

Abstract
Etiology of endometrial cancer (EMC) is not fully understood. Animal models with rapidly and spontaneously developing EMC will help explore mechanisms of cancer initiation and progression. Pten+/− mice are currently being used as a model to study EMC. These females develop atypical endometrial hyperplasia of which ~20% progresses to EMC. In addition, tumors develop in other organs, complicating the use of this model to specifically study EMC. Here we show that conditional deletion of endometrial Pten results in EMC in all female mice as early as one month of age with myometrial invasion occurring by three months. In contrast, conditional deletion of endometrial p53 had no phenotype within this time frame. While mice with endometrial Pten deletion had a lifespan of about five months, mice with combined deletion of endometrial Pten and p53 had a shorter lifespan with an exacerbated disease state. Such rapid development of EMC from homozygous loss of endometrial Pten suggests that this organ is very sensitive to this tumor suppressor gene for tumor development. All lesions at early stages exhibited elevated Cox-2 and phospho-Akt levels, hallmarks of solid tumors. More interestingly, levels of two microRNAs miR-199a* and miR-101a that post-transcriptionally inhibit Cox-2 expression, were downregulated in tumors in parallel with Cox-2 upregulation. This mouse model in which the loxP-Cre system has been used to delete endometrial Pten and/or p53 allows us to study in detail the initiation and progression of EMC. These mouse models have the added advantage since they mimic several features of human EMC.
Keywords: Endometrial cancer, mouse model, Pten, p53, Cox-2
INTRODUCTION
Endometrial cancer (EMC) is a common gynecological malignancy, affecting approximately 40,000 women and leading to about 7,000 deaths each year in the United States alone (1). The etiology of EMC is not yet fully understood, although there is evidence that endocrine and genetic factors contribute to its initiation and progression (1). EMC is categorized into two major types, Type I and II, with approximately 85% of EMCs classified as Type I. Type I EMC is divided into well, moderately and poorly differentiated grades, depending on the degree of solid tumor growth. Type II EMC, while uncommon, is more aggressive (1, 2).
In human Type I EMCs, the most common genetic mutations are detected in the Pten (phosphatase and tensin homolog) gene (1, 3). Pten mutations are observed in 30–80% of Type I EMCs and in ~20% of complex atypical hyperplasia (CAH), a precursor to Type I EMC. Mutations of p53 are also found in Type I EMC, but this alteration occurs in approximately 50% of poorly differentiated carcinomas and some moderately differentiated Type I EMCs, suggesting that p53 mutations are later events that contribute to progression of the disease (1). On the other hand, the majority of Type II EMCs which are more aggressive and less common mainly contain p53, but not Pten, mutations (1, 2).
The most widely used animal model for studying EMC is Pten heterozygous mice (3). While Pten homozygous mice are unavailable due to their embryonic lethality, all Pten heterozygous females develop atypical endometrial hyperplasia with 20% progressing to well-differentiated EMC by 10 months of age. The timing and incidence of hyperplasia and carcinoma vary from mouse to mouse in this model (3, 4). Furthermore, Pten heterozygous mice also develop other types of cancer, creating limitations to exclusively study endometrial cancer in this model. With respect to p53, both p53 heterozygous and homozygous mice develop many types of cancers, with most homozygous mice dying by 6 months of age due to development of widespread lymphoma (5–7). However, EMCs are rarely observed in p53 null mice (5–7). Therefore, endometrial-specific Pten and/or p53 deleted mice would be more preferred models to study endometrial-specific cancer.
Pten is a dual-specificity phosphatase with phosphatidylinositol-3,4,5-phosphate (PIP3) a major substrate. PIP3 is dephosphorylated to phosphatidylinositol-4,5-phosphate (PIP2) by Pten, an event that opposes phosphatidylinositol-3-kinase (PI3K) signaling (8). Loss of Pten function, resulting in stimulation of PI3K signaling, is widely found in many types of cancers. PI3K activates Akt, a serine-threonine kinase, and phosphorylated/activated Akt regulates a variety of target molecules that control cell survival and growth. It was recently shown that introducing Akt deficiency in Pten heterozygous mice impedes tumor development including that of EMC (9), suggesting pAkt to be an immediate downstream molecule of Pten. Cox-2 is a major target of Akt signaling in cancer, overexpressed in tumors and carcinomas of the colon, breast and lung (10). We and others have shown that human EMCs and endometria with hyperplasia express elevated Cox-2 (11–13). Moreover, Cox-2 expression is also elevated in human EMC cell lines harboring Pten mutations and activated Akt (14, 15). These studies collectively indicate that elevated pAkt and Cox-2 levels resulting from Pten mutations probably contribute to EMC development.
In this study, endometrial-specific Pten and/or p53 deletion were generated by crossing floxed Pten (PtenloxP/loxP) and/or floxed p53 (p53loxP/loxP) mice with mice expressing Cre under the regulation of the progesterone receptor promoter (PRcre/+) (16). We found that 100% of PtenloxP/loxP/PRcre/+ (Ptenpr−/−) and PtenloxP/loxP/p53loxP/loxP/PRcre/+ (Ptenpr−/−/p53pr−/−) mice develop in situ carcinoma as early as three weeks to one month of age. While the development of hyperplasia was similar between Ptenpr−/− and Ptenpr−/−/p53pr−/− mice, the loss of both Pten and p53 exacerbated the disease state and was associated with a shorter lifespan. In contrast, p53loxP/loxP/PRcre/+ (p53pr−/−) mice had apparently normal endometrial histology even through 5 months of age. We found that Cox-2 and phospho-Akt (pAkt) were upregulated in regions with hyperplasia and carcinoma in both Ptenpr−/− and Ptenpr−/−/p53pr−/− uteri. Additionally, microRNAs miR-199a* and miR-101a that are known to post-transcriptionally impede Cox-2 expression in the mouse uterus and human cancer cell lines (17), were downregulated in Ptenpr−/− and Ptenpr−/−/p53pr−/− uteri. These mouse models have provided valuable information on genetic, temporal and dynamic aspects of EMC initiation and progression. Our findings present an opportunity for further study, especially with regards to drug development focused at EMC treatment at early stages.
MATERIALS and METHODS
Mice
All mice were housed in the Institutional Animal Care Facility according to NIH and institutional guidelines for laboratory animals. PtenloxP/loxP mice were obtained from Jackson Laboratory (Bar Harbor, ME), and p53loxP/loxP mice were obtained from the Mouse Models of Human Cancers Consortium (MMHCC, NCI, Rockville, MD). PR-Cre (PRcre/+) (C57BL6/129SV) mice were obtained from John B. Lydon (Baylor College of Medicine, Houston, TX). PRcre/+ mice have Cre recombinase introduced into exon 1 of the PR gene (16). Therefore, Cre expression is regulated by the endogenous PR promoter. PR-Cre heterozygous (PRcre/+) mice are phenotypically indistinguishable from wild-type mice (16). PtenloxP/loxP, p53loxP/loxP and PRcre/+ mice were generated as described previously (16, 18, 19). PCR analysis of tail genomic DNA determined the genotypes of mutant mice. Standard breeding strategies were used to generate conditional deletion of Pten and/or p53 using transgenic mice (20). Bilateral ovariectomy was performed under appropriate anesthesia. Since the currently used Ptenpr−/−, p53pr−/−, and Ptenpr−/−/p53pr−/− mouse models are on mixed backgrounds, we consistently used littermate floxed (WT) mice in all of our experiments. This ensures that results obtained within each experimental set are meaningful.
Western blot analysis
Tissue samples were prepared as previously described (21). After measuring protein concentrations, supernatants mixed with SDS sample buffer were boiled for 5 min. The samples were run on 10% SDS-PAGE gels under reducing conditions and transferred onto nitrocellulose membranes. Membranes were blocked with 10% milk in TBST, and probed with antibodies to Pten (Cell Signaling, Danvers, MA), pAkt (Cell Signaling), p53 (Cell Signaling), Cox-2 (Cayman, Ann Arbor, MI) and actin (SantaCruz, Santa Cruz, CA) overnight at 4°C. After washing, blots were incubated in peroxidase-conjugated donkey-anti-goat IgG, donkey-anti-rabbit IgG or donkey-anti-mouse IgG (Jackson Immuno Research Laboratories, Inc, West Grove, PA). For 2nd antibody detection of p53, mouse-IgG Trueblot (eBioscience, San Diego, CA) was used to remove IgG signals. All signals were detected using chemiluminescent reagents (GE Healthcare, Piscataway, NJ). Actin served as a loading control.
Immunohistochemistry
Immunohistochemistry was performed as previously described by us (21). In brief, formalin-fixed paraffin-embedded sections (6 μm) were subjected to immunostaining using antibodies to cytokeratin 8 (Developmental Studies Hybridoma Bank, Iowa City, IA), Pten, pAkt, Cox-2, Ki-67 (Lab Vision Corporation, Fremont, CA) or α-SMA (Abcam, Cambridge, MA). After deparaffinization and hydration, sections were subjected to antigen retrieval by autoclaving in 10 mM sodium citrate solution (pH = 6) for 10 min. A Histostain-Plus kit (Invitrogen, Carlsbad, CA) was used to visualize antigens.
In situ hybridization
cDNA clones for Cox-2 have previously been described (22, 23). cDNA clones for p53 were generated by RT-PCR. Sense and antisense 35S-labeled cRNA probes were generated using Sp6 or T7 RNA polymerases. Frozen uterine sections were used for in situ hybridization. Uteri from 10-day old mice were placed inside a small groove made on a piece of kidney as a holding cassette since they are extremely tiny, and then snap-frozen for cryosectioning. In situ hybridization was performed as previously described (21). Sections hybridized with sense probes did not exhibit any positive signals and served as negative controls.
RT-PCR
RT-PCR was performed as previously described (21). Primers to detect p53 are 5′ ACAGGACCCTGTCACCGAGACC 3′ and 5′ GACCTCCGTCATGTGCTGTGAC 3′.
Northern hybridization
Northern blotting of miRNAs was performed as previously described (17). Total RNA (20 μg) was resolved through a 12.5% urea-polyacrylamide gel, transferred onto GeneScreen Plus membranes (PerkinElmer, Waltham, MA), and UV-cross-linked. Antisense oligonucleotide (IDT, Coralville, IA) was labeled with 32P with a StarFire labeling kit (IDT). Prehybridization, hybridization and washing were performed at 42°C.
RESULTS
PR-Cre efficiently deletes endometrial Pten and p53
Our objective was to study endometrial specific cancer by conditional deletion of Pten, p53 or both in the mouse endometrium. We therefore crossed Pten (PtenloxP/loxP), p53 (p53loxP/loxP) or Pten/p53 (PtenloxP/loxP/p53loxP/loxP) floxed mice with PRcre/+ mice. We have previously used PRcre/+ mice to delete endometrial genes to examine the roles of Hbegf and Bmp2 in pregnancy (20, 24). As shown in Supplemental Figure 1, endometrial Pten and p53 are deleted by PR-driven Cre activity as confirmed by conventional genotyping.
We next confirmed loss of Pten protein in Ptenpr−/− uterine lysates by Western blotting. As shown in Figure 1A upper panel, Pten levels were drastically reduced in Ptenpr−/− uteri from three weeks of age with concomitant increases in Akt phosphorylation (pAkt). This observation of Akt activation with the loss of Pten is consistent withprevious findings in other systems (25). We also used immunohistochemistry to monitor cell-specific downregulation of Pten and upregulation of Akt activation (Figure 1A lower panel). Pten levels were efficiently downregulated in uteri of Ptenpr−/− mice from as early as 10 days of age substantiating the Western blot results. While levels of pAkt were low to undetectable in wild-type uteri with normal levels of Pten, pAkt levels were remarkably upregulated in Ptenpr−/− uteri in both 10-day and 3-week old mice (Figure 1A lower panel). Similar analyses were carried out using Ptenpr−/−/p53pr−/− uteri. Western blotting and in situ hybridization showed decreased p53 expression in Ptenpr−/−/p53pr−/− uteri as expected (Figure 1B). Loss of both Pten and p53 in these mice was also accompanied by heightened levels of pAkt (Figure 1B, left panel). Loss of p53 in p53pr−/− uteri was confirmed by RT-PCR (Figure 1C). Collectively, PR-Cre efficiently deletes endometrial Pten and p53.
Figure 1. PR-Cre efficiently deletes endometrial Pten and p53.

A) Western blot analysis of Pten, pAkt, and total Akt in wild-type and Ptenpr−/− uteri (upper panel). Immunohistochemistry of Pten and pAkt in 10-day (10 d) and 3-week (3 wk) old Ptenpr−/− and wild-type uteri (lower panel). Bar, 200 μm. le, luminal epithelium; ge, glandular epithelium; s, stroma; myo, myometrium. B) Western blot analysis of p53, Pten, pAkt, and total Akt in wild-type and Ptenpr−/−/p53pr−/− uteri (left panel). Actin serves as a loading control. In situ hybridization of p53 in 3-week old wild-type and Ptenpr−/−/p53pr−/− uteri (right panel). Because of extremely tiny sizes of uteri, all wild-type and Ptenpr−/− uteri from 10-day 3-week old mice were placed into small grooves made in kidney slices to serve as cassettes for making frozen sections. Kid, kidney; Bar, 400 μm. C) RT-PCR of p53 in wild-type and p53pr−/− uteri. β-Actin is a house keeping gene.
Endometrial deletion of Pten induces endometrium-specific cancer
Our next objective was to examine whether endometrial deletion of Pten in mice results in EMC. To address this question, we examined histology of Ptenpr−/− and/or p53pr−/− uteri. Ptenpr−/− uteri showed endometrial epithelial hyperplasia as early as 10 days of age (Figure 2A). Staining with cytokeratin 8 (CK8), an epithelial cell-specific marker, showed luminal and glandular epithelial hyperplasia in Pten-deleted uteri from mice as young as 10 days. Hyperplasia progressed to carcinoma by one month of age with invasion into the myometrium occurring by 3 months (Figure 2A). Myometrial invasion was confirmed by α-smooth muscle actin (α-SMA) immunostaining (Supplemental Figure 2). Detailed characterization of tumor types and grades are shown (Table 1). These results showthat conditional deletion of endometrial Pten specifically results in EMC rapidly with 100% penetrance, a much more drastic phenotype than observed in mice heterozygous for Pten deletion.
Figure 2. Uteri from Ptenpr−/− and Ptenpr−/−/p53pr−/− show aberrant histology and their loss affects survival.

Histology of A) Ptenpr−/− and B) Ptenpr−/−/p53pr−/− uteri. Hematoxylin and Eosin (H & E) and cytokeratin 8 (CK8) staining of A) Ptenpr−/− and B) Ptenpr−/−/p53pr−/− uteri in mice at various ages (10 days, 3 weeks, 1 month, 2 months and 3 months). C) Effects of endometrial loss of Pten and/or p53 on survival. Wild-type, Ptenpr−/−, p53pr−/− and Ptenpr−/−/p53pr−/− mice were monitored for any sign of sickness for a total of 250 days. We sacrificed mice with any sign of sickness or discomfort and the day of sacrifice was recorded.
Table 1.
Endometrial cancers in mice with uterine loss of Pten and/or p53.
| Age (days) | Genotype | No. of Mice examined | Uterine histology | No. of mice with Indicated histology (%) |
|---|---|---|---|---|
| 10 | Pten +/+ | 6 | Normal epithelium (no lesion) | 6 (100%) |
| Pten pr−/− | 6 | Hyperplasia with cytological atypia | 6 (100%) | |
| Pten +/+/p53 +/+ | 6 | Normal epithelium (no lesion) | 6 (100%) | |
| Pten pr−/− p53 pr−/− | 6 | Hyperplasia with cytological atypia | 6 (100%) | |
| 21 | Pten +/+ | 8 | Normal epithelium (no lesion) | 8 (100%) |
| Pten pr−/− | 8 | CAH with acute inflammation | 8 (100%) | |
| Pten +/+/p53 +/+ | 8 | Normal epithelium (no lesion) | 8 (100%) | |
| Pten pr−/− p53 pr−/− | 8 | CAH with in situ carcinoma, Fallopian tube hyperplasia | 8 (100%) | |
| 30 | Pten +/+ | 8 | Normal epithelium (no lesion) | 8 (100%) |
| Pten pr−/− | 8 | CAH with in situ carcinoma | 8 (100%) | |
| Pten +/+/p53 +/+ | 8 | Normal epithelium (no lesion) | 8 (100%) | |
| Pten pr−/− p53 pr−/− | 8 | Grade II carcinoma, disease more severe than Pten pr−/− uteri | 7 (87.5%) | |
| Cribriform appearance of glands, invasion into myometrium | 1 (12.5%) | |||
| 60 | Pten +/+ | 8 | Normal epithelium (no lesion) | 8 (100%) |
| Pten pr−/− | 8 | Carcinoma | 4 (50%) 1 | |
| CAH with focal carcinoma | 1 (12.5%) | |||
| CAH with squamous metaplasia | 3 (37.5%) 2 | |||
| Pten +/+/p53 +/+ | 7 | Normal epithelium (no lesion) | 7 (100%) | |
| Pten pr−/− p53 pr−/− | 6 | In situ carcinoma with cribriform pattern without invasion | 3 (50%) | |
| Invasion into the myometrium with well to moderate differentiation | 3 (50%) | |||
| 90 | Pten +/+ | 9 | Normal epithelium (no lesion) | 8 (88.9%) |
| Hyperplasia | 1 (11.1%) | |||
| Pten pr−/− | 9 | Carcinoma | 8 (88.9%) 3 | |
| CAH with squamous metaplasia | 1 (11.1%) | |||
CAH, complex atypical hyperplasia;
One mouse showed invasion;
Two mice showed invasion.
One mouse showed extensive carcinoma;
one showed well differentiated adenocarcinoma with invasion into the serosa and two showed carcinoma with squamous metaplasia.
We next examined the effects of combined deletion of p53 and Pten. Histological analyses of PtenloxP/loxP/p53loxP/loxP/PRcre/+ (Ptenpr−/−/p53pr−/−) uteri after staining with H&E showed that Ptenpr−/−/p53pr−/− uteri were enlarged compared to those of Ptenpr−/−, with luminal and glandular hyperplasia (Figure 2B). Lesions were observed as early as 10 days of age similar to the time frame noted in Ptenpr−/− uteri (Figure 2B). Furthermore, these lesions progressed to in situ carcinoma by 3 weeks of age (Table 1). Uteri from wild-type mice did not exhibit any pathology. We next examined p53pr−/− uteri. Although loss of endometrial p53 expression was seen in mice as young as 3 weeks of age (Figure 1C), endometrial morphology and histology appeared normal through 5 months of age (Supplemental Figure 3 & Supplemental Table 1).
Endometrial deletion of both Pten and p53 advances mortality
In human, loss of Pten and p53 increases severity of EMC development when compared to those with only Pten mutation (1, 2). However, we observed similar histology between Ptenpr−/− and Ptenpr−/−/p53pr−/− uteri during EMC initiation. A close monitoring of conditionally deleted mice for sickness during the tenure of these experiments revealed that loss of both Pten and p53 affects their survival as early as two months of age, while loss of Pten alone does not affect longevity until around five months, with deletion of p53 alone affecting viability even later (Figure 2C). H & E staining of uterine sections from Ptenpr−/−/p53pr−/− uteri indicate that the cause of early death in these mice is due to excessuterine bleeding due to invasion of uterine blood vessels by tumor cells (Supplemental Figure 4). At sacrifice, extensive blood clots on the surface of the entire uterus were visible in these mice.
Endometrial deletion of Pten or of both Pten and p53 induces epithelial Cox-2 expression and proliferation
Cox-2 is a downstream target of pAkt signaling and associated with development of many types of cancers (14, 26). Thus, we examined Cox-2 expression in Ptenpr−/− uteri. As shown in Figure 3A, levels of Cox-2 protein increased in Ptenpr−/− uteri compared to wild-type uteri from 3 weeks of age. Interestingly, uterine levels of Cox-1 were very low to undetectable at these time points irrespective of genotype (Figure 3A). Levels of Cox-2 transcripts as determined by RT-PCR correlated well with Cox-2 protein levels (Figure 3B). We also used in situ hybridization (Figure 3C) and immunohistochemistry (Figure 3D) to determine the spatiotemporal expression of Cox-2. We observed increased Cox-2 mRNA and protein levels primarily in endometrial epithelia of Ptenpr−/− mice. As expected, Cox-2 expression was low to undetectable in wild-type uteri. This observation is similar to higher Cox-2 expression that is observed in human type I EMC (12, 13).
Figure 3. Heightened Cox-2 expression in Ptenpr−/− uteri with EMC.

A) Western blot analysis of Cox-2 and Cox-1 in Ptenpr−/− uteri. Actin serves as control. B) RT-PCR of Cox-2 in Ptenpr−/− uteri. β-Actin is a housekeeping gene. C) In situ hybridization of Cox-2 in Ptenpr−/− and wild-type uteri. Bar, 400 μm. le, luminal epithelium; ge, glandular epithelium; s, stroma; myo, myometrium; Kid, kidney. D) Immunohistochemistry of Cox-2 in uteri of 3-week old Ptenpr−/− and wild-type mice. Bar, 400 μm.
Heightened levels of Cox-2 protein and mRNA were also observed in Ptenpr−/−/p53pr−/− uteri (Figure 4A, B, C & D upper panel). In contrast, levels of Cox-2 transcripts remained very low and unaltered in endometria deleted with p53 alone (Supplemental Figure 5). Since Cox-2 is known to stimulate cell proliferation in other cancers, we performed Ki67 immunostaining in uterine sections of wild-type, Ptenpr−/− or Ptenpr−/−/p53pr−/− mice at three weeks of age. We observed increased Ki67-positive cells primarily in the epithelia of both Ptenpr−/− and Ptenpr−/−/p53pr−/− uteri compared with wild-type mice (Figure 4D lower panel). Since levels of Cox-2 expression were lower in Ptenpr−/−/p53pr−/− uteri of two month old mice, we also performed Ki67 immunostaining to determine the proliferation status (Figure 4D lower panel). We found a correlation between Cox-2 expression and proliferation index. Uteri with low Cox-2 expression at two months also had reduced Ki-67 staining. This is in contrast with the observed higher Cox-2 expression and Ki-67 staining in three week old tumors (Figure 4D lower panel). It is important to note, however, that immunostaining of Ki67 was noted at the leading edge of tumors in uteri of older mice. This agrees with previous studies showing that the middle of tumors is hypoxic and necrotic (27). These findings in our Ptenpr−/− and Ptenpr−/−/p53pr−/− mice are exciting and identify these two mouse models as suitable systems for studying human type I EMC.
Figure 4. Cox-2 expression and cell proliferation in Ptenpr−/−/p53pr−/− uteri.

A) Western blot analysis of Cox-2 and Cox-1 in Ptenpr−/−/p53pr−/− uteri. Actin serves as control. B) RT-PCR of Cox-2 in Ptenpr−/−/p53pr−/− uteri. β-Actin is a housekeeping gene. C) In situ hybridization of Cox-2 in Ptenpr−/−/p53pr−/− and wild-type uteri. kid, kidney. Bar, 400 μm. D) Immunohistochemistry of Cox-2 in 3-week old Ptenpr−/−/p53pr−/− and wild-type mice (upper panel). Immunohistochemistry of Ki67 in 3-week and 2 months old Ptenpr−/− and Ptenpr−/−/p53pr−/− and wild-type uteri (lower panels). Arrowheads indicated leading edge. Bar, 200 μm. le, luminal epithelium; ge, glandular epithelium; s, stroma; myo, myometrium; T, tumor.
Downregulation of miR-199a* and miR-101a correlates with heightened Cox-2 expression in endometrial carcinogenesis
microRNAs (miRNAs) are noncoding small RNAs (19–22 nucleotides) transcribed from genomic DNAs. These small RNAs influence posttranscriptional gene expression and impact various biological events (28, 29). Recent studies have shown differential expression of various miRNAs in certain cancers compared to normal tissues. This differential expression is thought to affect the tumor suppressor genes such as p27 and cyclin G1 (30, 31). We recently found that miR-199a* and miR-101a posttranscriptionally suppress Cox-2 expression in a human cancer cell line (17). We also observed that these miRNAs co-localize with Cox-2 in themouse uterus during implantation and regulate its protein levels at the implantation site (17). Thus, we examined whether expression levels of these two miRNAs are affected by endometrial carcinogenesis in Ptenpr−/− or Ptenpr−/−/p53pr−/− uteri. We observed that Cox-2 overexpression in Ptenpr−/− uteri (Figures 3 and 4) is indeed correlated with decreased levels of miR-199a* and miR-101a expression (Figure 5A). The expression of these miRNAs is also reduced in Ptenpr−/−/p53pr−/− uteri with elevated Cox-2 expression (Figure 5B). These data suggest that miR-199a* and miR-101a play roles in regulating Cox-2 expression in EMC.
Figure 5. Levels of miR-199a* and miR-101a expression are reduced in uteri with EMC.
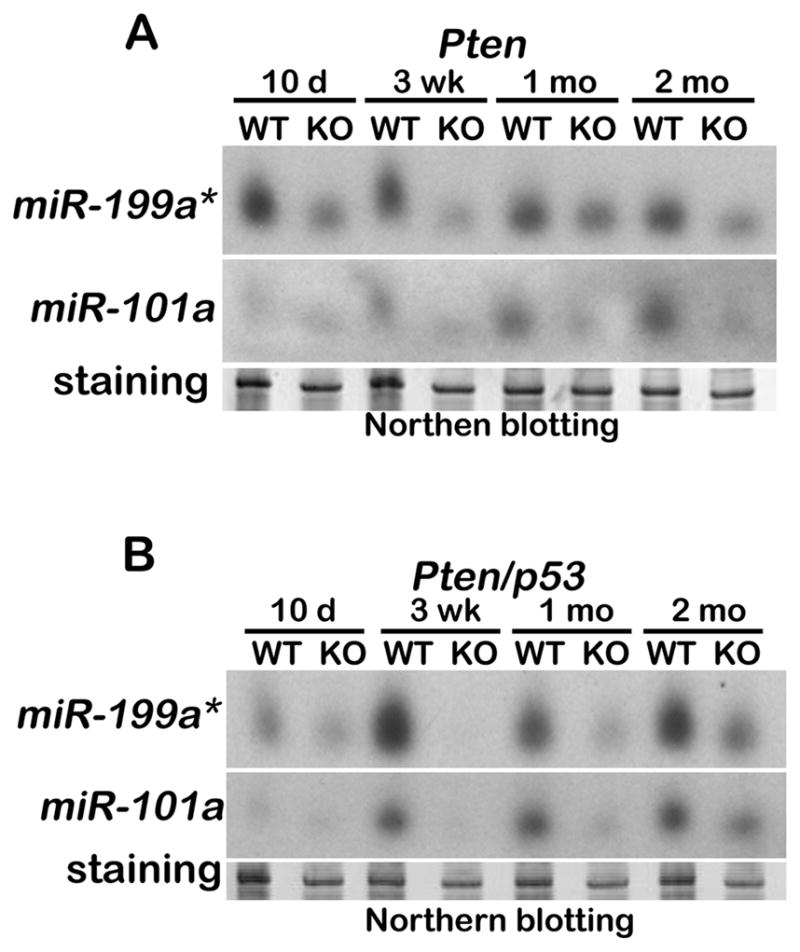
Northern blot analysis of A) Pten+/+ vs Ptenpr−/− uteri and B) Pten+/+/p53+/+ vs Ptenpr−/−/p53pr−/− uteri. Stained gels served as loading controls.
DISCUSSION
Mutations of tumor suppressor genes are linked to carcinogenesis in various organ systems. Unfortunately, systemic deletion of these genes in mice often results in embryonic lethality. The loxP-Cre system allows for tissue-specific and temporal deletion of tumor suppressor genes (32, 33). The goal of this study was to take advantage of the loxP-Cre system to generate conditional deletion of Pten and/or p53 in the uterus to study the initiation and progression of endometrial carcinoma. Since there is no endometrial-specific promoter identified as of yet, PR-Cre mice are being widely used to delete floxed genes in the uterus (16, 20, 24). It is to be noted, however, that PR is also expressed in tissues such as oviduct, ovary, mammary gland and pituitary. Our current study shows that endometrial cancer develops in Ptenpr−/− or Ptenpr−/−/p53pr−/− mice within three weeks to one month of age with no evidence of cancers in other tissues, including those that express PR. One reason for development of endometrial-specific cancer could perhaps be attributed to increased sensitivity of the uterus to Pten loss. Nonetheless, this endometrial-specific phenotype demonstrates the importance of Pten loss in the initiation of EMC. Studies in humans have shown that type I EMC originates mainly from the epithelial components and not the stroma; however PR is known to be expressed in both epithelia and stroma (1). Therefore, whether endometrial cancer in Ptenpr−/− or Ptenpr−/−/p53pr−/− mice originates from epithelial or stromal components remains to be determined. Since Pten is expressed primarily in endometrial epithelia during early neonatal development, it is speculated that cancer is initiated in this compartment due to the loss of Pten. Definitive answers to the specific roles of epithelial versus stromal Pten in endometrial cancer will require epithelial-specific loxP-Cre and stromal-specific loxP-Cre systems for deletion of Pten.
We observed that the combined loss of Pten and p53 resulted in a shorter lifespan compared to mice with single deletion of either Pten or p53. Previous reports suggested a role for p53 in advanced type I endometrial cancer in women (1). Therefore, we speculated that superimposing p53 mutation on Pten deletion would aggravate the disease condition. Our findings of shorter lifespan of Ptenpr−/−/p53pr−/− mice supports this hypothesis, with the cause of early death attributed to excessive uterine bleeding due to invasion of uterine blood vessels by invasive tumor cells. As a note, type II endometrial cancer, which comprises only 10–15% of all endometrial cancers, shows a high frequency of p53 mutations. This is contrary to our observation that uterine deletion of p53 does not cause cancer in mice (1). Therefore, understanding the role of p53 mutations on endometrial cancer requires further studies.
Our findings of deletion of both p53 and Pten aggravating the mortality rate are consistent with a similar study that used conditional deletion of Pten to induce prostate cancer (32). In this study, the investigators used Ptenloxp/loxp/probasin-Cre mice and showed that Pten deletion alone did not increase the incidence of mortality, but the combined deletion of both Pten and p53 did. Their explanation for this observation was that deletion of Pten alone resulted in p53 upregulation which protected cells from senescence (32). We also observed p53 upregulation primarily in endometrial epithelia of Ptenpr−/− mice (data not shown), consistent with their explanation. Another possibility is that PR-Cre driven deletion of Pten and p53 results in deleterious effects on immune cells. Studies have shown that T-cell or B-cell specific deletion of Pten results in T-cell lymphomas or defects in class switch recombination (reviewed in 34). It seems unlikely that local immune complications contribute to the progression of endometrial cancer in Ptenpr−/−/p53pr−/− mice since neither estrogen receptor nor PR is expressed in uterine lymphocytes, macrophages or natural killer cells (35, 36).
Since endometrial cancer is also known to be influenced by hormonal components (1), we ovariectomized Ptenpr−/−/p53pr−/− mice aged 3 weeks to evaluate the contribution of ovarian steroid hormones. We found that ovariectomizing these mice did not minimize tumor development when examined at 2 months (Supplemental Figure 6), suggesting limited contribution of ovarian steroid hormones to endometrial cancer progression induced by Pten and p53 mutations.
Cox-2 is thought to play an important role in carcinogenesis and is overexpressed in a number of solid tumors, including colorectal, breast and lungcancers (10). Treatments with nonsteroidal anti-inflammatory drugs (NSAIDs) that inhibit Cox activity have been shown to be effective in the chemoprevention of many types of solid tumors. In fact, even their daily consumption reduces the risk of certain cancers (37–40). Our study showing elevated Cox-2 expression at the early stages of hyperplasia and carcinoma in both Ptenpr−/− and Ptenpr−/−/p53pr−/− mice suggests that NSAIDs could be considered potential treatment options for type I EMC patients at early stages. Unfortunately, recent clinical studies show that prolonged use of highly selective Cox-2 inhibitors, such as Vioxx, leads to increased myocardial infarctions and stroke. However, NSAIDs like aspirin or naproxen show lesser side effects and are still being widely used to combat inflammatory diseases and to reduce the risk of developing cancers.
While Cox-2 expression was elevated in the early stages of hyperplasia and carcinoma, we were initially surprised to observe its gradual disappearance with cancer progression. However, it has been shown that Cox-2 is often associated with early stages of cancer in both liver and uterine cancers, and then is downregulated with tumor advancement (41, 42). In uterine cancers, levels of Cox-2 correlate with VEGF expression, implicating their roles in angiogenesis at early stages (42). These studies suggest the potential for NSAID treatment to prevent recurrence of endometrial cancers.
Our observations of increased levels of pAkt and Cox-2 at early stages of EMC suggest that the Pten-Akt-Cox-2 signaling axis is important for the initiation of tumor growth. The accelerated growth perhaps occurs due to increased cell proliferation that is evident from Ki67 staining even at three weeks of age. Our results showing decreased expression of miR-199a* and miR-101a with enhanced Cox-2 levels in Ptenpr−/− and Ptenpr−/−/p53pr−/− uteri also suggests their close relationship with Cox-2 status in the uterus as we have previously shown (17). However, it is not known whether Pten directly regulates the expression of these miRNAs or their downregulation is a consequence of the development of EMC with enhanced Cox-2 expression. Nonetheless, these mRNAs are potential targets for anti-cancer therapy, because of their role in downregulating Cox-2 levels.
In conclusion, the present study presents mouse models that rapidly and unfailingly produce EMC, many characteristics of which mimic human EMC. These models will help delineate other downstream signaling pathways critical to the initiation and progression of human EMC.
Supplementary Material
Acknowledgments
We thank Dr. Satoshi Umezawa (Musashino RedCross Hospital, Tokyo, Japan) for his scientific advice. This work was supported in part from the United States Public Health Service Grants, P01-CA-77839 (RND), VICC-Meharry U54CA091405-06 and HD12304 (SKD) and U01 CA105352-01/05 (FD), Vanderbilt-Ingram Cancer Center Support Grant, P30CA068485 and Gynecologic Cancer Foundation Awards/Ann Schreiber Ovarian Cancer Research Grant (TD). YH is supported by a Research Fellowship from the Japan Society for the Promotion of Science for Young Scientists. ST is supported by an NICHD Training Grant 2THD007043.
References
- 1.Di Cristofano AaE LH. Endometrial Carcinoma. Annu Rev Pathol Mech Dis. 2007;2:57–87. doi: 10.1146/annurev.pathol.2.010506.091905. [DOI] [PubMed] [Google Scholar]
- 2.Baergen RN, Warren CD, Isacson C, Ellenson LH. Early uterine serous carcinoma: clonal origin of extrauterine disease. Int J Gynecol Pathol. 2001;20(3):214–9. doi: 10.1097/00004347-200107000-00002. [DOI] [PubMed] [Google Scholar]
- 3.Podsypanina K, Ellenson LH, Nemes A, et al. Mutation of Pten/Mmac1 in mice causes neoplasia in multiple organ systems. Proc Natl Acad Sci U S A. 1999;96(4):1563–8. doi: 10.1073/pnas.96.4.1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Di Cristofano A, Pesce B, Cordon-Cardo C, Pandolfi PP. Pten is essential for embryonic development and tumour suppression. Nat Genet. 1998;19(4):348–55. doi: 10.1038/1235. [DOI] [PubMed] [Google Scholar]
- 5.Donehower LA, Harvey M, Slagle BL, et al. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature. 1992;356(6366):215–21. doi: 10.1038/356215a0. [DOI] [PubMed] [Google Scholar]
- 6.Harvey M, McArthur MJ, Montgomery CA, Jr, Bradley A, Donehower LA. Genetic background alters the spectrum of tumors that develop in p53-deficient mice. Faseb J. 1993;7(10):938–43. doi: 10.1096/fasebj.7.10.8344491. [DOI] [PubMed] [Google Scholar]
- 7.Jacks T, Remington L, Williams BO, et al. Tumor spectrum analysis in p53-mutant mice. Curr Biol. 1994;4(1):1–7. doi: 10.1016/s0960-9822(00)00002-6. [DOI] [PubMed] [Google Scholar]
- 8.Luo J, Manning BD, Cantley LC. Targeting the PI3K-Akt pathway in human cancer: rationale and promise. Cancer cell. 2003;4(4):257–62. doi: 10.1016/s1535-6108(03)00248-4. [DOI] [PubMed] [Google Scholar]
- 9.Chen ML, Xu PZ, Peng XD, et al. The deficiency of Akt1 is sufficient to suppress tumor development in Pten+/− mice. Genes Dev. 2006;20(12):1569–74. doi: 10.1101/gad.1395006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Turini ME, DuBois RN. Cyclooxygenase-2: a therapeutic target. Annu Rev Med. 2002;53:35–57. doi: 10.1146/annurev.med.53.082901.103952. [DOI] [PubMed] [Google Scholar]
- 11.Hasegawa K, Ohashi Y, Ishikawa K, et al. Expression of cyclooxygenase-2 in uterine endometrial cancer and anti-tumor effects of a selective COX-2 inhibitor. Int J Oncol. 2005;26(5):1419–28. [PubMed] [Google Scholar]
- 12.Nasir A, Boulware D, Kaiser HE, et al. Cyclooxygenase-2 (COX-2) expression in human endometrial carcinoma and precursor lesions and its possible use in cancer chemoprevention and therapy. In Vivo. 2007;21(1):35–43. [PubMed] [Google Scholar]
- 13.Tong BJ, Tan J, Tajeda L, et al. Heightened expression of cyclooxygenase-2 and peroxisome proliferator-activated receptor-delta in human endometrial adenocarcinoma. Neoplasia. 2000;2(6):483–90. doi: 10.1038/sj.neo.7900119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.St-Germain ME, Gagnon V, Mathieu I, Parent S, Asselin E. Akt regulates COX-2 mRNA and protein expression in mutated-PTEN human endometrial cancer cells. Int J Oncol. 2004;24(5):1311–24. [PubMed] [Google Scholar]
- 15.St-Germain ME, Gagnon V, Parent S, Asselin E. Regulation of COX-2 protein expression by Akt in endometrial cancer cells is mediated through NF-kappaB/IkappaB pathway. Mol Cancer. 2004;3:7. doi: 10.1186/1476-4598-3-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Soyal SM, Mukherjee A, Lee KY, et al. Cre-mediated recombination in cell lineages that express the progesterone receptor. Genesis. 2005;41(2):58–66. doi: 10.1002/gene.20098. [DOI] [PubMed] [Google Scholar]
- 17.Chakrabarty ATS, Daikoku T, Jensen K, Furneaux H, Dey SK. MicroRNA Regulation of Cyclooxygenase-2 during Embryo Implantation. Proc Natl Acad Sci U S A. 2007;104(38):15144–9. doi: 10.1073/pnas.0705917104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Flesken-Nikitin A, Choi KC, Eng JP, Shmidt EN, Nikitin AY. Induction of carcinogenesis by concurrent inactivation of p53 and Rb1 in the mouse ovarian surface epithelium. Cancer Res. 2003;63(13):3459–63. [PubMed] [Google Scholar]
- 19.Lesche R, Groszer M, Gao J, et al. Cre/loxP-mediated inactivation of the murine Pten tumor suppressor gene. Genesis. 2002;32(2):148–9. doi: 10.1002/gene.10036. [DOI] [PubMed] [Google Scholar]
- 20.Xie H, Wang H, Tranguch S, et al. Maternal heparin-binding-EGF deficiency limits pregnancy success in mice. Proc Natl Acad Sci U S A. 2007;104(46):18315–20. doi: 10.1073/pnas.0707909104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Daikoku T, Tranguch S, Trofimova IN, et al. Cyclooxygenase-1 is overexpressed in multiple genetically engineered mouse models of epithelial ovarian cancer. Cancer Res. 2006;66(5):2527–31. doi: 10.1158/0008-5472.CAN-05-4063. [DOI] [PubMed] [Google Scholar]
- 22.Daikoku T, Wang D, Tranguch S, et al. Cyclooxygenase-1 is a potential target for prevention and treatment of ovarian epithelial cancer. Cancer Res. 2005;65(9):3735–44. doi: 10.1158/0008-5472.CAN-04-3814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tan J, Paria BC, Dey SK, Das SK. Differential uterine expression of estrogen and progesterone receptors correlates with uterine preparation for implantation and decidualization in the mouse. Endocrinology. 1999;140(11):5310–21. doi: 10.1210/endo.140.11.7148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee KY, Jeong JW, Wang J, et al. Bmp2 is critical for the murine uterine decidual response. Mol Cell Biol. 2007;27(15):5468–78. doi: 10.1128/MCB.00342-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cantley LC, Neel BG. New insights into tumor suppression: PTEN suppresses tumor formation by restraining the phosphoinositide 3-kinase/AKT pathway. Proc Natl Acad Sci U S A. 1999;96(8):4240–5. doi: 10.1073/pnas.96.8.4240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pommery N, Henichart JP. Involvement of PI3K/Akt pathway in prostate cancer--potential strategies for developing targeted therapies. Mini Rev Med Chem. 2005;5(12):1125–32. doi: 10.2174/138955705774933356. [DOI] [PubMed] [Google Scholar]
- 27.Neeman M, Abramovitch R, Schiffenbauer YS, Tempel C. Regulation of angiogenesis by hypoxic stress: from solid tumours to the ovarian follicle. International journal of experimental pathology. 1997;78(2):57–70. doi: 10.1046/j.1365-2613.1997.d01-247.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Du T, Zamore PD. microPrimer: the biogenesis and function of microRNA. Development. 2005;132(21):4645–52. doi: 10.1242/dev.02070. [DOI] [PubMed] [Google Scholar]
- 29.Nilsen TW. Mechanisms of microRNA-mediated gene regulation in animal cells. Trends Genet. 2007;23(5):243–9. doi: 10.1016/j.tig.2007.02.011. [DOI] [PubMed] [Google Scholar]
- 30.le Sage C, Nagel R, Egan DA, et al. Regulation of the p27(Kip1) tumor suppressor by miR-221 and miR-222 promotes cancer cell proliferation. Embo J. 2007;26(15):3699–708. doi: 10.1038/sj.emboj.7601790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gramantieri L, Ferracin M, Fornari F, et al. Cyclin G1 is a target of miR-122a, a microRNA frequently down-regulated in human hepatocellular carcinoma. Cancer Res. 2007;67(13):6092–9. doi: 10.1158/0008-5472.CAN-06-4607. [DOI] [PubMed] [Google Scholar]
- 32.Chen Z, Trotman LC, Shaffer D, et al. Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature. 2005;436(7051):725–30. doi: 10.1038/nature03918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhou Z, Flesken-Nikitin A, Corney DC, et al. Synergy of p53 and Rb deficiency in a conditional mouse model for metastatic prostate cancer. Cancer Res. 2006;66(16):7889–98. doi: 10.1158/0008-5472.CAN-06-0486. [DOI] [PubMed] [Google Scholar]
- 34.Kishimoto H, Hamada K, Saunders M, et al. Physiological functions of Pten in mouse tissues. Cell structure and function. 2003;28(1):11–21. doi: 10.1247/csf.28.11. [DOI] [PubMed] [Google Scholar]
- 35.Henderson TA, Saunders PT, Moffett-King A, Groome NP, Critchley HO. Steroid receptor expression in uterine natural killer cells. The Journal of clinical endocrinology and metabolism. 2003;88(1):440–9. doi: 10.1210/jc.2002-021174. [DOI] [PubMed] [Google Scholar]
- 36.King A, Gardner L, Loke YW. Evaluation of oestrogen and progesterone receptor expression in uterine mucosal lymphocytes. Human reproduction (Oxford, England) 1996;11(5):1079–82. doi: 10.1093/oxfordjournals.humrep.a019300. [DOI] [PubMed] [Google Scholar]
- 37.Schildkraut JM, Moorman PG, Halabi S, Calingaert B, Marks JR, Berchuck A. Analgesic drug use and risk of ovarian cancer. Epidemiology. 2006;17(1):104–7. doi: 10.1097/01.ede.0000190538.55645.f8. [DOI] [PubMed] [Google Scholar]
- 38.Larsson SC, Giovannucci E, Wolk A. Long-term aspirin use and colorectal cancer risk: a cohort study in Sweden. Br J Cancer. 2006;95(9):1277–9. doi: 10.1038/sj.bjc.6603442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chan AT, Giovannucci EL, Meyerhardt JA, Schernhammer ES, Curhan GC, Fuchs CS. Long-term use of aspirin and nonsteroidal anti-inflammatory drugs and risk of colorectal cancer. Jama. 2005;294(8):914–23. doi: 10.1001/jama.294.8.914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Baron JA, Cole BF, Sandler RS, et al. A randomized trial of aspirin to prevent colorectal adenomas. N Engl J Med. 2003;348(10):891–9. doi: 10.1056/NEJMoa021735. [DOI] [PubMed] [Google Scholar]
- 41.Koga H, Sakisaka S, Ohishi M, et al. Expression of cyclooxygenase-2 in human hepatocellular carcinoma: relevance to tumor dedifferentiation. Hepatology (Baltimore, Md. 1999;29(3):688–96. doi: 10.1002/hep.510290355. [DOI] [PubMed] [Google Scholar]
- 42.Toyoki H, Fujimoto J, Sato E, Sakaguchi H, Tamaya T. Clinical implications of expression of cyclooxygenase-2 related to angiogenesis in uterine endometrial cancers. Ann Oncol. 2005;16(1):51–5. doi: 10.1093/annonc/mdi020. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.