, metastasis to lymph nodes and other organs is also a notable feature similar to the human disease (26). More recent studies have shown that the poorly-differentiated (PD) carcinomas in this model are neuro-endocrine(NE)-like carcinomas that are androgen-insensitive (lack AR) and express synaptophysin and belong to a distinct lineage from the PIN and well-differentiated (WD) and moderately differentiated (MD) adenocarcinoma lesions (27, 28). Furthermore, the incidence of these NE-carcinomas is mouse strain dependent: in the C57B/6 background, a lifetime incidence of ∼20% NE-carcinomas vs. in the FVB background, 87% NE-carcinoma incidence was recorded by as early as 16 weeks of age (28). In both strains these NE-carcinomas mostly arise in the ventral lobes instead of the DLP (27, 28). As is understood today, the TRAMP therefore represents two models of prostate carcinogenesis: One that approximates the androgen-driven glandular prostate epithelial cancer formation in the DLP and another that simulates the development of NE-carcinomas in the ventral prostate. The latter is also important for human PCa in that the number of NE-cells correlates with the stage, Gleason grades and survival in castration-recurrent PCa in the clinical cases (29, 30). The TRAMP is by far the most widely available and used transgenic model of PCa in cancer chemoprevention studies, for example (31-38).
In the present study, we evaluated the efficacy of MSeA and MSeC against the early stage lesion development in the TRAMP model and the associated biomarkers of cell proliferation and apoptosis. We tested the survival benefits of starting MSeA oral treatment at either PIN (10 weeks of age) or more advanced stages of lesions (16 weeks of age).
Materials and Methods
Selenium compounds
Methaneseleninic acid (same as MSeA, >95%, white powder) was purchased from Sigma Chemical Company (St. Louis, MO). Se-Methylselenocysteine was purchased from LKT Labs (St. Paul, MN). Stock solutions of each compound were prepared in water and filter-sterilized, and then stored in 1-ml aliquots in a -70 °C freezer as previously described (13). A fresh vial was thawed for each day's use.
Animal breeding
The animal use protocol was approved by the Institutional Animal Care and Use Committee of the University of Minnesota, and carried out at the Hormel Institute's AAALAC-accredited animal facility. The female heterozygous C57BL/TGN TRAMP mice, Line PB Tag 8247NG, were purchased from The Jackson Laboratory (Bar Harbor, ME), and were cross-bred with non-transgenic C57BL/6 breeder males. Mouse-tail DNA was isolated from the 3-week old pups for genotyping by PCR-based screening using the following primers (25).
Primer A, 5′CCGGTCGACCGGAAGCTTCCACAAGTGCATTTA 3′, Primer B, 5′AGGCATTCCACTGCTCCCATTCATC 3′; As a control for PCR efficiency, multiplex PCR using primers for mouse beta-globulin was done concurrently: Primer 1 (beta-globulin): 5′ CCAATCTGCTCACAGGATAGAGAGGGCAGG 3′ Primer 2, (beta-globulin): 5′ CCTTGAGGCTGTCCAAGTGATTCAGGCCATCG 3′.
Selenium treatments
Two sets of animal experiments were carried out. In the first experiment, we compared the effect of daily (Monday-Friday, 5 times per week) oral administration of MSeA and MSeC starting at 8 weeks of age on early stage disease burden and lesion progression as represented by the weight of genital urinary (GU) tract and prostate pathology profiles. Male C57BL6 TRAMP mice (N = 20 mice per group) received a daily oral dose of water (control), MSeA or MSeC delivered to the back of the tongue (5 times per week) at the dosage of 3 mg Se/kg body weight as we described recently for nude mice (13). Briefly, Se was delivered to the back of the tongue in a volume of 1 microliter per g body weight (e.g. for a mouse weighing 30 g, 30 μl were delivered via a disposable plastic tip from a 100 μl-Pipetman®). The mice ingested the small volume easily without physical irritation to the esophagus that might have resulted from intra-gastric gavages. The tight range of the measured tissue Se content within a group indicated good consistency of this manner of Se delivery (See ref. (13)). The dosing frequency simulated that used in the Clark study (7) and the SELECT study (9). Mice were given ad libitum access to purified AIN-93M diet pellets (Harlan Teklad, Madison, WI) and water.
At 18 and 26 weeks of age, blood was taken prior to sacrifice for serum preparation for IGF-I assay by a commercially available kit (See below). The lower genital urinary (GU) tract including seminal vesicle, prostate, testes and bladder was dissected and weighed. The dorsal-lateral prostate (DLP) was dissected under a dissecting scope on a bed of ice. Approximately half was fixed for 24 hours in 10%(v/v) neutral-buffer formalin (Fisher Scientific Company) and the other portion was frozen on dry ice and stored in -80 °C for Western-blot assay and other biochemical measurements. The fixed tissues were stored in 70% ethanol for preservation until processing for histology and immunohistochemistry. At processing time, the fixed tissues were dehydrated in ascending grades of ethanol and xylene, then embedded in paraffin. Sections (5 μm) were cut with a microtome and mounted on superfrost® /plus microscope slides (Fisher Scientific Company). Tissues were processed and stained with hematoxylin and eosin (H&E) for routine histopathologic evaluation.
In the second (long-term survival) experiment, the impact of oral administration of MSeA on overall and cancer-specific survival of the TRAMP mice was studied. Groups of TRAMP mice received a daily oral dose of 3 mg Se/kg body weight as MSeA beginning at 10 weeks (N=31) or 16 weeks of age (N=30) or received water as placebo (N=31). Mice were given ad libitum access to purified AIN-93M diet pellets and water. TRAMP mice were inspected daily and weighed individually once a week. At 16 weeks of age, TRAMP mice were palpated daily before Se or water oral treatment for the presence of abdominal mass (tumor). Mice that lost >25% body weight or were weak or moribund (palpable tumor size >3 cm) were euthanized. The GU was removed en bloc and weighed. Tumors were dissected and weighed. The organs were inspected for metastasis and if visible, the lesions were dissected, weighed and fixed in formalin for H&E confirmation of metastasis. Mice surviving to the predetermined termination age of 50 weeks were killed and necropsy performed as above.
To assess the long-term safety of MSeA oral administration, we gave wild-type male littermates the same dose of MSeA (N=10) starting at 10 weeks of age or water (N=9). Body weight and general health were monitored until 41 weeks of age. Blood was collected prior to euthanasia for serum preparation. Serum alanine aminotransferase (ALT) activity was measured as a marker of liver damage. GU tracts, lungs, kidneys, livers and spleens as well as any visible tumors were removed, weighed and fixed in 10% neutral-buffered formalin.
Histopathology
H& E stained sections of dorsolateral prostate were examined, in blinded fashion to the treatment status, by trained associate LW (DVM, PhD) and pathologist J. Liao (MD, Ph.D.), and classified (26) as (a) low grade prostatic intraepithelial neoplasia (LGPIN) having foci with two or more layers of atypical cells with elongated hyperchromatic nuclei and intact gland profiles (b) high grade prostatic intraepithelial neoplasia (HGPIN) having increased epithelial stratification, foci of atypical cells fill or almost fill the lumen of the ducts, enlarged diameter of the glands, distorted duct profiles, increase in nuclear pleomorphism, hyperchromatic nuclei and cribriform structures (c) well differentiated adenocarcinoma (WD-PCa) showing invasion of basement membrane, loss of intraductal spaces and increased quantity of small glands, (d) moderately differentiated adenocarcinoma (MD-PCa) showing total loss of intraductal spaces and relatively solid growth and (e) poorly differentiated carcinoma (PD-PCa) showing sheets of poorly differentiated cells with remnants of trapped glands. For each animal, the highest lesion grade observed for the DLP section or prostate tumors was used for comparison of lesion progression status among groups.
Immunohistochemical Analysis
Sections (5 μm) were cut from paraffin embedded DL-prostate tissues, dried and deparaffinized. Immunostaining was performed using antibodies for the proliferation marker protein Ki67 (NeoMarker), T-antigen transgene product (Becton-Dickinson) with 1:100 dilution. Synaptophysin antibody was purchased from BD Biosciences. Normal horse serum was used as negative control. The biotinylated secondary antibody used was rabbit anti-mouse antibody IgG (1:200 in 10% normal rabbit serum; DAKOCytomation). The slides were developed in diaminobenzidine (DAB) and counter stained with a weak solution of hematoxylin stain. The stained slides were dehydrated and mounted in permount. Images were captured and analyzed by ImagePro-plus software. Synaptophysin expression in selected carcinomas and prostate tissues was verified by immunobloting.
In situ apoptosis detection by TUNEL
Five-μm thick sections were processed for terminal deoxynucleotidyl transferase–mediated nick-end labeling (TUNEL) assay using a commercial kit (Calbiochem, Germany). The apoptosis was evaluated by counting the brown-colored positive cells as well as the total number of cells at 5 randomly selected fields at ×400 magnification. The apoptotic index (per ×400 microscopic field) was calculated as number of apoptotic cells ×100/ total number of cells.
Immunoblot analysis of tissue lysates
Western blot was carried out on pooled DLP tissues (N =9 or 10 per group) for the 18-week samples. The tissues were homogenized in non-denaturing lysis buffer [10 mmol/L Tris-HCl(pH 7.4),150 mmol/L NaCl, 1% Triton X-100, 1 mmol/L EDTA, 1 mmol/L EGTA, 0.2 mmol/L sodium orthovanadate, 0.5% NP40, 5ul/mL aprotinin, 10ul/mL PMSF]. Protein concentration in lysates was determined using Bio-Rad detergent-compatible protein assay kit (Bio-Rad Laboratories). For immunoblot analyses, 20 to 80 μg of protein per lysate was denatured in 2×SDS-PAGE sample buffer and subjected to SDS-PAGE on 10% or 12% Tris-glycine gel as needed. The separated proteins were transferred onto nitrocellulose membrane (Amersham Biosciences) followed by blocking with 5% non-fat milk powder (w/v) in TBS (10 mmol/L Tris,100 mmol/L NaCl, 0.1% Tween 20) overnight at 4 °C. Membranes were probed with different primary antibodies, including c-PARP, PCNA, cyclin E, IGF1R, P27, AKT, p-AKT, XIAP, and beta-actin. The membranes were then incubated with appropriate peroxidase-conjugated secondary antibodies (Cell Signaling Technology) and detected by enhanced chemofluorescence.
Caspase Activity Assay
Caspase-3/-7 activity was assessed via a fluorescence assay peptide substrate, DEVD-conjugated to the fluorescent reporter molecule 7-amino-4-trifluoromethyl coumarin (AFC)(39). Tissue lysates were the same as Western-blot sample. The kit was purchased from R&D, Minneapolis.
IGF-1 ELISA
DSL-10-29200 Mouse/Rat IGF-1 ELISA kits (Diagnostic Systems Laboratories, Inc., Webster, TX) were used to determine serum concentrations of IGF-1. The absorbance was read at 450 nm with dual wavelength correction at 620 nm and concentrations were determined using the standards provided in the kit. All IGF-1 samples were assayed in duplicate. Intra-assay coefficient of variation was 3%. Within each cohort, all samples were run on one plate.
Serum Alanine aminotransferase (ALT) assay
For each sample, 50 μL of Infinity™ ALT (GPT) Liquid Stable Reagent (Thermo Scientific) was warmed to 37°C for 5 minutes. Five μL of each sample was added and incubated at 37°C for an additional 30 seconds. The absorbance was read at 340 nm and was recorded every 60 seconds for 5 minutes to measure the change in absorbance over time. All samples were run in duplicate. Within sample coefficient of variation was ∼5%. Normal (Data-Trol N™) and Abnormal (Data-Trol A™) sera from Thermo Scientific were used as internal controls and concentrations are expressed as U/L calculated as ΔAbsorbance/minute × factor where factor = total volume in mL × 1000/6.3 × sample volume in mL × cuvette path length in cm. In this case Factor = 1746. As a reference, normal un-treated mice had serum ALT of ∼50 U/L (40).
Statistical analyses
Data are presented as means ± SEM. Results were analyzed by ANOVA followed by Neuman-Keuls tests to determine significance between specific groups. Survival curves (Kaplan-Meier) and percent with metastasis were analyzed by Chi-square analysis.
Results
Orally administered MSeA and MSeC decreased indices of prostate tumor burden
In the TRAMP model, the activation of probasin-driven T-Ag expression leads to the accelerated growth of the DLP and ventral prostate where most prostate lesions arise and of the lower genital urinary (GU) tract organs including seminal vesicle, testes and bladder in addition to the prostate gland (25). The weights of GU and DLP have therefore been used as quantitative indicators of tumor burden in early stage carcinogenesis (25, 26, 31). In the first experiment, we initiated daily oral treatment (5 times per week) of MSeA, MSeC or water (vehicle) at 8 weeks of age and euthanized mice at 18 weeks and up to 26 weeks of age. For reference values, we obtained baseline GU weight of 8-week old wild-type and TRAMP mice and 18-week old wild-type mice. The GU weights for the 8 week-old wild-type and TRAMP mice were the same (0.52 g vs. 0.53 g, N = 10 mice per group; column 1 and 2) (Fig. 1A). Over the next 10 weeks, the GU weight of the wild-type mice grew by 36% (from 0.52 g to 0.71 g, N = 10 mice, p < 0.01; column 3 vs. 1), whereas GU weight of the TRAMP mice increased by 136% (from 0.53 to 1.25 g, N =10, p < 0.001; column 4 vs. 2). The GU weight of TRAMP mice receiving MSeA or MSeC only increased by 60% and 49% (from 0.53 g to 0.85 g and 0.79 g, column 5 and 6 vs. 2), respectively. Overall, MSeA and MSeC treatment in the 10-week period resulted in a net inhibition of T-Ag driven expansion of GU weight by 76% and 87%, respectively (p<0.001 for each compound). There was no statistical difference between MSeA and MSeC treatments.
Fig. 1.
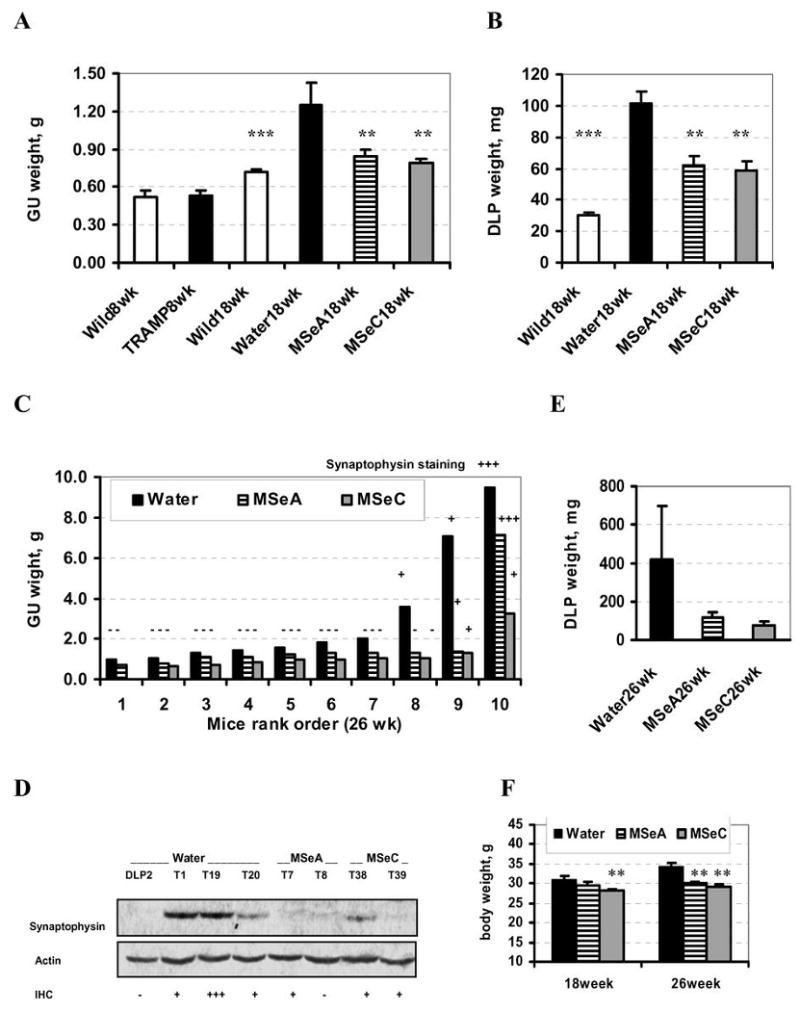
Effect of daily orally-administered MSeA and MSeC starting from 8 weeks of age on prostate carcinogenesis surrogate indicators (i.e., weight of genitourinary tract [GU] and weight of the dorsolateral prostate [DLP]) of TRAMP mice at 18 weeks and 26 weeks of age. A and B. The weight of (A) GU and (B) DLP at 18 weeks of age. Wild type littermates (Wild8wk) and TRAMP mice at 8 weeks of age (TRAMP8wk) provided baseline data on GU weight in A. N = 10 mice per group, except for the water-treated control TRAMP group (Water18wk) where one mouse with a 2.6 g prostate tumor was excluded. ** and *** indicate different from water-treated TRAMP group at P<0.01 and <0.001. C. The weight of GU of TRAMP mice in each group at 26 weeks of age presented in an ascending order. Extent of synaptophysin-staining by IHC in the prostate tissues/tumors was marked by − (negative), + (less than 5%) and +++ (greater than 20%). D. Immunoblot detection of synaptophysin expression in selected DLP and prostate tumor (T) samples from the different groups. The number in each sample designation corresponds to the animal No. from which the tissue/tumor was taken. E, the weight of DLP of water-, MSeA- or MSeC-treated TRAMP mice at 26-weeks of age. Water-group, N = 8, excluding 2 mice with large tumors; MSeA-group, N = 9, excluding one mouse with a large tumor, MSeC-group, N = 8, excluding one mouse with a large tumor. F. Body weight of TRAMP mice at 18 and 26 weeks of age. N=10 mice per group. ** indicate different from respective water-treated group at P<0.01.
The DL-prostate weight (Fig. 1B) of the TRAMP mice at 18 weeks (101 mg, N=9 mice, excluding 1 mouse with a tumor weighing 2.6 g (synaptophysin positive NE carcinoma); column 2) was 237% greater than that of the wild-type littermates (30 mg, N=10; column 1) (p < 0.001). The DLP weight of the TRAMP mice treated with MSeA or MSeC was significantly less than the water-treated mice, being 62 and 58 mg, respectively (p < 0.001 for each compound, columns 3 and 4 vs. 2). As a conservative estimate (excluding a 2.6 g tumor in the water-control group), the MSeA or MSeC treatment decreased the T-Ag driven DLP weight gain by 55% and 63%, respectively, after 10 weeks of administration.
At 26 weeks of age, the GU weight data were not normally distributed due to at least one very large tumor in each group and were therefore presented as ranking order, but were not analyzed by ANOVA (Fig. 1C). The mathematical mean GU weight of the water-, MSeA- and MSeC-treated TRAMP mice was 3.03 g (N = 10), 1.73 g (N = 10) and 1.21g (N = 9), respectively. The DLP weight of the water-, MSeA- or MSeC-treated mice was 424 mg (N = 8, excluding 2 mice with large tumors), 119 mg (N = 9, excluding one mouse with a large tumor) and 76 mg (N = 8, excluding one mouse with a large tumor), respectively (Fig. 1E). Together these data at 26 weeks of age indicated decreased growth of the DLP and GU organs by MSeA and MSeC treatment. Because of the demonstration of different lineages for NE-carcinomas (synaptophysin+, mostly arising from ventral prostate) and glandular epithelial lesions from the DLP (27, 28), we analyzed synaptophysin expression in the prostate tissue and tumor sections by immunohistochemistry staining (See supplementary Fig S1 for staining patterns) and immunobloting (Fig. 1D). As shown in Fig. 1C and D, synaptophysin(+) carcinomas represented the fast growing cancer in our model and their growth were seemingly inhibited by MSeA or MSeC.
The body weight of TRAMP mice at 18 weeks was not significantly affected by oral administration of 3 mg Se per kg body weight as MSeA, but was decreased by MSeC by 9.4% (Fig. 1F). At 26 weeks of age, the final body weight was significantly lower (compare 34.3 g in water-control TRAMP mice with 30.0 g of MSeA-treated mice and 29.0 g of MSeC-treated mice, respectively) (Fig. 1F). A portion of the body weight difference, however, was attributable to the heavier GU weight in the control mice at 26 weeks (Fig. 1C). These data indicated that the dosage level of 3 mg Se/kg was marginally tolerated by the TRAMP mice. Because of the complication of GU weight difference in TRAMP mice, the long-term safety issue of MSeA in wild type male mice was assessed in a second experiment along with TRAMP-mouse survival evaluation (See long-term experiment later).
MSeA and MSeC delayed the lesion progression in DL-prostate and the occurrence of poorly differentiation NE-carcinomas
As shown in Fig. 2A, at both 18 weeks and 26 weeks of age, the severity of DLP lesions (highest grade based on H&E for each mouse) showed that the MSeA or MSeC-treated groups had more mice with PIN lesions and fewer mice with more advanced poorly differentiated carcinomas (PD) than the control groups. These results suggest that both Se compounds when administered starting at 8 weeks of age inhibit lesion progression in the DLP from PIN and WD lesions to MD carcinomas (Fig,. 2A) and also decreased the emergence of PD carcinomas (Fig. 2A), which are mostly synaptophysin+ (Fig. 1C).
Fig 2.
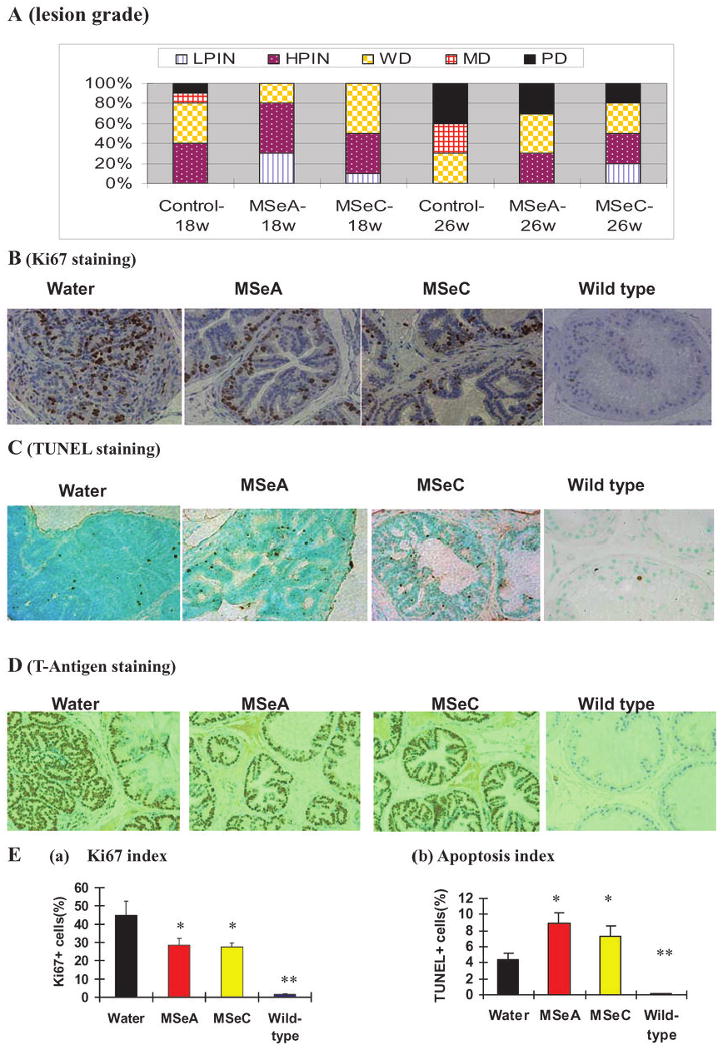
Effect of daily orally-administered MSeA and MSeC starting from 8 weeks of age on prostate carcinogenesis lesion progression (A), epithelial proliferation index (B and E.a; Ki67 staining), apoptosis (C and E.b; TUNEL staining) and T-antigen transgene expression (D) in the dorsolateral prostate of TRAMP mice at 18 weeks or 26 weeks of age. A. Lesion grade distribution of water-treated (control), MSeA- or MSeC-treated mice at 18 and 26 weeks of age. The highest lesion grade based on H&E staining in the DLP for each mouse was tabulated. N=10 mice per group. LPIN = low grade prostatic intraepithelial neoplasia, HPIN = high grade prostatic intraepithelial neoplasia, WD = well differentiated carcinoma with epithelial glandular features, MD = moderately differentiated carcinoma with epithelial glandular feature. PD = poorly differentiated carcinomas, mostly like synaptophysin expressing NE-carcinomas. B. Representative staining patterns of Ki67 in DLP tissues from water-treated, MSeA- or MSeC-treated TRAMP mice as well as from a wild type mice at 18 weeks of age. (×400). The overall estimated Ki67 indices for the different groups are shown in Ea based on quantification of five to 10 fields of each slide from each mouse. C. Representative staining patterns of TUNEL apoptosis in DLP tissues of water-treated, MSeA- or MSeC-treated TRAMP mice as well as from wild type mice at 18 weeks of age. (×400). The overall estimated TUNEL positive apoptosis indices for the different groups are shown in E.b based on quantification of five to 10 fields of each slide from each mouse. D. Representative staining patterns of T-antigen protein in DLP tissues from water-treated, MSeA- or MSeC-treated TRAMP mice as well as from wild type mice at 18 weeks of age (×400). Note nuclear staining in epithelial cells of TRAMP DLP, and a lack of staining in wild type DLP. * and ** indicate different from respective water-treated group at P<0.05 and <0.01.
Effect of MSeA and MSeC on the proliferation and apoptosis indices in DL-prostates
To provide a mechanistic explanation of the delayed DLP lesion progression, we next analyzed indices of epithelial proliferation and apoptosis focusing on the 18-week sample cohorts. Ki67 is a nuclear protein highly expressed in proliferating cells. IHC analysis of DLP (Fig. 2B) showed a virtual absence of Ki67-positive staining in wild-type mice and greatly increased Ki-67 index in TRAMP mice in the water-control group. TRAMP mice receiving either MSeA or MSeC treatment showed decreased Ki67 expression compared to the water group (t-test, p < 0.05) (Fig. 2E, graph a).
In vivo apoptotic response of MSeA- or MSeC-treated DL-prostate in the 18-week cohort was analyzed by TUNEL staining (Fig. 2C). Wild-type mice showed minimal apoptosis in the DLP, and TRAMP mice had much increased apoptosis (∼4.4%, Fig. 2E, graph b). The TUNEL-positive cells in MSeA- and MSeC-treated TRAMP mice were greater than in the water-treated TRAMP mice, being 8.9 % (t-test, p<0.01) and 7.4 % (t-test, p<0.05), respectively (Fig. 2E, graph b).
The expression of the oncogenic transgene T-Ag in the DLP was examined by immunohistochemistry staining (Fig. 2D). As expected, the wild type DLP was devoid of this transgene product. The epithelial expression of T-Ag in the TRAMP mice was evident and was not appreciably affected by either MSeA or MSeC treatment. These data support the involvement of anti-proliferative as well as pro-apoptotic effects of MSeA and MSeC in the suppression of DLP lesion growths in the TRAMP mice, without affecting transgene product expression.
Effect of MSeA or MSeC on selected proteins in cell proliferation and apoptosis
To begin to define potential in vivo molecular mediators or targets associated with the cell proliferation and apoptosis signaling responses in the DLP affected by Se treatments, we focused on the 18-week tissues, because these early samples were less complicated by the emergence of large tumors than at later time points. As shown in Fig. 3A, the abundance of CDK2 and CDK4, cyclin E, c-Jun and PCNA was greatly increased in the DLP of the TRAMP mice (lane 1) compared with the wild type mice (lane 4). MSeA and MSeC decreased the abundance of CDK2 and c-Jun and increased the abundance of CDK inhibitory protein p27Kip1, without appreciable effect on CDK4 and PCNA. MSeA treatment appeared to suppress cyclin E abundance (lane 2 vs. lane 1) whereas MSeC did not (lane 3 vs. lane 1).
Fig. 3.
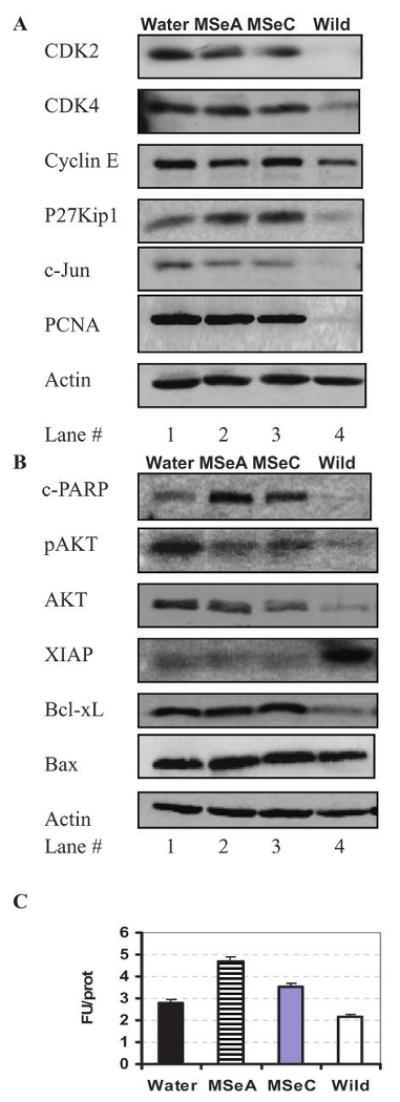
Analyses by Western blots of (A) cell cycle and (B) apoptosis regulatory proteins and assay of caspase-3/-7 enzyme activity (C) of the dorsolateral prostate of Water-treated, MSeA- or MSeC-treated TRAMP mice as well as from wild type mice at 18 weeks of age. DLP tissues were pooled from 9 (excluded a 2.6 g tumor in water treated group) -10 mice per group for protein extraction and immunoblot and for measurement of caspase-3-7 substrate hydrolysis activity. Error bars indicate variation of triplicate assay of each pooled sample.
As far as cell death was concerned, cleavage of PARP was barely detectable in the wild type DLP (lane 4) and was higher in the water-treated control TRAMP DLP (lane 1) (Fig. 3B). MSeA and MSeC-treated DLP contained elevated level of cleaved PARP in comparison with water-control group (lanes 2 and 3 vs. 1). Measurement of caspase-3/-7 enzymatic activity in the tissue lysate confirmed the increased activation of caspase-mediated execution in the MSeA- or MSeC-treated DLP (Fig. 3C). The abundance and phosphorylation level of the survival kinase AKT were lower in both Se-groups (lanes 2 and 3) in comparison with the water control group (lane 1) (Fig. 3B). The mitochondria integrity regulatory proteins Bax and Bcl-xL were not altered by the Se treatments. High expression of the caspase inhibitor protein XIAP was found to correlate with the low apoptosis in the wild type mice, in spite of low AKT and low Bcl-xL abundance in these mice (lane 4). These biochemical data support the possible involvement of CDK2, c-Jun and P27kip1 in regulating of DLP cell proliferation and of AKT inhibition in caspase-mediated apoptosis in vivo by MSeA or MSeC consumption.
MSeA and MSeC caused a significant decrease of serum IGF-1 levels in TRAMP mice
The IGF-1 axis plays a crucial role in prostate cancer progression (38) (41) and some studies suggest that serum IGF-1 level might be a better predictor of prostate cancer risk than serum prostate-specific antigen (PSA) (42, 43). We found that MSeA or MSeC treatment led to decreased serum IGF-1 in 18-week old TRAMP mice (Fig. 4A) (ANOVA, p<0.01), and 26-week old TRAMP mice (Fig. 4B) (ANOVA, p<0.05).
Fig. 4.
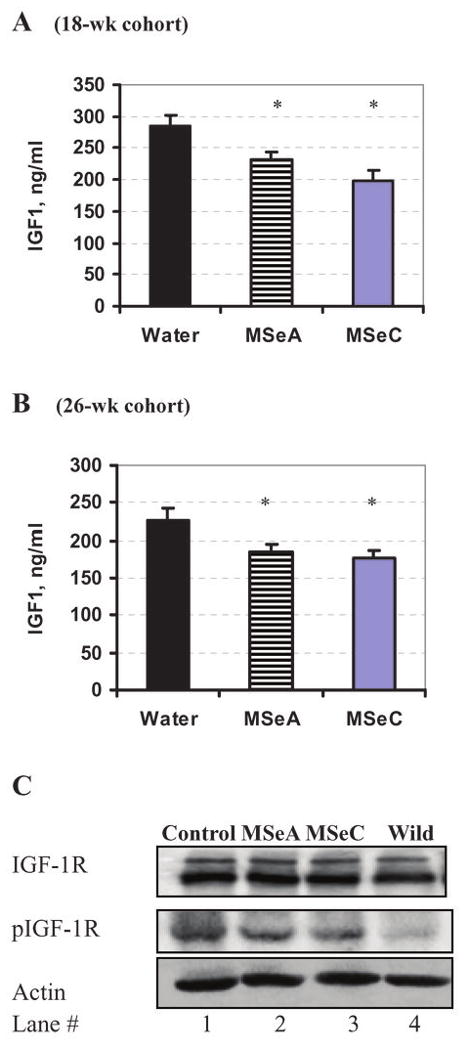
Effect of daily orally-administered MSeA and MSeC starting from 8 weeks of age on serum IGF-1 levels at (A) 18 weeks and (B) 26 weeks of age and on (C) the IGF-1R phosphorylation status of the dorsolateral prostate of TRAMP mice at 18 weeks of age detected by immunobloting. IGF-1 was detected by ELISA, n=10 mice per group. *Indicate different from respective water-group at P<0.05.
Western blot analyses of the DLP lysates did not reveal any change of IGF-1 receptor abundance resulting from Se treatment (Fig. 4C). The phosphorylation of IGF-1R was increased in the water-treated control TRAMP mice (lane 1) when compared to wild type mice (lane 4). The treatment by either Se compound decreased the phosphorylation modestly (lanes 2 and 3 vs. 1). These data suggest that MSeA and MSeC might inhibit IGF-1 axis at both the ligand and receptor phosphorylation level, upstream of AKT phosphorylation.
MSeA improved the overall and the cancer-specific survival of TRAMP mice
Prompted by the data from the above experiment supporting a similar in vivo inhibitory efficacy of MSeA and MSeC on early stage prostate lesion progression in DLP and the emergence of synaptophysin (+) NE-carcinomas, we next did a long-term experiment to establish the survival benefit of oral MSeA administration. Suspecting that the overall efficacy would depend on the stages of the disease when MSeA intervention was started, we therefore compared the MSeA oral treatment at 10 weeks (early PIN stage) vs. 16 weeks of age (PIN to organ confined NE-carcinomas).
Fig. 5A showed the Kaplan-Meier survival curves of TRAMP mice due to all causes of mortality (euthanasia or those found dead) among the three groups. The median survival time was 40.5, 40.5 and 45.0 weeks of age for the water group (N = 31), 16-week MSeA cohort (N = 30) and 10-week MSeA cohort (N = 31), respectively. Whereas nearly all water-treated TRAMP mice were dead by 50 weeks of age, approximately one third of mice treated with MSeA starting either at 10-weeks or 16-weeks of age survived to this age. Log rank tests show: water vs. 10-week MSeA cohort, p = 0.0225; water vs. 16- week MSeA cohort, p = 0.1793.
Fig. 5.
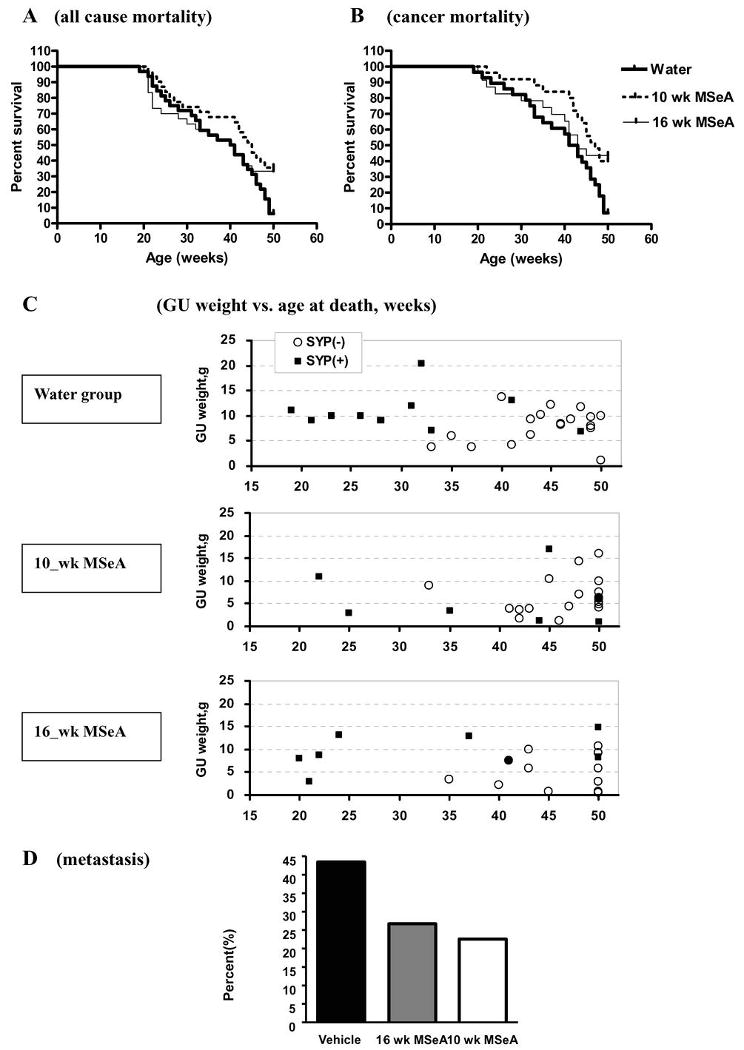
Effect of daily orally-administered MSeA starting from 10 weeks or 16 weeks of age on the long-term survival of TRAMP mice. A. Kaplan-Meier survival curves of all causes of mortality. Median survival time was 40.5, 40.5 and 45.0 weeks for the water-treated control mice (N =31), 16-week MSeA (N=30) and 10-week MSeA (N=31) groups, respectively. Two group log rank tests: water vs. 16 wk MSeA P = 0.1793; water vs. 10 wk MSeA, P=0.0225 and 16 wk vs. 10 wk MSeA P=0.4467. B. Kaplan-Meier survival curves with mice died of verifiable non-tumor causes before 30 weeks of age excluded (effective number of mice for plots: water-treated group, N = 28; 10 wk MSeA group, N=25; 16 wk MSeA group, N=23). Median survival time was 42, 43 and 47 weeks for the water-treated, 16 week MSeA and 10 week MSeA groups, respectively. Two group log rank tests: water vs. 16 wk MSeA, P=0.0385; water vs. 10 wk MSeA P=0.0078; and 16 wk MSeA vs. 10 wk MSeA, P=0.7269. C. Scatter-plots of the weight of GU at necropsy of the water-, 10-week and 16-week MSeA treated mice vs. age at death/euthanasia. Synaptophysin IHC staining status in prostate carcinoma/lesions was indicated (Syp(-), negative, empty circles; Syp(+), positive, solid squares). D. Metastasis incidence of the water- vs. 10-week and 16-week MSeA treated groups. When the MSeA groups were combined for comparison with the water group, Chi square = 4.51, 1 degree of freedom, 0.025< P <0.05.
There was some unexpected non-cancer-related mortality between 20-30 weeks in all 3 groups. Therefore, we excluded those mice that died of verifiable non-cancer causes before 30 weeks of age and re-plotted the survival curves (effective number of mice: water group, N = 28; 10-week MSeA cohort, N=25; 16-week MSeA cohort, N=23) (Fig. 5B). The median survival time of the corrected cancer-specific mortality was 42, 43 and 47 weeks of age for the water-, 16-week MSeA and 10-week MSeA cohorts, respectively. Nevertheless, a greater number of mice in both MSeA-treated groups than the control mice survived cancer-specific mortality to 50 weeks of age. Two group log rank tests showed: water vs. 10-week MSeA cohort, p = 0.0078; water vs. 16-week MSeA cohort, p = 0.0385; and 16-week vs. 10-week MSeA cohort, p = 0.7269. These data therefore support an improvement of the long-term survival of TRAMP mice against all-causes of mortality and cancer-specific mortality by intervening with MSeA at either 10 weeks or 16-weeks of age. The lack of improvement of median survival time by MSeA treatment starting at 16-weeks of age might indicate a decreased ability for MSeA to inhibit pre-existing aggressive early carcinomas in some mice at that age.
Indeed, when we plotted the GU weight of mice vs. the age at death/euthanasia according to the synaptophysin-staining status, it became apparent that the early cancer-death (<30 weeks) in the control mice was due to the formation of the aggressive synaptophysin(+) NE-carcinomas (Fig. 5C, water group) and the mice that were euthanized or died >40 weeks of age were due to enlarged seminal vesicles and their prostate lesions were mostly negative for synaptophysin-staining. When provided starting at 10-weeks of age, MSeA decreased and delayed not only death due to the synaptophysin(+) carcinomas, but also the death of mice bearing synaptophysin(-) staining lesions (Fig. 5C, 10_wk MSeA group). However, when started at 16 weeks, MSeA did not suppress the early death due to synaptophysin (+) carcinomas (20-24 weeks), but delayed the deaths due to the remaining synaptophysin (+) carcinomas and synaptophysin (-) prostate lesions (Fig. 5C, 16_wk MSeA group).
Consistent with improved long-term survival, the visible macro-metastasis to the lymph nodes, liver and lung were 43.8% (14/32) in the water group and 26.7% (8/30) and 22.6% (7/31) in the 16-week and 10-week MSeA cohorts, respectively (Fig. 5D). When the MSeA groups were combined for comparison with the water group, Chi square = 4.51, 1 degree of freedom, 0.025< P <0.05.
Lack of growth inhibition and liver damage by long term MSeA administration in wild type littermates
The safety of long-term MSeA treatment was a concern and therefore was simultaneously evaluated in wild-type littermates. The mice given daily orally MSeA treatment from 10 weeks of age until 41 weeks of age showed similar rate of body weight gain in comparison to the water-treated mice (Fig. 6A). MSeA treatment did not affect the weight of the GU when compared with the water control mice (Fig. 6B).
Fig. 6.
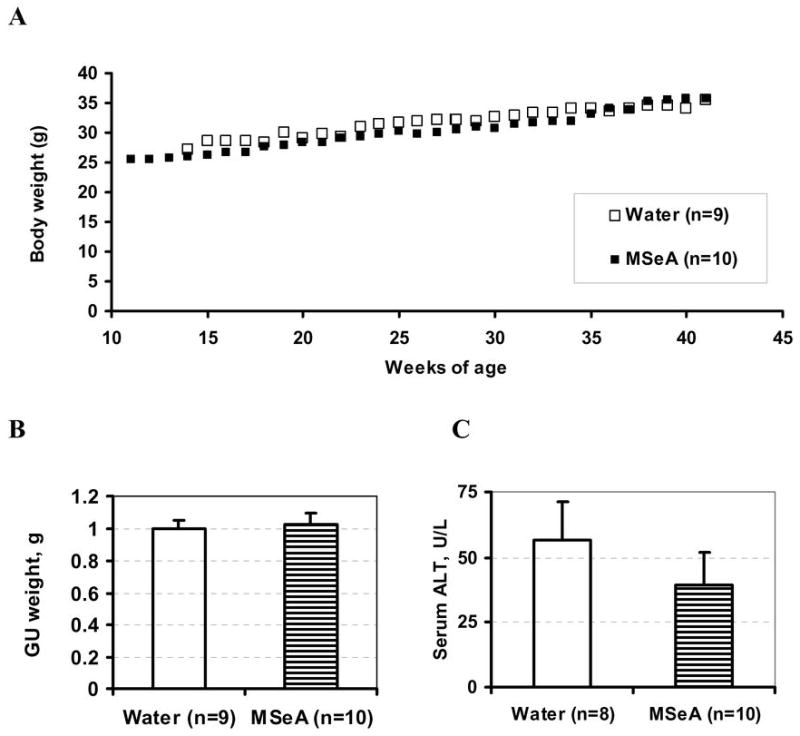
Lack of significant effect of orally administered MSeA from 10 weeks of age to 41 weeks of age on (A) body weight, (B) GU weight and (C) liver damage serum marker, ALT in wild type male littermates. Average body weights of water-treated (open symbols) and MSeA-treated (solid symbols) mice without error bars were plotted over time for clarity. No statistical significance for GU or ALT between the groups.
As a biochemical indicator of liver damage, we measured the serum ALT level of the non-TRAMP littermates. There was no significant difference between the water- and the MSeA-treated groups (Fig. 6C). These data support the well-tolerated nature of MSeA daily oral dosing regimen for long term chemoprevention use.
Discussion
The lack-luster performance of chemotherapy for advanced recurrent PCa that have failed surgical, hormone ablation and radiation therapies calls for alternative approaches to combat this disease. The long latency period and high prevalence of PCa in men make chemoprevention of prostate carcinogenesis a plausible and cost-effective means to deal with the problem at the root. Although Se has been implicated for its potential for PCa prevention (7, 8), there is a void of pertinent literature in preclinical models of prostate carcinogenesis to support their utility for prostate cancer chemoprevention. In retrospect, the paucity of such preclinical data was at least one reason that SeMet was not prevented from being chosen for the failed SELECT study (9, 10).
Here we have shown, to our knowledge for the first time, that daily oral administration of “second-generation” Se compounds MSeA and MSeC starting at 8 weeks of age (puberty) inhibit early stage prostate carcinogenesis as reflected by decreased GU and DLP weight at 18 weeks and 26 weeks of age (Fig. 1), delayed prostate lesion progression in the DLP (Fig. 2A) and slowed the emergence of PD synaptophysin(+) NE-carcinomas (Fig. 1C) in TRAMP mice. We further showed that long-term use of MSeA resulted in an improvement of the survival from cancer-specific and all causes of mortality (Fig. 5) with no appreciable adverse effects on body weight gain, GU weight or liver integrity (Fig. 6). Analyses of the synaptophysin expressing status of prostate lesions and carcinomas in the long-term experiment supported the inhibitory efficacy of MSeA against not only the very aggressive-growing synaptophysin(+) NE-carcinomas, but also deaths from the synpatophysin(-) DLP lesions and seminal vesicle hypertrophy (Fig. 5C). The daily oral Se dosing regimen was designed to mimic the frequency used in the Clark study (7), the SELECT study (9) and the lung cancer prevention trial with selenized yeast (44) and has been used by us in xenograft studies in athymic nude mice (13). Together these findings illustrate that these Se compounds may be effective chemopreventive agent against primary carcinogenesis in the TRAMP model of prostate NE-carcinomas as well as prostate glandular epithelial cancers and merit serious consideration for translational studies in the future.
A number of points are worthy of elaboration. First, the in vivo efficacy of MSeA and MSeC was indistinguishable at the dosage tested in the short term experiment (Fig. 1). The efficacy was associated with decreased prostate epithelial proliferation (Fig. 2B and 2E[a]), increased apoptosis (Fig. 2C and 2E[b]), and decreased circulating IGF-1 levels (Fig. 4). In our biomarker analyses (Fig. 3), many of the molecular changes identified in DLP were associated with caspase mediated apoptosis regulation such as PARP cleavage (Fig. 3B) and caspase-3/7 activity (Fig. 3C) and decreased expression and phosphorylation of AKT, a critical survival protein kinase (Fig. 3B). Anti-proliferation was associated with decreased CDK2, c-Jun and increased p27Kip1 (Fig. 3A). Subtle differences were occasionally seen for the analyzed biomarkers, such as cyclin E (Fig. 3A), lending some credence to the possibility of unique targets to each Se form. We have observed that while MSeA and MSeC were equally efficacious against the growth of human DU145 prostate cancer xenograft model in nude mice, but in the human PC-3 prostate xenograft model, MSeA was more efficacious than MSeC (13). Therefore, the patho-genetic background of the PCa cells may also affect the molecular and cellular targets of the different forms of Se and their in vivo efficacy. Although methylselenol has been suggested as a common mediator of the anti-cancer activity of MSeA and MSeC, further studies will be required to determine whether each Se involves unique in vivo molecular targets or cellular pathways in addition to shared ones through methylselenol.
In addition to molecular and morphological changes in DLP, our results showed the circulating IGF-1 was lower in the 18 week (P<0.01) and 26 week (P<0.05) MSeA- or MSeC-treated cohorts compared to untreated TRAMP mice (Fig. 4 A and B). Additionally, Se treatment of TRAMP mice modestly decreased phosphorylated IGF-1R in the DLP (Fig. 4C). The lower serum IGF-1 concentrations found in the treated mice suggest a possible systemic action of these Se forms to regulate prostate cancer genesis and progression. Epidemiological studies indicate IGF-1 axis signaling is de-regulated in clinical PCa, and that serum IGF-1 level could be a predictor of PCa risk (42, 43). Furthermore, IGF-1R has been reported to be over-expressed in prostate tumor specimens (45). Recent studies with an IGF-1R receptor neutralizing antibody have shown potent inhibition of in vivo prostate cancer growth alone or in combination with chemotherapy (46, 47). However, the issue with IGF-1/IGF-1R signaling in prostate cancer is rather complex as highlighted by a recent study by the Greenberg group that has shown that selective knockout of IGF-1R in the epithelial cells of TRAMP mice accelerates lesion progression to a more aggressive phenotype (48), contrary to an expected decreased tumorigenesis. It is possible that different thresholds exist for IGF-1R signaling therefore a complete IGF-1R knockout may not recapitulate the action of diminished IGF-1/IGF-1R signaling.
In the long-term experiment, starting MSeA treatment at 10 weeks of age, but not at 16 weeks of age improved the median survival time (40-41 weeks for control TRAMP mice) by 4-5 weeks against overall and cancer-specific mortality of TRAMP mice; yet both MSeA cohorts of mice outlived the water-treated TRAMP mice to 50 weeks of age (Fig. 5A and B). Although the results may appear paradoxical on first glance, it is likely that TRAMP mice dying early from cancer-related causes before the median survival time were due to early aggressive tumors. Indeed, these tumors were mostly synaptophysin+ NE-carcinomas (Fig. 5C). Starting MSeA after such tumors have formed, namely at 16 weeks of age, would be expected to be less effective than if MSeA was started at 10-weeks of age to prolong the life of the mice with these tumors, which was supported by data in Fig. 5C. This finding reaffirms the importance of early intervention of the carcinogenic processes of both NE-carcinomas as well as the slow-growing prostate glandular carcinomas by Se to achieve lasting preventive result. As a precedence, it has been demonstrated in a chemically-induced mammary carcinogenesis model that Se-enriched garlic given for 1 month duration right after a single carcinogen exposure was as effective as when the Se was provided throughout the post-initiation period of the study (49, 50), supporting “clonal deletion” of initiated cells or early precursor lesions for such permanent protection. We should remark that in the future, animal models that exclusively develop adenocarcinoma of the prostate should be evaluated for confirming the chemopreventive efficacy of these second generation Se forms without the complication of the NE-carcinomas.
The Se dosage and delivery regimen warrant further comments. The daily single oral dose of 3 mg Se per kg body weight of either MSeA or MSeC was shown to be tolerated by the TRAMP mice in this study (Fig. 1F; Fig. 6) and in nude mice in our previous studies (13). To provide the same Se intake through including these compounds in the diet would have required approximately 30 ppm (assuming each mouse weighing 30 g eats 3 g of diet per day). Experience indicates that such high dietary levels of Se will not be palatable to the mice and cause severe weight loss. In fact, a feeding study (unpublished data) with MSeA suggests 5 ppm as the maximally tolerated dietary level for the mice. We suggest that the bolus dosage may have the advantage of a transient elevation of the critical Se metabolites without a lasting effect on appetite and therefore more tolerable than when is chronically fed through the diet. In support of this speculation, we have documented a rapid peak spike of Se after an oral dose of MSeA (51).
In summary, our results indicate that the second generation Se compounds inhibit the PCa lesion progression in the DLP, delay the emergence of synaptophysin (+) NE-carcinomas, decrease visible metastasis to other organs and improve the survival of TRAMP mice, which represent a very aggressive PCa model. The findings in the present study together with our earlier findings in human prostate tumor xenograft in athymic nude mice (13) suggest that these Se compounds may prove to be effective dietary supplements for the chemoprevention of PCa.
Supplementary Material
Acknowledgments
We thank Nancy Mizuno and Emily Quealy for technical support for the genotyping of TRAMP mice; Dr. Olga Rogozina for advices on TRAMP colony maintenance. We thank Dr. Fred Bogott of the Austin Medical Center, Austin, MN for proof reading of the manuscript.
Funding supports: Grants from National Cancer Institute CA95642 (JL), CA126880 (JL and MPC), DOD WX8XWH-06-0292 (MPC), American Institute for Cancer Research (MPC) and Hormel Foundation.
Footnotes
Disclosure of Potential Conflicts of Interest: No potential conflicts of interest were disclosed.
References
- 1.Jemal A, Siegel R, Ward E, et al. Cancer statistics, 2008. CA Cancer J Clin. 2008;58(2):71–96. doi: 10.3322/CA.2007.0010. [DOI] [PubMed] [Google Scholar]
- 2.Petrylak DP. The current role of chemotherapy in metastatic hormone-refractory prostate cancer. Urology. 2005;65(5 Suppl):3–7. doi: 10.1016/j.urology.2005.03.053. discussion -8. [DOI] [PubMed] [Google Scholar]
- 3.Parnes HL, Thompson IM, Ford LG. Prevention of hormone-related cancers: prostate cancer. J Clin Oncol. 2005;23(2):368–77. doi: 10.1200/JCO.2005.08.027. [DOI] [PubMed] [Google Scholar]
- 4.Klein EA. Can prostate cancer be prevented? Nat Clin Pract Urol. 2005;2(1):24–31. doi: 10.1038/ncpuro0072. [DOI] [PubMed] [Google Scholar]
- 5.Li H, Stampfer MJ, Giovannucci EL, et al. A prospective study of plasma selenium levels and prostate cancer risk. J Natl Cancer Inst. 2004;96(9):696–703. doi: 10.1093/jnci/djh125. [DOI] [PubMed] [Google Scholar]
- 6.Brooks JD, Metter EJ, Chan DW, et al. Plasma selenium level before diagnosis and the risk of prostate cancer development. The Journal of urology. 2001;166(6):2034–8. [PubMed] [Google Scholar]
- 7.Clark LC, Combs GF, Jr, Turnbull BW, et al. Effects of selenium supplementation for cancer prevention in patients with carcinoma of the skin. A randomized controlled trial. Nutritional Prevention of Cancer Study Group. Jama. 1996;276(24):1957–63. [PubMed] [Google Scholar]
- 8.Clark LC, Dalkin B, Krongrad A, et al. Decreased incidence of prostate cancer with selenium supplementation: results of a double-blind cancer prevention trial. Br J Urol. 1998;81(5):730–4. doi: 10.1046/j.1464-410x.1998.00630.x. [DOI] [PubMed] [Google Scholar]
- 9.Lippman SM, Goodman PJ, Klein EA, et al. Designing the Selenium and Vitamin E Cancer Prevention Trial (SELECT) J Natl Cancer Inst. 2005;97(2):94–102. doi: 10.1093/jnci/dji009. [DOI] [PubMed] [Google Scholar]
- 10.Lippman SM, Klein EA, Goodman PJ, et al. Effect of selenium and vitamin E on risk of prostate cancer and other cancers: the Selenium and Vitamin E Cancer Prevention Trial (SELECT) Jama. 2009;301(1):39–51. doi: 10.1001/jama.2008.864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McCormick DL, Rao KV. Chemoprevention of hormone-dependent prostate cancer in the Wistar-Unilever rat. European urology. 1999;35(5-6):464–7. doi: 10.1159/000019880. [DOI] [PubMed] [Google Scholar]
- 12.Corcoran NM, Najdovska M, Costello AJ. Inorganic selenium retards progression of experimental hormone refractory prostate cancer. The Journal of urology. 2004;171(2 Pt 1):907–10. doi: 10.1097/01.ju.0000092859.16817.8e. [DOI] [PubMed] [Google Scholar]
- 13.Li GX, Lee HJ, Wang Z, et al. Superior in vivo inhibitory efficacy of methylseleninic acid against human prostate cancer over selenomethionine or selenite. Carcinogenesis. 2008;29(5):1005–12. doi: 10.1093/carcin/bgn007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lucia MS, Epstein JI, Goodman PJ, et al. Finasteride and high-grade prostate cancer in the Prostate Cancer Prevention Trial. J Natl Cancer Inst. 2007;99(18):1375–83. doi: 10.1093/jnci/djm117. [DOI] [PubMed] [Google Scholar]
- 15.Thompson IM, Goodman PJ, Tangen CM, et al. The influence of finasteride on the development of prostate cancer. The New England journal of medicine. 2003;349(3):215–24. doi: 10.1056/NEJMoa030660. [DOI] [PubMed] [Google Scholar]
- 16.Thompson IM, Tangen CM, Parnes HL, Lippman SM, Coltman CA., Jr Does the level of prostate cancer risk affect cancer prevention with finasteride? Urology. 2008;71(5):854–7. doi: 10.1016/j.urology.2008.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ip C. Lessons from basic research in selenium and cancer prevention. J Nutr. 1998;128(11):1845–54. doi: 10.1093/jn/128.11.1845. [DOI] [PubMed] [Google Scholar]
- 18.Lu J, Jiang C. Selenium and cancer chemoprevention: hypotheses integrating the actions of selenoproteins and selenium metabolites in epithelial and non-epithelial target cells. Antioxid Redox Signal. 2005;7(11-12):1715–27. doi: 10.1089/ars.2005.7.1715. [DOI] [PubMed] [Google Scholar]
- 19.Jiang C, Wang Z, Ganther H, Lu J. Caspases as key executors of methyl selenium-induced apoptosis (anoikis) of DU-145 prostate cancer cells. Cancer Res. 2001;61(7):3062–70. [PubMed] [Google Scholar]
- 20.Jiang C, Wang Z, Ganther H, Lu J. Distinct effects of methylseleninic acid versus selenite on apoptosis, cell cycle, and protein kinase pathways in DU145 human prostate cancer cells. Mol Cancer Ther. 2002;1(12):1059–66. [PubMed] [Google Scholar]
- 21.Dong Y, Zhang H, Hawthorn L, Ganther HE, Ip C. Delineation of the molecular basis for selenium-induced growth arrest in human prostate cancer cells by oligonucleotide array. Cancer Res. 2003;63(1):52–9. [PubMed] [Google Scholar]
- 22.Dong Y, Lee SO, Zhang H, Marshall J, Gao AC, Ip C. Prostate specific antigen expression is down-regulated by selenium through disruption of androgen receptor signaling. Cancer Res. 2004;64(1):19–22. doi: 10.1158/0008-5472.can-03-2789. [DOI] [PubMed] [Google Scholar]
- 23.Cho SD, Jiang C, Malewicz B, et al. Methyl selenium metabolites decrease prostate-specific antigen expression by inducing protein degradation and suppressing androgen-stimulated transcription. Mol Cancer Ther. 2004;3(5):605–11. [PubMed] [Google Scholar]
- 24.Hu H, Jiang C, Li G, Lu J. PKB/AKT and ERK regulation of caspase-mediated apoptosis by methylseleninic acid in LNCaP prostate cancer cells. Carcinogenesis. 2005;26(8):1374–81. doi: 10.1093/carcin/bgi094. [DOI] [PubMed] [Google Scholar]
- 25.Greenberg NM, DeMayo F, Finegold MJ, et al. Prostate cancer in a transgenic mouse. Proc Natl Acad Sci U S A. 1995;92(8):3439–43. doi: 10.1073/pnas.92.8.3439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kaplan-Lefko PJ, Chen TM, Ittmann MM, et al. Pathobiology of autochthonous prostate cancer in a pre-clinical transgenic mouse model. Prostate. 2003;55(3):219–37. doi: 10.1002/pros.10215. [DOI] [PubMed] [Google Scholar]
- 27.Huss WJ, Gray DR, Tavakoli K, et al. Origin of androgen-insensitive poorly differentiated tumors in the transgenic adenocarcinoma of mouse prostate model. Neoplasia (New York, NY. 2007;9(11):938–50. doi: 10.1593/neo.07562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chiaverotti T, Couto SS, Donjacour A, et al. Dissociation of epithelial and neuroendocrine carcinoma lineages in the transgenic adenocarcinoma of mouse prostate model of prostate cancer. The American journal of pathology. 2008;172(1):236–46. doi: 10.2353/ajpath.2008.070602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Segawa N, Mori I, Utsunomiya H, et al. Prognostic significance of neuroendocrine differentiation, proliferation activity and androgen receptor expression in prostate cancer. Pathology international. 2001;51(6):452–9. doi: 10.1046/j.1440-1827.2001.01226.x. [DOI] [PubMed] [Google Scholar]
- 30.Ahlgren G, Pedersen K, Lundberg S, Aus G, Hugosson J, Abrahamsson PA. Regressive changes and neuroendocrine differentiation in prostate cancer after neoadjuvant hormonal treatment. Prostate. 2000;42(4):274–9. doi: 10.1002/(sici)1097-0045(20000301)42:4<274::aid-pros4>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- 31.Gupta S, Ahmad N, Marengo SR, MacLennan GT, Greenberg NM, Mukhtar H. Chemoprevention of prostate carcinogenesis by alpha-difluoromethylornithine in TRAMP mice. Cancer Res. 2000;60(18):5125–33. [PubMed] [Google Scholar]
- 32.Gupta S, Adhami VM, Subbarayan M, et al. Suppression of prostate carcinogenesis by dietary supplementation of celecoxib in transgenic adenocarcinoma of the mouse prostate model. Cancer Res. 2004;64(9):3334–43. doi: 10.1158/0008-5472.can-03-2422. [DOI] [PubMed] [Google Scholar]
- 33.Adhami VM, Siddiqui IA, Ahmad N, Gupta S, Mukhtar H. Oral consumption of green tea polyphenols inhibits insulin-like growth factor-I-induced signaling in an autochthonous mouse model of prostate cancer. Cancer Res. 2004;64(23):8715–22. doi: 10.1158/0008-5472.CAN-04-2840. [DOI] [PubMed] [Google Scholar]
- 34.Raina K, Singh RP, Agarwal R, Agarwal C. Oral grape seed extract inhibits prostate tumor growth and progression in TRAMP mice. Cancer Res. 2007;67(12):5976–82. doi: 10.1158/0008-5472.CAN-07-0295. [DOI] [PubMed] [Google Scholar]
- 35.Narayanan BA, Narayanan NK, Pittman B, Reddy BS. Regression of mouse prostatic intraepithelial neoplasia by nonsteroidal anti-inflammatory drugs in the transgenic adenocarcinoma mouse prostate model. Clin Cancer Res. 2004;10(22):7727–37. doi: 10.1158/1078-0432.CCR-04-0732. [DOI] [PubMed] [Google Scholar]
- 36.Raina K, Blouin MJ, Singh RP, et al. Dietary feeding of silibinin inhibits prostate tumor growth and progression in transgenic adenocarcinoma of the mouse prostate model. Cancer Res. 2007;67(22):11083–91. doi: 10.1158/0008-5472.CAN-07-2222. [DOI] [PubMed] [Google Scholar]
- 37.Raina K, Rajamanickam S, Singh RP, Agarwal R. Chemopreventive efficacy of inositol hexaphosphate against prostate tumor growth and progression in TRAMP mice. Clin Cancer Res. 2008;14(10):3177–84. doi: 10.1158/1078-0432.CCR-07-5275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gupta S, Hastak K, Ahmad N, Lewin JS, Mukhtar H. Inhibition of prostate carcinogenesis in TRAMP mice by oral infusion of green tea polyphenols. Proc Natl Acad Sci U S A. 2001;98(18):10350–5. doi: 10.1073/pnas.171326098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jiang C, Hu H, Malewicz B, Wang Z, Lu J. Selenite-induced p53 Ser-15 phosphorylation and caspase-mediated apoptosis in LNCaP human prostate cancer cells. Mol Cancer Ther. 2004;3(7):877–84. [PubMed] [Google Scholar]
- 40.Lin JT, Kitzmiller TJ, Cates JM, Gorham JD. MHC-independent genetic regulation of liver damage in a mouse model of autoimmune hepatocellular injury. Laboratory investigation; a journal of technical methods and pathology. 2005;85(4):550–61. doi: 10.1038/labinvest.3700246. [DOI] [PubMed] [Google Scholar]
- 41.Kaplan PJ, Mohan S, Cohen P, Foster BA, Greenberg NM. The insulin-like growth factor axis and prostate cancer: lessons from the transgenic adenocarcinoma of mouse prostate (TRAMP) model. Cancer Res. 1999;59(9):2203–9. [PubMed] [Google Scholar]
- 42.Chan JM, Stampfer MJ, Giovannucci E, et al. Plasma insulin-like growth factor-I and prostate cancer risk: a prospective study. Science. 1998;279(5350):563–6. doi: 10.1126/science.279.5350.563. [DOI] [PubMed] [Google Scholar]
- 43.Chan JM, Stampfer MJ, Ma J, et al. Insulin-like growth factor-I (IGF-I) and IGF binding protein-3 as predictors of advanced-stage prostate cancer. J Natl Cancer Inst. 2002;94(14):1099–106. doi: 10.1093/jnci/94.14.1099. [DOI] [PubMed] [Google Scholar]
- 44.Karp DD. ECOG 5597: Phase III Chemoprevention Trial of Selenium Supplementation in Persons With Resected Stage I Non-Small-Cell Lung Cancer. Clin Adv Hematol Oncol. 2005;3(4):313–5. [Google Scholar]
- 45.Liao Y, Abel U, Grobholz R, et al. Up-regulation of insulin-like growth factor axis components in human primary prostate cancer correlates with tumor grade. Human pathology. 2005;36(11):1186–96. doi: 10.1016/j.humpath.2005.07.023. [DOI] [PubMed] [Google Scholar]
- 46.Wu JD, Odman A, Higgins LM, et al. In vivo effects of the human type I insulin-like growth factor receptor antibody A12 on androgen-dependent and androgen-independent xenograft human prostate tumors. Clin Cancer Res. 2005;11(8):3065–74. doi: 10.1158/1078-0432.CCR-04-1586. [DOI] [PubMed] [Google Scholar]
- 47.Wu JD, Haugk K, Coleman I, et al. Combined in vivo effect of A12, a type 1 insulin-like growth factor receptor antibody, and docetaxel against prostate cancer tumors. Clin Cancer Res. 2006;12(20 Pt 1):6153–60. doi: 10.1158/1078-0432.CCR-06-0443. [DOI] [PubMed] [Google Scholar]
- 48.Sutherland BW, Knoblaugh SE, Kaplan-Lefko PJ, Wang F, Holzenberger M, Greenberg NM. Conditional deletion of insulin-like growth factor-I receptor in prostate epithelium. Cancer Res. 2008;68(9):3495–504. doi: 10.1158/0008-5472.CAN-07-6531. [DOI] [PubMed] [Google Scholar]
- 49.Ip C, Lisk DJ, Thompson HJ. Selenium-enriched garlic inhibits the early stage but not the late stage of mammary carcinogenesis. Carcinogenesis. 1996;17(9):1979–82. doi: 10.1093/carcin/17.9.1979. [DOI] [PubMed] [Google Scholar]
- 50.Ip C, Dong Y. Methylselenocysteine modulates proliferation and apoptosis biomarkers in premalignant lesions of the rat mammary gland. Anticancer research. 2001;21(2A):863–7. [PubMed] [Google Scholar]
- 51.Hu H, Li GX, Wang L, Watts J, Combs GF, Jr, Lu J. Methylseleninic acid enhances taxane drug efficacy against human prostate cancer and down-regulates antiapoptotic proteins Bcl-XL and survivin. Clin Cancer Res. 2008;14(4):1150–8. doi: 10.1158/1078-0432.CCR-07-4037. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.