

Abstract
Background and purpose:
Potencies of compounds blocking KV11.1 [human ether-ago-go-related gene (hERG)] are commonly assessed using cell lines expressing the Caucasian wild-type (WT) variant. Here we tested whether such potencies would be different for hERG single nucleotide polymorphisms (SNPs).
Experimental approach:
SNPs (R176W, R181Q, Del187-189, P347S, K897T, A915V, P917L, R1047L, A1116V) and a binding-site mutant (Y652A) were expressed in Tet-On CHO-K1 cells. Potencies [mean IC50; lower/upper 95% confidence limit (CL)] of 48 hERG blockers was estimated by automated electrophysiology [IonWorks™ HT (IW)]. In phase one, rapid potency comparison of each WT-SNP combination was made for each compound. In phase two, any compound-SNP combinations from phase one where the WT upper/lower CL did not overlap with those of the SNPs were re-examined. Electrophysiological WT and SNP parameters were determined using conventional electrophysiology.
Key results:
IW detected the expected sixfold potency decrease for propafenone in Y652A. In phase one, the WT lower/upper CL did not overlap with those of the SNPs for 77 compound-SNP combinations. In phase two, 62/77 cases no longer yielded IC50 values with non-overlapping CLs. For seven of the remaining 15 cases, there were non-overlapping CLs but in the opposite direction. For the eight compound-SNP combinations with non-overlapping CLs in the same direction as for phase 1, potencies were never more than twofold apart. The only statistically significant electrophysiological difference was the voltage dependence of activation of R1047L.
Conclusion and implications:
Potencies of hERG channel blockers defined using the Caucasian WT sequence, in this in vitro assay, were representative of potencies for common SNPs.
This article is part of a themed section on QT safety. To view this issue visit http://www3.interscience.wiley.com/journal/121548564/issueyear?year=2010
Keywords: hERG, single nucleotide polymorphisms, pharmacology, electrophysiology, safety margins, QT interval prolongation, Torsades de Pointes
Introduction
Drug-induced prolongation of ventricular cell cardiac action potentials is manifested on an electrocardiogram as a prolongation of the QT interval, an effect that is associated with a potentially fatal cardiac arrhythmia called Torsades de Pointes (TdP). Evidence of a risk of TdP has led to the withdrawal of a number of medicines from the market (Shah, 2005) and to regulatory concern culminating in both pre-clinical and clinical guidance documents (Anon., 2005a,b;). In particular, the consequences for drug development of a compound prolonging the QT interval in man in the so-called ‘thorough QT/QTc study’ make it imperative that during the drug discovery phase, the risk of prolongation of the QT interval is minimized.
It is widely accepted that for most compounds the primary molecular mechanism for QT interval prolongation is direct inhibition of the rapid component of the delayed rectifier potassium current (IKR), which plays a key role in ventricular cell action potential repolarization. It is a logical undertaking, therefore, to test compounds for IKR inhibition early in the drug discovery process and, where possible, to eliminate any activity, by design. This is likely to be done using heterologous systems expressing a single genetic variant of the human ether-a-go-go-related gene (hERG) that encodes the pore-forming α sub-units of the channel complex carrying IKR. The channel is normally expressed from a clone containing the consensus wild-type (WT) Caucasian hERG sequence as used in the seminal work on this channel type (Trudeau et al., 1995; 1996;). These hERG-expressing cell lines or their membrane fragments can then be used in assays that directly or indirectly measure compound potency/affinity.
Several literature-based exercises have confirmed what is intuitively obvious about the relationship between plasma exposure levels for clinical efficacy, hERG potency and the risk of TdP. Namely, that in general, the larger the safety margin between systemic exposure in man and potency at hERG, the lower the risk of TdP (Cavero et al., 2000; Kang et al., 2001; Webster et al., 2002; Redfern et al., 2003; De Bruin et al., 2005). Furthermore, these authors have tried to quantify a suitable safety margin in order to provide a target value for those aiming to screen-out hERG activity. This is an important concept given that the pharmacophores for this channel type are promiscuous and that the physicochemical properties of compounds that inhibit the hERG channel match those often associated with favourable pharmacokinetic properties. In other words, designing hERG-inactive compounds with good drug-like properties will be difficult in some chemical areas, thus leading to a reliance on a wide safety margin to diminish the risk of hERG activity.
In applying this rational and quantitative approach to minimizing risk of QT interval prolongation, there is however an assumption. This is, that the pharmacology of the channel encoded by the Caucasian WT sequence is the same as that of genetic variants of the channel that may exist in clinical trial participants and will exist in large, post-marketing patient populations. Of particular concern is the possibility that a drug could be significantly more potent at hERG in a patient expressing a genetic variant of the WT channel, thus potentially posing an unexpectedly high risk of QT interval prolongation. The primary purpose of this study was therefore to determine whether the pharmacology of the channel was different when it was encoded by nine single nucleotide polymorphisms (SNPs) of hERG. The SNPs in this study are found in individuals with normal QT intervals in the absence of hERG-blocking drugs (i.e., they are not SNPs leading to the congenital form of long QT), although some of them have been linked with drug-induced long QT syndrome (Swan et al., 1999; Iwasa et al., 2000; Splawski et al., 2000; Larsen et al., 2001; Jongbloed et al., 2002; Yang et al., 2002; Ackerman et al., 2003; Paulussen et al., 2004; Aydin et al., 2005; Crotti et al., 2005; Mank-Seymour et al., 2006). One of the ‘SNPs’ studied is a three amino acid in-frame deletion (Delta187–189).
As part of this work, we also characterized the basic electrophysiological properties of the hERG SNP-encoded channels as any significant differences compared with WT would represent important information about the physiological consequences of a given SNP in the general population. This was also done in case any pharmacological differences were, in fact, indirect effects resulting from an alteration in, for example, channel inactivation (Zhang et al., 1999). Some of the data have already appeared in preliminary form (Männikköet al., 2006).
The data suggest that compound potencies defined using the Caucasian WT sequence, at least in this in vitro system, are representative of potencies for these relatively common SNPs.
Methods
Choice of compounds to determine pharmacology
To avoid generating data based on compounds with very similar structural and physiochemical properties, a panel of 48 compounds was selected in a way that ensured as much diversity as possible.
The starting point was the list of compounds reported by Redfern et al. (2003) to be hERG-encoded channel blockers from all the categories of torsadogenic risk. The remainder were chosen either because they are reported as having unusual mechanisms of action [fluvoxamine (Milnes et al., 2003) and d-norpropoxyphene (Ulens et al., 1999)] or, in the case of moxifloxacin, because it is frequently used as a positive control in the so-called ‘Thorough QT/QTc study’ in man.
We checked that the 48 compounds selected were diverse in terms of both their physicochemical properties and their structure. The former was determined by projecting the compounds into a drug-like chemical space representing molecular diversity, referred to as chemical global positioning system, and checking that their positioning was distributed in different areas of this reference system (Oprea and Gottfries, 2001). The drug-space map co-ordinates are scores, extracted from a principal component analysis (PCA) model of chemical properties describing size, lipophilicity, polarisability, charge, flexibility, rigidity and hydrogen bond capacity, and represent a diverse range of molecular structures for monocarboxylates, heteroaromatic compounds and α-amino acids. PCA enables the relationships between observations and variables to be uncovered and was used in this analysis to examine the relationship between the test compounds and their molecular properties. The principal components can be displayed graphically to uncover trends or groupings in either the observations (by plotting the principal component scores) or the variables (by plotting the principal component loadings). By plotting the scores of the first three principal components (Figure 1), it was possible to assess the chemical diversity of the test compounds with respect to the reference system. In this plot, the test compounds are scattered across the reference system, thus assuring good physicochemical diversity.’
Figure 1.
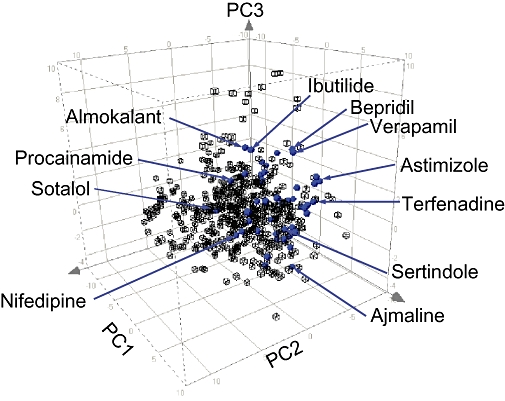
Physicochemical diversity of test compounds based on chemical global positioning system (ChemGPS) prediction. The plot shows the projected positions of the 48 test compounds (blue) relative to those of a reference set of 525 compounds (black) representing drug-like space using principal component analysis. The axes denote the scores of the first three principal components (PCs) from a model representing molecular parameters as size, polarity and flexibility as described by Oprea and Gottfries (2001). The three-dimensional plot illustrates that the 48 test compounds are distributed throughout the drug-like space and occupy the central area of the ChemGPS map. It was not desirable that any of the compounds should be positioned at the boundaries of this co-ordinate system, as this region was defined by molecules that contain drug-like fragments and extreme values of one or more of the chemical properties.
Structural diversity was assessed using Daylight's clustering package (Daylight Chemical Information Systems Inc., Aliso Veijo, CA, USA). This method encodes molecular structures into simple, unique linear strings that represent information about atoms, bonds, aromaticity, charge, stereochemistry and isotopic substitution (Weininger, 1988; Weininger and Weininger, 1989). Cluster analysis (using the Jarvis–Patrick method) was then performed on the string representations of the 48 molecules to determine any common structural denominators and any molecules similar to them. This analysis showed that all 48 molecules were structurally distinct. The measure of structural similarity was defined by the Tanimoto coefficient using a value <0.7 as the threshold for determining molecules as being structurally similar to one another.
Selection of SNPs to study
Published and AstraZeneca re-sequencing of KCNH2 (the gene encoding the hERG protein) identified numerous non-synonymous SNPs but many were only seen in single individuals. The SNPs selected for this study (Table 1) were detected in more than one individual, thus representing true polymorphisms within the general populations and not ‘singletons’ unique to an individual or family. In this respect, SNPs were selected based on an amalgamation of information from a number of sources:
Table 1.
Selection of SNPs showing:their allele frequencies in different ethnic populations, reference to the polymorphism report and comment on possible associations with cardiac adverse events
| SNP | Allele frequency (%) | Population | Reference | Location | Comments |
|---|---|---|---|---|---|
| R176W | <1 | White American | Ackerman et al. (2003) N-terminus | N-terminus | Also found in internal screening of 9 terodiline induced TdP cases (Ford et al. (2000). Found in LQTS patients (Laitinen et al., 2000) |
| R181Q | <1 | Black American | Ackerman et al. (2003) | N-terminus | |
| Del187-189 | <1 | Black American | Ackerman et al. (2003) N-terminus P347S | N-terminus | |
| P347S | <1 2 <1 | White American Caucasian Caucasian | Ackerman et al. (2003),Splawski et al. (2000) N-terminus | N-terminus | Identified in subject with cisapride/clarithromycin induced QT prolongation (Paulussen et al., 2004) |
| K897T | 16.5 4.2 4 3 | White American Black American Asian Hispanic | Ackerman et al. (2003) C-terminus | C-terminus | Associated with increased risk of cardiac mortality (Linna et al. (2006). Proposed to modify clinical expression of A1116V SNP (Crotti et al., 2005) |
| A915V | 2.2 | Asian | Ackerman et al. (2003) C-terminus P917L | C-terminus | |
| P917L | <1 <1 | White American Caucasian | Ackerman et al. (2003) C-terminus | C-terminus | Identified in LQTS population (Splawski et al., 2000) |
| R1047L | 3 1.8 <1 | Caucasian White American Black American | Ackerman et al. (2003) AstraZeneca, unpublished observation | C-terminus | Association with Dofetilide induced TdP described (Sun et al., 2004) |
| A1116V | <1 | Caucasian | AstraZeneca, unpublished observation | C-terminus | Also identified in case study of cardiac arrest caused by ventricular fibrillation (Crotti et al., 2005) |
LQTS, long QT syndrome; SNP, single nucleotide polymorphism; TdP, Torsades de Pointes; WT, wild-type.
AstraZeneca re-sequencing work to determine population KCNH2 variability using commercially available or appropriately consented European Caucasians (n = 130), African American (n = 20) and Japanese (n = 20) subject samples;
Literature reports;
Case reports of associations with QT prolongation or arrhythmia;
LQT databases (http://www.ssi.dk/graphics/html/lqtsdb/lqtsdb.htm, http://www.fsm.it/cardmoc/).
In order to validate the ability of the assay system to detect potency differences, a cell line expressing a channel mutated at Y652A was also produced. The mutation, which does not occur naturally, lies within the putative drug-binding site and has been shown to significantly reduce the IC50 of hERG blockers (Witchel et al., 2004).
Generation of SNP/WT-expressing cell lines
The hERG gene was cloned into the pTight expression vector (Clontech, Mountain View, CA, USA) to keep expression of the channel under tetracycline control. The mutations were introduced into the gene by standard polymerase chain reaction-based site-directed mutagenesis techniques (QuickChange, Stratagene, La Jolla, CA, USA) and confirmed by sequencing the entire gene in both directions.
Plasmid pTight-hERG was linearized by digestion with PvuI and ethanol-precipitated prior to transfection into Tet-On CHO K1 cells together with a linear hygromycin selection marker (Clontech) using the lipofectamine method (Invitrogen, Carlsbad, CA, USA). Cells were then incubated in presence of 0.6 mg·mL−1 hygromycin until cells in untransfected control flask had died (2–3 weeks). Cells were then incubated in the presence of 0.8 µg·mL−1 doxycyclin for 24–48 h to induce channel expression, which was confirmed electrophysiologically using IW (see below). Clonal cell lines were then created by dilution cloning and selected by measuring functional expression, again using IonWorks(tm) HT.
Pharmacological comparison
So that the pharmacological profile of each hERG SNP was based on a direct assessment of channel function, whole-cell voltage clamp electrophysiology was used. However, in order to support the volume of testing required, we used the IW automated, plate-based electrophysiology device (Schroeder et al., 2003) according to the method described by Bridgland-Taylor et al. (2006). In brief, for each experimental ‘Run’ of IonWorks™ HT, the device made perforated whole-cell recordings at ∼21°C, usually from more than 250 of the 384 wells in a PatchPlate™. The extracellular solution was Dulbecco's phosphate-buffered saline (PBS; Invitrogen), which contained (in mM): NaCl 137, KCl 2.7, Na2HPO4 8, KH2PO4 1.5, and to which was added 0.9 mM CaCl2 and 0.5 mM MgCl2. The ‘pipette’ solution was (in mM): KCl 140, EGTA 1, MgCl2 1 and HEPES 20 (pH 7.25–7.30 using 10 M KOH) plus 100 µg·mL−1 amphotericin B (Sigma-Aldrich, St Louis, MO, USA). After attainment of the whole-cell configuration, a pre-compound hERG current was evoked in each cell in the presence of PBS by the following voltage protocol: a 20 s period holding at −70 mV, a 160 ms step to −60 mV (to obtain an estimate of leak), a 100 ms step back to −70 mV, a 1 s step to +40 mV, a 2 s step to −30 mV and finally a 500 ms step to −70 mV. Test compounds, vehicle or 10 µM cisapride controls were then added to each well and after ∼3 min the voltage pulse was re-applied to generate a post-compound hERG current. In between the pre- and post-compound voltage pulses, there was no clamping of the membrane potential.
Leak subtraction was then applied to each current response based on a modified P/N subtraction method described by Bezanilla and Armstrong (1977). Pre- and post-scan hERG current magnitude was measured automatically from the leak subtracted traces by the IonWorks™ HT software by taking a 40 ms average of the current during the initial holding period at −70 mV (baseline current) and subtracting this from the peak of the tail current response. The acceptance criteria for the currents evoked in each well were: pre-scan seal resistance >60 MΩ, pre-scan hERG tail current amplitude >150 pA; post-scan seal resistance >60 MΩ. The degree of inhibition of the hERG current was assessed by dividing the post-scan hERG current by the respective pre-scan hERG current for each well.
Experimental design
Pharmacological evaluation was divided into two phases.
Phase 1
This phase was designed to allow a relatively rapid comparison of hERG-WT versus hERG-SNP pharmacology. It consisted of the following experimental design on each experimental day:
Runs 1 and 2: Test compounds 1 to 10 against WT
Runs 3 and 4: Test compounds 1 to 10 against SNP1
Runs 5 and 6: Test compounds 1 to 10 against SNP2
Runs 7 and 8: Test compounds 1 to 10 against SNP3
Using this design, the potency estimate for each of 10 compounds versus the WT channel could be compared with those obtained versus each of the three SNPs tested on that day. The data from the two runs was pooled for each clone. This design was then repeated for compounds 11 to 20, 21 to 30 etc., until all 48 compounds had been tested against SNPs1 to 3. The same approach was then used for SNPs 4 to 6 and SNPs 7 to 9, ultimately giving 432 potency comparisons (9 SNP vs. WT pairs for 48 compounds). The variability around the potency (IC50) estimate obtained for each compound SNP/WT combination was quantified as a lower and upper 95% confidence interval (see below). At the end of phase 1, any compound-SNP combinations where the relevant confidence interval for the SNP did not overlap with that for the WT were selected for re-testing in phase 2.
Phase 2
Great care was taken in phase 1 to ensure that sources of variability were minimized, for example, fresh cells were prepared immediately before the start of each run. We could not, however, completely rule out the time-dependent element of phase 1. Therefore, in phase 2, we sought to determine whether the apparent differences seen in the first phase comparisons were also observed using a within PatchPlate™ design. In this phase, a single run of IW was performed for each compound with an apparent potency difference in phase 1, with one half of the PatchPlate™ containing WT cells and the other the relevant SNP cells.
Preparation of compound plates
Phase 1
IW was set up such that each well of a 96-well plate containing the test compounds mapped to four wells of the 384-well PatchPlate™ in which the recordings were made. Each 96-well plate was made up of PBS containing 8 half log10-spaced concentrations of each test compound at threefold their final concentration; there were 10 different test compounds. This left 16 wells, eight of which contained 1% dimethyl sulphoxide (DMSO) and eight that contained 30 µM cisapride. Each well of a PatchPlate™ initially contained 3 µL of PBS and 3 µL of PBS containing a cell suspension of a single cell line at a concentration of 250 000 cells·mL−1. When 3 µL of test compound from the compound plate was added to each well of a PatchPlate™, this meant that the cells were exposed to the final compound test concentration, 0.33% DMSO or 10 µM cisapride, which gave full block of WT and SNP hERG currents.
Phase 2
This involved exactly the same procedure except that the compound test plate was made up of two identical halves. Each half was made up of PBS containing eight half log10-spaced concentrations of a single test compound at threefold its final concentration and this was repeated four times. For the remaining 16 wells of the half plate, eight contained 1% DMSO and eight contained 30 µM cisapride. Each well of a PatchPlate™ contained 3 µL of PBS and, for phase 2, one half of the PatchPlate™ had 3 µL of PBS containing a cell suspension of a hERG-SNP cell line and the other half a cell suspension of the WT-hERG cell line. Both cell suspensions were at a concentration of 250 000 cells·mL−1. Finally, when 3 µL of compound was added to each well of a PatchPlate™, this gave the final compound test concentration, 0.33% DMSO or 10 µM cisapride.
Data analysis and quantification
For each IW run, the hERG tail current amplitude in each cell in the presence of PBS was compared with that in the presence of test compound for the same cell. All the data were then scaled by defining the effect of 0.33% DMSO as 0% inhibition and the effect of the supramaximal blocking concentration of cisapride as 100% inhibition. As the contents of each well of the test plate were added to four wells of the PatchPlate™, there could be percentage inhibition data from between 0 and 4 cells for each test concentration. However, all the cell lines used in this study yielded successful recordings (tail current >150 pA) in >56% of the 384 wells available per PatchPlate™. Thus, with this success rate, the chances were that each concentration was tested, on average, in 2–3 cells. For phase 1, as two runs were done for each SNP-compound combination, the eventual data set for each compound was an 8-point, non-cumulative concentration-effect curve made up of, on average, data from 4–6 cells at each concentration. In phase 2, as only one compound was tested in a PatchPlate™ containing WT and a SNP cell line, each point of the non-cumulative curve came from 8–12 cells. In phase 2, the WT and SNP cell lines each had their own 0.33% DMSO and 10 µM cisapride data to define 0% and 100% inhibition respectively.
The pharmacological data were fitted with a custom-written Origin package to replicate the data using non-linear regression by means of a four-parameter logistic (curve top, slope and IC50 variable, curve bottom normalized to 0). The 95% confidence interval around the IC50 value was chosen to compare potencies between WT and SNP channels.
Electrophysiological comparison
Although IW is ideal for large-scale pharmacology studies, it is not an optimal method to assess detailed electrophysiological parameters. For this part of the study, conventional whole-cell electrophysiology was used. This has also already been described in depth by Bridgland-Taylor et al. (2006). Briefly, glass coverslips seeded with the same cells as used for the pharmacological assessments were placed at the bottom of a Perspex chamber containing bath solution at room temperature (∼21°C). The bath solution contained (in mM): NaCl 137, KCl 4, MgCl2 1, CaCl2 1.8, HEPES 10 and glucose 10 (pH 7.4 with 1 M NaOH). This chamber was fixed to the stage of an inverted, phase-contrast microscope and conventional ‘giga-seal’ whole-cell patch clamp recordings were made with pipettes made from borosilicate glass tubing (GC120F, Harvard Apparatus, Edenbridge, UK) using a P-87 micropipette puller (Sutter Instrument Co., Navato, CA, USA). The pipette solution was (in mM): KCl 130, MgCl2 1, HEPES 10, Na2ATP 5, EGTA 5 (pH 7.2 with 1 M KOH). The pipette was connected to the headstage of the patch clamp amplifier (Axopatch 200B, Axon Instruments, Foster City, CA, USA) via a silver/silver chloride wire. The headstage ground was connected to an earth electrode consisting of a silver/silver chloride pellet embedded in 3% agar made up with 0.9% NaCl. Pipette resistance was typically 1.5–4 MΩ. Following ‘break-in’ and appropriate adjustment of series resistance and capacitance controls, Clampex software (Axon Instruments) was used to set a holding potential and to deliver voltage protocols required to characterize the electrophysiological parameters of each hERG variant. The resulting current responses were analysed using Clampfit (Axon Instruments). All the current traces used to determine current-voltage (I–V) relationships were leak compensated offline. Series resistance compensation of ≥70% was applied. Currents that resulted in series resistance errors greater than 5 mV were discarded. The junction-potential error measured using the flowing 3 M KCl electrode method was found to be 4 mV. This small error was not corrected for. The holding voltage for every protocol was −80 mV.
It is well established that in terms of a simple model, the hERG-encoded channel can exist in
three distinct functional states [closed (C), open (O) and inactivated (I)] and cycles through these states during the cardiac action potential. Specifically, as shown next, it undergoes activation (a), inactivation (b), recovery from inactivation (c) and deactivation (d) during each cardiac cycle. We applied the relevant voltage protocols to measure the steady-state voltage dependence and the rate or rates of transitions at different voltages to look for any SNP-dependent differences.
Voltage dependence of activation was estimated using the activation I-V protocol as shown in the inset of Figure 4A. Membrane potential (Vm) was stepped to test voltages ranging from −60 mV to +50 mV for 4 s followed by a 4 s step to −50 mV to evoke a tail current. The peak of this tail current was plotted against test voltage and the data were fitted to a Boltzmann function to obtain voltage values where half of the channels are activated (V1/2) and for steepness of the curve (Vslope).
Figure 4.
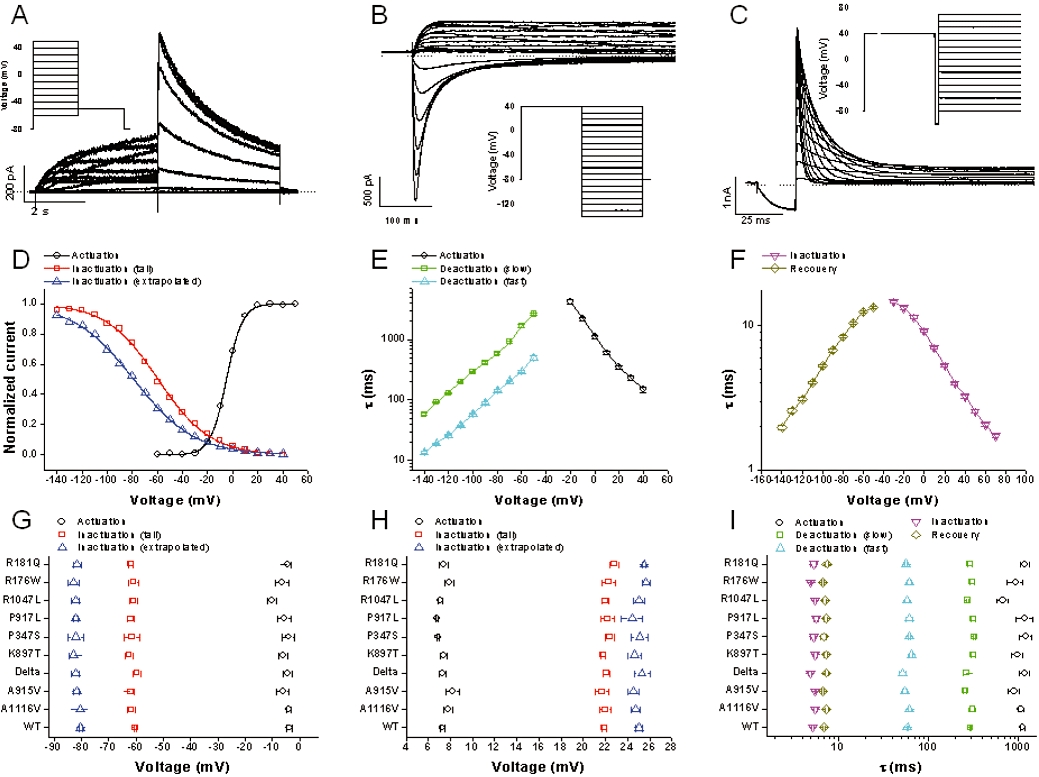
Electrophysiological properties. All data are from conventional whole cell patch clamp recordings. The top row shows typical wild-type (WT) current responses to the voltage protocols shown as insets. (A) Activation I–V protocol. (B) Fully activated I–V protocol. (C) Inactivation time constant protocol. Note that for B and C, currents are only shown for the second half of the voltage protocol. Analysis of the current responses to each protocol enabled electrophysiological parameters for WT and single nucleotide polymorphism (SNP) channels to be defined, as described in the Methods. The middle row shows electrophysiological parameters for WT human ether-ago-go-related gene. (D) Typical voltage-dependence of activation (○) and inactivation [peak (□) and extrapolated ( )] curves. (E) Mean voltage-dependence of the rate of activation (○), and slow (□) and fast (
)] curves. (E) Mean voltage-dependence of the rate of activation (○), and slow (□) and fast ( ) deactivation. (F) Mean voltage-dependence of the rate of inactivation (□) and recovery from inactivation (○). The bottom row compares mean ± standard error of the mean WT data with that for the SNPs, for all parameters. (G) V1/2 of activation (○) and inactivation [peak (□) and extrapolated (
) deactivation. (F) Mean voltage-dependence of the rate of inactivation (□) and recovery from inactivation (○). The bottom row compares mean ± standard error of the mean WT data with that for the SNPs, for all parameters. (G) V1/2 of activation (○) and inactivation [peak (□) and extrapolated ( )]. (H) Vslope of activation (○) and inactivation [peak (□) and extrapolated (
)]. (H) Vslope of activation (○) and inactivation [peak (□) and extrapolated ( )]. (I) Time constant of activation (○) and inactivation (
)]. (I) Time constant of activation (○) and inactivation ( ) at +10 mV, slow (□) and fast (
) at +10 mV, slow (□) and fast ( ) deactivation and recovery from inactivation (⋄) at −100 mV. The numerical values for all parameters are shown in Table 2.
) deactivation and recovery from inactivation (⋄) at −100 mV. The numerical values for all parameters are shown in Table 2.
Where Imin is the minimum current, Imax the maximum current and Vm the test voltage.
Voltage dependence of inactivation was estimated using the fully activated I–V protocol (inset in Figure 4B) where the channels are first fully activated and inactivated by holding Vm for 4 s at +40 mV before a 4 s step to test voltages ranging from −140 mV to +70 mV. The peak current at the test voltage gives an estimate of how many channels recover from inactivation at a given voltage. However, part of the recovered current is masked by closure of the channels, especially at the most negative voltages. This effect was adjusted for by fitting a two-exponential curve to the decaying phase of the current at the test voltage and extrapolating the current to the beginning of the pulse to calculate the extrapolated peak current (Smith et al., 1996; Vandenberg et al., 2006). The measured and extrapolated current amplitudes were then divided by the effective voltage (Vm–Vreversal) to obtain two conductance-voltage relationships. A Boltzmann curve was then fitted to these data to derive values for V1/2 and Vslope for inactivation.
The time constant of activation (τAct) was estimated by fitting a single exponential curve to the last 75% of the rising phase of the 4 s step of activation I–V protocol to avoid the effect of the delayed sigmoidal activation time-course on estimation of the time constant. Although other processes, such as inactivation, affect the time course of current development during the activation step, this protocol was chosen considering the volume of experimentation that would have been required to determine this parameter using a more sophisticated method. A small number of experiments suggested, however, that the activation time constants derived by this method were similar to those derived using an envelope of tails protocol (not shown). In addition, as the voltage dependence and time constant of inactivation was not found to be significantly different between WT and SNPs (see Results), the rate of activation should at least be comparable even if not precise in absolute terms.
The slow and fast time constants of channel deactivation (τDeactFast and τDeactSlow) were derived from the two exponential fit of the decaying phase of the current at the test voltage after full activation using the fully activated I–V protocol. The time constant of the recovery (τRec) from inactivation was also derived from the fully activated I–V protocol by fitting a single exponential curve on the rising phase of the current at the test voltage.
The inactivation time constant (τInact) was measured using a protocol where the channels were first fully activated and inactivated with a step to +40 mV (inset in Figure 4C). Vm was then stepped back briefly (for 20 ms) to −100 mV to remove channel inactivation without allowing the channel to close. Vm was then clamped at values ranging from −70 mV to +70 mV. The decaying phase of the current at the test voltage is well fitted by a single exponential curve and describes the time constant of inactivation.
Statistical analysis
Potency (IC50) data are expressed as mean; lower 95% confidence interval; upper 95% confidence interval. Other data are shown as mean ± standard error of the mean.
Activation V1/2 and Vslope were compared across the 10 variants using a one-way analysis of variance (anova), followed by pair-wise comparisons of each of the genetic variants versus the WT group using Dunnett's method (to adjust P values for the fact that this involves nine comparisons, and hence an increased chance of false positives).
For each assessment of time constant versus voltage relationships, a best straight line was fitted using log10 (time constant) as the response and voltage as the explanatory variable. This gave an estimate of the slope and an estimate of the intercept. The fitting was done for deactivation and recovery in the voltage range was from −140 mV to −60 mV, for activation over the range from −10 mV to +40 mV and for inactivation at voltages between −20 mV and +70 mV. For a given set of time constants, for example, τDeactslow, all of the slopes were compared across the 10 types/variants using a one-way anova, followed by pair-wise comparisons of each of the genetic variants versus the WT group using Dunnett's method. If the slopes do not vary significantly, it follows that the lines are parallel, that is, a constant separation. In this case, it is appropriate to test if this separation is significantly different from zero or not. As with the slopes, this is achieved using one-way anova, followed by pair-wise comparisons back to the WT group using Dunnett's method.
Materials
Drugs were either synthesized by and/or on behalf of AstraZeneca (Macclesfield, UK) or sourced from either Sigma-Aldrich or Apin Chemicals (Abingdon, UK) and dissolved in DMSO at a concentration 300-fold greater than the top test concentration. Compounds were diluted to the final test concentrations as previously explained.
Nomenclature
The ion channel nomenclature stipulated by Alexander et al. (2008) was used throughout.
Results
Pharmacological comparison
Validation of the test system to detect potency differences
Work of this type has usually been carried out using conventional electrophysiology techniques that involve fixing Vm to specific, experimenter-defined values throughout a recording. This contrasts with the automated electrophysiology performed by IonWorks™ HT, where voltage clamping only occurs just prior to and during both the pre- and post-compound assessment phase of the protocol. Thus, despite the fact that IW hERG potency values have been shown to correlate well with those measured conventionally (Kiss et al., 2003; Bridgland-Taylor et al., 2006), we sought to confirm that potency differences based on changes in hERG channel structure could be detected using our approach. A cell line was therefore established that expressed hERG channels containing a single amino acid change at a position known to be important for drug binding (Sanguinetti and Tristani-Firouzi, 2006). Specifically, a cell line expressing channels where the tyrosine at position 652 of the WT had been changed to alanine (Y652A), a modification that has been reported to decrease the potency of propafenone sixfold (Witchel et al., 2004). As shown in Figure 2, using the phase 2 experimental design, propafenone was 5.9-fold less potent in cells expressing the Y652A mutant (IC50 3.84 µM; 3.46, 4.26 µM) compared with WT (IC50 0.65 µM; 0.59, 0.75 µM). These data suggested that the IW methodology would be able to detect pharmacological differences should they occur.
Figure 2.
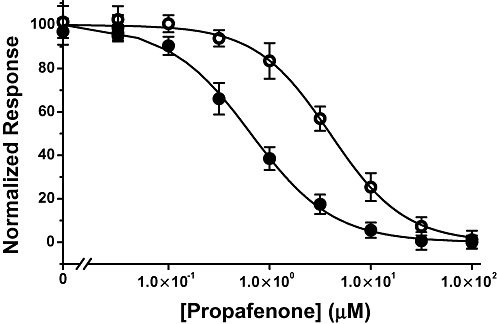
Effect of Y652A mutation on propafenone potency. Concentration-effect curves to propafenone against WT (•) or Y652A mutant (○) measured using IonWorks™ HT.
Phase 1. Having established that any differences could be detected using IW™ HT, we then progressed to the initial phase of the study that involved a relatively rapid assessment of potential SNP-based differences in pharmacology. Figure 3 shows typical concentration-effect data from phase 1 for six compounds with non-overlapping 95% confidence limits around the IC50 value. The whole dataset are summarized in detail in Table 2 and show that there are no very large IC50 differences, the biggest being 0.34 log units in phase 1: a 2.2-fold increase in sotalol potency for R176W compared with WT. There were, however, 77 instances out of the 432 comparisons where the 95% confidence limits around the IC50 for a SNP did not overlap with those of the WT; for 33 of these the compound was less potent as an inhibitor of the hERG SNP than the WT and for 44 instances the reverse was true. In order to rationally select comparisons for detailed assessment in phase 2, we selected these 77 comparisons where the 95% confidence intervals did not overlap.
Figure 3.
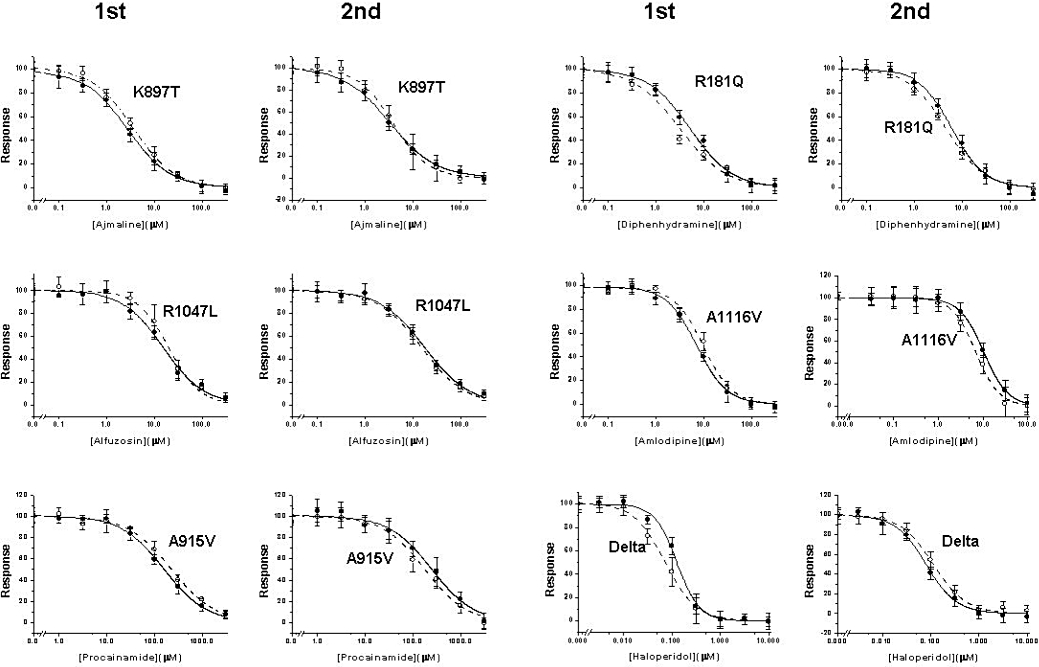
Typical concentration-effect curve data. The phase 1 and phase 2 data are shown for wild-type (WT) (•) or single nucleotide polymorphism (○) pairs, for six representative compounds. On three occasions (left panel), the difference detected in phase 1 was not evident in phase 2. On the right panel, diphenhydramine is more potent at R181Q than on WT when tested in phase 1 or 2 formats. In contrast, for amlodipine versus WT/A1116V and haloperidol versus WT/Delta187-189, the potency difference seen in phase 1 is reversed in phase 2.
Table 2.
Summary of WT versus SNP pharmacology
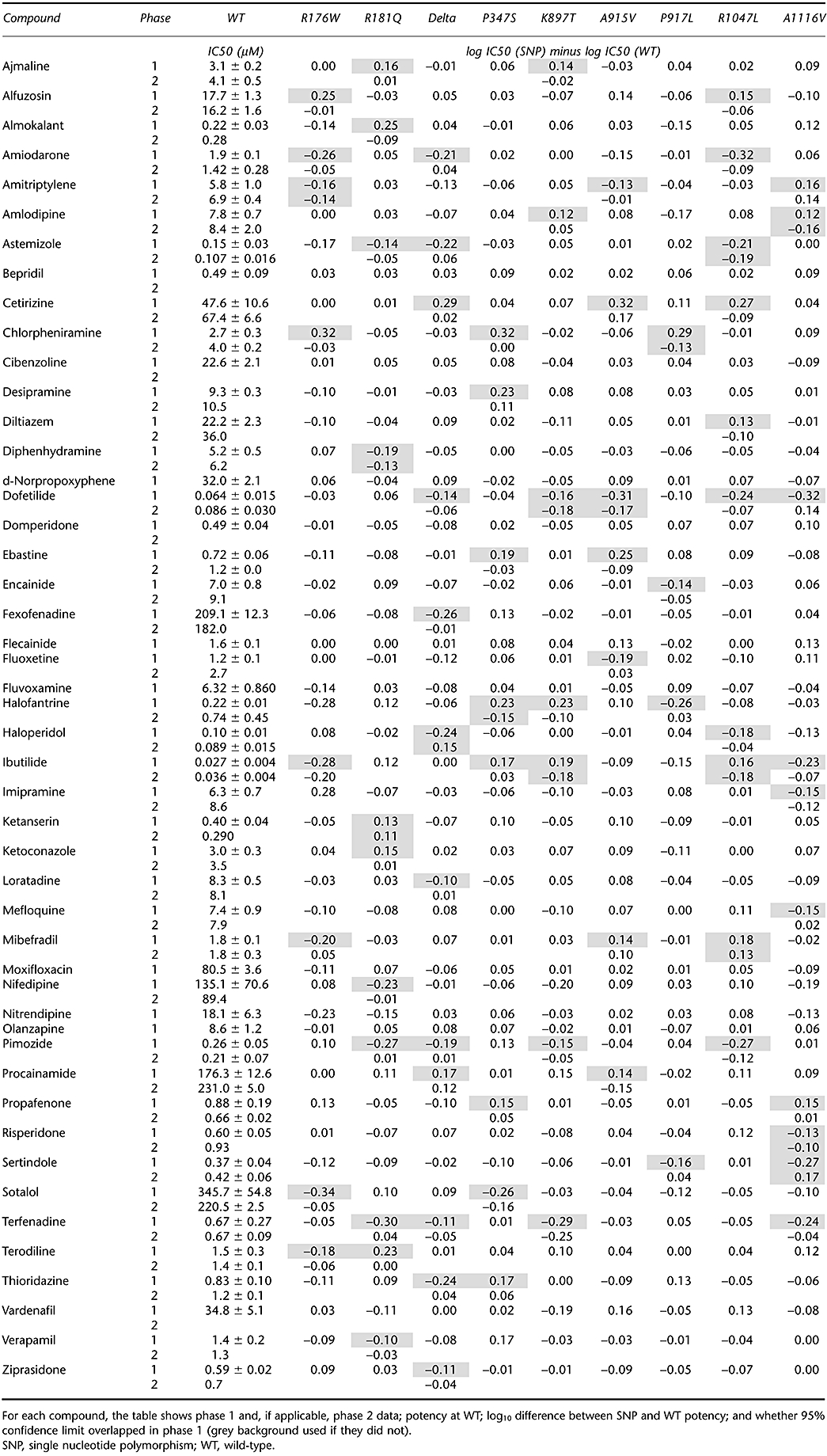 |
Phase 2. Even though there were 77 instances in phase 1 where the IC50 95% confidence limits did not overlap, the differences were small and some/all cases could have been the result of chance, particularly given the number of comparisons made. In addition, despite our best efforts, some differences could also have resulted from small, time-dependent variability in the assay. To mitigate possible time-dependent variations in IC50 comparisons, a within-PatchPlate™ comparison was made in phase 2 for all 77 examples. In this format, half of the cells on the PatchPlate™ expressed a SNP cell line and other half WT cells. Concentration-effect data from phase 2 are shown in Figure 3, next to phase 1 data for six compound-SNP pairs. Of the 77 differences seen in phase 1, 62 were not evident using this design. Although 15 examples remained where IC50 95% confidence intervals did not overlap, only eight of these showed potency ‘differences’ in the same direction as seen in phase 1; for the remaining seven, the differences were reversed. The data are summarized in Table 2.
Electrophysiological comparison
An extensive assessment of the electrophysiological parameters for WT and SNPs is summarized in Figure 4 and Table 3. However, the only statistically significant difference was in the voltage dependence of activation for R1047L (P < 0.01).
Table 3.
Summary of WT versus SNP electrophysiology
|
Activation |
Deactivation @−100mV |
Inactivation |
Vslope peak(mV) | V1/2 extrap(mV) | Vslope extrap(mV) |
Recovery |
Inactivation |
|||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| V1/2(mV) | Vslope(mV) | τ @ +10 mV(ms) | n | τFast(ms) | τSlow(ms) | V1/2 peak(mV) | τ @−100mV(ms) | n | τ @+10 mV(ms) | n | ||||
| WT | −3.8 ± 1.0 | 7.3 ± 0.2 | 1102 ± 65 | 49 | 58.8 ± 1.4 | 293 ± 8 | −60.1 ± 0.5 | 21.9 ± 0.2 | −80.3 ± 0.7 | 25.0 ± 0.3 | 5.2 ± 0.1 | 39 | 7.0 ± 0.2 | 32 |
| R176W | −6.5 ± 2.4 | 7.9 ± 0.4 | 933 ± 187 | 9 | 61.2 ± 3.4 | 306 ± 18 | −61.0 ± 1.7 | 22.3 ± 0.6 | −82.8 ± 2.0 | 25.7 ± 0.4 | 5.0 ± 0.1 | 7 | 6.8 ± 0.2 | 9 |
| R181Q | −4.3 ± 1.3 | 7.4 ± 0.4 | 1183 ± 143 | 14 | 56.9 ± 1.a7 | 290 ± 15 | −62.0 ± 0.9 | 22.8 ± 0.4 | −81.5 ± 1.4 | 25.5 ± 0.2 | 5.3 ± 0.4 | 8 | 7.5 ± 0.4 | 8 |
| Delta | −4.5 ± 1.6 | 7.3 ± 0.3 | 1173 ± 158 | 14 | 51.7 ± 3.9 | 267 ± 23 | −59.7 ± 1.4 | 22.1 ± 0.3 | −82.1 ± 1.4 | 25.3 ± 0.7 | 4.9 ± 0.2 | 5 | 7.4 ± 0.3 | 6 |
| P347S | −4.1 ± 2.3 | 6.9 ± 0.1 | 1205 ± 194 | 6 | 61.0 ± 2.2 | 323 ± 7 | −61.5 ± 3.0 | 22.4 ± 0.4 | −82.0 ± 2.9 | 25.1 ± 0.8 | 5.3 ± 0.3 | 6 | 6.9 ± 0.5 | 5 |
| K897T | −6.0 ± 1.6 | 7.4 ± 0.3 | 974 ± 141 | 11 | 65.1 ± 2.7 | 315 ± 16 | −62.6 ± 1.4 | 21.8 ± 0.2 | −82.7 ± 1.7 | 24.6 ± 0.6 | 5.6 ± 0.2 | 7 | 7.3 ± 0.3 | 10 |
| A915V | −6.1 ± 2.3 | 8.2 ± 0.6 | 892 ± 133 | 11 | 54.8 ± 2.9 | 257 ± 11 | −61.9 ± 1.2 | 21.7 ± 0.6 | −81.8 ± 0.9 | 24.5 ± 0.5 | 5.6 ± 0.1 | 8 | 6.7 ± 0.2 | 11 |
| P917L | −5.6 ± 2.3 | 6.8 ± 0.1 | 1179 ± 242 | 6 | 61.7 ± 5.9 | 317 ± 19 | −61.8 ± 1.3 | 22.2 ± 0.6 | −82.1 ± 0.7 | 24.4 ± 1.0 | 5.7 ± 0.3 | 7 | 7.3 ± 0.3 | 7 |
| R1047L | −10.2 ± 1.5* | 7.1 ± 0.2 | 671 ± 99 | 14 | 58.1 ± 3.0 | 272 ± 8 | −61.0 ± 1.4 | 22.0 ± 0.3 | −82.2 ± 1.3 | 25.0 ± 0.5 | 5.5 ± 0.2 | 10 | 7.1 ± 0.3 | 13 |
| A1116V | −3.9 ± 0.8 | 7.8 ± 0.4 | 1064 ± 75 | 15 | 62.0 ± 3.1 | 310 ± 28 | −61.7 ± 1.5 | 22.0 ± 0.5 | −80.4 ± 1.7 | 24.7 ± 0.4 | 5.6 ± 0.2 | 6 | 7.4 ± 0.3 | 10 |
P < 0.01.
For WT and each SNP, the table shows the mean ± standard error of the mean of the parameters that quantify basic channel electrophysiology.
SNP, single nucleotide polymorphism; WT, wild-type.
Discussion and conclusions
This is the first study to systematically and extensively explore the pharmacology of hERG SNPs and also their electrophysiological properties. The main conclusion is that the in vitro pharmacology of the hERG-encoded channel is not significantly affected by any of the nine common SNPs investigated in the IW test system. This confirms existing observations about single SNP-compound combinations. Thus, the potency of dofetilide was unaffected by the R1047L polymorphism (Sun et al., 2004); activity of prucalopride unchanged by K897T (Chapman and Pasternack, 2007); cisapride potency unaffected by four different SNPs, including K897T and R1047L (Anson et al., 2004); cisapride, clarithromycin, sulfamethoxazole and trimethoprim activity unchanged by P347S (Saenen et al., 2007).
It could be argued that this is not surprising given that the amino acid change for each SNP is not located in or near the putative drug binding site for low molecular weight compounds (Mitcheson and Perry, 2003). However, there are data suggesting that amino acid changes remote from drug binding sites can affect potassium channel pharmacology. For example, an SNP (Q9E) in MiRP1, a putative β sub-unit of the native channel complex carrying IKR, was reported to increase the potency of clarithromycin as a hERG channel blocker (Abbott et al., 1999) and a similar observation was made by Sesti et al. (2000) for the T8A SNP of MiRP1 using sulfamethoxazole. In addition, the P532L or R578K SNPs in the C-terminal region of the KV1.5 α sub-unit result in a significant reduction of both quinidine and propafenone potency (Simard et al., 2005). More broadly, other changes occurring remote from ion channel drug binding sites have been suggested to modify their pharmacology. For example, an association between the hERG-encoded channel and a putative binding partner (KCR1) was reported to decrease the potency of sotalol, quinidine and dofetilide (Kupershmidt et al., 2003) while phosphorylation of the α-sub unit of the IKS channel complex significantly decreased the IC50 of quinidine (Yang et al., 2003). Finally, an SNP without effect on the electrophysiology of NaV1.5 (S524Y) has been shown to affect use-dependent block by quinidine and flecainide (Shuraih et al., 2007). Based on these findings, the considerable evidence of allosteric interactions in the ion channel field and the implications for drug safety of SNP-based pharmacological differences, we felt this was a topic worthy of rigorous examination.
Although it cannot be assumed that our findings will apply to all SNPs of the type studied here, we can be confident in the validity of our conclusions for the nine SNPs investigated. Firstly, we opted to use a functional assay to replicate, as far as possible, physiological channel behaviour. For the volume of testing required, this necessitated the use of an automated method but we confirmed that this approach was able to detect pharmacological differences. Specifically, a binding-site mutant reported to cause a sixfold decrease in the potency of propafenone when measured using conventional electrophysiology (Witchel et al., 2004) was investigated using the same compound with the IW method. We found that in the Y652A mutant, propafenone was 5.9-fold less potent than in WT, the IC50 decreasing from 0.65 µM to 3.84 µM (see Figure 2). The compound test set was designed to ensure we used drugs that were chemically and structurally diverse, ensuring that any conclusions would be likely to be valid across a wide area of drug-like chemical space. Also, the WT IC50 values of hERG block reported in this study are similar to the values reported for these compounds previously (Redfern et al., 2003).
Even though the data suggest no pharmacological differences large enough to evoke concerns about use of the Caucasian WT sequence for screening purposes, do the data support any real potency differences, however small? The fact that only 15 of the 77 ‘differences’ in phase 1 were seen in phase 2, all smaller than 1.5-fold and that seven of these were in the opposite direction, strongly suggests that there was no real SNP-dependent potency variability. In support of this, there was no trend for compounds to be consistently more or less active against a particular SNP.
In the context of drug discovery, our findings strongly suggest that safety margins based on compound potency at the Caucasian WT variant of hERG will be representative of what is likely to happen when a drug is eventually prescribed to large and genetically diverse patient groups. This suggests that the association of some of the nine SNPs with an increased QT/TdP risk in man is not a direct consequence of greater pharmacological sensitivity.
We also found that the basic biophysical properties of the SNP channels were very similar to WT channel. The only significant difference we found was a hyperpolarizing shift in the voltage dependence of activation of R1047L. This would increase the current through hERG channels rather that suppress the repolarizing current. Our findings differ from those published on R1047L where the voltage dependence of activation is unaffected (Anson et al., 2004), or shifted in a depolarizing direction (Sun et al., 2004). Our results also differ from published data in relation to the deactivation rate of R176W (Fodstad et al., 2006), changes in inactivation and recovery kinetics of P347S (Saenen et al., 2007), any of the diverging parameters of K897T (Paavonen et al., 2003; Anson et al., 2004) or the depolarizing shift in voltage dependence of A1116V activation (Crotti et al., 2005). These inconsistencies highlight the difficulty of convincingly determining small differences in electrophysiological properties that can vary depending, perhaps, on differences in experimental design such as the host cell used.
Future work
Our work is based on the pharmacology of heterologously expressed α-sub-units only. We cannot be sure that in the native channel protein complex none of the SNPs would change the pharmacology of IKR. This is a relevant point given that this study focuses on the type 1a hERG transcript, yet there is evidence that the native channels may also be made up of α-sub-units coded for by the N-terminus-truncated (type 1b) transcript (Jones et al., 2004; Sale et al., 2008). The fourfold decrease in E4031 potency and the biophysical differences reported by Sale et al. (2008) comparing hERG 1a/b with hERG 1a alone are particularly interesting in the context of this work. Related to this, we are studying only channels containing four identical α-sub-units and cannot say whether channels comprising, for example, two WT and two SNP sub-units would have an altered pharmacology. The only piece of relevant data suggests this is unlikely to be the case, as in a presumed co-expression of WT with a trafficking mutant (A561P), clobutinol's potency was unaffected (Bellocq et al., 2004).
Finally, although the IW automated system enables the volume of compound testing reported here, it can only operate at room temperature. Bearing in mind the reported effects of temperature on hERG pharmacology (Kirsch et al., 2004) and electrophysiology (Vandenberg et al., 2006), we cannot exclude the possibility that at physiological temperatures, SNP-related pharmacological and/or electrophysiological differences could emerge.
Acknowledgments
We would like to thank Dr Martin Main for his critical evaluation of the manuscript and Dr J Bright for his statistical input.
Glossary
Abbreviations:
- DMSO
dimethyl sulphoxide
- hERG
human ether-ago-go-related gene
- IKR
delayed rectifier potassium current
- IW
IonWorks™ HT
- SNP
single nucleotide polymorphism
- TdP
Torsades de Pointes
- WT
wild-type
Conflict of interest
The authors state no conflict of interest.
References
- Abbott GW, Sesti F, Splawski I, Buck ME, Lehmann MH, Timothy KW, et al. MiRP1 forms IKr potassium channels with hERG and is associated with cardiac arrhythmia. Cell. 1999;97:175–187. doi: 10.1016/s0092-8674(00)80728-x. [DOI] [PubMed] [Google Scholar]
- Ackerman MJ, Tester DJ, Jones GS, Will ML, Burrow CR, Curran ME. Ethnic differences in cardiac potassium channel variants: implications for genetic susceptibility to sudden cardiac death and genetic testing for congenital long QT syndrome. Mayo Clin Proc. 2003;78:1479–1487. doi: 10.4065/78.12.1479. [DOI] [PubMed] [Google Scholar]
- Alexander SP, Mathie A, Peters JA. Guide to receptors and channels (GRAC), 3rd edition. Br J Pharmacol. 2008;153(Suppl 2):S1–S209. doi: 10.1038/sj.bjp.0707746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anon. The clinical evaluation of QT/QTc interval prolongation and proarrhythmic potential for non-antiarrhythmic drugs. 2005a. ICH E14 CHMP/ICH/2/04. [PubMed]
- Anon. The nonclinical evaluation of the potential for delayed ventricular repolarisation (QT interval prolongation) by human pharmaceuticals. 2005b. ICH S7B CHMP/ICH/423/02. [PubMed]
- Anson BD, Ackerman MJ, Tester DJ, Will ML, Delisle BP, Anderson CL, et al. Molecular and functional characterization of common polymorphisms in hERG (KCNH2) potassium channels. Am J Physiol Heart Circ Physiol. 2004;286:H2434–H2441. doi: 10.1152/ajpheart.00891.2003. [DOI] [PubMed] [Google Scholar]
- Aydin A, Bahring S, Dahm S, Guenther UP, Uhlmann R, Busjahn A, et al. Single nucleotide polymorphism map of five long-QT genes. J Mol Med. 2005;83:159–165. doi: 10.1007/s00109-004-0595-3. [DOI] [PubMed] [Google Scholar]
- Bellocq C, Wilders R, Schott JJ, Louerat-Oriou B, Boisseau P, Le Marec H, et al. A common antitussive drug, clobutinol, precipitates the long QT syndrome 2. Mol Pharmacol. 2004;66:1093–1102. doi: 10.1124/mol.104.001065. [DOI] [PubMed] [Google Scholar]
- Bezanilla F, Armstrong CM. Inactivation of the sodium channel. I. Sodium current experiments. J Gen Physiol. 1977;70:549–566. doi: 10.1085/jgp.70.5.549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bridgland-Taylor MH, Hargreaves AC, Easter A, Orme A, Henthorn DC, Ding M, et al. Optimisation and validation of a medium-throughput electrophysiology-based hERG assay using IonWorks HT. J Pharmacol Toxicol Methods. 2006;54:189–199. doi: 10.1016/j.vascn.2006.02.003. [DOI] [PubMed] [Google Scholar]
- Cavero I, Mestre M, Guillon JM, Crumb W. Drugs that prolong QT interval as an unwanted effect: assessing their likelihood of inducing hazardous cardiac dysrhythmias. Expert Opin Pharmacother. 2000;1:947–973. doi: 10.1517/14656566.1.5.947. [DOI] [PubMed] [Google Scholar]
- Chapman H, Pasternack M. The action of the novel gastrointestinal prokinetic prucalopride on the hERG K(+) channel and the common T897 polymorph. Eur J Pharmacol. 2007;554:98–105. doi: 10.1016/j.ejphar.2006.10.019. [DOI] [PubMed] [Google Scholar]
- Crotti L, Lundquist AL, Insolia R, Pedrazzini M, Ferrandi C, De Ferrari GM, et al. KCNH2-K897T is a genetic modifier of latent congenital long-QT syndrome. Circulation. 2005;112:1251–1258. doi: 10.1161/CIRCULATIONAHA.105.549071. [DOI] [PubMed] [Google Scholar]
- De Bruin ML, Pettersson M, Meyboom RH, Hoes AW, Leufkens HG. Anti-hERG activity and the risk of drug-induced arrhythmias and sudden death. Eur Heart J. 2005;26:590–597. doi: 10.1093/eurheartj/ehi092. [DOI] [PubMed] [Google Scholar]
- Fodstad H, Bendahhou S, Rougier JS, Laitinen-Forsblom PJ, Barhanin J, Abriel H, et al. Molecular characterization of two founder mutations causing long QT syndrome and identification of compound heterozygous patients. Ann Med. 2006;38:294–304. doi: 10.1080/07853890600756065. [DOI] [PubMed] [Google Scholar]
- Ford GA, Wood SM, Daly AK. CYP2D6 and CYP2C19 genotypes of patients with terodiline cardiotoxicity identified through the yellow card system. Br J Clin Pharmacol. 2000;50:77–80. doi: 10.1046/j.1365-2125.2000.00230.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwasa H, Itoh T, Nagai R, Nakamura Y, Tanaka T. Twenty single nucleotide polymorphisms (SNPs) and their allelic frequencies in four genes that are responsible for familial long QT syndrome in the Japanese population. J Hum Genet. 2000;45:182–183. doi: 10.1007/s100380050207. [DOI] [PubMed] [Google Scholar]
- Jones EM, Roti Roti EC, Wang J, Delfosse SA, Robertson GA. Cardiac IKr channels minimally comprise hERG 1a and 1b subunits. J Biol Chem. 2004;279:44690–44694. doi: 10.1074/jbc.M408344200. [DOI] [PubMed] [Google Scholar]
- Jongbloed R, Marcelis C, Velter C, Doevendans P, Geraedts J, Smeets H. DHPLC analysis of potassium ion channel genes in congenital long QT syndrome. Hum Mutat. 2002;20:382–391. doi: 10.1002/humu.10131. [DOI] [PubMed] [Google Scholar]
- Kang J, Wang L, Chen XL, Triggle DJ, Rampe D. Interactions of a series of fluoroquinolone antibacterial drugs with the human cardiac K+ channel hERG. Mol Pharmacol. 2001;59:122–126. doi: 10.1124/mol.59.1.122. [DOI] [PubMed] [Google Scholar]
- Kirsch GE, Trepakova ES, Brimecombe JC, Sidach SS, Erickson HD, Kochan MC, et al. Variability in the measurement of hERG potassium channel inhibition: effects of temperature and stimulus pattern. J Pharmacol Toxicol Methods. 2004;50:93–101. doi: 10.1016/j.vascn.2004.06.003. [DOI] [PubMed] [Google Scholar]
- Kiss L, Bennett PB, Uebele VN, Koblan KS, Kane SA, Neagle B, et al. High throughput ion-channel pharmacology: planar-array-based voltage clamp. Assay Drug Dev Technol. 2003;1:127–135. doi: 10.1089/154065803321537845. [DOI] [PubMed] [Google Scholar]
- Kupershmidt S, Yang IC, Hayashi K, Wei J, Chanthaphaychith S, Petersen CI, et al. The IKr drug response is modulated by KCR1 in transfected cardiac and noncardiac cell lines. FASEB J. 2003;17:2263–2265. doi: 10.1096/fj.02-1057fje. [DOI] [PubMed] [Google Scholar]
- Laitinen P, Fodstad H, Piippo K, Swan H, Toivonen L, Viitasalo M, et al. Survey of the coding region of the HERG gene in long QT syndrome reveals six novel mutations and an amino acid polymorphism with possible phenotypic effects. Hum Mutat. 2000;15:580–581. doi: 10.1002/1098-1004(200006)15:6<580::AID-HUMU16>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- Larsen LA, Andersen PS, Kanters J, Svendsen IH, Jacobsen JR, Vuust J, et al. Screening for mutations and polymorphisms in the genes KCNH2 and KCNE2 encoding the cardiac hERG/MiRP1 ion channel: implications for acquired and congenital long Q-T syndrome. Clin Chem. 2001;47:1390–1395. [PubMed] [Google Scholar]
- Linna EH, Perkiomaki JS, Karsikas M, Seppanen T, Savolainen M, Kesaniemi YA, et al. Functional significance of KCNH2 (HERG) K897T polymorphism for cardiac repolarization assessed by analysis of T-wave morphology. Ann Noninvasive Electrocardiol. 2006;11:57–62. doi: 10.1111/j.1542-474X.2006.00083.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mank-Seymour AR, Richmond JL, Wood LS, Reynolds JM, Fan YT, Warnes GR, et al. Association of torsades de pointes with novel and known single nucleotide polymorphisms in long QT syndrome genes. Am Heart J. 2006;152:1116–1122. doi: 10.1016/j.ahj.2006.08.020. [DOI] [PubMed] [Google Scholar]
- Männikkö R, Harmer AR, Overend G, Perrey C, Valentin JP, Morten J, et al. Pharmacological characterisation of 9 single nucleotide polymorphisms (SNPs) of the hERG-encoded K channel. Proc Physiol Soc. 2006;8:PC10. [Google Scholar]
- Milnes JT, Crociani O, Arcangeli A, Hancox JC, Witchel HJ. Blockade of hERG potassium currents by fluvoxamine: incomplete attenuation by S6 mutations at F656 or Y652. Br J Pharmacol. 2003;139:887–898. doi: 10.1038/sj.bjp.0705335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitcheson JS, Perry MD. Molecular determinants of high-affinity drug binding to hERG channels. Curr Opin Drug Discov Devel. 2003;6:667–674. [PubMed] [Google Scholar]
- Oprea TI, Gottfries J. Chemography: the art of navigating in chemical space. J Comb Chem. 2001;3:157–166. doi: 10.1021/cc0000388. [DOI] [PubMed] [Google Scholar]
- Paavonen KJ, Chapman H, Laitinen PJ, Fodstad H, Piippo K, Swan H, et al. Functional characterization of the common amino acid 897 polymorphism of the cardiac potassium channel KCNH2 (hERG) Cardiovasc Res. 2003;59:603–611. doi: 10.1016/s0008-6363(03)00458-9. [DOI] [PubMed] [Google Scholar]
- Paulussen AD, Gilissen RA, Armstrong M, Doevendans PA, Verhasselt P, Smeets HJ, et al. Genetic variations of KCNQ1, KCNH2, SCN5A, KCNE1, and KCNE2 in drug-induced long QT syndrome patients. J Mol Med. 2004;82:182–188. doi: 10.1007/s00109-003-0522-z. [DOI] [PubMed] [Google Scholar]
- Redfern WS, Carlsson L, Davis AS, Lynch WG, MacKenzie I, Palethorpe S, et al. Relationships between preclinical cardiac electrophysiology, clinical QT interval prolongation and torsade de pointes for a broad range of drugs: evidence for a provisional safety margin in drug development. Cardiovasc Res. 2003;58:32–45. doi: 10.1016/s0008-6363(02)00846-5. [DOI] [PubMed] [Google Scholar]
- Saenen JB, Paulussen AD, Jongbloed RJ, Marcelis CL, Gilissen RA, Aerssens J, et al. A single hERG mutation underlying a spectrum of acquired and congenital long QT syndrome phenotypes. J Mol Cell Cardiol. 2007;43:63–72. doi: 10.1016/j.yjmcc.2007.04.012. [DOI] [PubMed] [Google Scholar]
- Sale H, Wang J, O'Hara TJ, Tester DJ, Phartiyal P, He JQ, et al. Physiological properties of hERG 1a/1b heteromeric currents and a hERG 1b-specific mutation associated with Long-QT syndrome. Circ Res. 2008;103:e81–e95. doi: 10.1161/CIRCRESAHA.108.185249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanguinetti MC, Tristani-Firouzi M. hERG potassium channels and cardiac arrhythmia. Nature. 2006;440:463–469. doi: 10.1038/nature04710. [DOI] [PubMed] [Google Scholar]
- Schroeder K, Neagle B, Trezise DJ, Worley J. Ionworks HT: a new high-throughput electrophysiology measurement platform. J Biomol Screen. 2003;8:50–64. doi: 10.1177/1087057102239667. [DOI] [PubMed] [Google Scholar]
- Sesti F, Abbott GW, Wei J, Murray KT, Saksena S, Schwartz PJ, et al. A common polymorphism associated with antibiotic-induced cardiac arrhythmia. Proc Natl Acad Sci USA. 2000;97:10613–10618. doi: 10.1073/pnas.180223197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah RR. Drugs, QT interval prolongation and ICH E14: the need to get it right. Drug Saf. 2005;28:115–125. doi: 10.2165/00002018-200528020-00003. [DOI] [PubMed] [Google Scholar]
- Shuraih M, Ai T, Vatta M, Sohma Y, Merkle EM, Taylor E, et al. A common SCN5A variant alters the responsiveness of human sodium channels to class I antiarrhythmic agents. J Cardiovasc Electrophysiol. 2007;18:434–440. doi: 10.1111/j.1540-8167.2007.00777.x. [DOI] [PubMed] [Google Scholar]
- Simard C, Drolet B, Yang P, Kim RB, Roden DM. Polymorphism screening in the cardiac K+ channel gene KCNA5. Clin Pharmacol Ther. 2005;77:138–144. doi: 10.1016/j.clpt.2004.10.008. [DOI] [PubMed] [Google Scholar]
- Smith PL, Baukrowitz T, Yellen G. The inward rectification mechanism of the hERG cardiac potassium channel. Nature. 1996;379:833–836. doi: 10.1038/379833a0. [DOI] [PubMed] [Google Scholar]
- Splawski I, Shen J, Timothy KW, Lehmann MH, Priori S, Robinson JL, et al. Spectrum of mutations in long-QT syndrome genes. KVLQT1, hERG, SCN5A, KCNE1, and KCNE2. Circulation. 2000;102:1178–1185. doi: 10.1161/01.cir.102.10.1178. [DOI] [PubMed] [Google Scholar]
- Sun Z, Milos PM, Thompson JF, Lloyd DB, Mank-Seymour A, Richmond J, et al. Role of a KCNH2 polymorphism (R1047 L) in dofetilide-induced Torsades de Pointes. J Mol Cell Cardiol. 2004;37:1031–1039. doi: 10.1016/j.yjmcc.2004.09.001. [DOI] [PubMed] [Google Scholar]
- Swan H, Viitasalo M, Piippo K, Laitinen P, Kontula K, Toivonen L. Sinus node function and ventricular repolarization during exercise stress test in long QT syndrome patients with KvLQT1 and hERG potassium channel defects. J Am Coll Cardiol. 1999;34:823–829. doi: 10.1016/s0735-1097(99)00255-7. [DOI] [PubMed] [Google Scholar]
- Trudeau MC, Warmke JW, Ganetzky B, Robertson GA. hERG, a human inward rectifier in the voltage-gated potassium channel family. Science. 1995;269:92–95. doi: 10.1126/science.7604285. [DOI] [PubMed] [Google Scholar]
- Trudeau MC, Warmke JW, Ganetzky B, Robertson GA. hERG sequence correction. Science. 1996;272:1087. [PubMed] [Google Scholar]
- Ulens C, Daenens P, Tytgat J. Norpropoxyphene-induced cardiotoxicity is associated with changes in ion-selectivity and gating of hERG currents. Cardiovasc Res. 1999;44:568–578. doi: 10.1016/s0008-6363(99)00258-8. [DOI] [PubMed] [Google Scholar]
- Vandenberg JI, Varghese A, Lu Y, Bursill JA, Mahaut-Smith MP, Huang CL. Temperature dependence of human ether-a-go-go-related gene K+ currents. Am J Physiol Cell Physiol. 2006;291:C165–C175. doi: 10.1152/ajpcell.00596.2005. [DOI] [PubMed] [Google Scholar]
- Webster R, Leishman D, Walker D. Towards a drug concentration effect relationship for QT prolongation and torsades de pointes. Curr Opin Drug Discov Devel. 2002;5:116–126. [PubMed] [Google Scholar]
- Weininger D. SMILES, a chemical language and information system. 1. Introduction to methodology and encoding rules. J Chem Inf Comput Sci. 1988;28:31–36. [Google Scholar]
- Weininger D, Weininger JL. Algorithm for generation of unique SMILES notation. J Chem Inf Comput Sci. 1989;29:97–101. [Google Scholar]
- Witchel HJ, Dempsey CE, Sessions RB, Perry M, Milnes JT, Hancox JC, et al. The low-potency, voltage-dependent hERG blocker propafenone – molecular determinants and drug trapping. Mol Pharmacol. 2004;66:1201–1212. doi: 10.1124/mol.104.001743. [DOI] [PubMed] [Google Scholar]
- Yang P, Kanki H, Drolet B, Yang T, Wei J, Viswanathan PC, et al. Allelic variants in long-QT disease genes in patients with drug-associated torsades de pointes. Circulation. 2002;105:1943–1948. doi: 10.1161/01.cir.0000014448.19052.4c. [DOI] [PubMed] [Google Scholar]
- Yang T, Kanki H, Roden DM. Phosphorylation of the IKs channel complex inhibits drug block. Novel mechanism underlying variable antiarrhythmic drug actions. Circulation. 2003;108:132–134. doi: 10.1161/01.CIR.0000082708.86266.B8. [DOI] [PubMed] [Google Scholar]
- Zhang S, Zhou Z, Gong Q, Makielski JC, January CT. Mechanism of block and identification of the verapamil binding domain to hERG potassium channels. Circ Res. 1999;84:989–998. doi: 10.1161/01.res.84.9.989. [DOI] [PubMed] [Google Scholar]