

Abstract
We propose a systematic approach for a better understanding of how HIV viruses employ various combinations of mutations to resist drug treatments, which is critical to developing new drugs and optimizing the use of existing drugs. By probabilistically modeling mutations in the HIV-1 protease or reverse transcriptase (RT) isolated from drug-treated patients, we present a statistical procedure that first detects mutation combinations associated with drug resistance and then infers detailed interaction structures of these mutations. The molecular basis of our statistical predictions is further studied by using molecular dynamics simulations and free energy calculations. We have demonstrated the usefulness of this systematic procedure on three HIV drugs, (Indinavir, Zidovudine, and Nevirapine), discovered unique interaction features between viral mutations induced by these drugs, and revealed the structural basis of such interactions.
Keywords: Bayesian model selection, free energy calculation, Markov chain Monte Carlo, molecular dynamics, mutation interactions
HIV drug-resistance, which is caused by mutations of viral proteins that disrupt the drugs’ binding but do not affect the viral survival, is a major hurdle that hinders a successful treatment of AIDS (1, 2). Due to the high rate and low fidelity of HIV replication, resistant strains quickly become dominant in a viral population under the selection pressure of a drug. By sequencing viral strains in the treated-patient isolates, genotypic data have been accumulated for the drugs targeting two viral enzymes, protease and reverse transcriptase, that are essential to the virus’s replication. Because each mutation of the viral protein is not equally important for drug resistance, the observed, complicated mutation patterns are difficult to interpret (3, 4) and are limited in helping physicians design the best therapeutic regimen for a patient (5) (Fig. 1 A).
Fig. 1.

The posterior probabilities for each mutation to be associated interactively with indinavir treatment. The Upper shows the posterior probabilities using prior one, which assumes that it is equally likely (1/3) for a mutation to be unassociated, individually associated, and interactively associated with the drug treatment. The Lower shows the posterior probabilities using a more stringent prior (prior two) assuming that only two makers are expected to be associated with the drug, either individually or interactively.
In past decades, many statistical learning methods (3, 4, 6 7–8) have been employed to help predict phenotypes from genotypes. There are also rule-based systems that infer drug-resistance levels from sequence information such as the Stanford University HIV Drug Resistance Database (Stanford HIVdb). However, these methods provide little insight on the genetic and molecular basis of drug resistance and often give inconsistent results when analyzing the same input mutation data (4, 6).
In the present study, we investigated the problem of mutation interactions of the HIV induced by a certain drug treatment. Using a unique probabilistic model, we first detect resistant mutation combinations (9) and infer the interaction dependence structure of these combinations. Then, we use molecular dynamics (MD) simulations to reveal the molecular basis of how these mutations interact with each other to interfere with the drugs’ binding. We have shown that our procedure is applicable to different antiretroviral drugs treating different types of HIV infection by analyzing the sequence mutations induced by three different drug treatments: a protease inhibitor (indinavir), a nucleoside analog reverse-transcriptase inhibitor (zidovudine), and a nonnucleoside reverse-transcriptase inhibitor (nevirapine). We have rediscovered the majority of known resistant mutations to the three drugs (10) and uncovered several interacting structures for these mutations. Particularly, for protease we have discovered a conditional independence structure among the mutations M46I, I54V, and V82A that is consistent with several previous experimental results (3, 5, 6, 11 12 13–14) but has not been documented in the literature. Our MD simulations and free energy analyses have further confirmed and provided the molecular basis and implication of this conditional independence.
Results
Analytical Pipeline for Studying HIV Mutation Data.
We first design a Bayesian variable partition (BVP) model, a generalization of the “Bayesian epistasis association mapping” (BEAM) model in Zhang and Liu (9), to select mutations that are associated with drug resistance. Next, we design a recursive model selection (RMS) procedure that recursively partitions a set of mutation positions into three subsets so that the three sets of variables either follow a chain-dependence structure, or a “V” structure (see Methods section) to infer the dependence structure among the interacting mutation positions found by the BVP model. Finally, we illustrate the molecular basis of the mutation patterns predicted by BVP and RMS by using molecular dynamics simulations and inhibitor-residue free energy decomposition analyses.
Complex Interaction Patterns for Drug Resistance of Indinavir.
The data contain 949 HIV-1 (type B) protease sequences from indinavir-treated patients (indinavir is the only PI in their therapy) and 4,146 sequences (HIV-1 type B) from untreated patients. HIV-1 protease has 99 amino acids and each position has mutations in the dataset. Any combination of mutations among these 99 positions may be related to the virus’ drug resistance capability. Our goal is to find those positions that are either independently or interactively associated with the indinavir treatment.
Fig 1 shows the posterior probabilities for each marker to be associated interactively with the indinavir treatment based on the BVP model under two different prior distributions. We can see that the results are insensitive to the priors. Nine out of the 10 positions with high posterior probabilities of interaction (i.e., 10, 24, 32, 46, 54, 71, 73, 82, and 90) are on the drug resistance mutation list (5) updated in spring 2008 (Fig S1). The only one not on the list is position 47, which is well-known to be associated with indinavir drug resistance when combined with position 32 (3). We have found 17 mutation patterns (out of a total of 2099 possibilities) that are associated with indinavir treatment with a posterior probability > 0.0001 (this cutoff is much higher than the equally likely probability 1/2099). Table S1 in SI Text tabulates these patterns and their respective posterior probabilities. Phenotypic data from Stanford HIVdb provides confirming evidence for the configurations of the top interaction pattern {24,32,46,54,82} (SI Text). Many of these mutations are well-known for their drug resistance effects. For example, it is known that the mutations of V82A\F\T or L90M are necessary but not sufficient for measurable resistance to indinavir (11).
Dependence Structure of Interaction for Drug Resistance of Indinavir.
We applied the RMS procedure to infer the detailed dependence structure among the interacting positions 10, 24, 32, 46, 47, 54, 71, 73, 82, and 90 (Fig. 2). Two marginally independent interaction groups were found with high confidence: one is composed mainly of 46, 54, and 82; and the other of 73 and 90 (more details are given in SI Text). Interestingly, we found a strong conditional independence structure in group {46,52,82}. Given the amino acid at position 82, mutations at 46 and 54 are mutually independent. The data did not provide strong enough information regarding the structures for other variables (mutations) in this group, for example, 24, 32, and 47. For the second group, 73 and 90 strongly interact with each other.
Fig. 2.

Detection of a detailed mutation interaction structure for resisting indinavir. Positions 46 and 54 are conditionally independent given position 82, denoted as 42⊥54|82. The ? indicates where we are not able to confidently infer the dependence structure (SI Text).
In the study of Zhang et al. (13), a rebound in virus levels in plasma following the initial sharp decline at the beginning of indinavir therapy was found to be associated with a sequential acquisition of mutations at the protease positions of 46 → 82 → 54. We further searched the Stanford HIVdb, and found that 112 patients from the treated group (HIV-1, main group, and subtype B) had indinavir as their only PI in their therapy and also have detailed mutation records (more than one complete protease sequences) during the course of therapy. Among them 53.6% (60 patients) have at least one of the mutations at positions 46, 54, and 82. We observed no patient with the single mutation V82I, with the single mutation at 54, or with the double mutations at 46 and 54. Among the 21 patients who have all three mutations, only six of them have detailed mutation records such that we can tell the exact order of sequential acquisition of these three mutations. Four out of the six have the order 46 → 82 → 54, one has the order 82 → 46 → 54, and another has the order 82 → 54 → 46. Whereas all these observed orders are consistent with our inferred conditional independence structure, the non-observed orders, 46 → 54 → 82 and 54 → 46 → 82, are not. This suggests that the conditional independence is a direct consequence of sequential acquisition of the three mutations.
Molecular Basis of Interacting Mutations Revealed by MD Simulations and Free Energy Calculations.
To further investigate the molecular implication of the mutation interactions within the {46,54,82} group, and within the {73,90} group, we conducted MD simulations to analyze the binding free energies of the protease/indinavir complexes (SI Text). The free energy decomposition analyses for the wild-type and ten mutant proteases (Table 1) show that the drug resistant mutations primarily affect the van der Waals interactions between indinavir and the protease. Most of the mutations in the {46,54,82} group show positive relative binding free energies, that is, decrease of indinavir’s binding affinity.
Table 1.
The binding free energies and the energy components calculated by MM/GBSA (kcal/mol)
| No. | ΔEvdw | ΔEele | ΔGGB | ΔGSA | ΔEele + ΔGGB | ΔEvdw + ΔGSA | ΔGcal | ΔΔGcal* |
| WT | −80.00 ± 0.16 † | −25.59 ± 0.35 | 33.97 ± 0.35 | −9.87 ± 0.06 | 8.37 ± 0.00 | −89.87 ± 0.22 | −81.50 ± 0.46 | 0.00 |
| Group 1 | ||||||||
| M46I | −77.43 ± 1.85 | −24.01 ± 0.39 | 34.46 ± 0.37 | −10.32 ± 0.64 | 10.44 ± 0.76 | −87.74 ± 1.21 | −77.30 ± 0.45 | 4.20 |
| I54V | −82.99 ± 0.26 | −25.38 ± 0.21 | 36.13 ± 0.16 | −9.25 ± 0.57 | 10.74 ± 0.37 | −92.24 ± 0.83 | −81.50 ± 0.46 | 0.00 |
| V82A | −75.98 ± 1.27 | −24.67 ± 0.10 | 34.54 ± 0.13 | −9.56 ± 0.00 | 9.87 ± 0.23 | −85.54 ± 1.27 | −75.67 ± 1.50 | 5.83 |
| M46I/I54V | −83.26 ± 1.05 | −22.01 ± 1.23 | 34.19 ± 1.44 | −9.98 ± 0.06 | 12.18 ± 0.21 | −93.24 ± 1.11 | −81.06 ± 0.90 | 0.44 |
| M46I/V82A | −70.84 ± 2.86 | −24.92 ± 1.87 | 33.96 ± 1.40 | −9.67 ± 1.43 | 9.04 ± 0.47 | −80.52 ± 4.29 | −71.48 ± 4.76 | 10.02 |
| I54V/82A | −71.84 ± 0.64 | −19.62 ± 0.92 | 31.08 ± 1.06 | −9.82 ± 0.01 | 11.46 ± 0.13 | −81.66 ± 0.63 | −70.20 ± 0.77 | 11.30 |
| M46I/I54V/V82A | −79.58 ± 0.91 | −19.09 ± 0.17 | 29.89 ± 0.41 | −8.92 ± 0.59 | 10.79 ± 0.24 | −88.50 ± 0.33 | −77.71 ± 0.08 | 3.79 |
| Group 2 | ||||||||
| G73S | −78.79 ± 0.50 | −24.76 ± 0.51 | 33.26 ± 0.14 | −10.36 ± 0.67 | 8.50 ± 0.37 | −89.15 ± 0.17 | −80.65 ± 0.54 | 0.85 |
| L90M | −80.56 ± 0.32 | −27.13 ± 0.33 | 35.59 ± 0.06 | −9.78 ± 0.06 | 8.46 ± 0.39 | −90.33 ± 0.38 | −81.87 ± 0.00 | −0.37 |
| G73S/L90M | −81.31 ± 0.37 | −22.89 ± 0.19 | 33.01 ± 0.05 | −9.80 ± 0.00 | 10.12 ± 0.24 | −91.11 ± 0.38 | −80.99 ± 0.14 | 0.51 |
*ΔΔG cal is the difference between the binding free energy of the mutated complex and that of the wild-type.
†Standard deviations were estimated from two block average values.
Among the three single mutations, two of them (M46I and V82A) substantially increase the indinavir binding free energies (-77.30 ± 0.45 and -75.67 ± 1.50), whereas I54V does not impair the binding. This result is consistent with our observation in the Stanford HIVdb. Among the 112 patients who have more than one mutation record during their indinavir therapy, 10 have the single mutation at 46, 10 have the single mutation at 82, and zero have the single mutation at 54.
Among the double mutations, M46I/V82A and I54V/V82A severely impair the binding of indinavir whereas M46I/I54V does not significantly weaken the binding of indinavir. Incidentally, among the 112 patients in the Stanford HIVdb, 11 have double mutations at positions 46 and 82, eight at positions 54 and 82, and zero at positions 46 and 54. It appears that 46 and 54 cannot interact to resist to indinavir without the mutation at 82. The observations that double mutations M46I/V82A and I54V/V82A are the two strongest resistant mutants may have important implications for improving the potency of indinavir to combat resistance. If we can decrease the interaction between V82 and a derivative of indinavir without affecting the total binding affinity of the inhibitor, the resistant effects of 46 and 54 will be reduced, as well. This example highlights the usefulness of our approach for uncovering the interaction structure between mutations in developing potent drugs.
The triple mutation M46I/I54V/V82A impairs the binding of indinavir. As mentioned before, these three mutations occur sequentially in specific orders. Because single mutation at 54 is not able to resist indinavir, the first mutation has to be at either 46 or 82 so that the mutant virus can have a better chance to survive the attack of indinavir. If the first mutation occurs at 46, the second mutation has to be at 82 because the double mutations at 46 and 54 cannot resist to indinavir, as well. If the first mutation is at 82, the subsequent mutation can be at either 46 or 54. We observed exactly these (and only these) three possible orders (46 → 82 → 54, 82 → 46 → 54 and 82 → 54 → 46) in the Stanford HIVdb database. Our energy calculation and probabilistic modeling are all consistent with this sequential acquisition observation (Fig. 3).
Fig. 3.

Energetic and structural insight of the resistance mechanism. A1: The difference between each residue’s contribution to the interaction with indinavir in (A1) the M46I/I54V and the M46I; (A2) the I54V/V82A and the V82A. ΔΔG was calculated by subtracting each residue’s interaction energy in the single mutant (e.g., M46I) from the double mutant (M46I/I54V). Residues with absolute value greater than 0.75 kcal/mol are labeled. Structural distributions of important residues in Fig 3 A1 and A2 are shown in B1 and B2, resp. The protease is shown in Blue Strand and indinavir in Green Stick. Residues with negative and positive ΔΔG’s, which represent residues contributing more and less favorably to binding with indinavir in the double mutant (e.g., M46I/I54V) than in the single mutant (e.g., M46I) resp., are shown in Red and Green CPK models, resp. The favorable residues to the binding of indinavir to the M46I/54V mutant are shown as the Red CPK model and those of the unfavorable residues as the Green CPK model. Alignment of the average structure of the double and single mutant complexes (C1) between M46I/54V and M46I mutated; (C2) between I54V/V82A and V82A. The average structure was obtained by averaging the 125 snapshots taken from 0.5–3.0 ns MD simulations. The double (e.g., M46I/I54V) and single (e.g., M46I) protease mutants are shown in Blue and Green strands, resp. Indinavirs bound to the double (e.g., M46I/I54V) and single (e.g., M46I) are shown in Red and Green Sticks, resp. The Pink Arrow shows the configurational change of indinavir in the two complexes. The cooperation between, for example, V54 and A82 significantly changes the active site’s conformation that further enhances resistance caused by the mutation at position 82 alone. The conformational change is manifested in the alignment of the average structures of the double (e.g., I54V/V82A) and single (e.g., V82A) mutant complexes.
Compared to the protease with a single mutation M46I, the additional mutation I54V makes more residues contributing favorably to the indinavir binding (nine vs. five). From Fig. 3 B1 and C1 we can see that these nine favorable residues spread around the binding pocket, and thus enhance the binding of indinavir right in the pocket and block the function of protease. However, with V82A, the additional mutation I54V does not make indinavir interact more or less favorably with residues (seven vs. seven), which may superficially suggest that I54V would not affect the resistance caused by V82A. However, we can see from Fig. 3 C2 that the seven favorable residues cluster tightly at one side of the binding pocket and the seven unfavorable ones at the other side. We speculate that such an uneven or asymmetric distribution of favorable/unfavorable residues may have pushed indinavir aside from blocking the binding pocket and thus reduced the potency of the drug.
The resistance caused by 70 and 90 cannot be explained by the binding free energy analysis that is consistent with observations made in the previous experiments (15, 16), suggesting that the group {73,90} may follow a different resistant mechanism rather than impairing the binding affinity (SI Text).
Two Drugs Attacking Reverse Transcriptase.
HIV-1 RT is a heterodimer consisting of p66 and p51 subunits. The p66 subunit is composed of all 560 amino acids of RT whereas p51 subunit is composed of the first 440 amino acids. RT is critical for RNA-dependent DNA polymerization and DNA-dependent DNA polymerization. We analyzed drug resistant mutation data of two drugs targeting RT: Zidovudine, a nucleoside analog reverse transcriptase inhibitor (NRTI), and Nevirapine, a non-nucleoside reverse transcriptase inhibitor (NNRTI).
Zidovudine is not designed to bind with RT and block the function of RT (unlike indinavir and nevirapine in the following) but rather to compete with natural dNTPs for incorporation into the newly synthesized DNA chains where it causes chain termination. Therefore, we cannot investigate its structural basis of resistant mutations by using MD simulations and free energy decompositions. To date, three biochemical mechanisms of NRTI drug resistance have been uncovered or proposed (3, 17). These different resistance mechanisms seem to correlate with different sets of mutations in RT (17), but further biochemical investigations are needed to confirm which mechanism corresponds to which independent mutation set (SI Text). Unlike NRTIs, NNRTIs bind to a hydrophobic pocket in RT close to the active site and their binding can block the catalytic activity of RT. The RT mutations resistant to NNRTIs often occur in the hydrophobic binding pocket to deteriorate the inhibitors’ binding.
We have analyzed two RT-related datasets in the Stanford HIVdb by using our statistical procedure: for zidovudine, 339 HIV-1 type B RT sequences from zidovudine-treated patients and 2187 sequences (HIV-1 type B) from untreated patients contain mutations at each position of the 190aa-long polypeptide sequences (from position 31 to 220 of RT); for nevirapine, 380 RT sequences from nevirapine-treated patients and 1622 RT sequences from untreated patients (both HIV-1 type C) correspond to the same 190aa-long region as in the zidovudien data. Any combination of mutations among these 190 positions may be related to the virus’ drug resistance capability. Our goal is to find those positions that are either independently or interactively associated with each of the treatments.
Interaction Patterns for Drug Resistance of Zidovudine.
Fig. S3A shows the interactively associated mutations the BVP model found, all of which are on the drug resistance mutation list.Table S2 shows all the mutation interaction patterns we found. The top three have a posterior probability > 0.25. We have also checked the detailed configurations of the top interaction patterns with the phenotypic data [fold resistance from the Stanford HIVdb (Table S4), which provide confirming evidence for the significant ones (after Bonferroni corrections).
As shown in Fig. S3B, the RMS procedure decomposed the set of interacting mutations {41,67,70,210,215,219} into three independent groups: {41,210,215} for group one, {67,219} for group two, and 70 for group three. For group one, it has been observed that mutations between M41L, L210W, and T215Y/F tend to occur together (3, 18 19–20). We also inferred that L210W appears after T215Y/F, which is consistent with crystallographic studies. The aromatic side chain of Trp 210 can stabilize the interaction of Phe/Tyr215 with the dNTP-binding pocket (19). For group two, it has been observed earlier that these two mutations usually occur together (3) The finding that position 70 is independent of the others suggests that the R → K reversion of residue 70 may represent a compensatory mechanism allowing a functional rearrangement of the dNTP-binding pocket in the mutated RT (19).
No Interactions Among Nevirapine-Resistant Mutations.
Our analyses of the nevirapine data suggested that the interactions among nevirapine-resistant mutations are very weak. As shown in Fig. S4A, the posterior probabilities for mutations 103, 181, 188, and 190 to interact are reasonably high under one prior distribution, whereas these probabilities diminished to near zero when another prior is used. Fig. S4B shows the total posterior probability for a mutation to resist to the drug, indicating that the results from using the two priors are consistent. Six mutations, 103, 106, 135, 181, 188, and 190, have posterior probabilities > 0.99 under both prior distributions. All but mutation 135 are on the drug resistant mutation list (5, 20). Some other positions with slightly lower posterior probabilities are also of interest. For example, it was known that K101E causes low-level resistance to each of the NNRTIs (3). The independent effects of the mutations 103, 106, 181, 188, and 190 are further confirmed by using the RMS procedure.
We have conducted molecular dynamics simulations and free energy calculations for the single mutations we found for nevirapine. The predicted binding free energies and the corresponding energy components for the wild-type and five mutated RT/nevirapine complexes are shown in Table 2. The nevirapine/RT residue interactions in each of the mutated complexes and the wild-type complex were decomposed and compared systematically in Fig. S2. Interestingly, we found that K103N mutation does not significantly change the binding mode of nevirapine in the active site of RT, which is consistent with previous studies (21). For the other mutations, the loss of the binding of the mutated residue is an important contributor to the loss of the binding free energies of nevirapine (SI Text).
Table 2.
The binding free energies and the energy components calculated by MM/GBSA (kcal/mol)
| ΔEvdw |
ΔEele |
ΔGGB |
ΔGSA |
ΔEele + ΔGGB |
ΔEvdw + ΔGSA |
ΔGcal |
ΔΔGcal* |
|
| Wild | −41.90 ± 0.04 † | −3.75 ± 0.50 | 17.62 ± 0.23 | −4.92 ± 0.03 | 13.88 ± 0.27 | −46.81 ± 0.01 | −32.94 ± 0.26 | 0.00 |
| G190A | −39.67 ± 0.27 | −1.82 ± 0.48 | 16.47 ± 0.42 | −4.90 ± 0.02 | 14.64 ± 0.06 | −44.57 ± 0.25 | −29.92 ± 0.18 | 3.02 |
| K103N | −42.57 ± 0.13 | −3.59 ± 0.13 | 18.83 ± 0.02 | −4.87 ± 0.01 | 15.24 ± 0.15 | −47.46 ± 0.14 | −32.22 ± 0.29 | 0.72 |
| V106A | −41.31 ± 0.27 | −5.31 ± 0.95 | 18.25 ± 0.49 | −4.87 ± 0.02 | 12.94 ± 0.46 | −46.18 ± 0.26 | −33.24 ± 0.72 | −0.30 |
| Y181C | −41.53 ± 0.56 | −6.47 ± 0.46 | 19.73 ± 0.22 | −4.92 ± 0.02 | 13.62 ± 0.24 | −46.45 ± 0.58 | −33.19 ± 0.34 | −0.25 |
| Y188C | −39.42 ± 0.27 | −4.28 ± 0.92 | 19.21 ± 0.73 | −5.11 ± 0.00 | 14.92 ± 0.18 | −44.53 ± 0.27 | −29.61 ± 0.09 | 3.33 |
*ΔΔG cal is the difference between the binding free energy of the mutated complex and that of the wild-type.
†Standard deviations were estimated from two block average values.
Discussion
We have proposed a unique procedure that combines Bayesian statistical modeling with molecular dynamic simulations to investigate complex interactions of drug resistance mutations of the HIV-1 protease and reverse transcriptase. The interacting mutations we have inferred, solely based on the data of treated and untreated HIV-1 sequences isolated from AIDS patients, agree very well with the drug resistance mutation list (updated spring 2008) (5). More importantly, our method can also delineate the complicated interactions among these mutations, revealing independent groups (related with different resistant mechanisms) and conditional independence relationship (indicating sequential occurrence of mutations). The follow-up MD simulations and free energy analyses reveal that mutations at positions 46, 54, and 82 of the protease directly affect the binding of indinavir, whereas mutations at 73 and 90 do not, and the additional mutation I54V neutralizes the resistance caused by M46I while amplifying the one caused by V82A.
Most published works (7, 8, 23) attempted to predict phenotype (e.g., fold change) from genotype by using genotype-phenotype data from Stanford HIVdb. The phenotype data, unfortunately, were measured in vitro. Due to complex disease progression and other pharmacokinetic factors, the fold change measured in vitro does not necessary imply virologic failure in vivo (3). In contrast, our Bayesian method is not designed to predict phenotypes, but constructed to detect mutation patterns associated with drug treatment by using only the genotype-treatment data.
Among all the published methods (7, 8, 23, 24) that are related to our study, the one by Haq et al.(24) is most closely related. They attempted to achieve a similar goal by using similar datasets. Technically, their method tests individually all two-way and three-way mutation interactions for associations with drug treatment, and then selects significant terms to fit a full log-linear model with up to three-way interactions. They found many significant two- and three-way interactions, and a log-linear model with 15 positions. Although many of their findings are consistent with ours, Haq et al. (23) did not aim to and could not pin down the detailed interaction structures as we reported here that can lead to testable biological hypotheses (12, 13) and are to be verified biophysically. The lack of such an interaction structure, generally, makes it difficult to interpret the results. In addition, their exhaustive search and model building strategies may both be expensive to scale up and tend to miss high-order dependence structures (9) that are critical in revealing the molecular basis of drug resistance (e.g., the order of mutations of positions {46,54,82} to cause resistance).
There are still many complications that have not been considered in our current model. For example, our HIV data are from all over the world (downloaded from the Stanford HIVdb). It is possible that there are multiple subpopulations in both treated and untreated populations. Thus, population structure and possibly other factors may bias our statistical analysis, which is why it is important to conduct the follow-up molecular dynamic computations. Another issue is the quasi-species nature of HIV-1. The HIV-1 population within an individual consists of innumerable variants and minor variants that often go undetected (3). It is possible that our data underrepresented those minor variants.
Furthermore, HIV-1 drug resistance can be not only acquired (developing in a person receiving antiretroviral treatment) but also transmitted (occurring because a virus with drug-resistance mutations was transmitted to a drug-naive person) (14). In recent years, the transmitted resistance occurrence has been increasing due to scaled-up antiretroviral treatments. In Europe, North America, and Brazil, it has been reported that the prevalence of drug resistance ranges from 5–15% in newly diagnosed individuals (14). Because our untreated sequence data were collected from 1982 to 2005, it is possible that there are several transmitted drug resistant sequences in the untreated group that may affect both the sensitivity of our BVP algorithm and the power of our Bayesian model structure inference method. Because there are many antiretroviral drugs, cross-resistance is a severe and practical problem (3).
Nevertheless, this proof-of-concept study has demonstrated that the insights obtained from MD simulations guided by the Bayesian inference can shed light on how to improve the potency of drugs to combat resistance. We believe that this procedure can be generalized and applied to study drug resistance in other infectious diseases, antibiotics, or cancer cells.
Methods and Materials
Bayesian Variable Partition Model.
Suppose there are N t sequences in the drug-treated sample and N u sequences in the untreated sample. Each sequence is of p-residues long, and residue type X j at position j can be one of L j-possible amino acids. The dataset consists of observations on the status (or response) variable Y of each sequence, that is , zero if it is from an untreated person and one if from a treated person, and its p, “explanatory” variables, X 1,…,X p (i.e., the sequence). The N t sequences are assumed to be independent and identically distributed (IID) observations of the variables X 1,…,X p from the treated population and the N u sequences are IID observations of these variables from the untreated population.
The Bayesian variable partition (BVP) model seeks to partition the p variables into three groups: G 0 for variables unlinked to the response variable Y,G 1 for variables associated independently with Y, and G 2 for variables jointly associated with Y. Let the vector I = (I 1,…,I p) indicate memberships so that I j = k if X j is in group k. The BVP model postulates that, for individual i,
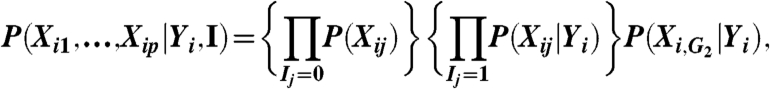 |
where we define X i,G2 = {X ij∶I j = 2}. Let (X,Y) be the observed data with N = N t + N u including both treated and untreated. We have the joint posterior distribution:
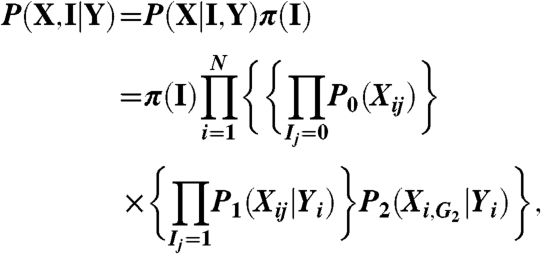 |
assuming that the partition indicator I and Y are mutually independent a priori. Note that I is the same for all individuals and π(I) is its prior distribution. We model P 0(X ij) by a multinomial distribution (it is independent of Y i because the variable is in group zero), denoted as multinom (θ j), with θ j following a Dirichlet distribution a priori. Similarly, we model P 1(X ij|Y i) by multinom (θ j,Yi) with θ j,Yi following a Dirichlet prior and model P 2(X i,G2|Y i) by multinom (ΘG2,Yi). For Y i = 1 (treated), the dimension of ΘG2,1 is equal to the cardinality of the support of X i,G2 and ΘG2,1 follows a Dirichlet prior.
Ideally, all mutation positions among the untreated sequences (Y
i = 0) should be mutually independent. Complications may arise, however. We thus introduce a model indicator variable J
un (same for all untreated individuals) so that the independence prior model 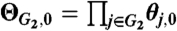 holds only when J
un = 0, with θ
j,0 following a Dirichlet distribution; ΘG2,0 is fully saturated as ΘG2,1 when J
un = 1, following a full Dirichlet distribution. We observed that J
un = 0 in most cases, that is, the mutations in G
2 are mutually independent for untreated individuals. Conditional on I and J
un, we can integrate out all the multinomial parameters so as to have the posterior distribution of (I,J
un). A Markov chain Monte Carlo (MCMC) algorithm (9) can be designed to sample from this posterior distribution so as to infer which variables are associated with the treatment status. More details on BVP can be found in SI Text.
holds only when J
un = 0, with θ
j,0 following a Dirichlet distribution; ΘG2,0 is fully saturated as ΘG2,1 when J
un = 1, following a full Dirichlet distribution. We observed that J
un = 0 in most cases, that is, the mutations in G
2 are mutually independent for untreated individuals. Conditional on I and J
un, we can integrate out all the multinomial parameters so as to have the posterior distribution of (I,J
un). A Markov chain Monte Carlo (MCMC) algorithm (9) can be designed to sample from this posterior distribution so as to infer which variables are associated with the treatment status. More details on BVP can be found in SI Text.
Recursive Model Selection.
In the above BVP model, variables in G 2 are not given any simplifying dependence structure, which in statistical term means that a “fully saturated” model was used. However, in practice, often a much more desirable and simpler model that takes advantage of conditional independence relationships among the variables can fit the data well. A possible approach is to infer a complete Bayesian network for all the variables in G 2. But this is computationally expensive and tends to over fit the limited amount of data. Our strategy is to first infer among two classes of cruder models, that is, the chain-dependence model and the V-dependence model, and then recursively apply this strategy until the data do not support more detailed models.
We say that a group of variables X G follow a chain-dependence model if the index set G can be partitioned into three subgroups A, B, and C such that X A and X C are independent given X B, such as X A → X B → X C. Only set C is allowed to be empty, in which case this model degenerates to the saturated model. Under the chain-dependence model, we can decompose the joint distribution of X G as: P(X G) = P(X A)P(X B|X A)P(X C|X B) (Fig. S5A). We say that X G follow a V-dependence model if X A and X C are mutually independent, that is, P(X G) = P(X A)P(X C)P(X B|X A,X C). In this case, X B can be viewed as “children” of X A and X C (Fig. S5B). Although these models are not fully identifiable, RMS attempts to land in the best equivalent class of models.
We define a model indicator I CV, which is equal to one for the chain-dependence model and zero for the V-dependence model. We let Π denote the set partition, that is, indicating which indices in G belong to which subset. In SI Text, we detailed the model likelihoods for the two competing models conditional on the partition Π, that is, P(D|Π,I CV = 1) and P(D|Π,I CV = 0), where D denotes all the data. Assuming an equal prior probability for I CV, we have that:
| [1] |
Here P(D|Π,I CV = 1) and P(D|Π,I CV = 0) can be computed, respectively, by using formulas (S5) and (S9) of SI Text. An MCMC algorithm is designed to simulate from (1) and to find the optimal model type and variable partition. The procedure is applied recursively until only single-variable nodes are available. We applied RMS to both treated data and untreated data separately. Fig. 2 illustrates the structure we found in the treated data (Fig. S7 shows the details of recursion). In contrast, we could not find an unambiguous structure in the untreated data.
Supplementary Material
Acknowledgments.
This work was supported in part by the National Institutes of Health Grant R01-HG02518-02 and R01GM085188, the National Science Foundation (NSF) Grant DMS-0706989, and NSF Physics Frontiers Center-sponsored Center for Theoretical Biological Physics (CTBP) Grants PHY-0216576 and PHY-0225630. MD simulations were performed on the Linux cluster in the CTBP at University of California, San Diego. T. H. was supported by a CTBP postdoctoral scholarship.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/cgi/content/full/0907304107/DCSupplemental.
References
- 1.Lengauer T, Sander O, Sierra S, Thielen A, Kaiser R. Bioinformatics prediction of HIV coreceptor usage. Nat Biotechnol. 2007;25:1407–1410. doi: 10.1038/nbt1371. [DOI] [PubMed] [Google Scholar]
- 2.Lengauer T, Sing L. Bioinformatics-assisted anti-HIV therapy. Nat Rev Microbiol. 2006;4:790–797. doi: 10.1038/nrmicro1477. [DOI] [PubMed] [Google Scholar]
- 3.Shafer RW. Genotypic testing for Human Immunodeficiency Virus type 1 drug resistance. Clin Microbiol Rev. 2002;15:247–277. doi: 10.1128/CMR.15.2.247-277.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liu TF, Shafer RW. Web resources for HIV type 1 genotypic-resistance test interpretation. Clin Infect Diseases. 2006;42:1608–1618. doi: 10.1086/503914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Johnson VA, et al. Update of the drug resistance mutations in HIV-1: Spring 2008. Top HIV Med. 2008;16:62–68. doi: 10.1007/s11750-007-0034-z. [DOI] [PubMed] [Google Scholar]
- 6.Ravela J, et al. HIV-1 protease and reverse transcriptase mutation patterns responsible for discordances between genotypic drug resistance interpretation algorithms. J Acq Immun Def Synd. 2003;33:8–14. doi: 10.1097/00126334-200305010-00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Beerenwinkel N, et al. Diversity and complexity of HIV-1 drug resistance: A bioinformatics approach to predicting phenotype from genotype. P Natl Acad Sci USA. 2002;99:8271–8276. doi: 10.1073/pnas.112177799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rhee SY, et al. Genotypic predictors of human immunodeficiency virus type 1 drug resistance. P Natl Acad Sci USA. 2006;103:17355–17360. doi: 10.1073/pnas.0607274103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang Y, Liu JS. Bayesian inference of epistatic interactions in case-control studies. Nat Genet. 2007;39:1167–1173. doi: 10.1038/ng2110. [DOI] [PubMed] [Google Scholar]
- 10.Rhee SY, et al. Human immunodeficiency virus reverse transcriptase and protease sequence database. Nucleic Acids Res. 2003;31:298–303. doi: 10.1093/nar/gkg100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Condra JH, et al. In vivo emergence of HIV-1 variants resistant to multiple protease inhibitors. Nature. 1995;374:569–571. doi: 10.1038/374569a0. [DOI] [PubMed] [Google Scholar]
- 12.Condra JH, et al. Genetic correlatets of in vivo viral resistance to indinavir, a Human Immunodeficiency Virus type 1 protease inhibitor. J Virol. 1996;70:8270–8276. doi: 10.1128/jvi.70.12.8270-8276.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang YM, et al. Drug resistance during indinavir therapy is caused by mutations in the protease gene and in its Gag substrate cleavage sites. J Virol. 1997;71:6662–6670. doi: 10.1128/jvi.71.9.6662-6670.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shafer RW, et al. HIV-1 protease and reverse transcriptase mutations for drug resistance surveillance. AIDS. 2007;21:215–223. doi: 10.1097/QAD.0b013e328011e691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu FL, Boross PI, Wang YF, Tozser J, Louis JM, et al. Kinetic, stability, and structural changes in high-resolution crystal structures of HIV-1 protease with drug-resistant mutations L241, 150V, and G73S. J Mol Bio. 2005;354:789–800. doi: 10.1016/j.jmb.2005.09.095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mahalingam B, Wang YF, Boross PI, Tozser J, Louis JM, et al. Crystal structures of HIV protease V82A and L90M mutants reveal changes in the indinavir-binding site. Eur J of Biochem. 2004;271:1516–1524. doi: 10.1111/j.1432-1033.2004.04060.x. [DOI] [PubMed] [Google Scholar]
- 17.Sluis-Cremer N, Arion D, Parniak MA. Molecular mechanisms of HIV-1 resistance to nucleoside reverse transcriptase inhibitors (NRTIs) CMLS, Cell Mol Life S. 2000;57:1408–1422. doi: 10.1007/PL00000626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Harrigan PR, et al. Significance of amino acid variation at human immunodeficiency virus type 1 reverse transcriptase residue 210 for zidovudine susceptibility. J Virol. 1996;70:5930–5934. doi: 10.1128/jvi.70.9.5930-5934.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hooker DJ, et al. An in vivo mutation from Leucine to tryptophan at position 210 in human immunodeficiency virus type 1 reverse transcriptase contributes to high-level resistance to 3′-azido-3′-deoxythymidine. J Virol. 1996;70:8010–8018. doi: 10.1128/jvi.70.11.8010-8018.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yahi N, Tamalet C, Tourres C, Tivoli N, Fantini J. Mutation L210W of HIV-1 reverse transcriptase in patients receiving combination therapy: Incidence, association with other mutations, and effects on the structure of mutated reverse transcriptase. J Biomed Sci. 2000;7:507–513. doi: 10.1007/BF02253366. [DOI] [PubMed] [Google Scholar]
- 21.Deeks SG. Nonnucleoside reverse transcriptase inhibitor resistance. J Acq Immun Def Synd. 2001;26:S25–33. doi: 10.1097/00042560-200103011-00004. [DOI] [PubMed] [Google Scholar]
- 22.Hsiou Y, et al. The Lys103Asn mutation of HIV-1 RT: a novel mechanism of drug resistance. J Mol Biol. 2001;309:437–445. doi: 10.1006/jmbi.2001.4648. [DOI] [PubMed] [Google Scholar]
- 23.Saigo H, Uno T, Tsuda K. Mining complex genotypic features for predicting HIV-1 drug resistance. Bioinformatics. 2007;23:2455–2462. doi: 10.1093/bioinformatics/btm353. [DOI] [PubMed] [Google Scholar]
- 24.Haq O, Levy RM, Morozov AV, Andrec M. Pairwise and higher-order correlations among drug-resistance mutations in HIV-1 subtype B protease. BMC Bioinformatics. 2009;10(Suppl 8):S10. doi: 10.1186/1471-2105-10-S8-S10. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.