

Abstract
Neurofibrillary tangles, principally composed of bundles of filaments formed by the microtubule-associated protein Tau, are a hallmark of a group of neurodegenerative diseases such as Alzheimer disease. Polyanionic cofactors such as heparin can induce Tau filament formation in vitro. Here we quantitatively characterize the interaction between recombinant human Tau fragment Tau244–372 and heparin (average molecular mass = 7 kDa) as well as heparin-induced fibril formation by using static light scattering, isothermal titration calorimetry, turbidity assays, and transmission electron microscopy. Our data clearly show that at physiological pH, heparin 7K, and human Tau244–372 form a tight 1:1 complex with an equilibrium association constant exceeding 106 m−1 under reducing conditions, triggering Tau fibrillization. In the absence of dithiothreitol, heparin shows a moderate binding affinity (105 m−1) to Tau244–372, similarly triggering Tau fibrillization. Further fibrillization kinetics analyses show that the lag time appears to be approximately invariant up to a molar ratio of 2:1 and then increases at larger ratios of heparin/Tau. The maximum slope representing the apparent rate constant for fibril growth increases sharply with substoichiometric ratios of heparin/Tau and then decreases to some extent with ratios of >1:1. The retarding effect of heparin in excess is attributed to the large increase in ionic strength of the medium arising from free heparin. Together, these results suggest that the formation of the 1:1 complex of Tau monomer and heparin plays an important role in the inducer-mediated Tau filament formation, providing clues to understanding the pathogenesis of neurodegenerative diseases.
Keywords: Biophysics, Diseases/Alzheimer Disease, Diseases/Amyloid, Methods/Calorimetry, Protein/Thermodynamics, Heparin Binding, Protein Misfolding, Tau Protein
Introduction
Microtubule-associated protein Tau is the major protein subunit of neurofibrillary tangles, one of the major neuropathological hallmarks of a group of neurodegenerative diseases such as Alzheimer disease (1, 2). Neurofibrillary tangles are mainly composed of bundles of Tau in the form of paired helical filaments, straight filaments, or twisted ribbons (3). Because the accumulation of Tau aggregates correlates well with nerve cell loss and the severity of dementia, formation of a neurofibrillary tangle is considered to be involved in the pathways leading to neuronal death (4–6), and the characterization of factors involved in Tau fibrillization is of great importance to clarify the etiology of such neurodegenerative diseases.
Tau is among one of the largest proteins without recognizable secondary structure and adopts a natively unfolded structure in solution (7). It binds to microtubules through four repeat domains in its C-terminal part (8). The repeat domain of Tau forms the core of paired helical filaments in Alzheimer disease and also assembles more readily than full-length Tau into bona fide paired helical filaments in vitro (9, 10).
To mimic Tau fibrillization in vivo, polyanionic cofactors such as heparin have been used to induce Tau filament formation in vitro (1, 11–14). It has been shown that Tau and heparan sulfate (HS)2 coexist in nerve cells with overt neurofibrillary lesion (12). The molecular basis for such an inducement process remains unknown. However, the discovery that other polyanions such as RNA (15) or arachidonic acid (16) can also cause Tau filament formation indicates that Tau aggregation is rather influenced by electrostatics than by the precise nature of the negatively charged polymers (17). The kinetics for inducer-mediated Tau filament formation can be characterized by a lag period, followed by a period of exponential growth and an asymptotic approach to equilibrium (13, 18–20). The filamentous structures induced by heparin are structurally similar to those found in Alzheimer disease (1), and thus heparin-induced Tau aggregation serves as a good model for pathological Tau fibrillization.
In the present study, we quantitatively characterized the interaction between recombinant human Tau fragment Tau244–372 and heparin as well as heparin-induced fibril formation of Tau244–372 by using several biophysical methods, such as static light scattering, isothermal titration calorimetry (ITC), turbidity assays, and transmission electron microscopy. Our data demonstrated that at physiological pH and temperature, human Tau244–372 did form a 1:1 complex with heparin. Heparin shows a strong binding affinity to Tau244–372 in the presence of dithiothreitol (DTT), triggering Tau fibrillization under such reducing conditions.
EXPERIMENTAL PROCEDURES
Materials
Heparin (average molecular mass of 7 kDa) was obtained from Sigma-Aldrich. DTT was obtained from Ameresco (Solon, OH). All other chemicals used were made in China and were of analytical grade. All of the reagent solutions were prepared in 10 mm HEPES buffer (pH 7.4) containing 100 mm NaCl.
Plasmids and Proteins
The cDNA encoding human Tau fragment Tau244–372 was amplified using the plasmid for human Tau40 (kindly provided by Dr. Michel Goedert) as a template. The PCR-amplified Tau244–372 was subcloned into pRK172 vector. Recombinant Tau244–372 was expressed in Escherichia coli and purified to homogeneity by SP Sepharose chromatography as described (21). Purified Tau protein was analyzed by SDS-PAGE with one band. The concentration of human Tau fragment was determined according to its absorbance at 214 nm with a standard calibration curve drawn by bovine serum albumin.
Turbidity Assays
Samples of 160 μm Tau244–372 were incubated at 37 °C in 10 mm HEPES buffer (pH 7.4) containing heparin at different concentrations (20–640 μm) and 100 mm NaCl in the presence and absence of 1 mm DTT or in 10 mm HEPES buffer (pH 7.4) containing 160 μm heparin and NaCl at different concentrations (0–500 mm) in the presence of 1 mm DTT. The solutions with a volume of 250 μl were placed into a well of a 96-well plate followed by monitoring the turbidity at 350 nm using a SpectraMax 190 microplate reader (Molecular Devices, Sunnyvale, CA). Each sample was run in triplicate or quadruplicate. The profile of absorbance versus time was analyzed as follows. A seventh order polynomial was fitted to all of the data {t, A350}from replicates at each heparin concentration. The maximum slope of the best fit polynomial (dP/dt)max, the value of t corresponding to maximum slope of the polynomial t*, and the value of the polynomial at this time P(t*) were evaluated. A straight line with slope (dP/dt)max was drawn through {t*, P(t*)}, and the value of t at which this line crosses the t axis (P = 0) is designated the lag time.
Transmission Electron Microscopy
The formation of filaments by human Tau fragment was confirmed by electron microscopy of negatively stained samples. Sample aliquots of 10 μl were placed on copper grids and left at room temperature for 1–2 min, rinsed with H2O twice, and then stained with 2% (w/v) uranyl acetate for another 1–2 min. The stained samples were examined using an H-8100 transmission electron microscope (Hitachi, Tokyo, Japan) operating at 100 kV or an FEI Morgagni transmission electron microscope operating at 80 kV.
Isothermal Titration Calorimetry
ITC experiments on the interaction of heparin with Tau244–372 in the absence of DTT were carried out at 37.0 °C using a VP-ITC titration calorimetry (MicroCal, Northampton, MA). Freshly purified wild-type Tau244–372 and heparin were thoroughly dialyzed against 10 mm HEPES buffer (pH 7.4) containing 100 mm NaCl. A solution of 40 μm Tau protein was loaded into the sample cell (1.43 ml), and a solution of 500 μm heparin was placed in the injection syringe (280 μl). A titration experiment consisted of 13 consecutive injections of 20-μl volume and 40-s duration each, with a 4-min interval between injections, and the 14th injection of 20-μl volume and 40-s duration, with an 8-h interval. Dilution heat of heparin was measured by injecting heparin solution into buffer alone and was subtracted from the experimental curves prior to data analysis. The resulting data (injections 1–13) were fitted to a single set of identical sites model using the MicroCal ORIGIN software supplied with the instrument, and the standard molar enthalpy change for the binding, ΔbHm0, the binding constant, Kb, and the binding stoichiometry, n, were thus obtained. The standard molar free energy change, ΔbGm0, and the standard molar entropy change, ΔbSm0, for the binding reaction were calculated by the fundamental equations of thermodynamics (22).
Calorimetric data of injection 14 was integrated of the heat power peak of the reaction.
Composition Gradient Static Light Scattering
Tau244–372 and heparin were thoroughly dialyzed against 10 mm HEPES buffer (pH 7.4) containing 100 mm NaCl and 1 mm DTT. Immediately before light scattering measurement, both protein and heparin solutions were filtered through a 0.02-μm filter (Anotop 10 Plus, Whatman) and centrifuged at 5000 rpm and 4 °C for 30 min to remove microscopic bubbles. The concentration of Tau244–372 in solution A (wA,load) and the concentration of heparin in solution B (wB,load), determined in an Optilab rEX differential refractometer (Wyatt, Santa Barbara, CA), were 0.55 and 0.42 g/liter, respectively. The specific refractive increment of Tau (A in equations below) is dñ/dwA = 0.185 cm3/g (23). The specific refractive increment of heparin (B in equations below) was determined experimentally. A calibration curve of refractive index versus heparin concentration was constructed, and the best fit value of the slope dñ/dwB = 0.126 cm3/g is in agreement with the published values (23, 24). A composition gradient was created using the Calypso system (Wyatt, Santa Barbara, CA), in which the fraction of solution A (fA) is decreased from 1 to 0 in steps of 0.08, and the total concentrations of Tau (A) and heparin (B) in each of these mixtures is given by the following equations.
These solution mixtures were pumped in sequence into parallel flow cells in which the static light scattering intensity and refractive index of each mixture were measured simultaneously in a DAWN-EOS light scattering photometer (Wyatt) and an Optilab rEX differential refractometer (Wyatt). The raw data obtained from each instrument were respectively converted into values of the normalized scattering intensity R/Kopt and fA as described (25, 26).
The equilibrium constant for formation of a 1:1 complex of Tau (A) and heparin (B) is defined as follows,
 |
where cAB, cA, and cB, respectively, denote the molar concentrations at equilibrium of the complex, free A, and free B. The conservation of mass is given by the following,
 |
 |
where wA,tot and wB,tot, respectively, denote the total w/v concentrations of Tau and heparin, and MA and MB, respectively, denote the molar masses of Tau and heparin. Given the values of wA,tot, wB,tot, MA, MB, and KAB, Equations 5–7 may be solved analytically to yield the equilibrium values of cA, cB, and cAB.
The normalized scattering intensity is given by the following equations (27).
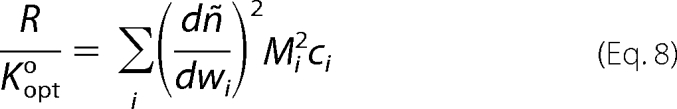 |
 |
In the above equations, dñ/dwi, Mi, and ci, respectively, denote the specific refractive increment, molecular weight, and molar concentration of the ith scattering species, ñ0 is the refractive index of buffer, λ0 is the wavelength in vacuum of incident light (here 690 nm), and NA is Avogadro's number. To facilitate modeling of scattering in mixtures containing scattering species of significantly different refractive increment, we scale the above equations as follows. We define a new optical constant,
 |
where dñ/dwprotein is set equal to the average for polypeptides, 0.185 cm3/g (23). Then Equation 3 may be rewritten as follows.
 |
 |
Gi2 is thus a measure of the scattering power of a given substance relative to that of protein. The specific refractive increments of Tau (A) and heparin (B) are as follows: dñ/dwA = 0.185 cm3/g (23); dñ/dwB = 0.126 cm3/g. The refractive increment of the 1:1 complex AB is a mass average (25).
Thus GA = 1, GB = 0.681, and GAB = (MAGA + MBGB)/MAB.
The experimentally determined dependence of R/Kopt upon fA was modeled using Equations 3–7 to calculate the equilibrium values of cA, cB, and cAB and then using Equation 11 with the values of GA, GB, and GAB given above to calculate R/Kopt. The values of MA, MB, and log10 KAB were allowed to vary to achieve a best fit via nonlinear least squares.
RESULTS
Interaction between Tau and Heparin under Reducing Conditions
The experimentally measured dependence of R/Kopt on fA is plotted in Fig. 1. The dependence calculated using Equation 13 with the best fit parameter values given below is plotted together with the data. Here we quantitatively characterize the interaction between recombinant human Tau fragment Tau244–372 and heparin in the presence of DTT using static light scattering. The best fit parameter values with 95% confidence limits of estimate were: MA = 14,100 ± 700 g mol−1; MB = 7200 ± 800 g mol−1; log KAB = 6.5 (−1, + indeterminate). Our data clearly indicated that at physiological pH, heparin and human Tau244–372 form a tight 1:1 complex with an equilibrium association constant exceeding 106 m−1 under reducing conditions.
FIGURE 1.

Light scattering measurements of the binding of heparin to Tau244–372 in the presence of DTT and at 37.0 °C. Shown is the experimentally measured composition dependence of light scattering intensity (solid circles), plotted with dependence calculated as described in text using the following best fit parameter values and 95% confidence limits of estimate: MA = 14,100 ± 700 g mol−1; MB = 7200 ± 800 g mol−1; log KAB = 6.5 (−1, + indeterminate).
Interaction between Tau and Heparin under Oxidative Conditions
ITC provides a direct route to the complete thermodynamic characterization of noncovalent, equilibrium interactions (28, 29), and DTT concentrations as low as 1 mm can cause severe base-line artifacts caused by background oxidation during the titration. Therefore ITC was used to measure the binding affinity of heparin to Tau protein in the absence of DTT. ITC profiles for the binding of heparin 7K to wild-type Tau244–372 at 37.0 °C in 10 mm HEPES buffer (pH 7.4) containing 100 mm NaCl are shown in Fig. 2. Fig. 2A shows the raw ITC curve resulting from the injections of heparin into a solution of wild-type Tau244–372. The titration curve shows that heparin binding to Tau244–372 is exothermic, resulting in negative peaks in the plots of power versus time. Fig. 2B shows the plot of the heat evolved per mol of heparin added, corrected for the heat of heparin dilution, against the molar ratio of heparin to Tau244–372. The thermodynamic parameters for the binding between heparin and Tau244–372 (ΔbHm0 = −1.44 ± 0.12 kcal mol−1, n = 1.08 ± 0.05, Kb = (4.05 ± 1.44) × 105 m−1, ΔbSm0 = 30.28 ± 1.08 cal mol−1 K−1, and ΔbGm0 = −7.95 ± 0.22 kcal mol−1) were obtained by fitting the data to a single set of identical sites model, indicating that one heparin bound to one Tau244–372 with a moderate affinity in the absence of DTT. The formation of the heparin-Tau complex was driven by a large favorable entropy increase with a small favorable enthalpy increase under such oxidative conditions. Our data clearly indicated that at physiological pH, heparin and human Tau244–372 formed a 1:1 complex with an equilibrium association constant of approximately 105 m−1 under oxidative conditions.
FIGURE 2.

ITC measurements of the binding of heparin to Tau244–372 and the fibrillation of Tau244–372 induced by heparin at 37.0 °C. A represents typical calorimetric titration of Tau244–372 (40 μm) with heparin (500 μm) in the absence of DTT. B shows the plots of the heat evolved (kcal) per mol of heparin added, corrected for the heat of heparin, against the molar ratio of heparin to Tau244–372. The data (solid squares) were fitted to a single set of identical sites model, and the solid line represented the best fit. C represents typical calorimetric curve for the fibrillation of Tau244–372 induced by heparin as a function of time following the binding of heparin to Tau244–372, and the inset shows the heat effect of the fibrillation of Tau244–372. D represents the transmission electron micrographs of Tau244–372 fibrils formed in the calorimeter cell. The scale bar represents 200 nm.
Heparin-mediated Tau Filament Formation under Reducing Conditions
Tau244–372 was incubated with various concentrations of heparin in the presence of 1 mm DTT and monitored by turbidity assays. Fig. 3 shows the dependence of turbidity changes for Tau244–372 filament formation at 37.0 °C on the molar ratio of heparin to Tau protein. The molar ratio of heparin to Tau was 1:8, 1:6, 1:4, 1:2, 1:1, 2:1, and 4:1, respectively. The kinetic curves were analyzed as described above (Fig. 3), and the lag time and maximum slope of these curves were obtained. As shown in Figs. 1 and 3, at physiological pH, heparin and human Tau244–372 formed a tight 1:1 complex under reducing conditions, triggering Tau fibrillization. Further fibrillization kinetics analyses showed that the lag time appeared to be approximately invariant up to a molar ratio of 2:1 and then got longer at larger ratios of heparin/Tau in the presence of DTT (Fig. 4B). The maximum slope representing the apparent rate constant for fibril growth increased sharply with substoichiometric ratios of heparin/Tau and then decreased to some extent with ratios of >1:1 in the presence of DTT (Fig. 4A).
FIGURE 3.

The dependence of turbidity changes for Tau244–372 filament formation at 37.0 °C on the molar ratio of heparin to Tau protein. The concentrations of Tau244–372 and DTT were 160 μm and 1 mm, respectively. The molar ratio of heparin to Tau244–372 was 1:8 (A), 1:6 (B), 1:4 (C), 1:2 (D), 1:1 (E), 2:1 (F), and 4:1 (G), respectively. A seventh order polynomial was fitted to all data (open circles) from replicates at each heparin concentration. The kinetic curves were analyzed as described under “Experimental Procedures.”
FIGURE 4.

Kinetic analyses for filament formation of Tau244–372 as a function of the molar ratio of heparin to Tau protein. Maximum slope (A) and lag time (B) changes for filament formation of Tau244–372 are shown. The data are expressed as the means ± S.D. (n = 3–4). The temperature was 37.0 °C.
Heparin-mediated Tau Filament Formation under Oxidative Conditions
To monitor the fibrillization of Tau244–372 in the absence of DTT, injection 14 had an 8-h interval after 13 consecutive injections. The ITC curve of the fibrillization of Tau244–372 was shown in Fig. 2C. The curve represented heat of the reaction as a function of time by integrating the power change (ΔP) peak of the reaction shown in the inset box, corrected by the reference. This represented the kinetics of the reaction via the enthalpy production, indicating the high accuracy and low noise of the measurement even without smoothing of the data (Fig. 2C, inset). The calorimetric data between 60 and 540 min clearly confirmed that heparin mediated Tau filament formation under oxidative conditions (Fig. 2C). Meanwhile the transmission electron microscopy image of Tau244–372 aggregates formed in the ITC cell showed typical long and branched fibrils, further confirming the fibrillization of Tau244–372 induced by heparin (Fig. 2D).
Characterization of Morphology of Human Tau Samples
Transmission electron microscopy was used to study the morphology of human Tau samples incubated under different conditions. For wild-type Tau244–372, the addition of DTT had no significant effect on the morphology of Tau samples, and long and branched fibrils as well as straight filaments were observed in both samples (Fig. 5), indicating that heparin could induce Tau filament formation under reducing or oxidative conditions.
FIGURE 5.

Transmission electron micrographs of Tau244–372 samples at physiological pH. The micrographs shown were created in the presence of 1 mm DTT (A and B) and in the absence of DTT (C and D) after incubation for 12 h. Straight filaments formed by Tau244–372 in the presence of DTT are shown in the inset of B. The concentration of Tau244–372 was 80 μm and the molar ratio of heparin to Tau244–372 was 1:2. A 2% (w/v) uranyl acetate solution was used to negatively stain the fibrils. The scale bars in B–D represent 100 nm, and that in A represents 500 nm.
DISCUSSION
Tau monomers do not assemble spontaneously under physiological conditions over experimentally tractable time periods (20), and Tau fragments cannot assemble spontaneously to form amyloid filaments over a period of days when the concentration is lower than 150 μm (30, 31). The pathological aggregation of Tau is a nucleation-elongation reaction that involves the formation of β-structure containing some hexapeptide motifs of the repeat domain (21). The “driving force” for filament formation by the Tau repeat domain involves both the formation of an intermolecular disulfide bond (10) and noncovalent interactions such as hydrogen bonds, electrostatic interactions, and hydrophobic interactions (17). Then the elongation can proceed from the nuclei (10).
In the present study, Tau filaments formed within hours when the Tau fragment was incubated with heparin. Polyanionic cofactors, such as heparin and thiazine red, cause structural and conformational changes of Tau protein (17, 32, 33), and then the kinetic and thermodynamic barriers for Tau fibrillization can be overcome (1, 34). Although heparin is not a perfect model for HS, which coexists with Tau protein in nerve cells (12), both heparin and HS induce formation of Tau filaments with very similar morphologies (35). Moreover, the addition of heparin into full-length human Tau protein induces the assembly of Tau into filaments that closely resemble those observed in Alzheimer disease brain tissue (12). Therefore, this study has implications for the putative role of endogenous HS proteoglycans in the formation of neurofibrillary tangles in Alzheimer disease. The extension of the present study of the characterization of heparin binding to human Tau fragment to the quantitative characterization of HS binding to full-length human Tau protein should lead to a better understanding of how HS proteoglycans mediate Tau filament formation in the physiological environment in Alzheimer disease brain.
So far, our experiments were conducted with human Tau fragment, but because filaments in Alzheimer disease contain full-length Tau protein, it is important to demonstrate the behavior of full-length Tau bound by heparin. The human Tau fragment used in this study encompasses the four microtubule-binding repeats, which does exclude a number of domains that could potentially interact with heparin. The main difference is that full-length Tau forms filaments in the presence of heparin much more slowly than Tau244–372 induced by heparin (36). However, we did observe straight filaments formed by Tau244–372 induced by heparin (Fig. 5), similar to those formed by more complete Tau proteins (12) and by full-length human Tau (Fig. 6) induced by heparin. Our additional ITC experiments (Fig. 7) indicated that in the absence of DTT, heparin bound to full-length human Tau protein with a moderate affinity (2.38 × 105 m−1) at the first binding site followed by one weak binding event (2.80 × 103 m−1). Clearly, the binding affinity of heparin to full-length human Tau at the first binding site closely resembles that of heparin to Tau244–372. Based on our results and the recently published data (17), we suggest that heparin binds to full-length human Tau mainly through its segment 244–372, forming the 1:1 complex and triggering Tau fibrillization.
FIGURE 6.

Transmission electron micrograph of full-length human Tau sample at physiological pH in the presence of 2 mm DTT after incubation for 72 h. The concentration of full-length human Tau protein was 20 μm, and the molar ratio of heparin to Tau was 1:1. A 2% (w/v) uranyl acetate solution was used to negatively stain the fibrils. The scale bar represents 200 nm.
FIGURE 7.

ITC measurements of the binding of heparin to full-length Tau protein at 37.0 °C. A represents typical calorimetric titration of full-length Tau (40 μm) with heparin (1.5 mm) in the absence of DTT. The first injection (0.5 μl) was followed by 19 injections of 2 μl. B shows the plots of the heat evolved (kcal) per mol of heparin added, corrected for the heat of heparin, against the molar ratio of heparin to full-length Tau. The data (solid squares) were fitted to a two sequential binding sites model and the solid line represents the best fit. The ITC machine used here was an iTC200 titration calorimetry (MicroCal).
Neuronal cells normally have a reducing environment maintained by an excess of glutathione (1, 8). In this study, DTT, a strong reducing agent, was used to mimic the reducing environment present in normal neuronal cells. Our data here clearly showed that at physiological pH, heparin 7K and human Tau fragment formed a tight 1:1 complex under physiological reducing conditions, triggering Tau fibrillization. High affinity binding of heparin to Tau protein under reducing conditions provides the necessary charge compensation to allow fibril growth (17). An oxidative (without DTT present plus air oxidation) experimental condition is closer to the physiological environment in Alzheimer disease brain (31, 37). Our ITC data demonstrated that heparin bound to Tau fragment with a moderate binding affinity under pathological oxidative conditions, similarly triggering Tau fibrillization. Kardos and co-workers (38) have reported the first ITC study of amyloid formation, e.g. the seed-dependent extension of β2-microglobulin amyloid fibrils under acidic conditions. Here, we report for the first time a direct thermodynamic study of the interaction between amyloidogenic protein and inducer as well as inducer-triggered fibril formation using ITC.
Our time-dependent turbidity measurements revealed that the lag time did not vary significantly, but the apparent rate constant for the growth of fibrils increased sharply as the molar ratio of heparin to Tau increased up to 1:1 under reductive conditions, suggesting that heparin plays a regulatory role mainly in the elongation process of Tau fibrillization. When the molar ratio of heparin to Tau exceeded unity, the lag time increased, and the apparent rate constant of fibril growth decreased to some extent. The retarding effect of excess heparin is attributed to a large increase in ionic strength of the medium arising from excess unbound heparin (13, 39), which was confirmed by control experiments at fixed heparin concentrations and increasing salt concentrations (supplemental Fig. S1).
Based on our kinetic and thermodynamic data and the recently reported results (17, 33, 34, 39, 40), we propose a valuable hypothesis for heparin-mediated Tau filament formation that the first step of kinetics involves binding of heparin to the natively unfolded Tau protein, forming a 1:1 complex and inducing structural and conformational changes of Tau protein, and then the nuclei are formed, which is the rate-limiting step, followed by the step of fibril elongation at physiological pH (Fig. 8). The hypothesis proposed here is different from the previous hypothesis (33) in the following aspects. First, we propose that the binding of heparin (or another polyanion) to monomeric Tau is a prerequisite for the growth of Tau fibrils. Second, we speculate that the binding of heparin to Tau monomer induces conformational changes in Tau, as well as reducing the large net positive charge borne by Tau protein, thus reducing the activation energy required to add Tau to the end of a growing Tau fiber and accelerating fibril growth. Therefore the formation of a 1:1 complex of Tau monomer and heparin plays an important role in the inducer-mediated Tau filament formation, providing clues to understanding the pathogenesis of neurodegenerative diseases.
FIGURE 8.

Hypothetical model for heparin-mediated Tau filament formation at physiological pH. The first step is the interaction of heparin (red branched structure) with the natively unfolded Tau protein (ribbon diagram in color), forming a 1:1 complex and inducing structural and conformational changes of Tau protein, and then the nuclei are formed, which is the rate-limiting step, followed by the step of fibril elongation. Regions of β-structure are shown in blue.
Supplementary Material
Acknowledgments
We sincerely thank Dr. Michel Goedert (Laboratory of Molecular Biology, Medical Research Council, Cambridge, UK) for kindly providing the plasmid for human Tau40 and Prof. Guang-Fu Yang (College of Chemistry, Central China Normal University, Wuhan, China) for technical assistance with iTC200.
This work was supported by National Key Basic Research Foundation of China Grant 2006CB910301, National Natural Science Foundation of China Grants 30770421 and 30970599, and Chinese 111 Project Grant B06018. This work was also supported in part by National Institutes of Health grants through the Intramural Research Program of the NIDDK.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Fig. S1.
- HS
- heparan sulfate
- DTT
- dithiothreitol
- ITC
- isothermal titration calorimetry.
REFERENCES
- 1.Kuret J., Chirita C. N., Congdon E. E., Kannanayakal T., Li G., Necula M., Yin H., Zhong Q. (2005) Biochim. Biophys. Acta 1739, 167–178 [DOI] [PubMed] [Google Scholar]
- 2.Skovronsky D. M., Lee V. M., Trojanowski J. Q. (2006) Annu. Rev. Pathol. 1, 151–170 [DOI] [PubMed] [Google Scholar]
- 3.Braak H., Braak E. (1997) Neurobiol. Aging 18, 351–357 [DOI] [PubMed] [Google Scholar]
- 4.Goedert M. (1993) Trends Neurosci. 16, 460–465 [DOI] [PubMed] [Google Scholar]
- 5.Gómez-Isla T., Hollister R., West H., Mui S., Growdon J. H., Petersen R. C., Parisi J. E., Hyman B. T. (1997) Ann. Neurol. 41, 17–24 [DOI] [PubMed] [Google Scholar]
- 6.Aoyagi H., Hasegawa M., Tamaoka A. (2007) J. Biol. Chem. 282, 20309–20318 [DOI] [PubMed] [Google Scholar]
- 7.Mandelkow E., Song Y. H., Schweers O., Marx A., Mandelkow E. M. (1995) Neurobiol. Aging 16, 347–354 [DOI] [PubMed] [Google Scholar]
- 8.Rosenberg K. J., Ross J. L., Feinstein H. E., Feinstein S. C., Israelachvili J. (2008) Proc. Natl. Acad. Sci. U.S.A. 105, 7445–7450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wille H., Drewes G., Biernat J., Mandelkow E. M., Mandelkow E. (1992) J. Cell Biol. 118, 573–584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Friedhoff P., von Bergen M., Mandelkow E. M., Davies P., Mandelkow E. (1998) Proc. Natl. Acad. Sci. U.S.A. 95, 15712–15717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pérez M., Valpuesta J. M., Medina M., Montejo de Garcini E., Avila J. (1996) J. Neurochem. 67, 1183–1190 [DOI] [PubMed] [Google Scholar]
- 12.Goedert M., Jakes R., Spillantini M. G., Hasegawa M., Smith M. J., Crowther R. A. (1996) Nature 383, 550–553 [DOI] [PubMed] [Google Scholar]
- 13.Friedhoff P., Schneider A., Mandelkow E. M., Mandelkow E. (1998) Biochemistry 37, 10223–10230 [DOI] [PubMed] [Google Scholar]
- 14.Chirita C. N., Kuret J. (2004) Biochemistry 43, 1704–1714 [DOI] [PubMed] [Google Scholar]
- 15.Kampers T., Friedhoff P., Biernat J., Mandelkow E. M., Mandelkow E. (1996) FEBS Lett. 399, 344–349 [DOI] [PubMed] [Google Scholar]
- 16.Wilson D. M., Binder L. I. (1997) Am. J. Pathol. 150, 2181–2195 [PMC free article] [PubMed] [Google Scholar]
- 17.Sibille N., Sillen A., Leroy A., Wieruszeski J. M., Mulloy B., Landrieu I., Lippens G. (2006) Biochemistry 45, 12560–12572 [DOI] [PubMed] [Google Scholar]
- 18.Ferrone F. (1999) Methods Enzymol. 309, 256–274 [DOI] [PubMed] [Google Scholar]
- 19.Necula M., Kuret J. (2004) Anal. Biochem. 333, 205–215 [DOI] [PubMed] [Google Scholar]
- 20.Chirita C. N., Congdon E. E., Yin H., Kuret J. (2005) Biochemistry 44, 5862–5872 [DOI] [PubMed] [Google Scholar]
- 21.Barghorn S., Biernat J., Mandelkow E. (2005) Methods Mol. Biol. 299, 35–51 [DOI] [PubMed] [Google Scholar]
- 22.van Holde K. E., Johnson W. E., Ho P. S. (2006) Principles of Physical Biochemistry, pp. 91–93, Pearson Prentice Hall, Upper Saddle River, NJ [Google Scholar]
- 23.Theisen A., Deacon M. P., Johann C., Harding S. E. (2000) Refractive Increment Data-book: For Polymer and Biomolecular Scientists, p. 55, Nottingham University Press, Nottingham, UK [Google Scholar]
- 24.Pavlov G., Finet S., Tatarenko K., Korneeva E., Ebel C. (2003) Eur. Biophys. J. 32, 437–449 [DOI] [PubMed] [Google Scholar]
- 25.Attri A. K., Minton A. P. (2005) Anal. Biochem. 346, 132–138 [DOI] [PubMed] [Google Scholar]
- 26.Kameyama K., Minton A. P. (2006) Biophys. J. 90, 2164–2169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stacey K. A. (1956) Light Scattering in Physical Chemistry, Academic Press, New York [Google Scholar]
- 28.Liang Y., Du F., Sanglier S., Zhou B. R., Xia Y., Van Dorsselaer A., Maechling C., Kilhoffer M. C., Haiech J. (2003) J. Biol. Chem. 278, 30098–30105 [DOI] [PubMed] [Google Scholar]
- 29.Xu H., Liang Y., Zhang P., Du F., Zhou B. R., Wu J., Liu J. H., Liu Z. G., Ji L. N. (2005) J. Biol. Inorg. Chem. 10, 529–538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Goux W. J., Kopplin L., Nguyen A. D., Leak K., Rutkofsky M., Shanmuganandam V. D., Sharma D., Inouye H., Kirschner D. A. (2004) J. Biol. Chem. 279, 26868–26875 [DOI] [PubMed] [Google Scholar]
- 31.Schweers O., Mandelkow E. M., Biernat J., Mandelkow E. (1995) Proc. Natl. Acad. Sci. U.S.A. 92, 8463–8467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Paudel H. K., Li W. (1999) J. Biol. Chem. 274, 8029–8038 [DOI] [PubMed] [Google Scholar]
- 33.Congdon E. E., Kim S., Bonchak J., Songrug T., Matzavinos A., Kuret J. (2008) J. Biol. Chem. 283, 13806–13816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Carlson S. W., Branden M., Voss K., Sun Q., Rankin C. A., Gamblin T. C. (2007) Biochemistry 46, 8838–8849 [DOI] [PubMed] [Google Scholar]
- 35.Hasegawa M., Crowther R. A., Jakes R., Goedert M. (1997) J. Biol. Chem. 272, 33118–33124 [DOI] [PubMed] [Google Scholar]
- 36.Barghorn S., Mandelkow E. (2002) Biochemistry 41, 14885–14896 [DOI] [PubMed] [Google Scholar]
- 37.Smith C. D., Carney J. M., Starke-Reed P. E., Oliver C. N., Stadtman E. R., Floyd R. A., Markesbery W. R. (1991) Proc. Natl. Acad. Sci. U.S.A. 88, 10540–10543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kardos J., Yamamoto K., Hasegawa K., Naiki H., Goto Y. (2004) J. Biol. Chem. 279, 55308–55314 [DOI] [PubMed] [Google Scholar]
- 39.Jeganathan S., von Bergen M., Mandelkow E. M., Mandelkow E. (2008) Biochemistry 47, 10526–10539 [DOI] [PubMed] [Google Scholar]
- 40.von Bergen M., Barghorn S., Müller S. A., Pickhardt M., Biernat J., Mandelkow E. M., Davies P., Aebi U., Mandelkow E. (2006) Biochemistry 45, 6446–6457 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.