

Summary
Recent studies suggest that p53 binds predominantly to consensus sites composed of two decameric half-sites with zero spacing in vivo. Here we report the crystal structure of the p53 core domain bound to a full consensus site as a tetramer at 2.13Å resolution. Comparison with previously reported structures of p53 dimer:DNA complexes and a chemically trapped p53 tetramer:DNA complex reveals that DNA binding by the p53 core domain is a cooperative self-assembling process accompanied by structural changes of the p53 dimer and DNA. Each p53 monomer interacts with its two neighboring subunits through two different protein-protein interfaces. The DNA is largely B-form and shows no discernible bend, but the central base-pairs between the two half sites display a significant slide. The extensive protein-protein and protein-DNA interactions explain the high cooperativity and kinetic stability of p53 binding to contiguous decameric sites and the conservation of such binding-site configuration in vivo.
Introduction
P53 is essential for the prevention of cancer development; its function is inactivated by mutations and signal-dependent modifications in a majority of human cancers (Horn and Vousden, 2007; Vogelstein et al., 2000). While p53 may suppress tumor formation and progression through multiple mechanisms (Green and Kroemer, 2009; Suzuki et al., 2009), its primary function is to regulate transcription programs of cell growth control (Vousden and Prives, 2009). In responses to oncogenic stresses, p53 binds specific DNA targets and regulates the expression of genes involved in cell cycle arrest, DNA repair or apoptosis. Like many eukaryotic transcription factors, p53 has a modular structure with multiple domains, including a core DNA-binding domain (residues ∼94-292) and a tetramerization domain (residues 325-356). Both domains are structurally folded and are connected by a flexible linker (residues 293-324). Conversely, the N- and C-terminal regions (residues 1-93 and 357-393, respectively) of p53 are largely unstructured. These regions harbor the transactivation domain (TAD) and a variety of motifs that can modulate the function of p53 through signal-specific posttranslational modifications and interactions with other proteins (Joerger and Fersht, 2008).
More than 80% missense mutations of p53 found in human cancers are located in the core domain (Olivier et al., 2002). Many of these mutations disrupt DNA binding directly or reduce the folding stability of the core domain. These observations suggest that DNA binding is important to p53 function. Earlier studies indicated that p53 binds two decameric sites RRRCWWGYYY (R=A, G; W=A, T; Y=C, T), separated by 0-13 base pairs (el-Deiry et al., 1992; Funk et al., 1992). Further structural and biochemical analyses show that p53 binds each decameric half site as a dimer and the two dimers from the two half sites constitute the tetramer (or dimer of dimer) (Ho et al., 2006; Kitayner et al., 2006; Klein et al., 2001; Malecka et al., 2009; Rippin et al., 2002). The flexible linker between the tetramerization domain and the DNA-binding domain can apparently accommodate the variable spacing between the two half sites. This model of DNA binding is consistent with the fact that p53 functions as a tetramer. It also suggests that the structure of the p53 tetramer on DNA varies depending on the DNA sequences it binds.
With new technologies capable of identifying binding sites of transcription factors at a genome-wide scale, recent studies reveal that p53 binds mostly to two decameric sites with no spacing in vivo (Smeenk et al., 2008; Wei et al., 2006). In these studies, more than 80% p53-binding sequences identified by ChIP-on-chip or ChIP-PET (ChIP coupled with paired-end ditag sequencing) contain such a configuration. Considering the background of antibody-based pull-down and other factors (e.g. recruited to DNA by partner proteins), the percentage could be even higher so that p53 binds almost exclusively to elements made of contiguous decameric repeats. We will refer to such sites as the consensus p53 site throughout the text. It is important to point out that while the configuration of the p53 consensus site is conserved, its sequence could vary in different promoters for specific gene regulation (Ma et al., 2007). The conservation of the binding-site configuration suggests that the structure of the p53 tetramer bound to DNA is well defined and that the stereo-specific structure may be important for function. The specific arrangement of binding sites in a composite element is often conserved for protein-protein interactions between neighboring transcription factors that bind DNA cooperatively (Chen, 1999). These protein-protein interactions are usually mediated by DNA-proximal domains, including the DNA-binding domain (Chen, 1999; Chen et al., 1998; Wu et al., 2006). Indeed, in vitro binding assays show that p53 or its core domain binds contiguous decameric sites cooperatively and any base insertion in between disrupts the cooperativity, however, the structural basis and functional significance of this cooperativity is not fully understood (Balagurumoorthy et al., 1995; Kitayner et al., 2006; McLure and Lee, 1998; Weinberg et al., 2004).
The structure of the p53 core domain and its complexes with DNA has been extensively characterized (Cho et al., 1994; Joerger and Fersht, 2008). These studies have revealed the details of protein folding, DNA recognition, and potential mechanisms of some cancer-associated mutations. Protein-protein interaction between p53 core domains in higher-order p53 complexes has also been a subject of experimental and modeling studies (Cho et al., 1994) (Ho et al., 2006) (Kitayner et al., 2006; Klein et al., 2001; Ma et al., 2005; Rippin et al., 2002). These studies have identified a functionally important dimer interface between two p53 core domains bound to the decameric half site. In one study (Kitayner et al., 2006), two such dimers, each bound to a separate DNA molecule, stack end-to-end along the DNA axis. As such, a p53 tetramer is formed in the crystal lattice. However, the two p53 dimers showed little interaction with each other because the two decameric half-sites are separated by two base pairs and the axes of the two DNA molecules are off-set. More recently, the crystal structure of a chemically trapped p53 tetramer bound to DNA has been solved (Malecka et al., 2009). In this complex, the p53 core domain is covalently linked to each quarter site of a full p53-binding element that contains no spacing between the two decameric half sites. The structure showed that the two p53 dimers could bind the adjacent decameric half sites without significant DNA bending (Ho et al., 2006; Nagaich et al., 1999; Pan and Nussinov, 2007). The structure also revealed an interaction interface between p53 dimers bound to the contiguous decameric half sites. However, as discussed below, the dimer-dimer interactions observed in the chemically trapped complex are likely affected by the covalent crosslinking and may differ significantly from those in the native complex.
In this study, we have solved the crystal structure of a p53 core domain tetramer assembled on a full consensus site. A striking feature of the complex is that the four p53 core domains form an enclosed structure to bind a large region of DNA. The stability of the complex is enhanced by the extensive and interconnected protein-protein and protein-DNA interactions of the core domain, which would function together with the tetramerization domain in the full protein to enhance DNA binding. The structure reveals a novel dimer-dimer interface in the p53 core tetramer and suggests a highly cooperative self-assembling mechanism for its formation. The dimer-dimer interface is absent in previous p53 complexes bound to short DNA and significantly different and smaller in the chemically crosslinked complex. These findings explain, at least partly, the zero spacing in the consensus p53-binding sites observed in vivo. The structural features observed in the naturally assembled p53 core domain tetramer complex underlie the high cooperativity and kinetic stability of DNA binding by p53 to contiguous decameric sites. These structural insights will help to understand and further study the function of p53 and its role in tumor suppression.
Results
Crystallization and structure determination
It is well known that p53 and its core domain can bind contiguous decameric sites cooperatively (Balagurumoorthy et al., 1995; McLure and Lee, 1998). Recent data further show that p53 binds predominantly to such sites in vivo (Smeenk et al., 2008; Wei et al., 2006). However, crystallization of the corresponding higher-order p53:DNA complexes has not been achieved despite extensive efforts (Zhao et al., 2001). The p53 core domain is a monomer in solution but forms a tetramer upon binding to DNA. Such a dynamic and multi-body binding reaction may present a challenge to crystallization. One way to overcome this difficulty is to covalently link the p53 core domain to DNA, which has led to the crystallization of a p53 core domain tetramer chemically trapped on DNA (Malecka et al., 2009). To crystallize a naturally assembled complex, we took a more traditional approach by screening DNA fragments of different lengths and overhang sequences under various conditions. These efforts have led to the crystallization of the human p53 core domain (residues 92 – 292) bound to a full consensus p53 site as a tetramer (Figure 1a). For simplicity we will refer the core domain tetramer as the tetramer throughout the text here. The structure was solved by the molecular replacement method using the structure of the p53 core domain as a partial search model (Kitayner et al., 2006). The statistics of data collection and model refinement are listed in Table 1.
Figure 1. Overall structure of the p53 core domain bound to DNA as a tetramer.
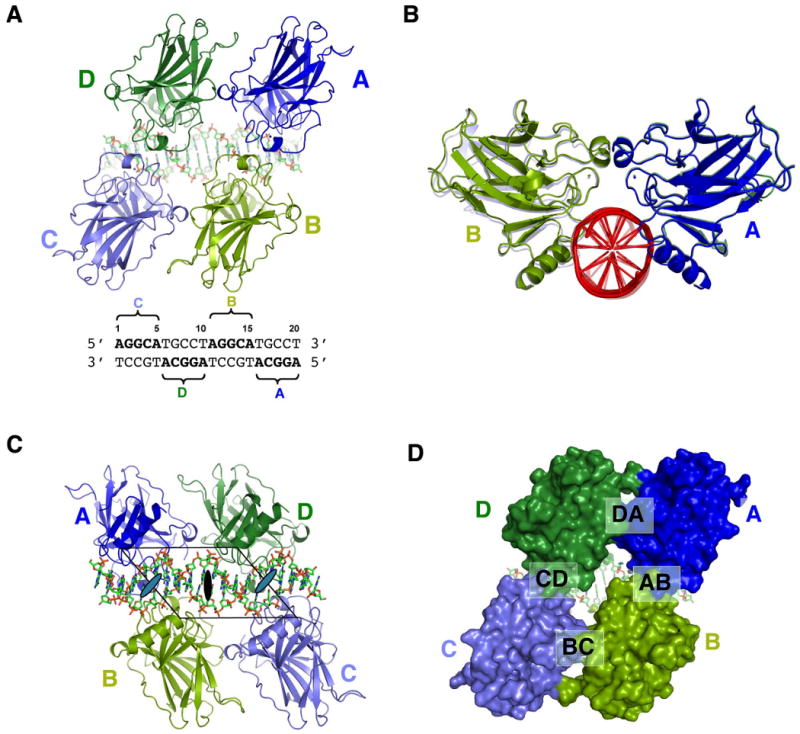
(a) The tetramer viewed from the protein side. The four monomers are colored in blue (A), light green (B), light blue (C) and green (D). The same color scheme is used throughout the illustration unless indicated otherwise. The DNA is in stick model with its sequence shown below. The four pentameric motifs (quarter site) and their corresponding monomers are indicated in the sequence; (b) A view of the tetramer along the DNA axis. This view shows that the tetramer has a planer structure wherein the A-B dimer (front) and C-D dimer aling almost perfectly along the DNA axis; (c) The tetramer viewed from the DNA side. The parallelogram is shown together with the global two fold axis (dark oval) and the two local dyad aces (gray ovals). (d) A surface model of the tetramer view in the same orientation as (a). The four protein-protein interfaces are indicated.
Table 1. Statistics of crystallographic analysis.
| Data Set | |
|---|---|
| Resolution (Å) | 50-2.13 (last bin 2.23Å -2.13Å) |
| 1Rsym | 0.138 (0.554) |
| 2Completeness (%) | 99.5 (95.7) |
| I/σ(I) | 17.74 (2.00) |
| Redundancy | 7.0 (5.4) |
| Refinement | |
| Resolution (Å) | 48.52-2.15 (last bin 2.23Å -2.13Å) |
| 3,4R-factor (%) | 0.2161 (0.2556) |
| 3,4Rfree (%) | 0.2395 (0.2770) |
| r.m.s deviations | |
| Bond lengths (Å) | 0.006 |
| Bond angles (º) | 1.034 |
| Average B-Factor (Å2) | 32.57 |
Rsym= |I• 〈I〉 |/ I, where I is the observed intensity, 〈I〉 is the statistically weighted average intensity of multiple observations of symmetry-related reflections.
Number in parentheses is for the outer shell (last bin).
Rwork= ‖Fo| - |Fc‖/ |Fo| where Fo and Fc are the observed and calculated structure factor amplitudes, respectively.
Rfree is calculated for 5% of the data that was withheld from refinement.
Overall structure of the tetramer complex
The asymmetric unit of the crystal contains a fully assembled tetramer:DNA complex, wherein each monomer occupies one of the four identical pentameric motifs, also referred to as the quarter site (Figure 1a). The tetramer has a planar structure with the shape of a parallelogram (Figure 1a,b). One set of the parallel sides, which align with the major groove axis of each decameric half site, is formed by protein-protein contacts between monomer pairs of A-B and C-D (Figure 1c). The other set of the parallel sides, which align with the DNA axis, is formed by protein-protein contacts between the monomer pairs of D-A and B-C (Figure 1a). There is no direct contact between monomer pairs A-C and B-D. A large solvent channel is present at the center of the tetramer leading to the surface of the DNA (Figure 1d). The angle between the D-A contact and the A-B contact is about 45°, which is approximately the same as the crossing angle between the major groove and the DNA axis. The tetramer has a global two-fold axis at the center and two local dyads between A-B and C-D, while monomer pairs A-D and B-C are related by translation of one turn of DNA along the DNA axis. All three dyad axes are perpendicular to, and intercepting with, the DNA axis. Overall, the geometry and symmetry of the tetramer has an exquisite match with that of the DNA, such that the p53 tetramer form a plane of protein to bind the minor groove face of the p53 site (Figure 1a,b) while leaving the major groove side of the DNA largely exposed (Figure 1c). Despite the perfectly palindromic DNA sequence and the four identical core domains, the packing interactions experienced by the A-B dimer and C-D dimer are different. The crystal environments of interface D-A and B-C (see Figure 1d) are also very different. As a result, some local structural details at the interface of D-A and B-C are different (data not shown).
Structure of the p53 core domain
With the high-resolution data, we did not use any non-crystallographic symmetry constraints during the late stages of the refinement. Nevertheless, the overall structures of the four monomers are highly similar to each other and to the previously determined structures. For example, backbone superposition of 200 Cα atoms between monomer pairs of A-B and A-D gives an rmsd of 0.136Å and 0.416Å, respectively. A similar superposition with previously determined human p53 core domain structures 1TSR and 2ATA gives an rmsd of 0.601Å and 0.480Å, respectively (Cho et al., 1994; Kitayner et al., 2006). These observations suggest that the beta-sandwich fold of the p53 core domain is relatively stable in different complexes. However, loops emanating from the immunoglobulin core do show some variations in different structures. Most notable is the L1 loop (residue 117-122), whose conformation was shown to be variable in previous studies (Cho et al., 1994; Ho et al., 2006; Kitayner et al., 2006; Malecka et al., 2009). In the present structure this loop shows two distinct conformations. In monomers B and D bound to the two inner quarter sites (Figure 1a), the L1 loop shows well-defined electron density and tucks in the major groove (Figure 2a). In monomers A and C bound to the two outer quarter sites, the electron density of the L1 loop is less well-defined, but the backbone density suggests that it adopts an outward projection away from the major groove (Figure 2a). However, this interpretation is complicated by the fact that the DNA ends, to which the L1 loop interacts with, are partially disordered (see Supplemental Figure 1 below). The N-terminal tail of the p53 core domain (residues 92-100) also shows conformational variations in different structures, ranging from completely disordering in some structures to partial ordering in others. In the present structure, this region of the p53 core domain shows well-defined electron density and forms a major part of the dimer-dimer interface (discussed further below) (Figure 2b)
Figure 2. Structure of the p53 core domain in the tetramer.
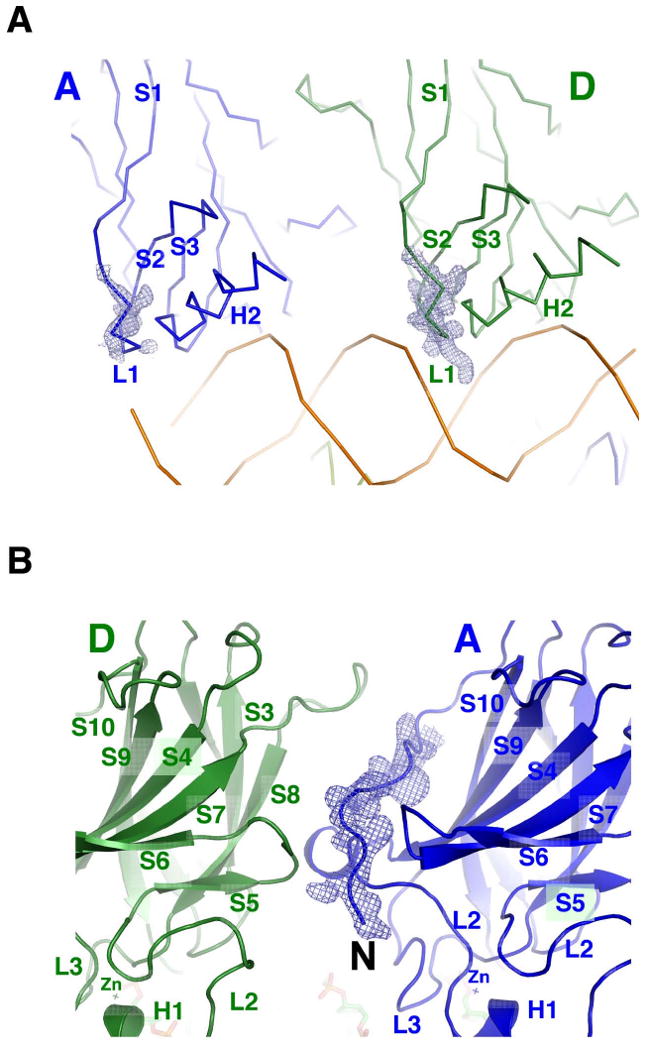
(a) Structural variation of the L1 loop in the tetramer is shown by the comparison between monomer A (left) and monomer D (right). The electron density (sigma-a weighted, 3fo2fc, contour level at 1 e/Å3) of the L1 loop of monomer D is well defined and is pointed into the major groove, whereas that of monomer A is partially disordered with a trajectory away from the major groove. This view is the same as Figure 1c; (b) The N-terminal tail has well-defined electron density. The secondary structural elements, including the zinc clusters, are indicated for both monomers (A and D). This view is the same as Figure 1a.
Structure of the DNA
In contrast to other p53:DNA complex crystals (Cho et al., 1994; Ho et al., 2006; Kitayner et al., 2006; Malecka et al., 2009), the DNA molecules in the present crystal do not pack end-to-end to form a pseudo-continuous helix. Rather, it is embedded in the protein complex and makes few crystal contacts. Modeling analyses suggest that the DNA has to bend to accommodate the binding of four p53 core domains to two contiguous decameric sites (Ho et al., 2006; Nagaich et al., 1999; Pan and Nussinov, 2007). We therefore did not use any DNA constraints in the structure refinement. As shown in Figure 3, the DNA has well-defined electron density and shows no discernible deformation, although the ends of the DNA appear to be partially disordered (Supplemental Figure 1a and b). Detailed analysis using the program 3DNA shows that the tetramer-bound DNA is largely B-form and straight (Lu and Olson, 2003). However, the center of the DNA does show some unusual structural features. The base-pair step between Thy10/Ade and Ade11/Thy has a large slide (2.8 Å), negative roll (-9.0°) and twist (49.6°) (Table 2). These local base-pair step parameters differ significantly (by more than two standard deviations) from their corresponding value found in standard B-DNA. Thus, the middle of the p53 site does undergo significant structural changes upon the binding of p53.
Figure 3. Structure of the DNA in the tetramer.
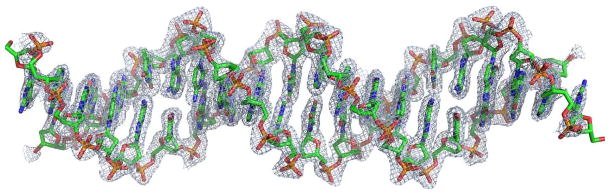
The sigma-a weighted 3fo2fc density at 3 e/Å3 shows that the structure of the DNA is well defined except for the two ends. The DNA does not show significant bend and other major deformations from the standard B-form.
Table 2. Local base-pair step parameters.
| Step | Shift | Slide | Rise | Tilt | Roll | Twist |
|---|---|---|---|---|---|---|
| 2 GG/CC | 0.77 | 0.71 | 2.46 | -8.95 | 5.61 | 29.81 |
| 3 GC/GC | 0.76 | -0.48 | 3.25 | 4.01 | 1.37 | 30.13 |
| 4 CA/TG | 0.29 | 0.02 | 3.86 | 0.80 | 8.16 | 42.65 |
| 5 AT/AT | -0.02 | -0.33 | 2.96 | 0.29 | 4.81 | 23.49 |
| 6 TG/CA | -0.31 | 0.14 | 3.70 | -2.32 | 4.05 | 41.93 |
| 7 GC/GC | -0.32 | 0.06 | 3.36 | -2.11 | -3.95 | 37.59 |
| 8 CC/GG | -0.94 | -0.01 | 3.35 | -3.35 | 3.72 | 32.07 |
| 9 CT/AG | 0.20 | 0.00 | 3.22 | 2.14 | 1.68 | 31.94 |
| 10 TA/TA | 0.02 | 2.79 | 3.25 | 0.50 | -9.00 | 49.55 |
| 11 AG/CT | -0.16 | 0.12 | 3.35 | -1.65 | 2.58 | 28.36 |
| 12 GG/CC | 0.78 | 0.17 | 3.24 | 3.08 | 5.97 | 32.70 |
| 13 GC/GC | -0.04 | -0.02 | 3.43 | 0.91 | -4.95 | 40.06 |
| 14 CA/TG | 0.46 | 0.05 | 3.55 | 0.30 | 6.04 | 42.40 |
| 15 AT/AT | 0.21 | -0.39 | 2.89 | 1.66 | 3.34 | 22.59 |
| 16 TG/CA | -0.49 | 0.24 | 3.68 | -1.47 | 6.00 | 44.27 |
| 17 GC/GC | -0.51 | -0.64 | 3.41 | -3.13 | 3.93 | 30.11 |
| 18 CC/GG | -0.73 | 1.18 | 2.43 | 9.00 | 7.90 | 31.68 |
| ave. | -0.00 | 0.21 | 3.26 | -0.02 | 2.78 | 34.78 |
| s.d. | 0.52 | 0.79 | 0.39 | 3.80 | 4.68 | 7.62 |
The table was generated using the program of 3DNA (Lu and Olson, 2003). The definition of base-pair step parameters follows the convention by Olson et al. (Olson et al., 2001). The base pairs at the ends of the DNA were not included in the analysis. The central base pairs and its significantly deviated parameters are highlighted in bold.
Protein-DNA interactions
The tetramer buries a total of 4002Å2 solvent accessible surface area at the protein-DNA interface. At each quarter site (5′-AGGCA-3′), the immunoglobulin-like core domain clamps down the phosphate backbone of the complementary strand (3′-TCCGT-5′) from the major and minor grooves. At the major groove side, residues from the L1 loop (Lys120 and Ser121), strand β10 and helix H2 (Arg273, Ala276, Cys277, and Arg280) interact with DNA bases and backbone, whereas residues from the L3 loop (Ser241 and Arg248) contact DNA from the minor groove side. Most of the interactions are similar to that seen in the monomeric and dimeric p53:DNA complexes, including some water-mediated interactions (Cho et al., 1994; Ho et al., 2006; Kitayner et al., 2006). For example, three water molecules in the major groove mediate an extensive network of hydrogen bonds at the protein-DNA interface (Figure 4a). The positions of these water molecules and their interactions with protein and DNA are nearly identical to that seen in the p53 dimer:DNA complexes (Kitayner et al., 2006) (Figure 4a, superimposed with 2ATA.pdb). These structural similarities not only demonstrate the importance of water-mediated interactions but also the conserved nature of DNA binding by β10 and H2. By contrast, DNA contacts made by the L1 loop and the L3 loop show notable variations between different monomers. In monomers B and D, which bind the two inner quarter sites, the L1 loop tucks into the major groove. Lys120 makes a pair of hydrogen bonds with N7 of the second guanine and O6 of the third guanine, respectively, while Ser121 makes a hydrogen bond to the first adenine the quarter site (5′-AGGCA-3′) (Figure 4b). The L1 loop in monomers A and C is less well defined but seems poised to interact with DNA outside the core region (see Figure 2a above). Arg248 of monomers B and D inserts into the minor groove and makes a water-mediated hydrogen bond to N3 of the fifth adenine from the neighboring quarter site (Figure 4c). Arg248 of monomers A and C is also similarly positioned toward the minor groove, but its guanidinium group flips out to interact with the DNA backbone (Figure 4d). These structural variations may reflect the adaptability of DNA binding by p53 to different sequences (Cho et al., 1994; Ho et al., 2006; Kitayner et al., 2006; Malecka et al., 2009).
Figure 4. Conserved and variable protein-DNA interactions in the tetramer.
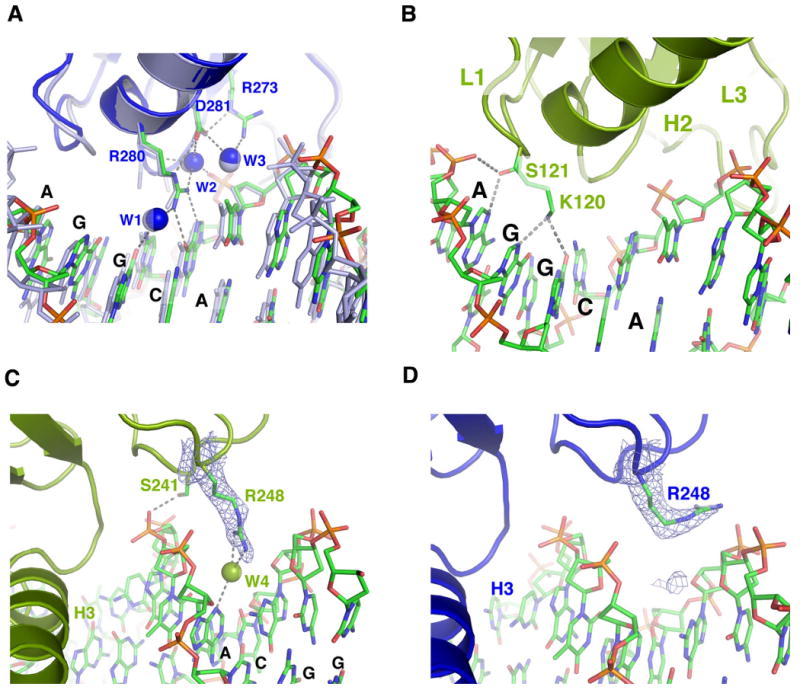
(a) A network of interactions between β10, H2, ordered water molecules (w1, w2 and w3) and DNA is shown for monomer A (blue). This protein-DNA interaction network is also seen in monomers B, C and D (not shown) and in the p53 dimer:DNA complex (2ATA.pdb, gray) (Kitayner et al., 2006); (b) DNA binding interactions by the L1 loop in monomer D. The same interactions are also seen in monomer B but not in monomers A and C. (c) Arg248 of monomers B and D (not shown) adopts an extended conformation in the minor groove and makes a water-mediated hydrogen bond to N3 of the fifth adenine from the neighboring quarter site. (d) Arg248 of monomers A and C (not shown) flips out of the minor groove to interact with the DNA backbone. The electron density of Arg248 in (c) and (d) is calculated from simulated annealing omit map and contoured at 3 e/Å3 level.
Protein-protein interactions
The tetramer contains four protein-protein interaction interfaces that bury a total of 3346Å2 of solvent accessible area (Figure 1d). The interfaces between A-B and C-D are identical to each other and are referred to as the dimer interface. This interface is mediated by helix H1, loops L2 and L3 from symmetry-related monomers within each half site, which has 776Å2 of buried solvent accessible area. The detailed protein-protein interactions are similar to those seen in a number of p53 dimer:DNA complexes (Ho et al., 2006; Kitayner et al., 2006) as well as in the chemically trapped tetramer (Malecka et al., 2009) (data not shown). The interfaces between D-A and B-C are identical to each other and are referred to as the dimer-dimer interface. The buried solvent accessible surface area of this interface is 897Å2. The dimer-dimer interface is asymmetric and involves two different protein surfaces on each side. Using the D-A interface as an example, strands β3, β8, the β5-β6 loop, and the β7-β8 loop of monomer D constitute one side of the interface whereas the N-terminal tail, the L2 loop, and the β6-β7 loop of monomer A form the other side of the interface (Figure 5a).
Figure 5. The dimer-dimer interface.
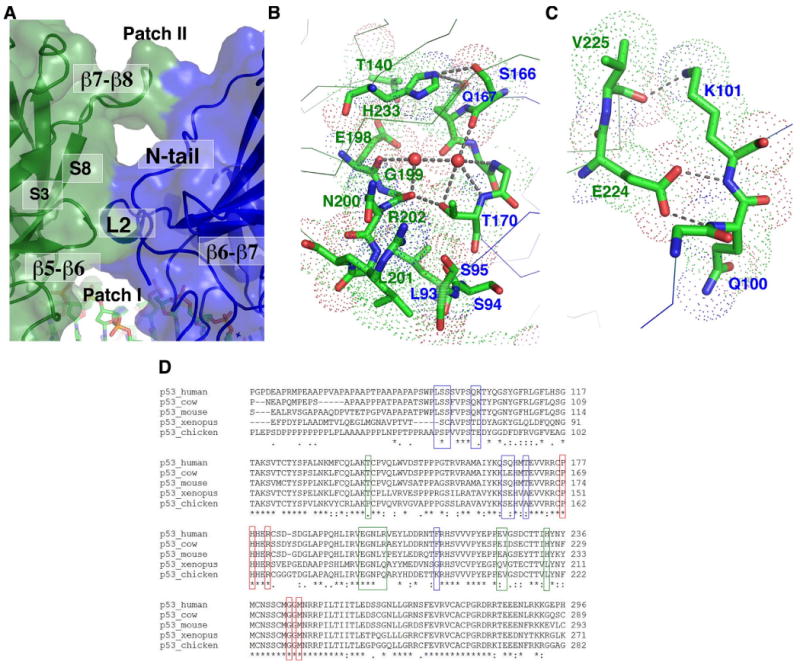
(a) Structural elements from monomer D (green) and monomer A (blue) forming the dimer-dimer interface are shown in a transparent surface model. The two patches of the interface (Patch I and II) are separated by a large solvent cavity; (b) The detailed interactions at Patch I viewed from above the dimer-dimer interface (Patch II is removed in this view for clarity). The van der Waals spheres of interface residues are shown as dotted spheres and are labeled by the color of their respective monomers (green: monomer D; blue: monomer A). The hydrogen bonds mediated by two ordered water molecules (red spheres) at the interface are also indicated; (c) The detailed interactions at Patch II viewed from the side the dimer-dimer interface (the same view as Figure 1a); (d) Sequence alignment of p53 from several representative species. Red boxes indicate residues at the dimer interface. Blue boxes indicate residues at the dimer-dimer interface on the 5′ side (monomers A and C). Green boxes indicate residues at the dimer-dimer interface on the 3′ side (monomers B and D).
The interface is made of discontinuous foci of protein-protein contacts and a number of ordered water molecules. A striking feature of the dimer-dimer interface is the large number of hydrogen bonds and van der Waals contacts made by protein main chain atoms. One patch of this interface (patch I) is formed by Leu93, Ser94, Ser95, Ser166, Gln167, Thr170, and Phe212 of monomer A and Leu201, Gly199, Asn200, Arg202, His233, Thr140, and Glu198 of monomer D (Figure 5b). Here, Leu93 of monomer A contacts the main chain of Gly199 of monomer D, Leu201 of monomer D contacts the main chain of Leu93, Ser94, Ser95 of Monomer A, Thr170 of monomer A forms a hydrogen bond with the carbonyl of Gly199 of monomer D, and His233 of monomer D contacts the backbone of Ser166 and Gln167 of monomer A. Two ordered water molecules also bridge a number of main chain carbonyls and amides together through hydrogen-bond interactions. Similarly at another patch of the interface (Patch II), Glu224 of monomer D forms a pair of hydrogen bonds with the main chain amide of Gln100 and Lys101 of monomer A, and reciprocally, Lys100 of monomer A donates a hydrogen bond to the carbonyl of Val225 of monomer D (Figure 5c). These structural features suggest that a major determinant of protein-protein interactions at the dimer-dimer interface is the shape complementarity. Most of the residues at the dimer-dimer interface are conserved across species (Figure 5d). For residues that show a certain degree of variation, modeling analyses suggest that their dimer-dimer interactions are maintained. For example, Arg202 of human p53 corresponds to a tyrosine in mouse, which is expected to make similar hydrogen bond and van der Waals contacts to the main chain of its dimer-dimer partner. Similarly, His233 of human p53 corresponds to a leucine residue in Xenopus and Chicken, which could also form van der Waals contacts to its dimer-dimer partner in the region at the end of β4 and the beginning of the L2 loop. These observations suggest that the observed dimer-dimer interface is a conserved structural feature of the p53 tetramer.
Comparison with other p53:DNA complexes
We superimposed the present structure with the chemically trapped tetramer (3EXJ.pdb) using one of the p53 core domains (monomer A) as the reference (Malecka et al., 2009). As shown in Figure 6a, although the core domain structure is nearly identical, the relative position of the four monomers within the tetramer is significantly different. A common feature of the two structures is that the four p53 core domains can bind the full p53 consensus site without bending the DNA. The dimer interface within each decameric half site is also the same, although the relative orientation between the two monomers in each dimer shows a notable rotational shift around the dyad axis (Figure 6a, the shift can be seen between monomer B and its counterpart in 3EXJ.pdb). The dimer-dimer interface, on the other hand, is very different between the two structures. In the chemically trapped tetramer, monomer D is shifted by 7Å with respect to its counterpart in the naturally assembled complex (Figure 6b, upper panel, viewed from the side of the dimer-dimer interface). Interestingly, when viewed from the top of the dimer-dimer interface (Figure 6b, lower panel), monomer D in the two complexes appears to be overlapping. The dimer-dimer interface in the chemically trapped tetramer is smaller (665Å2 as compared with 897Å2 observed here) and less complementary. This is also evident by the disordering of the N-terminal tail in the chemically trapped tetramer. By contrast, all the structural elements at the dimer-dimer interface are well ordered due to protein-protein interactions. Dimer-dimer interactions mediated by the N-terminal tail (residues 92-99) were all missing in the chemically trapped tetramer. The major dimer-dimer interface observed in the cross-linked complex is between the L2 loop of monomer A and the beta sheet of monomer D. This interface is completely shifted and different in the naturally assembled complex observed here (Supplemental Figure 2). The rotational shift observed in each dimer suggests that the assembly of the tetramer may require conformational changes in its subcomponents and that these structural changes may have been limited by the covalent crosslinking. To further address this question, we superimposed the tetramer with a dimer bound to DNA of the same decameric sequence (2ATA.pdb) (Kitayner et al., 2006). Again, we observed a structural shift within the dimer (Figure 6c, shown is the superposition of the A-B dimer using monomer A as the reference). This structural shift can be observed form both the side (Figure 6c) and above (Figure 6d) of the dimer-dimer interface. Using monomer C as the reference, we observed a similar structure shift in the C-D dimer (data not shown). These observations suggest that the assembly of the p53 core domain tetramer is not simply a rigid-body dock of two dimers bound to the adjacent decameric half sites, but rather involves significant remodeling of the dimer structure.
Figure 6. Structural comparison with other p53:DNA complexes.
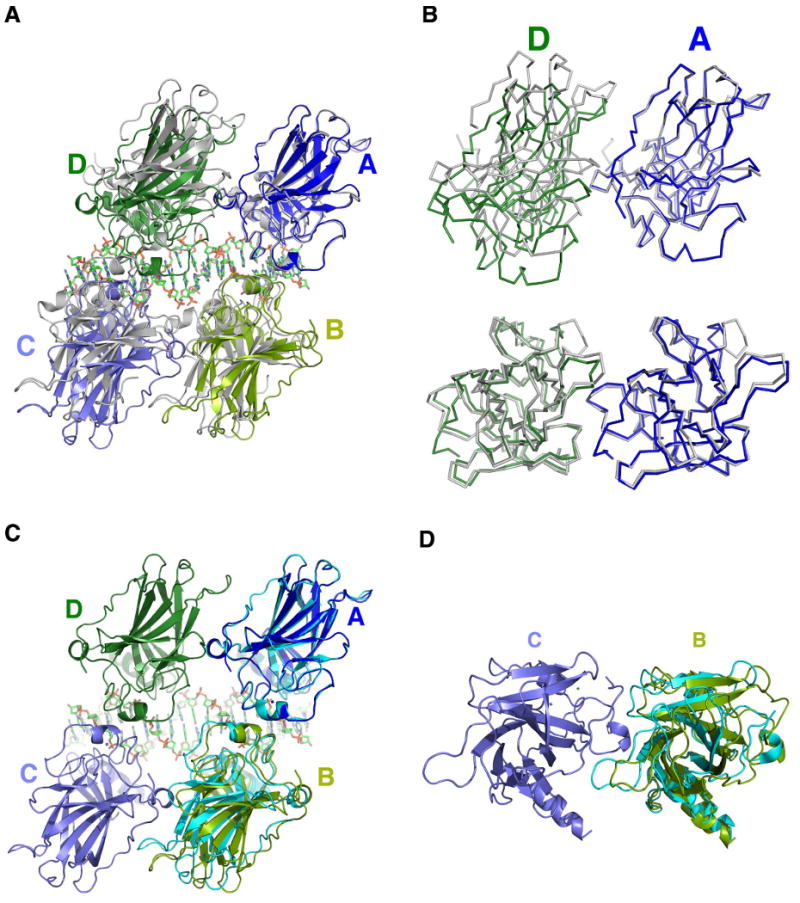
(a) The tetramer is superimposed with the chemically trapped complex (gray, 3EXJ.pdb) using the Cα of monomer A as the reference (Malecka et al., 2009). The DNA of 3EXJ.pdb is omitted for clarity; (b) A detailed comparison using Cα backbone overlay to show the structural shift at the dimer-dimer interface. Upper panel: viewed from the side of the dimer-dimer interface; lower panel: viewed from the top of the dimer-dimer interface; (c, d) The structure of a p53 dimer bound to DNA (cyan, 2ATA.pdb) is superimposed on the A-B dimer of the tetramer using the Cα of monomer A as the reference (Kitayner et al., 2006). The rotational shift of the dimer partner (monomer B) can be observed from the side of the dimer-dimer interface (c) as well as above the interface (d).
Discussion
Genome wide binding-site studies reveal that p53 binds mostly to contiguous decameric half sites in vivo (Smeenk et al., 2008; Wei et al., 2006), suggesting that p53 complexes bound to physiological sites have a well-defined structure near the DNA-binding core. This is in contrast to the previous view that p53 binds variable DNA sequences that would implicate a range of different higher-order structures. The specific assembly of the p53 tetramer on DNA must be important for function, thereby imposing a strong selection pressure on the binding site configuration. In this study, we have solved the high-resolution crystal structure of the p53 core domains bound to a full consensus site as a self-assembled tetramer. The most important finding from the present structural study is the identification of a novel dimer-dimer interface that underlies the assembly of the p53 core tetramer on contiguous decameric half sites. Moreover, comparison of the present structure with the previous structures reveals that the binding of p53 to its consensus sites is a highly cooperative self-assembling process that involves structural changes of protein and DNA. However, in contrast to the widely suggested model of DNA bending, our study reveals that it is the structure of the p53 dimer bound to each decameric half sites that undergoes the most significant structural changes, although the center of the DNA does display a subtle but significant distortion from the B-form conformation. Taken together, detailed analyses of the present structure and its comparison with other p53 complexes provide insight into the assembly mechanism and potential functions of the p53 core tetramer.
The overall structure of the core tetramer:DNA complex is similar to that described in various modeling analyses (Cho et al., 1994; Ho et al., 2006; McLure and Lee, 1998; Pan and Nussinov, 2007). The dimer interface within each decameric half site is also similar to that observed in a number of crystal structures and solution studies (Ho et al., 2006; Kitayner et al., 2006; Klein et al., 2001; Rippin et al., 2002). However, the dimer-dimer interface, which would account for the stringent conservation of zero spacing between the decameric half sites, is completely different from previously described. Although the general location of the dimer-dimer interface and some of the interacting residues were correctly located by modeling analyses, the detailed interactions could not be predicted with high confidence because the interface interactions involve mostly flexible loops. In the complexes studied by Kitayner et al., the two p53 dimers bound to discontinuous DNA duplexes make few contacts with each other (Kitayner et al., 2006). This is because the arrangement of the DNA molecules does not conform to the consensus configuration of the p53 binding site (Smeenk et al., 2008; Wei et al., 2006). In the chemically trapped p53 tetramer bound to DNA (Malecka et al., 2009), a much more extensive dimer-dimer interface is observed. This interface overlaps partially with that observed here in the naturally assembled complex, but the details of the protein-protein interactions differ significantly. Although we could not rule out the effect of different DNA sequences used in the two studies, the covalent modification is more likely the cause of the difference (see below). The dimer-dimer interface observed here has a larger contact area and better chemical and shape complementarity than that seen in the chemically trapped complex. Moreover, the N-terminal tail, which is predicted by modeling analyses to mediate dimer-dimer interactions (Cho et al., 1994; Ho et al., 2006; McLure and Lee, 1998; Pan and Nussinov, 2008), is disordered in the chemically trapped complex. In the present structure, this region is indeed located at the dimer-dimer interface and engages in extensive protein-protein interactions.
Structural comparison between the tetramer complex and its subcomponents (monomer and dimer complexes) suggests that the binding of the four p53 core domains to DNA is a self-assembling process. The protein-protein interactions observed in the tetramer are not merely incidental contacts between proteins that happen to bind nearby DNA sites, but rather as the result of active self-assembling accompanied by structural changes in proteins and DNA. This is most apparent from the structural comparison between the tetramer and the dimer. Such a self-assembling mechanism presents a challenge to modeling studies, as most of these conformational changes would be difficult to predict. In fact, most modeling analyses suggested that DNA must bend for two rigid p53 dimers to bind contiguous decameric half sites (Ho et al., 2006; Nagaich et al., 1999; Pan and Nussinov, 2007, 2008). Our studies here suggest that the conformational changes of the dimer account for a major part of the mechanism of tetramer formation on the consensus site. However, experimental studies in solution indicated that DNA bound by p53 was indeed bent (Nagaich et al., 1999). It is possible that the DNA bends at the junction between the p53 site and the flanking region. The simultaneous binding of the L1 loop and helix H2 in the major groove seems to create a strain in DNA, such that each p53 core domain has the potential to distort the DNA structure at the 5′ end of the quarter site. At the center of the DNA bound by p53, the two core domains apparently compensate for each other, leaving only localized structural deformation at the junction between the two half sites. At the two ends of the p53 site, however, a DNA bend may occur depending on the orientation and DNA contacts of the L1 loop, which is known to modulate the function of p53 in vitro and in vivo (Sykes et al., 2006; Tang et al., 2006; Zupnick and Prives, 2006). Consistent with this hypothesis, the DNA ends in the present crystal are partially disordered, probably as a result of the “push” by the L1 loop of monomer A and C, which shows a tendency to “spring out” of the major groove based on the partially observed density of the backbone. The self-assembling model also reveals a potential limitation of the chemical trapping strategy used in the study of the p53 tetramer (Malecka et al., 2009). This is because covalent crosslinking could interfere with the cooperation between protein-DNA interaction and protein-protein interaction. Although protein-DNA contacts and the dimer interface near the crosslinking site did not show substantial structural changes (Malecka et al., 2009), a small angular bias imposed by the crosslinking would translate into a larger displacement over a longer distance. The dimer-dimer interface, which is far from the covalent linking site, could therefore be significantly altered.
The tetramer structure explains why the p53 core domain, which is a monomer with low intrinsic DNA affinity, can bind the consensus site cooperatively with high affinity (Balagurumoorthy et al., 1995; McLure and Lee, 1998). Although DNA contacts by each subunit in the tetramer are similar to that of the isolated monomer, the DNA binding surfaces of the four monomers are connected by the protein-protein interactions, leading to a large combined DNA-binding surface and enhanced DNA affinity. The tetramer:DNA complex is also known to be kinetically stable (McLure and Lee, 1998), with its dissociation half-life of 15 minutes being much longer than most protein:DNA complexes, including the p53 core dimer bound to a decameric half, which has a half-life of only 30 seconds. The enclosed structure of the p53 tetramer may be responsible for such unusual kinetic stability. While DNA binding and protein-protein interactions can enhance the affinity of the dimer for the decameric half site (Klein et al., 2001), the dissociation of protein in such an open structure is still faster (Kitayner et al., 2006; McLure and Lee, 1998). In the tetramer, on the other hand, each subunit is trapped by DNA binding and protein-protein interactions with two neighboring monomers. Therefore, the dimer-dimer interface, which is supported by the contiguous configuration of the p53 consensus site, is important for the thermodynamic, as well as kinetic, stability of DNA binding by p53. The functional importance of this interface is also supported by the conservation of dimer-dimer interactions, however, the dimer-dimer interface seems to be less frequently mutated in cancers compared to some of the hot spots (Olivier et al., 2002). Two structural features of the dimer-dimer interface may partially account for this observation. First, the protein-protein interactions involve a large number of van der Waals contacts by the main chain atoms, which will be less sensitive to side chain changes from missense mutations. Second, the dimer-dimer interface contains multiple discontinuous foci of contacts. Similar protein-protein binding interfaces are also observed in a number of other higher-order transcription factor complexes (Chen et al., 1998; Wu et al., 2006). Functional disruption of such interfaces often requires simultaneous mutations of multiple residues (Chen et al., 1998; Wu et al., 2006).
P53 has a dedicated tetramerization domain near the C-terminus that plays a key role in cooperative DNA binding by p53. However, since the core DNA binding domain is linked to the tetramerization domain through a flexible linker (Tidow et al., 2007), the tetramerization domain itself is unlikely the factor that constrains the configuration of the p53 consensus site, although it could facilitate the assembly of the p53 core domain tetramer by bringing four monomers in proximity. The fact that the p53 core domain is flexibly linked to the rest of the p53 region at both its N- and C-termini suggest that the self-assembling of the core tetramer is likely maintained in the full-length protein. Interestingly, a p53 variant (p53β) lacking the C-terminal tetramerization domain (residues 331-393) was shown to regulate the expression of specific p53 target genes (Bourdon et al., 2005), suggesting that the presence or absence of the tetramerization domain can indeed affect the formation of the p53 core domain tetramer on selected target sites. Nonetheless, the function of p53β alone could not explain the evolution selection for the self-assembled p53 core domain tetramer and its required consensus site. Rather, tetramerization of the p53 core domain is an important function of the full-length native protein. In addition to enhancing DNA binding, the stereo-specific structure of the core domain tetramer can serve as a structural platform for the folding of other p53 domains and for the binding of proteins that interact with p53 and modulate its functions.
Experimental Procedures
Sample Preparation and Crystallization
The GST fusion system was used to affinity-purify the human p53 core domain (residues 92-292), as described previously (Lilyestrom et al., 2006). Briefly, the protein was expressed as GST-p53 fusion protein using the pGEX-2T vector, with a thrombin cleavage site between GST and p53. The fusion protein was first purified by glutathione affinity column. The GST was released by thrombin cleavage, and the p53 fragment was further purified by ion exchange and gel filtration chromatography (Amersham Biosciences, Piscataway, NJ). DNA was synthesized by Integrated DNA technologies (Coralville, IA) and purified as described previously (Chen et al., 1998). The DNA sequence is 5′-AGGCATGCCTAGGCATGCCT-3′, which is a reverse complement to itself. The protein/DNA complex was prepared by mixing protein and DNA at 4:1 molar ratio. Crystals were grown by the hanging drop method at 18 °C using a reservoir buffer of 50 mM bis-tris-propane (BTP)(pH 6.68), 142 mM NaCl, 100 mM ammonium acetate, 16% PEG4K, 9% glycerol. Crystals belong to the space group P212121 with cell dimensions of a = 65.084 Å, b = 93.713 Å, and c = 145.57 Å. The crystals diffract to 2.13Å. With four p53 core domains and one 20mer double stranded DNA in the asymmetric unit, the calculated Matthews coefficient is 2.18Å3/Dalton with 43.6% solvent.
Data collection, structure determination and analysis
Crystals were stabilized in the harvest/cryoprotectant buffer: 50 mM BTP (pH 6.68), 142 mM NaCl, 100 mM ammonium acetate, 30% PEG4K, 20% glycerol and flash frozen with liquid nitrogen for cryocrystallography. Data were collected at the ALS BL8.2.1, BL8.2.2 and BL5.0.2 beamlines at the Lawrence Berkeley National Laboratory.
Data were reduced with HKL2000 (Otwinowski and Minor, 1997). The structure of the p53-DNA complex was solved by the molecular replacement method using the coordinates of previously solved p53 core domain structure as a search model (Kitayner et al., 2006). Refinement, model building and analysis were carried out with CNS (Brunger et al., 1998), CCP4 (1994), Phenix (Adams et al., 2002) and O (Jones et al., 1991). All residues have backbone and angles in the “allowed” region of a Ramachandran plot, with 95.8% in the most favored region. The statistics of the crystallographic analysis are presented in Table 1. Figures illustrating structure were prepared with PyMol (DeLano Scientific).
Supplementary Material
Acknowledgments
The authors would like to thank Dr, Xiaojiang Chen for providing the expression vector of the human p53 core domain and for helpful discussion, and Dr. James C. Stroud for help with DNA structural analysis, Michael Philips for proof reading the manuscript. The authors would also like to thank ALS BCSB staff members Corie Ralston, Peter Zwart, and Kevin Royal for help with data collection. This work is supported by a NIH postdoctoral fellowship to Y. C. and NIH grants to L. C.
The coordinates and structural factors have been deposited to RCSB Protein Data Bank under accession code of 3KMD.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- The CCP4 suite: programs for protein crystallography. Acta crystallographica. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- Adams PD, Grosse-Kunstleve RW, Hung LW, Ioerger TR, McCoy AJ, Moriarty NW, Read RJ, Sacchettini JC, Sauter NK, Terwilliger TC. PHENIX: building new software for automated crystallographic structure determination. Acta crystallographica. 2002;58:1948–1954. doi: 10.1107/s0907444902016657. [DOI] [PubMed] [Google Scholar]
- Balagurumoorthy P, Sakamoto H, Lewis MS, Zambrano N, Clore GM, Gronenborn AM, Appella E, Harrington RE. Four p53 DNA-binding domain peptides bind natural p53-response elements and bend the DNA. Proceedings of the National Academy of Sciences of the United States of America. 1995;92:8591–8595. doi: 10.1073/pnas.92.19.8591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourdon JC, Fernandes K, Murray-Zmijewski F, Liu G, Diot A, Xirodimas DP, Saville MK, Lane DP. p53 isoforms can regulate p53 transcriptional activity. Genes & development. 2005;19:2122–2137. doi: 10.1101/gad.1339905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, et al. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta crystallographica. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- Chen L. Combinatorial gene regulation by eukaryotic transcription factors. Current opinion in structural biology. 1999;9:48–55. doi: 10.1016/s0959-440x(99)80007-4. [DOI] [PubMed] [Google Scholar]
- Chen L, Glover JN, Hogan PG, Rao A, Harrison SC. Structure of the DNA-binding domains from NFAT, Fos and Jun bound specifically to DNA. Nature. 1998;392:42–48. doi: 10.1038/32100. [DOI] [PubMed] [Google Scholar]
- Cho Y, Gorina S, Jeffrey PD, Pavletich NP. Science. Vol. 265. New York, N.Y: 1994. Crystal structure of a p53 tumor suppressor-DNA complex: understanding tumorigenic mutations; pp. 346–355. [DOI] [PubMed] [Google Scholar]
- el-Deiry WS, Kern SE, Pietenpol JA, Kinzler KW, Vogelstein B. Definition of a consensus binding site for p53. Nature genetics. 1992;1:45–49. doi: 10.1038/ng0492-45. [DOI] [PubMed] [Google Scholar]
- Funk WD, Pak DT, Karas RH, Wright WE, Shay JW. A transcriptionally active DNA-binding site for human p53 protein complexes. Molecular and cellular biology. 1992;12:2866–2871. doi: 10.1128/mcb.12.6.2866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green DR, Kroemer G. Cytoplasmic functions of the tumour suppressor p53. Nature. 2009;458:1127–1130. doi: 10.1038/nature07986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho WC, Fitzgerald MX, Marmorstein R. Structure of the p53 core domain dimer bound to DNA. The Journal of biological chemistry. 2006;281:20494–20502. doi: 10.1074/jbc.M603634200. [DOI] [PubMed] [Google Scholar]
- Horn HF, Vousden KH. Coping with stress: multiple ways to activate p53. Oncogene. 2007;26:1306–1316. doi: 10.1038/sj.onc.1210263. [DOI] [PubMed] [Google Scholar]
- Joerger AC, Fersht AR. Structural biology of the tumor suppressor p53. Annual review of biochemistry. 2008;77:557–582. doi: 10.1146/annurev.biochem.77.060806.091238. [DOI] [PubMed] [Google Scholar]
- Jones TA, Zou JY, Cowan SW, Kjeldgaard Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr A. 1991;47(Pt 2):110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- Kitayner M, Rozenberg H, Kessler N, Rabinovich D, Shaulov L, Haran TE, Shakked Z. Structural basis of DNA recognition by p53 tetramers. Molecular cell. 2006;22:741–753. doi: 10.1016/j.molcel.2006.05.015. [DOI] [PubMed] [Google Scholar]
- Klein C, Planker E, Diercks T, Kessler H, Kunkele KP, Lang K, Hansen S, Schwaiger M. NMR spectroscopy reveals the solution dimerization interface of p53 core domains bound to their consensus DNA. The Journal of biological chemistry. 2001;276:49020–49027. doi: 10.1074/jbc.M107516200. [DOI] [PubMed] [Google Scholar]
- Lilyestrom W, Klein MG, Zhang R, Joachimiak A, Chen XS. Crystal structure of SV40 large T-antigen bound to p53: interplay between a viral oncoprotein and a cellular tumor suppressor. Genes & development. 2006;20:2373–2382. doi: 10.1101/gad.1456306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu XJ, Olson WK. 3DNA: a software package for the analysis, rebuilding and visualization of three-dimensional nucleic acid structures. Nucleic acids research. 2003;31:5108–5121. doi: 10.1093/nar/gkg680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma B, Pan Y, Gunasekaran K, Venkataraghavan RB, Levine AJ, Nussinov R. Comparison of the protein-protein interfaces in the p53-DNA crystal structures: towards elucidation of the biological interface. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:3988–3993. doi: 10.1073/pnas.0500215102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma B, Pan Y, Zheng J, Levine AJ, Nussinov R. Sequence analysis of p53 response-elements suggests multiple binding modes of the p53 tetramer to DNA targets. Nucleic acids research. 2007;35:2986–3001. doi: 10.1093/nar/gkm192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malecka KA, Ho WC, Marmorstein R. Crystal structure of a p53 core tetramer bound to DNA. Oncogene. 2009;28:325–333. doi: 10.1038/onc.2008.400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLure KG, Lee PW. How p53 binds DNA as a tetramer. The EMBO journal. 1998;17:3342–3350. doi: 10.1093/emboj/17.12.3342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagaich AK, Zhurkin VB, Durell SR, Jernigan RL, Appella E, Harrington RE. p53-induced DNA bending and twisting: p53 tetramer binds on the outer side of a DNA loop and increases DNA twisting. Proceedings of the National Academy of Sciences of the United States of America. 1999;96:1875–1880. doi: 10.1073/pnas.96.5.1875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olivier M, Eeles R, Hollstein M, Khan MA, Harris CC, Hainaut P. The IARC TP53 database: new online mutation analysis and recommendations to users. Human mutation. 2002;19:607–614. doi: 10.1002/humu.10081. [DOI] [PubMed] [Google Scholar]
- Olson WK, Bansal M, Burley SK, Dickerson RE, Gerstein M, Harvey SC, Heinemann U, Lu XJ, Neidle S, Shakked Z, et al. A standard reference frame for the description of nucleic acid base-pair geometry. Journal of molecular biology. 2001;313:229–237. doi: 10.1006/jmbi.2001.4987. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z, Minor W. Processing of X-ray Diffraction Data Collected in Oscillation Mode. In: Carter CWJ, Sweet RM, editors. Methods in Enzymology. New York: Academic Press; 1997. pp. 307–326. [DOI] [PubMed] [Google Scholar]
- Pan Y, Nussinov R. Structural basis for p53 binding-induced DNA bending. The Journal of biological chemistry. 2007;282:691–699. doi: 10.1074/jbc.M605908200. [DOI] [PubMed] [Google Scholar]
- Pan Y, Nussinov R. p53-Induced DNA bending: the interplay between p53-DNA and p53-p53 interactions. The journal of physical chemistry. 2008;112:6716–6724. doi: 10.1021/jp800680w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rippin TM, Freund SM, Veprintsev DB, Fersht AR. Recognition of DNA by p53 core domain and location of intermolecular contacts of cooperative binding. Journal of molecular biology. 2002;319:351–358. doi: 10.1016/S0022-2836(02)00326-1. [DOI] [PubMed] [Google Scholar]
- Smeenk L, van Heeringen SJ, Koeppel M, van Driel MA, Bartels SJ, Akkers RC, Denissov S, Stunnenberg HG, Lohrum M. Characterization of genome-wide p53-binding sites upon stress response. Nucleic acids research. 2008;36:3639–3654. doi: 10.1093/nar/gkn232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki HI, Yamagata K, Sugimoto K, Iwamoto T, Kato S, Miyazono K. Modulation of microRNA processing by p53. Nature. 2009;460:529–533. doi: 10.1038/nature08199. [DOI] [PubMed] [Google Scholar]
- Sykes SM, Mellert HS, Holbert MA, Li K, Marmorstein R, Lane WS, McMahon SB. Acetylation of the p53 DNA-binding domain regulates apoptosis induction. Molecular cell. 2006;24:841–851. doi: 10.1016/j.molcel.2006.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Y, Luo J, Zhang W, Gu W. Tip60-dependent acetylation of p53 modulates the decision between cell-cycle arrest and apoptosis. Molecular cell. 2006;24:827–839. doi: 10.1016/j.molcel.2006.11.021. [DOI] [PubMed] [Google Scholar]
- Tidow H, Melero R, Mylonas E, Freund SM, Grossmann JG, Carazo JM, Svergun DI, Valle M, Fersht AR. Quaternary structures of tumor suppressor p53 and a specific p53 DNA complex. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:12324–12329. doi: 10.1073/pnas.0705069104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature. 2000;408:307–310. doi: 10.1038/35042675. [DOI] [PubMed] [Google Scholar]
- Vousden KH, Prives C. Blinded by the Light: The Growing Complexity of p53. Cell. 2009;137:413–431. doi: 10.1016/j.cell.2009.04.037. [DOI] [PubMed] [Google Scholar]
- Wei CL, Wu Q, Vega VB, Chiu KP, Ng P, Zhang T, Shahab A, Yong HC, Fu Y, Weng Z, et al. A global map of p53 transcription-factor binding sites in the human genome. Cell. 2006;124:207–219. doi: 10.1016/j.cell.2005.10.043. [DOI] [PubMed] [Google Scholar]
- Weinberg RL, Veprintsev DB, Fersht AR. Cooperative binding of tetrameric p53 to DNA. Journal of molecular biology. 2004;341:1145–1159. doi: 10.1016/j.jmb.2004.06.071. [DOI] [PubMed] [Google Scholar]
- Wu Y, Borde M, Heissmeyer V, Feuerer M, Lapan AD, Stroud JC, Bates DL, Guo L, Han A, Ziegler SF, et al. FOXP3 controls regulatory T cell function through cooperation with NFAT. Cell. 2006;126:375–387. doi: 10.1016/j.cell.2006.05.042. [DOI] [PubMed] [Google Scholar]
- Zhao K, Chai X, Johnston K, Clements A, Marmorstein R. Crystal structure of the mouse p53 core DNA-binding domain at 2.7 A resolution. The Journal of biological chemistry. 2001;276:12120–12127. doi: 10.1074/jbc.M011644200. [DOI] [PubMed] [Google Scholar]
- Zupnick A, Prives C. Mutational analysis of the p53 core domain L1 loop. The Journal of biological chemistry. 2006;281:20464–20473. doi: 10.1074/jbc.M603387200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.