

Abstract
The Ca2+-binding protein recoverin is thought to regulate rhodopsin kinase and to modulate the lifetime of the photoexcited state of rhodopsin (Rh*), the visual pigment of vertebrate rods. Recoverin has been postulated to inhibit the kinase in darkness, when Ca2+ is high, and to be released from the disk membrane in light when Ca2+ is low, accelerating rhodopsin phosphorylation and shortening the lifetime of Rh*. This proposal has remained controversial, in part because the normally rapid turnoff of Rh* has made Rh* modulation difficult to study in an intact rod. To circumvent this problem, we have made mice that underexpress rhodopsin kinase so that Rh* turnoff is rate limiting for the decay of the rod light response. We show that background light speeds the decay of Rh* turnoff, and that this no longer occurs in mice that have had recoverin knocked out. This is the first demonstration in an intact rod that light accelerates Rh* inactivation and that the Ca2+-binding protein recoverin may be required for the light-dependent modulation of Rh* lifetime.
Introduction
The bleaching of rhodopsin in a vertebrate rod activates a phosphodiesterase (PDE6), which hydrolyzes cGMP and decreases the opening probability of cGMP-dependent channels in the rod outer segment (Fain, 2003). The closing of these channels produces a decrease in inward current and lowers the outer segment concentration of Ca2+, which is thought to act as a second messenger regulating sensitivity during light adaptation (Pugh et al., 1999; Fain et al., 2001). One possible target for Ca2+-dependent sensitivity regulation was first identified by Kawamura (1993), who showed that the Ca2+-binding protein recoverin/S-modulin can regulate the phosphorylation of rhodopsin. Further experimentation confirmed the Ca2+-dependent regulation of rhodopsin kinase by recoverin (C.-K. Chen et al., 1995; Klenchin et al., 1995). It is thought that in high Ca2+ in darkness, recoverin inhibits kinase activity probably sterically (Ames et al., 2006; Higgins et al., 2006), decreasing the rate of rhodopsin phosphorylation and extending rhodopsin lifetime; in low Ca2+ in the light, recoverin is released and may even diffuse out of the outer segment (Strissel et al., 2005), removing the inhibition and accelerating phosphorylation.
Some observations seem to support this proposed role for Ca2+ and recoverin. An early step in transduction is apparently regulated by Ca2+ concentration (Matthews, 1997), perhaps by modulating the rate of rhodopsin phosphorylation (Matthews et al., 2001). Extrinsic recoverin introduced into the outer segments of rods prolongs flash response recovery (Gray-Keller et al., 1993; Erickson et al., 1998), and the knocking out of recoverin does just the opposite, accelerating flash response decay (Makino et al., 2004). Other observations are less easily reconciled with a role of Ca2+ or recoverin in modulation of Rh* lifetime. The Ca2+ affinity for recoverin is rather low, with an EC50 of about 3 μm (C.-K. Chen et al., 1995; Klenchin et al., 1995). It is unclear whether the light-dependent change of Ca2+ in a mouse rod (Woodruff et al., 2002) is large enough to produce a significant change in recoverin activity. Phosphorylation of rhodopsin seems insensitive to Ca2+ in permeabilized rods (Otto-Bruc et al., 1998), and the acceleration of the flash response decay in recoverin knock-out rods now seems more consistent with an effect of recoverin on PDE activity than rhodopsin phosphorylation (Krispel et al., 2006).
The principal difficulty with studying recoverin and modulation of Rh* lifetime in intact rods is that PDE turnoff is normally rate limiting for the decay of the WT mouse rod light response (Krispel et al., 2006) (see also Tsang et al., 2006), so that the normally much faster decay of Rh* is not easily observable. We sought to overcome this difficulty by underexpressing rhodopsin kinase at a level low enough to make Rh* turnoff the slowest step in response decay. We show that when kinase is underexpressed in this way, response recovery can be accelerated by background light, and this effect disappears when recoverin is inactivated in the genome. This is to our knowledge the first direct evidence for modulation of Rh* lifetime in intact rods.
Materials and Methods
Animals.
The transgenic construct used for the generation of the R9AP95 mouse line was described by Krispel et al. (2006). The RGS9-1 level in retinal extract from heterozygous R9AP95 animals is approximately threefold that of nontransgenic controls and approaches sixfold in the homozygous R9AP95 background. Retinal morphology appeared normal in R9AP95 animals at 12 month of age (data not shown). The Bark/RK transgenic founders were generated by pronuclear injections into C57BL/6 × BALB/c embryos of a construct consisting of a 4 kb mouse opsin promoter, a mutant rhodopsin kinase (RK) cDNA, and a mouse protamine polyadenylation signal (see Fig. 3A). The mutant RK cDNA was provided by James Inglese of the National Institutes of Health (NIH) Chemical Genomics Center (Rockville, MD) and was a chimeric RK/GRK2 cDNA carrying a stretch of the GRK2 (Bark1) sequence in the vicinity of RK autophosphorylation sites (Palczewski et al., 1995). Eight founders were identified by PCR amplification of a 450 bp product from tail snip DNA by means of two primers: RH1.1: 5′-TCA GTG CCT GGA GTT GCG CTG TGG and RKb1: 5′-TCG TCG GCC GTG TCG TAG TCC TCG. PCR conditions used were as follows: 94°C for 3 min followed by 35 cycles of 94°C for 1 min, 60°C for 1 min, and 72°C for 1 min, and a final extension at 72°C for 10 min. Two lines, 4 and 7, were found to express the transgene in rods at lower levels homogeneously across the retinas. By crossing them into a rhodopsin kinase knock-out (RK−/−) background, the resulting lines, which we call Bark/RK4 and Bark/RK7, expressed only mutant RK in rods. The recoverin knock-out (Rv−/−) animals were kindly provided by Jeannie Chen of the University of Southern California, genotyped by PCR, and verified by western blotting (data not shown). PCR primers used for genotyping are Rec1.5: CGA GCT TCT GCG TGG GCT, Rec7: CCA TGG GGA ACA GCA AGA GCG, and Neo2: TCG CCG CTC CCG ATT CGC AGC GC. The presence of a 320 bp product indicates the WT allele when Rec1.5 and Rec7 is used. The presence of a 275 bp product indicates the targeted allele when Rec1.5 and Neo2 is used. All animals used in the study were pigmented. All studies were performed in accordance to the rules and regulations of the NIH guidelines for research animals and approved by the Institutional Animal Care and Use Committees of Virginia Commonwealth University and the University of California, Los Angeles.
Figure 3.

Generation of BarkRK4 and BarkRK7 transgenic mouse lines. A, Transgenic construct carrying a cDNA called Bark/RK was generated as described in Materials and Methods. Bark/RK, previously named RK/GRK2 (Palczewski et al., 1995), is a mutant GRK1 with 13 aa (underlined) derived from Bark1 around its major autophosphorylation sites (asterisk). B, Determination of GRK1 levels by quantitative Western blotting in BarkRK4 (3) and BarkRK7 (2) mouse lines, versus nontransgenic wild-type control (1). Shown here is a representative Western blot image of GRK1, Bark/RK, and RGS9-1. The mutant Bark/RK has slower mobility during electrophoresis. The level of mutant GRK in BarkRK4 and BarkRK7, is ∼15% and ∼60% of the control level, respectively (n = 3).
Western blotting.
Retinal extracts (10 μg) were resolved by 12% SDS-PAGE and transferred onto nitrocellulose membranes. The membranes were blocked with 10% dry milk in TBST buffer containing 25 mm Tris, pH 7.5, 137 mm NaCl, and 0.05% Tween-20. To detect endogenous and mutant RK, monoclonal G8 antibody (MA1-720, Fisher Scientific) was used at 1:5000 dilution. To detect RGS9-1, rabbit CT318 polyclonal antibody (kindly provided by Mel Simon of the California Institute of Technology, Pasadena, CA) was used at 1:5000 dilution. Species-specific secondary antibodies were used at 1:25,000 dilution, and the signal was detected by enhanced chemiluminescence with the SuperSignal West Dura extended duration substrate kit (Thermo Fisher Scientific). Signals were captured and quantified in the Kodak IS440 imaging system with accompanying 1D image analysis software.
Histology.
Animals were raised in 12/12 light/dark cycles. Enucleated eyeballs were fixed overnight in 2% paraformaldehyde/2.5% glutaraldehyde in 0.1 m cacodylic acid, pH 7.4. After removing the cornea and lens, the eyecup was postfixed in 1% osmium tetroxide in 0.1 m cacodylic acid at room temperature for 120 min. The eyecup was gradually dehydrated in ethanol and impregnated and hardened in Spurr reagent at 70°C (Electron Microscopy Sciences). Gross retinal morphology was assessed in 990 nm semithin sections followed by staining with 0.15% Toluidine Blue in 0.5% sodium borate.
Immunohistochemistry.
Animals were killed by CO2 inhalation. The eyeballs were enucleated in room light and a small incision was made in the cornea. Eyeballs were then immersion-fixed in 4% paraformaldehyde in PBS at 4°C for 12–18 h. After removal of the cornea and lens, the resulting eyecups were cryoprotected in cold 30% sucrose in PBS, embedded in TBS (Triangle Biomedical Sciences), and sectioned at −20°C at 20 μm thickness. The sections were blocked for 60 min at room temperature with 10% goat serum and 0.3% Triton X-100 in 1× PBS (PBT) and incubated for 4–8 h at room temperature with antibodies against rhodopsin kinase (G8, used at 1:100) and cone arrestin (VCU011, raised against peptide sequence, CEEFMQHNSQTQS, derived from mouse cone arrestin, used at 1:5000). Alexa488-conjugated goat anti-mouse antibody and Alexa563-conjugated goat anti-rabbit antibody (both at 1:2000 dilution; Invitrogen) were used to localize RK and cone arrestin, respectively. Fluorescent images were captured with a Zeiss LSM510Meta confocal microscope at the shared EM and microscopy facility of the Department of Anatomy and Neurobiology, Virginia Commonwealth University. Unmodified files were viewed in a separate computer with the LSM Image Browser program (Carl Zeiss Microimaging), and images were exported as *.tif files to Adobe Photoshop for cropping. No post hoc image processing was performed during or after image acquisition.
Suction-electrode recordings.
Methods for recording responses of mouse rods have been given previously (Dizhoor et al., 2008; Woodruff et al., 2008). Animals between 2 and 6 months of age were dark adapted typically for 5 h but for at least 3 h in a light-tight box. Rods were perfused at 37°C with DMEM (D-2902, Sigma Chemical), supplemented with 15 mm NaHCO3, 2 mm Na succinate, 0.5 mm Na glutamate, 2 mm Na gluconate, and 5 mm NaCl, bubbled with 5% CO2, pH 7.4. Data were filtered at 30 Hz (8 pole, Bessel) and sampled at 100 Hz. Flashes of 500 nm light 20 ms in duration were attenuated to different light levels by absorptive neutral density filters. At dim intensities, 10–20 individual responses presented at 5 s intervals were averaged to obtain mean flash responses. At medium intensities 5–10 responses were averaged, and the interflash interval was increased to 10 s. At bright intensities above saturation for the rods, only 3–5 responses were averaged, and the interflash interval was increased to 15–20 s. A 500 nm light was also used for steps of light and backgrounds. Other information about the details of response presentation are given in the figure legends. The value of τD was measured as in Woodruff et al. (2008) by giving a series of 5 flashes at each of between 4 and 7 intensities chosen for each rod to fall within one and a half log units above the flash intensity that just produced saturation of that rod's response amplitude. Flash intensities were in the range of 159–3250 photons · μm−2. The time in saturation (Tsat) was measured as the time from the beginning of the flash to the time at which the mean circulating current recovered to 25% of its dark-adapted value. The value of τD was then calculated as the best-fitting slope of Tsat versus the natural logarithm of the flash intensity. Unless otherwise stated, errors are given as SEMs. Curve fitting and plotting of data were done with the program Origin (OriginLab).
Results
Responses of rhodopsin kinase heterozygotes
In our first attempt to investigate the consequences of reducing kinase expression, we recorded from rods of mice heterozygous for the rhodopsin kinase gene (RK+/−). The heterozygous mice expressed rhodopsin kinase at a level of ∼40–50% of normal (Chen et al., 1999). Rod responses declined somewhat more slowly in RK+/− rods than in WT rods (Fig. 1A,B) (Chen et al., 1999), and the integration time was somewhat longer (Table 1). There was little or no effect on the dark-adapted sensitivity of the response, measured either as SfD (in pA · photon−1 · μm2) or as the half-saturation constant I0. The small differences in Table 1 for these two measures of sensitivity are unlikely to be significant, given the variation we and others have observed in the physiological parameters of mouse rods even from the same strain (Woodruff et al., 2008).
Figure 1.

Effect on response waveform of reducing kinase expression to 40% of normal in RK+/− animals. A–D, Averaged suction-electrode recordings of currents to 20 ms flashes delivered at t = 0 for 10 rods from two WT mice (A); 22 rods from 3 RK+/− mice heterozygous for the rhodopsin kinase gene (B); 13 rods from 2 R9AP95 mice, which overexpress the GAP complex by approximately sixfold (see Materials and Methods) (C); and 19–28 rods from two mice which are both RK+/− and R9AP95 (D). Flash intensities in units of photons · μm−2 were as follows: A: 4, 17, 43, 159, 453, 1120; B: 4, 17, 43, 159, 453, 863, 1871; C: 17, 43, 159, 453, 646, 863, 1120, 1871; D: 4, 17, 43, 159, 453, 1120.
Table 1.
Kinetic and sensitivity parameters of rods
| Animal line | rmax (pA) | SfD (pA photon−1 · μm2) | I0 (photons · μm−2) | ti (ms) | τD (ms) |
|---|---|---|---|---|---|
| WT (21) | 14.5 ± 0.7 | 0.34 ± 0.13 | 27 ± 1 | 262 ± 15 | 185 ± 11 |
| RK+/− (13) | 15.0 ± 1.4 | 0.33 ± 0.03 | 30 ± 1 | 386 ± 34 | 217 ± 15 |
| R9AP95 (12) | 11.5 ± 1.1 | 0.06 ± 0.01 | 105 ± 1 | 109 ± 15 | 54 ± 3 |
| RK+/−:R9AP95 (14) | 16.1 ± 1.0 | 0.20 ± 0.02 | 50 ± 1 | 192 ± 17 | 73 ± 4 |
| Bark/RK4 (18) | 13.3 ± 0.8 | 0.32 ± 0.04 | 28 ± 1 | 396 ± 37 | 141 ± 8 |
| Bark/RK7 (21) | 15.0 ± 0.6 | 0.36 ± 0.03 | 29 ± 1 | 444 ± 19 | 212 ± 10 |
| Bark/RK7:R9AP95 (18) | 11.2 ± 0.6 | 0.18 ± 0.01 | 48 ± 1 | 470 ± 47 | 149 ± 19 |
| Bark/RK7:Rv−/− (15) | 10.9 ± 0.7 | 0.38 ± 0.04 | 24 ± 1 | 349 ± 43 | 213 ± 23 |
All values are mean ± SEM. Values of rmax (maximum response amplitude) were determined cell by cell from responses to saturating flashes; SfD (dark-adapted flash sensitivity), by dividing the peak amplitude of the mean dim-flash response for each cell by the flash intensity; I0 (the intensity required to produce a half-maximal response), from the fit of response-intensity data for each cell to a Boltzmann function in the program Origin; ti (the integration time), from the time integral of the mean dim-flash response for each cell divided by the peak amplitude of the response; and τD (the Pebberberg constant) for dark-adapted rods as described in Materials and Methods.
If the slowing of kinase activity were sufficient to make the rate of decay of Rh* rate limiting for the declining phase of the light response, overexpression of the GTPase accelerating protein (GAP) should have little or no effect on the time course of response decline, since it should make no difference whether the PDE6 turned off at its normal rate or at a rate even faster than normal. We therefore mated RK−/− mice with animals that were RK+/+ but overexpressed the RGS9 anchoring protein (R9AP) and the other GAP complex proteins (RGS9-1 and Gβ5). The line we used (R9AP95) was constructed in the same way as previously (see Materials and Methods and Krispel et al., 2006), and the homozygous R9AP95 animals overexpressed RGS9-1 approximately six times greater than normal (data not shown). Recordings from control R9AP95 rods showed that the decay of the response was greatly accelerated compared to WT (Fig. 1C), and there was also a severalfold decrease in sensitivity (Table 1), presumably the result of the much shorter lifetime of activated PDE in these rods. The dominant time constant of recovery τD (Table 1) was smaller than previously reported for rods overexpressing GAP proteins by fourfold [line 2 of Krispel et al. (2006)], but the decay of the response is so rapid in these rods (and the time in saturation so short) that the determination of τD may have been influenced by the change in outer segment free-Ca2+ during the time course of the measurement (see Woodruff et al., 2002).
In rods that were both RK+/− and R9AP95 (Fig. 1D), responses decayed considerably more rapidly than in rods from mice that were RK+/− with normal levels of GAP proteins (Fig. 1B), though more slowly than animals that were R9AP95 but expressed normal levels of rhodopsin kinase (Fig. 1C). The simplest explanation for this finding is that rhodopsin kinase was not sufficiently underexpressed in RK+/− rods to make Rh* turnoff rate limiting for the decay of the response. The value of τD was larger for RK+/−:R9AP95 rods than for rods from mice that were R9AP95 alone (Table 1), but it was much smaller than in WT rods.
To explore in more detail the effects of underexpression of rhodopsin kinase and overexpression of the GAP proteins on the time course of the light response, we measured cell by cell the time constant τREC by fitting a single-exponential decay function to the declining phase of the response (see for example Krispel et al., 2006). For responses to bright flashes, we were careful to fit only that part of the waveform below 0.5 of the maximum response amplitude (rmax), to avoid the nonlinearity that occurs near saturation (Woodruff et al., 2008). Furthermore, only responses to flashes below 103 photons · μm−2 were included in our analysis, since responses to brighter flashes often showed long tails of recovery that could not be fit to a single exponential. The results of these fits are given in Figure 2. The value of τREC is uniformly greater in RK+/− rods (□) than in WT rods (•) and much smaller in R9AP95 rods with overexpressed GAP proteins (▵). When the RK+/− and R9AP95 genotypes were combined, the values of τREC were much smaller than in rods that were RK+/− alone, indicating that acceleration of the turnoff of PDE accelerated the declining phase of the light response even though rhodopsin kinase had been underexpressed. It is of some interest that the values of τREC in the RK+/−:R9AP95 rods were uniformly greater than in R9AP95 animals with normal levels of kinase. One possible explanation for this finding is that in the R9AP95 animals, PDE turnoff is so rapid that Rh* turnoff becomes rate limiting for the decline of the response, as Krispel et al. (2006) previously hypothesized for rods with lesser levels of GAP overexpression (see also Burns and Pugh, 2009). The rate of turnoff (and the values of τREC and τD) would then be slower in RK+/−:R9AP95 rods than in R9AP95 rods because the rate of Rh* turnoff would be slower when kinase expression was reduced in the RK heterozygotes.
Figure 2.

Value of τREC as function of flash intensity. The declining phase of the flash response was fitted cell by cell with a single exponential decay function; for responses to bright light, only that part of the response which fell below 0.5 of rmax was used to avoid saturation nonlinearly. Data points give mean ± SEM from the following mouse lines (with the number of rods in parentheses): WT (22), RK+/− (11), R9AP95 (12), RK+/−:R9AP95 (11), and the Bark/RK4 line of the RK/Bark chimera (15).
Bark/RK chimeric mice
Since the results in Figure 1 indicate that the level of rhodopsin kinase expression in RK+/− rods is not low enough to make the rate of Rh* turnoff rate limiting for the declining phase of the flash response, we attempted to reduce rhodopsin kinase activity and/or expression levels further by injecting several RK transgenes, hoping that in some of the lines of animals we made, the expression level of the transgene would be significantly lower than normal. Expression levels with this method are whimsical, dependent upon the copy number and the position in the genome where each transgene is inserted, and we were unable to achieve a significantly reduced level of expression with a normal wild-type RK transgene despite considerable effort. We were however fortunate to generate animals significantly underexpressing rhodopsin kinase after injecting a transgene for Bark/RK, a rhodopsin kinase mutant in which 13 aa surrounding the major autophosphorylation sties of GRK1 (Fig. 3A) are changed to those of a related kinase, the β-adrenergic receptor kinase (GRK2 or Bark1). The substitution of these amino acids had relatively little effect on the activity of the kinase, producing a fourfold elevation in Km for ATP, a 1.5-fold increase of Km for Rh*, and a twofold reduction in Vmax in the mutant RK when assayed in vitro (data not shown) (see Palczewski et al., 1995).
By crossing the transgenic animals into the RK−/− background, we were able to characterize two resulting lines: Bark/RK4 and Bark/RK7, with expression levels of the mutant rhodopsin kinase at ∼60% and 15% of the normal RK level (Fig. 3B). In the Bark/RK4 line, the mutant kinase is correctly targeted to the outer segment (Fig. 4A). It also rescues the outer segment morphological defects associated with rhodopsin kinase deletion (Fig. 4B), indicating that the chimeric kinase is functional inside mouse rods. The morphology and abundance of the rods was normal (Fig. 4B), and this was true also of the Bark/RK7 retinas, which were identical in appearance to Bark/RK4 and also rescued the outer segment defects produced by RK deletion (data not shown), even though the level of rhodopsin kinase expression was a factor of 4 lower.
Figure 4.

Normal outer segment morphology in Bark/RK4 mouse retinas. A, Immunohistochemistry showing that mutant GRK1 (green) is expressed only in rods and is targeted to the outer segment (OS) layer in Bark/RK4 animals. Cones, marked by cone arrestin (mCAR, red), do not express the mutant kinase. Blue signals are DAPI stains of outer nuclear layer (ONL) nuclei. IS, Inner segment; OPL, outer plexiform layer. B, Semithin retinal sections showing normal morphology in Bark/RK4 and WT animals as compared to the shortened OS layer found in RK−/− animals. All animals were reared in 12/12 cyclic light. INL, Inner nuclear layer; IPL, inner plexiform layer. Scale bars equal 10 μm.
Recordings from the Bark/RK4 line
Because the protein we have expressed is a chimera of Bark and RK and shows some differences with the native kinase, we sought first to establish whether responses from rods with Bark/RK differed in their sensitivity or response kinetics from rods expressing normal rhodopsin kinase. We began by recording from rods from the Bark/RK4 line of mice, which express the Bark/RK protein at a level of ∼60% of normal (Fig. 3B). The results in Table 1 show that Bark/RK4 rods have nearly normal circulating current and a sensitivity similar to RK+/− animals. The integration time is somewhat longer than in WT, but values of τREC are very similar to those in WT animals (Fig. 2). Although we cannot exclude the possibility that the Bark/RK protein produced some change in the rate or extent of phosphorylation of rhodopsin, the evidence in Table 1 and Figure 2 together with the biochemical and morphological evidence in Figures 3 and 4 indicates that this change is likely to be small and that the Bark/RK protein functions much like normal rhodopsin kinase.
Recordings from rods of Bark/RK7 mice
Since the results from RK+/− rods in Figure 1 indicate that underexpression of RK by 40–50% is not enough to make rhodopsin turnoff rate limiting for the decline of the light response, there seemed little point in pursuing recordings from rods from the Bark/RK4 line, which express the Bark/RK at an even higher level. We therefore turned instead to rods from the Bark/RK7 line, in which the kinase is expressed only at ∼15% of normal (Fig. 3B). The records in Figure 5A (and the data in Table 1) are representative of 90% of the rods from which we recorded. In a small sample of ∼10% of the cells, responses decayed much more slowly than normal. These responses resembled those from RK−/− rods, which completely lack rhodopsin kinase (Chen et al., 1999), suggesting that in a small proportion of the Bark/RK7 rods, there was little or no expression of the chimeric protein. These rods were excluded from our analysis.
Figure 5.
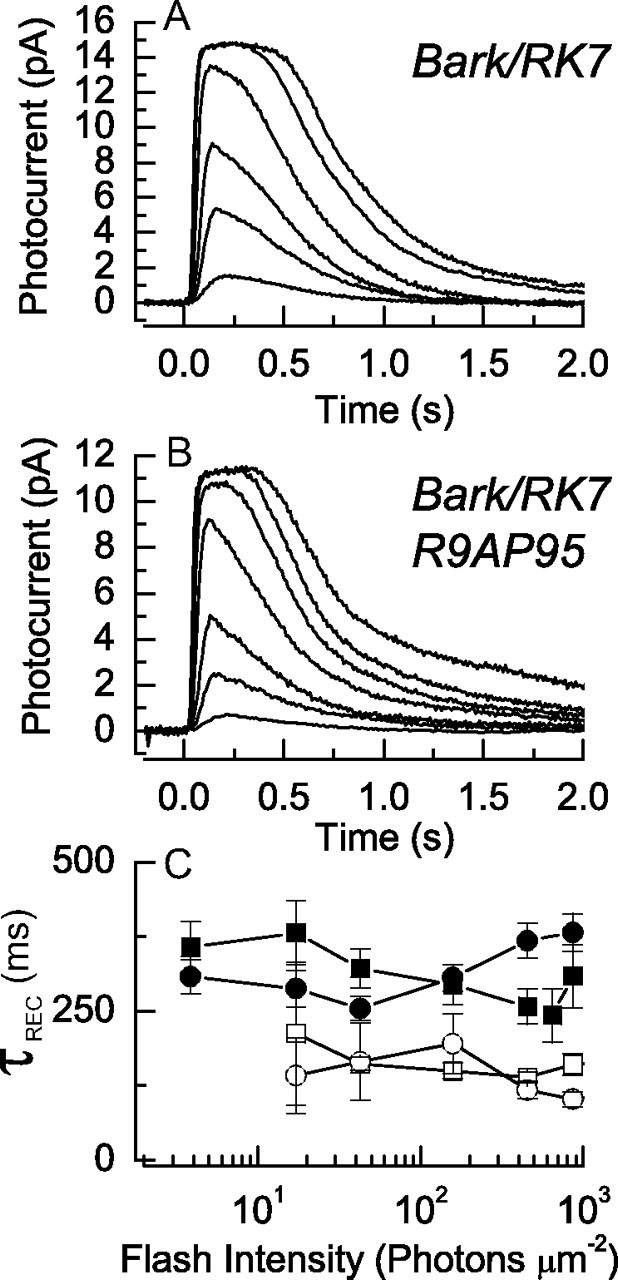
Effect of reducing kinase expression to ∼15% of normal on response waveform in Bark/RK7 animals. A, B, Averaged suction-electrode recordings of currents of 22 rods from two Bark/RK7 mice (A) and 19 rods from three Bark/RK7:R9AP95 mice (B). Intensities of 20 ms flashes in photons · μm−2 delivered at t = 0 were as follows: A, 4, 17, 43, 159, 453, 863; and B, 4, 17, 43, 159, 453, 863, 1120. C, τREC as a function of light intensity (as in Fig. 2) for dark-adapted Bark/RK7 rods (•) and Bark/RK7:R9AP95 rods (■); and for Bark/RK7 rods (○) and Bark/RK7:R9AP95 rods (□) in the presence of a steady background light of 438 photons · μm−2 · s−1.
We next repeated the experiment of Figure 1, mating Bark/RK7 mice with animals that were R9AP95 and overexpressed the GAP proteins. Rods from these animals (Fig. 5B) had a nearly twofold smaller sensitivity compared to rods in mice which were Bark/RK7 alone, which we attribute to the decrease in the lifetime of the PDE. There was, however, no significant change in the time course of decay of the response. The integration times showed no significant difference (see Table 1), and values of τREC for Bark/RK7 and Bark/RK7:R9AP95 also showed no systematic differences (Fig. 5C), in striking contrast to those for RK+/− and RK+/−:R9AP95 rods (Fig. 2). We conclude that in rods from Bark/RK7 mice, the expression level of rhodopsin kinase is sufficiently low to make the decay of Rh* the determining process for the decline of the light response, at least for flashes in the absence of a background light.
The effect of background light on responses of rods from Bark/RK7 mice
We then asked whether the rate of Rh* could be modulated in background illumination. Dark-adapted rods from Bark/RK7 mice (Fig. 6A) were exposed to a steady background light of 438 photons · μm−2 · s−1, and responses were recorded to a series of flash intensities (Fig. 6B). Responses were smaller and less sensitive, as is typical for mouse rods exposed to background illumination (Mendez et al., 2001; Makino et al., 2004; Woodruff et al., 2008). They also showed a more rapid decay. This can be seen more easily in Figure 6C, where we show recordings from parts A and B normalized to their maximum amplitudes at the same flash intensity of 159 photons · μm−2 in the presence (gray trace) and absence (black trace) of the background light. The much more rapid decline in the presence of the background indicates that rhodopsin is being phosphorylated and its activity extinguished more rapidly in the background than when the cell was dark adapted. Figure 6D provides a similar comparison but for responses at a flash intensity of 453 photons · μm−2. The acceleration in the time course of decay can also be seen from the systematic decrease in the values of τREC as a function of light intensity for Bark/RK7 rods, which we give in Figure 5C as the open circles.
Figure 6.

Effect of background light on time course of decay of responses of rods from Bark/RK7 mice. A, B, Suction-electrode recordings of currents averaged from 22 rods of two Bark/RK7 mice in the absence (A) and presence (B) of a background light of 438 photons · μm−2 · s−1. Intensities of 20 ms flashes in photons · μm−2 delivered at t = 0 were as follows: A, 4, 17, 43, 159, 453, 863, 1870; and B, 43, 159, 453, 863, 1871. C, Comparison of waveforms of responses from Bark/RK7 rods for flash of 159 photons · μm−2. Responses of A and B have been normalized rod by rod to the maximum amplitude of the response and then averaged. Black trace is from data in A (dark adapted), gray trace from data in B (in presence of background). D, As in C but for flash of 453 photons · μm−2.
As a further control that rhodopsin turnoff was rate limiting in the experiment of Figure 6, we exposed rods to background illumination from mice that were Bark/RK7 and also R9AP95, that is that underexpressed the kinase and overexpressed the GAP proteins. In Figure 7 we show responses to a series of flashes, whose intensities are given as the numbers in each of the panels to the left in units of photons · μm−2. Each panel shows responses to this same flash intensity in darkness (black traces) and in the presence of a moderate intensity background light (gray traces). In the panels to the right, we have normalized cell by cell the responses on the left by the peak amplitude of the response to each of the flashes and then averaged the normalized responses. The results are similar to those in Figure 6: background light accelerates the return of the current to its resting level. This can also be seen from the values of τREC as a function of light intensity, given in Figure 5C as the filled squares in dark-adapted rods and open squares in the presence of the background.
Figure 7.

Effect of background light on time course of decay of responses of rods from Bark/RK7:R9AP95 mice. Left, Suction-electrode recordings of currents averaged from 19 rods of 3 Bark/RK7:R9AP95 mice. All responses are to 20 ms flashes delivered at t = 0 in absence (black traces) and presence (gray traces) of steady background light of 438 photons · μm−2 · s−1. Intensities of flashes (IF) increased from top to bottom and are given in each panel in units of photons · μm−2. Right, Responses in left panels have been normalized cell by cell to the peak amplitude of the response and then averaged. Note more rapid decay of gray traces recorded in background light.
Role of recoverin in mediating modulation of Rh* lifetime
Since the small-molecular-weight Ca2+-binding protein recoverin has been shown to modulate the catalytic rate of rhodopsin kinase, we asked whether the acceleration of the time course of decay produced by background light in rods of Bark/RK7 mice could be affected by removal of the recoverin gene. We therefore mated Bark/RK7 mice with Rv−/− mice in which the gene for recoverin had been knocked out (Makino et al., 2004). The results of those recordings are given in Figure 8, in a format identical to the one for Figure 6. Responses from dark-adapted rods (Fig. 8A) are similar to those from Bark/RK7 mice containing the recoverin gene (Fig. 6A), though there are small differences in sensitivity and a modest reduction both in integration time (Table 1) and in τREC (data not shown). Background lights produce a characteristic decrease in sensitivity and response amplitude in the Bark/RK7:Rv−/− rods (Fig. 8B), but when responses were normalized and their waveforms compared at the same flash intensities in the presence and absence of the background (Fig. 8C,D), there was no apparent acceleration of response decay. Similar results were observed at all the flash intensities at which this comparison was made. In this same series of experiments, we recorded responses to a single dim and a single bright flash intensity at a series of different background levels from 13 to 438 photons · μm−2 · s−1, and the responses at each of the flash intensities were normalized and superimposed (data not shown). There was again little or no change in the time course of decay. The absence of recoverin greatly reduced or eliminated the light-induced acceleration of Rh* turnoff.
Figure 8.

Effect of background light on time course of decay of responses of rods from Bark/RK7:Rv−/− mice. A, B, Suction-electrode recordings of currents averaged from 8–11 rods of four Bark/RK7:Rv−/− mice in the absence (A) and presence (B) of a background light of 438 photons · μm−2 · s−1. Intensities of 20 ms flashes in photons · μm−2 delivered at t = 0 were as follows: A, 4, 17, 43, 159, 453, 863, 1870; and B, 17, 43, 159, 453, 863, 1870. C, Comparison of waveforms of responses from Bark/RK7:Rv−/− rods for flash of 159 photons · μm−2. Responses of A and B have been normalized rod by rod to the maximum amplitude of the response and then averaged. D, As in C but for flash of 453 photons · μm−2.
Discussion
Our experiments show that if rhodopsin kinase expression is sufficiently reduced, the overexpression of GAP proteins no longer accelerates the decay of the photoreceptor light response (Fig. 5) as it has been shown to do in WT rods (Krispel et al., 2006). Since underexpression of rhodopsin kinase would decrease the rate of rhodopsin phosphorylation, which is known to be a necessary step in the turning off of Rh* (J. Chen et al., 1995; C.-K. Chen et al., 1999), the simplest explanation of our result is that underexpression of the kinase makes the rate of phosphorylation and decay of Rh* rate limiting for the decay of the rod light response. On this assumption, our data show that the rate of Rh* turnoff is accelerated in background light (Figs. 6, 7), consistent with previous observations that an early step in response turnoff is under control of outer segment Ca2+ concentration (Matthews, 1997; Matthews et al., 2001). Furthermore, the observation that this effect disappears when the gene for recoverin is inactivated (Fig. 8) supports the original proposal of Kawamura (1993), that the Ca-binding protein recoverin/S-modulin regulates the rate of rhodopsin phosphorylation.
Since we were unable to produce animals with underexpression of normal rhodopsin kinase, our results are from mice expressing a chimeric protein having 13 aa of Bark1 (Fig. 3A). This chimeric kinase can however phosphorylate rhodopsin in vitro with kinetic parameters only slightly different from those of the normal protein (Palczewski et al., 1995). It can moreover be correctly transported to the outer segments (Fig. 4A) and prevent the morphological anomalies that are produced by the absence of a normal rhodopsin kinase gene (Fig. 4B). Rods from Bark/RK4 mice expressing the chimeric protein at ∼60% of the level of WT protein respond to light much like WT mice with a somewhat greater integration time but similar values for exponential time constants of decay (Fig. 2). Since the kinetic parameters even of WT rod responses show considerable variability (see for example Woodruff et al., 2008), the comparison of responses from animals of different genetic backgrounds can never be entirely convincing. Although we do not exclude the possibility that the rate or the sites of rhodopsin phosphorylation were subtly altered in rods expressing the chimeric kinase, it seems nevertheless unlikely that such differences would have been large enough to alter our principal conclusions.
One puzzling feature of our data is the discrepancy between value of τREC in Figure 5C for Bark/RK7 mice, which is of the order of 350–400 ms for dark-adapted responses, and the mean dark-adapted value of τD for these rods, which is only approximately half as great (212 ms, Table 1). There is a similar discrepancy for the RK+/− and Bark/RK7:R9AP95 rods. This was unexpected, since the values of τREC and τD for mouse rods are normally quite close (see, for example, Woodruff et al., 2008, their supplemental Fig. 1), probably because both τREC and τD are usually determined by the same process, namely the GAP-mediated decay of PDE activity (Krispel et al., 2006).
In mice in which the rate of rhodopsin phosphorylation has been reduced, τREC is likely to be influenced by the rate of decay of Rh*, which we have made artificially slow by underexpressing the kinase. The value of τD in these rods might nevertheless be shorter, since to measure τD, we exposed the rods to saturating light, which produces a decrease in Ca2+ concentration in the rod outer segment. This Ca2+ decrease might decrease the lifetime of Rh*, accounting for the reduced value of τD. If this were true, we might expect the discrepancy to be eliminated in rods lacking recoverin, since the Ca2+-dependent acceleration in Rh* turnoff would no longer occur; but that is not what we observed. Our data show that responses of Bark/RK7:Rv−/− to dim lights turn off only marginally faster than responses of Bark/RK7 rods (compare Figs. 6A and 8A), even though the values of τD are nearly identical (Table 1) and approximately a factor of 2 smaller than the τREC's of either receptor. The reason for this is unknown but suggests that effects of recoverin deletion may be more complicated than present models presuppose.
Although our experiments do not permit us to decide what determines the limiting time constant of decay τD in Bark/RK7 or Bark/RK7:Rv−/− rods, in Bark/RK7:R9AP95 rods the picture is somewhat clearer. Here the value of τD is significantly larger than in rods that are R9AP95 and either RK+/− or RK+/+ (see Table 1), showing (1) that the rate of turnoff of Rh* is rate limiting in Bark/RK7:R9AP95 mice and (2) that the lifetime of Rh* is longer in Bark/RK7 than in RK+/− or RK+/+, even after exposure to saturating light. This is presumably because the kinase concentration is lower in the Bark/RK7 rods. The small changes in kinetic parameters of the Bark/RK enzyme as determined in vitro may also play some role in reducing the Rh* phosphorylation rate, but the extent of this effect cannot be determined from our experiments and would in any case have little effect on the conclusions of our study.
It is of some interest that in rods in which RGS9-1 and the other GAP proteins are overexpressed by sixfold but kinase concentration is normal, the mean limiting time constant τD is 54 ms (Table 1), significantly lower than the limiting time constant of any rods previously recorded (Krispel et al., 2006; Woodruff et al., 2008). This result is consistent with the prediction of a recent model of PDE turnoff (Burns and Pugh, 2009), which indicates that the time constant of decay of WT mouse rod Rh* is smaller than originally thought (Krispel et al., 2006) and likely to be 53 ms or less. In recent experiments (in preparation), we have shown that both τD and τREC can decrease even further when R9AP95 rods are exposed to background light. This may reflect a light-dependent reduction in Rh* lifetime, but we cannot at present exclude the possibility that it is caused instead by modulation of PDE turnoff (Woodruff et al., 2008), and that both Rh* and PDE are extinguished with similar time constants in R9AP95 rods. Future experiments may allow us to decide between these alternative explanations.
We were surprised by the small effect of kinase underexpression on the sensitivity of the rod response; consideration of the data in Table 1 shows that there was little or no difference in the dark-adapted sensitivities or half-saturation constants for WT, RK+/−, Bark/RK4, and Bark/RK7 rods, despite the sevenfold difference in kinase expression among these animal strains. Since activation of transducin by Rh* is thought to proceed at a steady rate of the order of a few hundred activated transducins per second (Leskov et al., 2000; Burns and Pugh, 2009), a decrease in kinase concentration should decrease the rate of phosphorylation and increase the lifetime of Rh*. This should make the rod more sensitive. A similar observation was made by Krispel et al. (2006), who increased kinase expression and found this also to have no appreciable effect on the values of dim flash sensitivity and half-saturation constant. Although complete deletion of rhodopsin kinase can increase sensitivity by twofold to threefold (Chen et al., 1999; Mendez et al., 2000), the sensitivity of the rod is apparently unaffected by kinase concentration over a rather wide range; the reasons for this are presently unknown.
We have shown that when recoverin is inactivated in Bark/RK7 rods, background light no longer accelerates the decay of the response (Fig. 8). These experiments provide support for the notion that recoverin/S-modulin plays a role in the modulation of Rh* (Kawamura, 1993), but that is unlikely to be the only function of this protein. The knocking out of recoverin alters response waveform even in rods with normal kinase expression (Makino et al., 2004), which may indicate that recoverin can also contribute to modulation of PDE or GAP function. Furthermore, recoverin has been shown to be present in the rod inner segment and to enhance signal transfer between rods and rod bipolar cells (Sampath et al., 2005). Further experiments may help clarify the role of this enigmatic protein in the physiology of the photoreceptor.
Footnotes
This work was supported by National Institutes of Health Grants EY01844 (to G.L.F.) and EY013811 (to C.-K.C.). We thank the transgenic mouse core facility at Virginia Commonwealth University for generating transgenic mouse lines. We also thank Alapakkam Sampath and Daniel Tranchina for reading an earlier version of this manuscript.
References
- Ames JB, Levay K, Wingard JN, Lusin JD, Slepak VZ. Structural basis for calcium-induced inhibition of rhodopsin kinase by recoverin. J Biol Chem. 2006;281:37237–37245. doi: 10.1074/jbc.M606913200. [DOI] [PubMed] [Google Scholar]
- Burns ME, Pugh EN., Jr RGS9 concentration matters in rod phototransduction. Biophys J. 2009;97:1538–1547. doi: 10.1016/j.bpj.2009.06.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C-K, Inglese J, Lefkowitz RJ, Hurley JB. Ca(2+)-dependent interaction of recoverin with rhodopsin kinase. J Biol Chem. 1995;270:18060–18066. doi: 10.1074/jbc.270.30.18060. [DOI] [PubMed] [Google Scholar]
- Chen C-K, Burns ME, Spencer M, Niemi GA, Chen J, Hurley JB, Baylor DA, Simon MI. Abnormal photoresponses and light-induced apoptosis in rods lacking rhodopsin kinase. Proc Natl Acad Sci U S A. 1999;96:3718–3722. doi: 10.1073/pnas.96.7.3718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Makino CL, Peachey NS, Baylor DA, Simon MI. Mechanisms of rhodopsin inactivation in vivo as revealed by a COOH-terminal truncation mutant. Science. 1995;267:374–377. doi: 10.1126/science.7824934. [DOI] [PubMed] [Google Scholar]
- Dizhoor AM, Woodruff ML, Olshevskaya EV, Cilluffo MC, Cornwall MC, Sieving PA, Fain GL. Night blindness and the mechanism of constitutive signaling of mutant G90D rhodopsin. J Neurosci. 2008;28:11662–11672. doi: 10.1523/JNEUROSCI.4006-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erickson MA, Lagnado L, Zozulya S, Neubert TA, Stryer L, Baylor DA. The effect of recombinant recoverin on the photoresponse of truncated rod photoreceptors. Proc Natl Acad Sci U S A. 1998;95:6474–6479. doi: 10.1073/pnas.95.11.6474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fain GL. Sensory transduction. Sunderland, MA: Sinauer; 2003. [Google Scholar]
- Fain GL, Matthews HR, Cornwall MC, Koutalos Y. Adaptation in vertebrate photoreceptors. Physiol Rev. 2001;81:117–151. doi: 10.1152/physrev.2001.81.1.117. [DOI] [PubMed] [Google Scholar]
- Gray-Keller MP, Polans AS, Palczewski K, Detwiler PB. The effect of recoverin-like calcium-binding proteins on the photoresponse of retinal rods. Neuron. 1993;10:523–531. doi: 10.1016/0896-6273(93)90339-s. [DOI] [PubMed] [Google Scholar]
- Higgins MK, Oprian DD, Schertler GF. Recoverin binds exclusively to an amphipathic peptide at the N terminus of rhodopsin kinase, inhibiting rhodopsin phosphorylation without affecting catalytic activity of the kinase. J Biol Chem. 2006;281:19426–19432. doi: 10.1074/jbc.M602203200. [DOI] [PubMed] [Google Scholar]
- Kawamura S. Rhodopsin phosphorylation as a mechanism of cyclic GMP phosphodiesterase regulation by S-modulin. Nature. 1993;362:855–857. doi: 10.1038/362855a0. [DOI] [PubMed] [Google Scholar]
- Klenchin VA, Calvert PD, Bownds MD. Inhibition of rhodopsin kinase by recoverin. Further evidence for a negative feedback system in phototransduction. J Biol Chem. 1995;270:16147–16152. doi: 10.1074/jbc.270.27.16147. [DOI] [PubMed] [Google Scholar]
- Krispel CM, Chen D, Melling N, Chen YJ, Martemyanov KA, Quillinan N, Arshavsky VY, Wensel TG, Chen C-K, Burns ME. RGS expression rate-limits recovery of rod photoresponses. Neuron. 2006;51:409–416. doi: 10.1016/j.neuron.2006.07.010. [DOI] [PubMed] [Google Scholar]
- Leskov IB, Klenchin VA, Handy JW, Whitlock GG, Govardovskii VI, Bownds MD, Lamb TD, Pugh EN, Jr, Arshavsky VY. The gain of rod phototransduction: reconciliation of biochemical and electrophysiological measurements. Neuron. 2000;27:525–537. doi: 10.1016/s0896-6273(00)00063-5. [DOI] [PubMed] [Google Scholar]
- Makino CL, Dodd RL, Chen J, Burns ME, Roca A, Simon MI, Baylor DA. Recoverin regulates light-dependent phosphodiesterase activity in retinal rods. J Gen Physiol. 2004;123:729–741. doi: 10.1085/jgp.200308994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews HR. Actions of Ca2+ on an early stage in phototransduction revealed by the dynamic fall in Ca2+ concentration during the bright flash response. J Gen Physiol. 1997;109:141–146. doi: 10.1085/jgp.109.2.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews HR, Cornwall MC, Crouch RK. Prolongation of actions of Ca2+ early in phototransduction by 9-demethylretinal. J Gen Physiol. 2001;118:377–390. doi: 10.1085/jgp.118.4.377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendez A, Burns ME, Roca A, Lem J, Wu LW, Simon MI, Baylor DA, Chen J. Rapid and reproducible deactivation of rhodopsin requires multiple phosphorylation sites. Neuron. 2000;28:153–164. doi: 10.1016/s0896-6273(00)00093-3. [DOI] [PubMed] [Google Scholar]
- Mendez A, Burns ME, Sokal I, Dizhoor AM, Baehr W, Palczewski K, Baylor DA, Chen J. Role of guanylate cyclase-activating proteins (GCAPs) in setting the flash sensitivity of rod photoreceptors. Proc Natl Acad Sci U S A. 2001;98:9948–9953. doi: 10.1073/pnas.171308998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otto-Bruc AE, Fariss RN, Van Hooser JP, Palczewski K. Phosphorylation of photolyzed rhodopsin is calcium-insensitive in retina permeabilized by alpha-toxin. Proc Natl Acad Sci U S A. 1998;95:15014–15019. doi: 10.1073/pnas.95.25.15014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palczewski K, Ohguro H, Premont RT, Inglese J. Rhodopsin kinase autophosphorylation. Characterization of site-specific mutations. J Biol Chem. 1995;270:15294–15298. doi: 10.1074/jbc.270.25.15294. [DOI] [PubMed] [Google Scholar]
- Pugh EN, Jr, Nikonov S, Lamb TD. Molecular mechanisms of vertebrate photoreceptor light adaptation. Curr Opin Neurobiol. 1999;9:410–418. doi: 10.1016/S0959-4388(99)80062-2. [DOI] [PubMed] [Google Scholar]
- Sampath AP, Strissel KJ, Elias R, Arshavsky VY, McGinnis JF, Chen J, Kawamura S, Rieke F, Hurley JB. Recoverin improves rod-mediated vision by enhancing signal transmission in the mouse retina. Neuron. 2005;46:413–420. doi: 10.1016/j.neuron.2005.04.006. [DOI] [PubMed] [Google Scholar]
- Strissel KJ, Lishko PV, Trieu LH, Kennedy MJ, Hurley JB, Arshavsky VY. Recoverin undergoes light-dependent intracellular translocation in rod photoreceptors. J Biol Chem. 2005;280:29250–29255. doi: 10.1074/jbc.M501789200. [DOI] [PubMed] [Google Scholar]
- Tsang SH, Woodruff ML, Chen C-K, Yamashita CY, Cilluffo MC, Rao AL, Farber DB, Fain GL. GAP-independent termination of photoreceptor light response by excess γ subunit of the cGMP-phosphodiesterase. J Neurosci. 2006;26:4472–4480. doi: 10.1523/JNEUROSCI.4775-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodruff ML, Sampath AP, Matthews HR, Krasnoperova NV, Lem J, Fain GL. Measurement of cytoplasmic calcium concentration in the rods of wild-type and transducin knock-out mice. J Physiol. 2002;542:843–854. doi: 10.1113/jphysiol.2001.013987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodruff ML, Janisch KM, Peshenko IV, Dizhoor AM, Tsang SH, Fain GL. Modulation of phosphodiesterase6 turnoff during background illumination in mouse rod photoreceptors. J Neurosci. 2008;28:2064–2074. doi: 10.1523/JNEUROSCI.2973-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]