

Abstract
Antiviral immunity is triggered by immunorecognition of viral nucleic acids. The cytosolic helicase RIG-I is a key sensor of viral infections and is activated by RNA containing a triphosphate at the 5′end. The exact structure of RNA activating RIG-I remains controversial. Here we established a chemical approach for 5′triphosphate oligoribonucleotide synthesis and found that synthetic single-stranded 5′triphosphate oligoribonucleotides were unable to bind and activate RIG-I. Conversely, the addition of the synthetic complementary strand resulted in optimal binding and activation of RIG-I. Short double strand conformation with base pairing of the nucleoside carrying the 5′triphosphate was required. RIG-I activation was impaired by a 3′overhang at the 5′triphosphate end. These results define the structure of RNA for full RIG-I activation and explain how RIG-I detects negative strand RNA viruses which lack long double-stranded RNA but do contain panhandle blunt short double-stranded 5′triphosphate RNA in their single-stranded genome.
Keywords: 5′-triphosphate RNA, immunorecognition of RNA virus, RIG-I
Antiviral innate and antigen-specific immunity is triggered by immunorecognition of viral nucleic acids. The receptor repertoire of the innate immune system is genetically programmed to detect a wide range of viral infections and activate a set of genes including type I interferons (IFNs), which confer antiviral activity to the host (Samuel, 2001). Long double-stranded RNA is widely accepted as a virus-associated nucleic acid pattern normally absent in mammalian cells. Receptors proposed to detect long double-stranded RNA include double-stranded RNA-activated protein kinase “PKR” (Manche et al., 1992) as well as the two helicases MDA-5 (Gitlin et al., 2006; Kang et al., 2002; Kato et al., 2008; Kato et al., 2006; Takahasi et al., 2008; Yoneyama et al., 2004) and RIG-I (Yoneyama et al., 2004). RIG-I is a key sensor of negative strand RNA viruses (Habjan et al., 2008; Plumet et al., 2007; Takeuchi and Akira, 2008). The presence of a triphosphate group at the 5′end of RNA (5′triphosphate RNA) is another molecular feature that is recognized by the innate immune system (Hornung et al., 2006; Kim et al., 2004; Nallagatla et al., 2007; Pichlmair et al., 2006). A triphosphate is initially present at the 5′end of any viral or self RNA molecule generated by RNA polymerases; however, in eukaryotes, RNA is processed and modified by cleavage, capping, and base modifications in the nucleus before the RNA is released to the cytoplasm. Most studies agree on the fact that single-stranded 5′triphosphate RNA is sufficient to bind to and activate RIG-I (Cui et al., 2008; Hornung et al., 2006; Pichlmair et al., 2006; Plumet et al., 2007; Saito et al., 2008; Takahasi et al., 2008). Recent work suggests that long (>300 bp) double-stranded RNA can substitute for the presence of a 5′triphosphate group (Kato et al., 2008). However, in the life cycle of negative strand RNA viruses, such double-stranded RNA is absent (Pichlmair et al., 2006; Weber et al., 2006), and 5′triphosphate single-stranded RNA seems the prime RIG-I ligand. Here we generated fully synthetic well-characterized 5′triphosphate RNA and to our surprise found that 5′triphosphate single-stranded RNA is not a ligand for RIG-I, but that a 5′triphosphate short blunt end double strand RNA structure as contained in the panhandle of negative strand viral genomes confers full RIG-I ligand activity to a RNA molecule.
Results
Synthetic 5′triphosphate single-stranded RNA is not sufficient to activate RIG-I
A 24mer RNA oligonucleotide with 5′-G (3P-G) was designed for which self-complementarity and thus secondary structure formation (intra- or intermolecular double strand formation) was predicted to be absent (see Suppl. Table 1). A triphosphate group was covalently attached to the 5′end of the corresponding synthetic oligonucleotide by using a previously established method (Ludwig, 1989). Purity of RNA oligonucleotides was confirmed using HPLC (High-performance liquid chromatography) and MALDI-ToF-MS (matrix-assisted laser desorption ionization mass spectrometry) (exemplary spectra for “3P-A” are shown in Fig. 1A-C). The same sequence (ivt3P-G) as well as a positive control oligonucleotide (IVT2, 30mer, Suppl. Table 1) were generated by in vitro transcription. The RIG-I activity of RNA oligonucleotides was examined in primary human monocytes (Hornung et al., 2006). Unexpectedly, synthetic 3P-G showed no IFN-α induction (Fig. 1D). Polyacrylamide gel analysis revealed that unlike synthetic 3P-G, ivt3P-G presented as two major bands (Fig. 1E, lane 5 versus lane 1). It has been reported that during IVT reactions RNA template dependent RNA transcription (Cazenave and Uhlenbeck, 1994; Triana-Alonso et al., 1995) can lead to complementary side products and to double-stranded RNA products which where originally designed to be single-stranded. Indeed, the addition of a fully synthetic complementary single strand (not containing a triphosphate group, AS G24) to synthetic 3P-G (3P-G+AS G24) led to full RIG-I ligand activity (Fig. 1D).
Fig. 1. Fully synthetic 5′triphosphate single-stranded RNA is not sufficient to activate RIG-I.
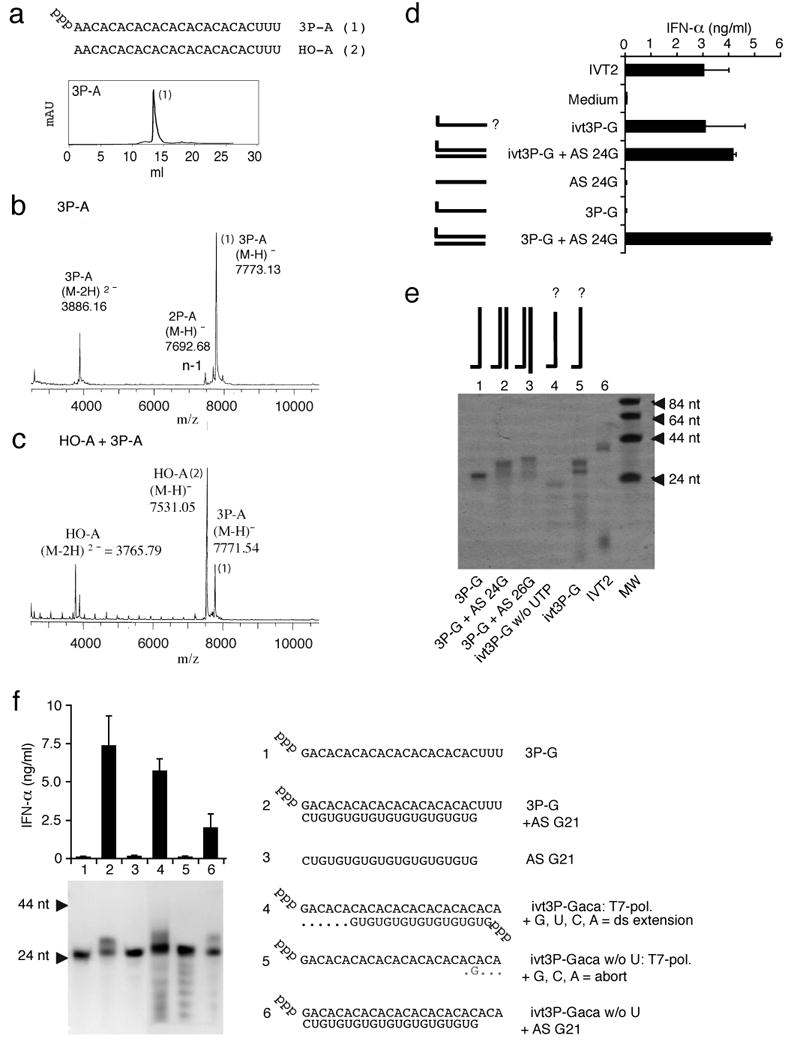
A: Reverse Phase HPLC analysis of 3P-A = pppAAC ACA CAC ACA CAC ACA CAC UUU after deprotection under standard ACE deprotection conditions (pH=3.8, 60°C in 30 min). B: MALDI-ToF analysis of 3P-A: calculated molecular weight of 3P-A is 7770; the peak at 3886 represents the double charged peak (z=2). C: MALDI-ToF analysis of 3P-A mixed with HO-A. The difference between the molecular weight of the product 3P-A (1) and the educt HO-A (2) is 240 which corresponds to the molecular weight of an additional triphosphate group (H3P3O9). D: Purified monocytes were stimulated with the indicated single-stranded or double-stranded synthetic or in vitro transcribed RNA oligonucleotides. The questionmark indicates that the 3′end of this in vitro transcribed RNA is not molecularly defined. IFN-α production was analysed 24 hours after stimulation. Data from four independent donors are depicted as mean values ± SEM. E: Indicated RNA stimuli (see suppl. Table 1) were analysed on a denaturing 12 % polyacrylamide gel (containing 50% urea w/v) and stained with methylene blue detecting single-stranded and double-stranded RNA. ivt3P-G without U was generated by in vitro transcription in the absence of the nucleotide UTP. F: Purified monocytes were stimulated with the indicated single-stranded or double-stranded synthetic or in vitro transcribed RNA oligonucleotides. IFN-α production was analysed 24 hours after stimulation. Data from four independent donors are depicted in the bar graph as mean values ± SEM. RNAs were analysed on a denaturing 12 % polyacrylamide gel (containing 50 % urea w/v) and stained with methylene blue.
We then changed the sequence of 3P-G to 3P-Gaca by replacing the three 3′UUU to 3′ACA (see Fig. 1F right panel). The in vitro transcription of 3P-Gaca requires only three nucleotides G, C and A but not U in the in vitro reaction mix. Therefore, double strand formation can only occur in the presence of U. Consistent with the results obtained by using defined synthetic RNAs, we found that ivt3P-Gaca strongly induced IFN-α, whereas ivt3P-Gaca without U (absence of U in reaction mix, no double strand expected) showed no IFN-α induction (Fig. 1F). However, the addition of the 21mer complementary strand (AS G21) restored IFN-α inducing activity of ivt3P-Gaca without U. In order to retrieve complementary species in the ivt3P-Gaca reaction mix, digestion with RNase T was performed (Suppl. Fig. 1). RNase T cleaves after G in single-stranded RNA. Under native cleaving conditions (37°C), only a small fraction of small RNA species (<10 nt) disappeared from the reaction mix (Suppl. Fig. 1, upper right panel, ivt3P-Gaca). In contrast almost all RNA species with the exeption of the main band disappeared when digestion was performed under denaturating conditions (lower right panel). These results strongly suggested that the RNA mix contains substantial amounts of G-rich RNA that are not encoded by the T7 promoter containing DNA template. The protection of the G-rich species from digestion under native conditions imply the presence of double stranded RNA and therefore complementary RNA, which cannot be cleaved by RNase T. In order to identify the immunostimulatory RNA species of the ivt3P-Gaca reaction, we extracted RNA from different bands of the PAGE (Suppl. Fig. 2). Although the diffuse band (25-35nt) above the main band contained almost the whole IFN-α inducing activity, RNA retrieved from the main band was nearly inactive. Although RNA from the band 3 (10-15 nt) had no IFN-α inducing activity by itself, it was as active as ivt3P-Gaca when added to ivt3P-Gaca without U (Suppl. Fig. 2A-C). Reverse cloning of RNA and sequencing revealed the presence of sequences generated by self-coding intramolecular 3′-extension (Triana-Alonso et al., 1995), which form blunt ended double-stranded RNA (Suppl. Fig. 2F). In addition, complementary poly-GU sequences were found in both band 1 (e.g. a 17mer was found, 7) and band 3 (9-12mer) (Suppl. Fig. 2E). As the sequences start with a G (Suppl. Fig. 2E, 7-12), sequences were generated by transcript-coded “de novo” synthesis (Cazenave and Uhlenbeck, 1994; Triana-Alonso et al., 1995). Thus, the use of synthetic 5′triphosphate RNA revealed that a single strand is not sufficient to support RIG-I activation; furthermore in the case of IVT the formation of unexpected transcripts and not the intended single-stranded RNA are responsible for RIG-I activation.
RIG-I activation requires a short double stranded RNA with a 5′triphosphate
Next the length of the strand complementary to 3P-G (AS G24) was decreased from 24 down to 13 nucleotides (3P-G with complementary AS G24 to AS G13, see Fig. 2A, and Suppl. Table 1). We found that with the sequence 3P-G, AS G21 was the minimal tolerated length of a complementary strand (Fig. 2A). AS G23 together with 3P-G (one nucleotide 3′ overhang at the non-triphosphate end) consistently showed slightly higher activity than AS G24 (no overhang at the non-triphosphate end); the same was seen for AS A23 and AS A24 with 3P-A (Fig. 2B), suggesting that blunt end formation at the non-triphosphate end is not preferred.
Fig. 2. RIG-I activation requires a short double strand and prefers 5′-adenosine.

Purified monocytes were stimulated with the indicated single-stranded or double-stranded synthetic RNA oligonucleotide. IFN-α production was analysed 24 hours after stimulation. Data from n independent donors are depicted as mean values ± SEM, n as indicated. A: 3P-A was hybridized with synthetic antisense single-stranded RNA with different lengths generating double-stranded RNA with blunt end carrying the triphosphate group. B: 23mer (AS G23 and AS A23) with a blunt triphosphate end and a 3′overhang at the non-triphosphate end. C: Comparison of RIG-I ligand activity of 3P-G to the other three synthetic variants (3P-A, 3P-C, 3P-U) which were hybridized with the corresponding synthetic 24mer (AS-A24, AS-C24, AS-U24) or used as single strands. D: IFN-α inducing activity of 3P-A+AS A24 and 3P-G+AS G24.
To analyze the contribution of the 5′-nucleoside of the triphosphate strand we compared the RIG-I ligand activity of 3P-G to the other three synthetic variants (3P-A, 3P-C, 3P-U). Interestingly, all 5′-nucleosides induced substantial amounts of IFN-α, with a preference for 5′-adenosine and 5′-guanosine with 5′-adenosine being slightly but significantly higher than 5′-guanosine (Fig. 2C and 2D). The response was weakest for a 5′-cytidine. Furthermore, elongation of the complementary strand at the non-triphosphate end by one or two nucleosides did not reduce but rather increased the IFN-α inducing activity for both 3P-G and 3P-A (Fig. 3A).
Fig. 3. Blunt end at the triphosphate end but not at the non-triphosphate end contributes to RIG-I ligand activity, and 5′monophosphate does not substitute for 5′triphosphate.

Purified monocytes were stimulated with the indicated single-stranded or double-stranded synthetic RNA oligonucleotides. IFN-α production was analysed 24 hours after stimulation. Data from four independent donors are depicted as mean values ± SEM. 3P-G and 3P-A were hybridized with corresponding antisense strands with different lengths and positions: A: The use of 25mer (AS G24+A and AS A24+A) and 26mer (AS G24+2A and AS A24+2A) results in a mononucleotide or dinucleotide 5′ overhang at the non-triphosphorylated end. B: The use of 25mer (AS G25 and AS A25) and 26mer (AS G26 and AS A26) results in a mononucleotide or dinucleotide 3′ overhang at the triphosphate end. C: The use of 19mer, 21mer and 23mer single-stranded antisense RNA (AS19, AS21, AS23) results in a 5′overhang at the triphosphate end (-5nt, -3nt, -1nt). D: IFN-α-inducing activity of single-stranded 5′monophosphate RNA (P-A, P-G, P-U, P-C) and synthetic 5′triphosphate single-stranded RNA (3P-A) and combinations with complementary strands of different lengths are compared.
Unlike the non-triphosphate end, elongation at the triphosphate end reduced RIG-I ligand activity (Fig. 3B and suppl. Fig. 3B). RIG-I ligand activity was specifically sensitive to shortening of the complementary strand resulting in a 5′overhang at the triphosphate end (3P-G+AS 23, 3P-G+AS 21, 3P-G+AS 19, Fig. 3C). Next we compared synthetic 5′monophosphate single-stranded RNA (P-A, P-G, P-U, P-C) to synthetic 5′triphosphate single-stranded RNA (3P-A) and varied the length of the complementary strand in order to study the contribution of blunt end (Fig. 3D). Compared to the 5′triphosphate version of the same sequence we found no considerable IFN-α induction by 5′monophosphate blunt end double-stranded RNA (P-N +AS N24; N=A,G,U,C) or 5′monophosphate RNA with a two nucleotide 3′overhang (P-N+AS N26) or a 5′overhang (P-N+AS23) at the monophosphate end (Fig. 3D).
RIG-I is activated by double stranded blunt end 5′triphosphate RNA with bulges as contained in viral panhandle structures
Additional sequences were examined with different degrees of secondary structure formation. 3P-GFP2 and 3P-GFP3 represents variations of 3P-GFP1 (Hornung et al., 2006). 3P-GFP1 forms a 4 bp hairpin and 3P-GFP3 forms a 6 bp hairpin. 3P-GFP2 is not self-complementary. Consistent with the data obtained with 3P-G and 3P-A, none of the additional sequences by itself induced IFN-α in primary human monocytes; addition of the complementary strand to 3P-GFP2 resulting in a blunt end double-stranded RNA induced a full IFN-α response (Fig. 4A). Next, 3P-GFP2 was hybridized with complementary RNA of different lengths and at different positions (Fig. 4B). In agreement with the results obtained with 3P-G and 3P-A, the activity of 3P-GFP2 required a minimal length of 19 base pairs double strand as long as the triphosphate end was blunt. Elongation of the complementary strand at the triphsophate end by three nucleosides (3′-overhang) strongly reduced the IFN-α inducing activity. Shortening of the complementary strand at the triphsophate end by one, two or three nucleosides (5′-overhang) abolished the activity. Next, the impact of mismatches in short double-stranded RNA on RIG-I activity was analyzed. A mismatch at the first position at the triphosphate end abolished the activity, whereas a wobble base pair at the first position or a mismatch at the second position were partially tolerated (Fig. 4C). Furthermore, a 3 nt bulge loop starting at position 8 from the triphosphate end was well tolerated (Fig. 4C). Such bulge loop structures are contained in predicted panhandle structures of (-)ssRNA virus genomes (Suppl. Fig. 4). We generated in vitro-transcribed RNA corresponding to the predicted Rabies genome panhandle. In this construct 40 nt of the 5′ end of the Rabies genome is connected by a 5 nt loop to the 40 nt 3′end of the Rabies genome (resulting in a blunt end). A second RNA transcript was prepared (Rabies panhandle mismatch), identical to the first transcript except for an additional 3′ overhang and two mismatched nucleotides at the triphosphorylated end (no blunt end, see Suppl. Table 1). PAGE purification of the two RNA transcripts was used to isolate the correct RNA molecules from the RNA molecules generated by in vitro transcription. The blunt end Rabies panhandle RNA was more potent than the RNA that contained the 5′mismatches and the 3′overhang (Fig. 4D). The residual activity of the 3′overhang RNA may be due to partial contamination with blunt ended byproducts. Thus, double stranded blunt end 5′triphosphate RNA with bulges as contained in viral panhandle structures is recognized by RIG-I.
Fig. 4. RIG-I is not activated by highly structured 3P-ssRNA and tolerates central but not distal mismatches of 3P-dsRNA.

Purified monocytes were stimulated with the indicated single-stranded or double-stranded synthetic RNA oligonucleotides. IFN-β production was analysed 24 hours after stimulation. Data from four independent donors are depicted as mean values ± SEM.
Analysis of A) related 3P-RNAs (3P-GFP1 (Hornung et al., 2006), and variations thereof), with gradual stability of secondary structure, B) 3P-GFP2 hybridized with corresponding antisense strands with different lengths and positions as indicated, C) 3P-GFP2 hybridized with corresponding antisense strands with single or multiple mismatched nucleotides at different positions as indicated, D) PAGE-purified in vitro transcripts corresponding to the predicted panhandle structure of Rabies virus genome with blunt end (match) and with 3′overhang of a poly A tail and a mismatched (2 nt) triphosphorylated end (mismatch).
Synthetic short double-stranded 5′triphosphate RNA is recognized by RIG-I but not MDA-5
Mouse embryonic fibroblasts (MEFs) prepared from mice deficient of RIG-I (Ddx58-/-), MDA-5 (Ifih1-/-) or IPS-1 (Mavs-/-) were stimulated with blunt end short double-stranded RNA (3P-GFP2 + AS GFP2 5′24) or with short double-stranded RNA containing a 3 bp overhang at the non-triphosphate end (3P-GFP2 + AS GFP2 5′21) or the triphosphate end (3P-GFP2 + AS GFP2 3′21). Consistent with the results obtained in human monocytes, MEFs produced IFN-β in response to the blunt end double-stranded RNA (3P-GFP2 + AS GFP2 5′24) or the double-stranded RNA with the overhang at the non-triphosphate end (3P-GFP2 + AS GFP2 5′21); almost no activity was seen in response to double-stranded RNA with the overhang at the triphosphate end (3P-GFP2 + AS GFP2 3′21) (Fig. 5). For all three double-stranded RNA molecules, IFN-β production required the presence of RIG-I and IPS-1, but not MDA-5.
Fig. 5. Synthetic 3P-dsRNA is recognized by RIG-I.

Mouse embryonic fibroblasts (MEFs) with indicated genotype were transfected with indicated 3P-dsRNA. Ifih1(+/+)and Mavs (+/+)represent wild type MEFs with the same passage number as the corresponding mutant MEFs Ifih1 (-/-) and Mavs (-/-), respectively. Triplicate data of a representative experiment are depicted as mean values ± SEM.
IFN-α induction correlates with induction of ATPase activity of purified RIG-I
Next we studied whether IFN-α induction by RNA correlates with the induction of RIG-I ATPase activity in vitro. First we analyzed ATPase activity in response to direct interaction of purified human RIG-I protein isolated from HEK293T cells with different single-stranded and double-stranded RNA ligands. Single-stranded RNA (all open symbols) irrespectively of 5′ triphosphate or 5′monophosphate did not induce ATPase activity (Fig. 6A). Double-stranded RNA molecules (all black symbols) showed an EC50 in the range of 15 nM to 600 nM depending on the composition of the double strand and the configuration of the 5′end (Fig. 6A). Double-stranded RNA with 5′monophosphate or RNA without 5′phosphate (P–A+AS A24, HO-A+AS A24) showed a 35- and 20-fold higher EC50 (lower ATPase activity) than double-stranded RNA with a 5′triphosphate (Fig. 6A). The 5′triphosphate double-stranded RNA molecules which induced substantial amounts of IFN-α in monocytes (3P–A+AS A24, 3P-A+AS A23, 3P-A+AS A21, see Suppl. Fig. 3A) reached their EC50 at 95 % to 99 % lower concentrations than dsRNA ligands that weakly induced IFN-α (3P-A+AS A34, 3P-A+AS23, 3P-A+ASA19) (Fig. 6B, compare to Suppl. Fig. 3A and 3B). The ATPase activities of different 5′triphosphate RNAs with different 5′bases (3P-A+AS A24, 3P-G+AS G24, 3P-U+AS U24, 3P-C+AS C24, Fig. 6C) reflected the observed IFN-α inducing activity (A, G>U>C, see Fig. 2C). These data demonstrate that the IFN-α inducing activity of RIG-I ligands correlates with ATPase activity.
Fig. 6. IFN-α inducing activity of RIG-I RNA ligands correlates with RIG-I ATPase activity and with RIG-I binding affinity.
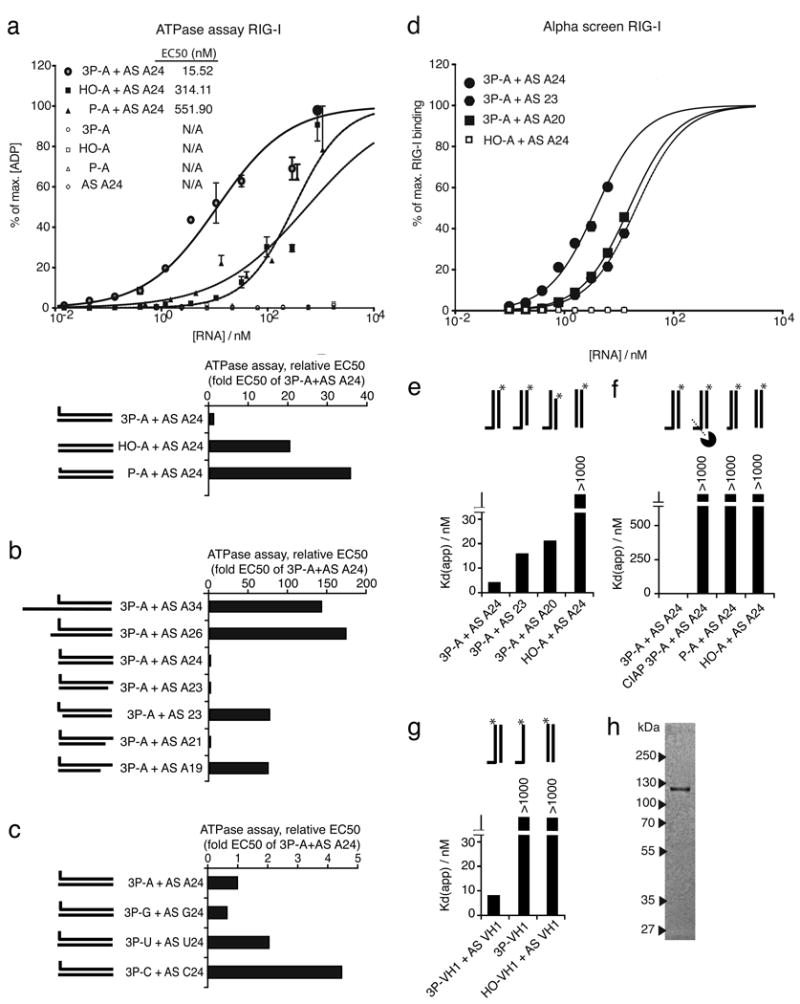
A: For the ATPase assay purified RIG-I protein was incubated with increasing amounts of indicated RNA molecules (from 10-9 nM to 1800 nM) and the release of ADP was analysed after 30 min at 37°C by a FRET-based competitive immunoassay. The percentage of ADP release is plotted against the decadic logarithm of the concentration of indicated RNAs. Triplicate data of a representative experiment are depicted as mean values ± SEM. Half effective concentration (EC50) was determined by statistical analysis (non-linear regression). Low EC50 represents high RIG-I ATPase activity. B: The EC50 of the synthetic 3P-A hybridized with the indicated antisense RNAs are compared. C: The EC50 of 5′triphosphate double-stranded RNA with different 5′bases (A, G, U, C) are compared. D-G: Alphascreen: Purified (His6)-tagged RIG-I was incubated with different RNA molecules bearing a biotin (*) on the 5′end of the antisense strand (at the non-triphosphate end of the double-stranded RNA; D-F) or at the 3′ end of the 3P-bearing strand (G). (His6)-tagged RIG-I protein was bound to Ni-chelate beads (donor); biotinylated RNA was bound to streptavidine beads (acceptor). The resulting fluorescence correlates with the number and proximity of interacting donor - acceptor pairs. Concentration of indicated RNAs is plotted against the percentage of maximum binding to RIG-I. The dissociation constant Kd(app) is calculated by statistical analysis (non-linear regression). E, F and G: Kd(app) of non-modified, monophosphate and triphosphate RNAs hybridized to indicated antisense RNAs of different lengths are compared. F: CIAP 3P-A + AS A24 was incubated with active alkaline phosphatase; the other stimuli were incubated with heat-inactivated active alkaline phosphatase. H: Purified (His6)-tagged RIG-I protein was analysed by SDS-PAGE and Coomassie Blue staining.
High affinity binding of RNA to RIG-I requires double-stranded RNA with a 5′-triphosphate
Using a homogenous ligand interaction assay we analysed the affinity of different RNA molecules to RIG-I. We found that binding of RNA to RIG-I strictly depended on the presence of a triphosphate at the 5′end and on the presence of short double-stranded RNA (Fig. 6D, E and 6G). The dissociation constant of 5′-triphosphate RNA (3P-A+AS A24 and 3P-VH1 + AS VH1) was between 4 nM and 9 nM. The Kd(app) of corresponding non-modified RNA, 5′monophosphate RNA, dephosphorylated 3P-RNA (HO-A+AS A24, P-A+AS A24, 3P-A+AS A24 CIAP; HO-VH1+ AS VH1) or single-stranded 3P-RNA (3P-VH1) was above the detection limit of this assay but at least higher than 1000 nM (Fig. 6D, 6E, 6F and 6G). Consistent with the magnitude of IFN-α inducing activity in monocytes, the magnitude of RIG-I binding of IFN-α inducing (3P-A+AS A24) and of non-IFN-α-inducing (3P-A+AS23, 3P-A+AS A20, compare Fig. 6C, and Suppl. Fig. 3A) 5′-triphosphate RNA differed 4- to 5-fold (Fig. 6E). For these studies we used purified (His6)-tagged RIG-I protein analyzed by SDS-PAGE and Coomassie Blue staining (Fig. 6H). These results demonstrate that RIG-I activity of RNA is associated with high affinity binding but that binding to RIG-I is not sufficient for RIG-I activation.
Discussion
Well-characterized RNA molecules are crucial for a sound analysis of RNA functions. Here we made use of a protocol for a complete chemical synthesis of 5′triphosphate RNA. By using these chemically defined RNA molecules we identified the exact molecular structure for RIG-I binding and activation (Fig. 7). We demonstrate that i) the presence of a 5′triphosphate is required for full activation of RIG-I, and that the 5′triphosphate can not be replaced by 5′monophosphate or no 5′OH without loosing substantial IFN-α inducing activity; ii) that the recognition of 5′triphosphate requires a short double strand spanning at least 19 nucleotides and encompassing the 5′nucleotide carrying the triphosphate; single-stranded RNA is not sufficient to support recognition of 5′triphosphate RNA; iii) a blunt end at the triphosphate end but not at the non-triphosphate end of the RNA supports recognition by RIG-I, and a 3′overhang at the 5′triphosphate end decreases and any 5′overhang at the 5′triphosphate end abolishes the activity. Structure dependent IFN-α inducing activity of chemically-defined RNA molecules was confirmed in the mouse system, and required RIG-I and IPS-1 but not MDA-5. RIG-I binding and RIG-I ATPase activity were structure dependent, with the triphosphate being strictly required for effective RIG-I binding of short RNA molecules, and the ATPase activity being strongly dependent on the presence of a blunt end formation at the triphosphate end. Our data on RIG-I binding and RIG-I ATPase activity are in agreement with the results obtained by Cui and colleagues (Cui et al., 2008) but inconsistent with Takahashi and colleagues (Takahasi et al., 2008) which may partially be due to the different nature of the RNA ligand used.
Fig. 7. Molecular structure of RIG-I ligand.

Optimal RIG-I ligand activity requires short double-stranded RNA containing at least one 5′triphosphate. The nucleoside carrying the 5′triphosphate needs to be part of the Watson-Crick base pairing (GU wobble base pairing less efficient) and pppA, pppG and pppU are preferred over pppC. Although overhangs at the non-triphosphorylated end of the double-stranded RNA have no major impact on RIG-I activity, a 3′overhang at the 5′triphosphorylated end impairs RIG-I activity.
So far, in the literature most studies agree on the fact that single-stranded 5′triphosphate RNA is sufficient to bind to and activate RIG-I (Cui et al., 2008; Hornung et al., 2006; Pichlmair et al., 2006; Plumet et al., 2007; Saito et al., 2008; Takahasi et al., 2008). Of note, these studies including our own previous work used in vitro transcription to generate 5′triphosphate RNA without analyzing the purity and identity of those RNA molecules. However, it has been observed that during in vitro transcription with T7 RNA polymerase self-coded 3′-extension of run-off transcripts produces aberrant products including size-variable extensions with complementarity to the correct transcript (Cazenave and Uhlenbeck, 1994; Triana-Alonso et al., 1995). In addition to our chemical approach to 5′triphosphate RNA we established a protocol for in vitro transcription that avoids such RNA template dependent RNA synthesis, thereby confirming that unintended formation of double-stranded RNA in fact is the cause of RIG-I activity of in vitro transcribed RNA. Reverse cloning and sequencing of RNA species confirmed that transcript-coded “de novo” synthesis and self-coding intramolecular 3′-extension occur and can form RNA duplexes that are consistent with the structural requirements for RNA RIG-I ligands (blunt end, short double strand, 5′triphosphate) responsible for the RIG-I stimulating activity of in vitro transcribed RNA. It is interesting to note that the use of undefined RNA molecules generated by in vitro transcription has led to confusion in the past. In the seminal work on RNA interference by Fire and Mello (Fire et al., 1998), only the use of gel-purified RNA finally provided the evidence that only antisense but not sense RNA silences the sense strand, ending the uncertainty on this in the literature and paving the way for RNAi. Another intriguing parallel to RNAi is that it now turns out that short double-stranded RNA is sufficient for the recognition of 5′triphophate RNA by RIG-I, and that long double-stranded RNA, initially thought to be required, is not. In the RNAi field, the possibility to use synthetic short double-stranded RNA (siRNA) boosted the applicability of this approach. Likewise, the possibility to use short double-stranded RNA molecules instead of long double-stranded RNA to provoke the full spectrum of antiviral activities via RIG-I may facilitate the therapeutic application. In addition, because the structural requirements are characterized on a molecular level, functional activities of both RNAi and RIG-I can be combined in one RNA molecule.
Our results support the view that recognition of viral infection by RIG-I will always require the presence of double-stranded RNA, at least of short double-stranded RNA. Double-stranded RNA is not only present in double strand RNA viruses but substantial amounts of cytosolic double-stranded RNA are also produced during the replicative life cycle of positive single strand RNA viruses (Weber et al., 2006). In agreement with the requirement of double-stranded RNA for RIG-I recognition, double strand and positive single strand RNA viruses indeed are recognized by RIG-I (Kato et al., 2006; Loo et al., 2008). However, at first sight, this seemed less clear for RIG-I-mediated detection of negative single strand RNA viruses (Kato et al., 2006; Loo et al., 2008) for which no double-stranded RNA can be detected (Pichlmair et al., 2006; Weber et al., 2006) but still viral genomic single-stranded RNA activates RIG-I (Hornung et al., 2006; Pichlmair et al., 2006). However, the antibody used to demonstrate the absence of double-stranded RNA (Pichlmair et al., 2006; Weber et al., 2006) is limited to the detection of double-stranded RNA longer than 40 bases (Bonin et al., 2000). Our analysis of sequence data revealed that genomes of negative strand viruses known to activate RIG-I contain 5′and 3'sequences that form a short double strand with a perfect blunt end (panhandle) and with a 5′adenosine carrying the triphosphate group. For Influenza virus, such panhandle structures (Hofacker et al., 2004) were shown to serve as a RNA transcription initiation site for the viral RNA polymerase complex (Portela and Digard, 2002). Of note mismatches and bulge loops as present in the panhandle of negative strand RNA viruses are tolerated by RIG-I as demonstrated in our study. Furthermore, triphosphate-A (pppA) is the most abundant first nucleotide in genomes of RNA viruses (exceptions are Ebola: pppU; and Hepatitis C: pppG).
Genomic single-stranded RNA of negative strand RNA viruses is thought to be encapsidated by structural proteins. However, recent data (Strahle et al., 2007) suggest that activation of RIG-I by Sendai virus is due to contamination with defective viral genomes (DI-H4) which occur during infection and do not show encapsidation and therefore can form panhandle structures in vivo. It is well accepted that negative strand RNA viruses for replication require a highly conserved promoter at both ends of the genome. For Vesicular Stomatitis Virus (VSV) it was shown that during replication, artificially introduced extra nucleotides at the 5′end of the VSV (-)ssRNA genome are eliminated whereas extra nucleotides at the 3′end are not tolerated at all (Pattnaik et al., 1992). Thus, blunt end 5′triphosphate short double strand RNA formation, the consequence of the requirement of two conserved promoters, represents a negative strand virus-associated molecular pattern detected by RIG-I.
Short (23mer and longer) blunt end double-stranded RNA without triphosphate was proposed to be the key feature for RIG-I activation (Marques et al., 2006). Although our data are in agreement with the importance of a blunt end for RIG-I activation, we show that full RIG-I activation requires the additional presence of a 5′triphosphate; furthermore, a blunt end at the non-triphosphate end is not required. Our data are also partially in agreement with the notion by Takahasi and colleagues that a longer 3′overhang decreases the activity of a 25mer double-stranded RNA oligonucleotide (Takahasi et al., 2008). On the other hand Takahasi and colleagues (Takahasi et al., 2008) suggested that blunt end 5′monophosphate double-stranded RNA is sufficient for full RIG-I activation, a conclusion that is not supported by our findings. However, 5′monophosphate may be sufficient to support partial activation of RIG-I, but on a lower level.
Kato and colleagues (Kato et al., 2008) reported the activation of RIG-I by “short” double-stranded RNA without the need of a 5′triphosphate or blunt end conformation in order to activate RIG-I. In our study, the term short double-stranded RNA refers to RNA molecules of approximately 20 bp which is much shorter than the lengths of “short” double-stranded RNA reported earlier (300 and 2000 bp) (Kato et al., 2008). This may explain why no 5′ triphosphate dependency was seen by this group. Another group confirmed the 5′ triphosphate requirement, yet suggested that single-stranded homopolyuridine or homopolyriboadenine motifs, as they are present in genomes of RNA viruses, constitute a key structural element of RIG-I recognition (Saito et al., 2008). Although the 3'sequence under certain circumstances may contribute to RIG-I activity, our results provide no indication that homopolyuridine or homopolyriboadenine motifs are in fact required for full binding and activation of RIG-I by 5′triphosphate RNA. Of note, since in vitro transcription was used in this study self-coded 3′-extension of RNA transcripts (fold back) and transcript coded de novo synthesis might have been responsible for the production of aberrant dsRNA and thus RIG-I activation (Cazenave and Uhlenbeck, 1994; Triana-Alonso et al., 1995).
Interestingly, data from Gondai and colleagues support the requirement of a double-stranded 5′ triphosphate structure even for in vitro transcribed RNA with regards to RIG-I activation (Gondai et al., 2008). In their study the introduction of increasing numbers of guanosines at the 5′end of in vitro transcribed shRNA reduced the IFN induction by shRNA. By introducing guanosines at the 5′end, overhangs of the 5′triphosphate end are generated leading to the disruption of the double strand conformation resulting in a loss of RIG-I activity.
The identification of the exact molecular structure required for RIG-I activation now allows generation of defined synthetic 5′triphosphate RNA ligands for RIG-I with reproducible and predictable induction of potent antiviral responses. Synthetic 5′triphosphate RNA has a number of advantages over 5′triphosphate RNA generated by in vitro transcription: First, purity and identity of synthetic 5′triphosphate RNA is independent of the respective sequence. Second, unlike with in vitro transcription the synthetic approach allows to modify the base composition at the 5′triphosphate end of the RNA as exemplified by defining adenosine or guanosine as the preferred 5′nucleosides. Third, large scale synthesis based on a process that includes standard RNA chemistry with all possibilities of additional modifications can be used. Fourth, the biological activity of synthetic 5′triphosphate RNA is superior to in vitro transcribed RNA. These advantages of the synthetic 5′triphosphate RNA will greatly improve the development of such RNA oligonucleotides for the treatment of viral infection and cancer.
Experimental procedures
Chemical synthesis of triphosphate oligoribonucleotides
Oligoribonucleotides containing a free 5′-OH terminus were synthesized on an ABI 392 synthesizer using commercial 5′-Silyl 2′ ACE protected amidites from Dharmacon. Solid phase triphosphorylation of oligoribonucleotides was performed using an improved version of the protocol developed by Ludwig and Eckstein (Ludwig, 1989) (unpublished data). After internucleotide phosphate and base deprotection, 5′- triphosphorylated 2′-ACE protected derivates were purified by RP-HPLC, followed by removal of the 2′-OH protecting groups under standard deprotection conditions at pH=3.8, 60°C. MALDI-ToF analysis was performed by Metabion (Martinsried, Germany).
Monophosphate RNA, non-modified RNA and in vitro transcribed RNA
Monophosphate RNA and non-modified RNA oligoribonucleotides (ORN) were synthesized by commercial providers (Metabion, Martinsried, Germany and Biomers, Ulm, Germany, respectively). The sequences are listed in Suppl. Table 1. ivt3P-G and ivt3P-Gaca were generated by in vitro transcription (IVT) with a commercial In vitro T7-Transcription Kit (Epicentre). For generation of DNA template dependent in vitro transcribed RNA, the T7-promoter region CAGTAATAGGACTCACTATAG was hybridized with the promoter+template strand (ivt3P-G: 5′- AAA GTG TGT GTG TGT GTG TGT GTC TAT AGT GAG TCG TAT TAC TG -3′; ivt3P-Gaca: 5′-TGT GTG TGT GTG TGT GTG TGT GTC TAT AGT GAG TCG TAT TAC TG-3′; IVT2: 5′- GTC GTC GTC GTC GTC GTC GTC GTC GTC GTC TAT AGT GAG TCG TAT TAC TG -3′) and directly used as a template for the in vitro transcription reaction. For generation of ivt3P-G without Uridine (ivt3P–G w/o U) and ivt3P-G without Uridine (ivt3P-Gaca w/o U) the in vitro transcription was performed in the absence of UTP. Single bands from ivt3P-Gaca and in vitro transcripts corresponding to predicted panhandle structures of Rabies virus genomes were PAGE purified: briefly, the 3P-RNA with estimated size was eluted from a 50% urea PAGE-gel slice by incubation in 0.3 M sodiumacetate (pH 5.4) and subsequent precipitation in 70% ethanol. RNaseT (Fermentas) digestion under native conditions was performed according to the manufacturers protocol. RNaseT digest under denaturating conditions occured 30 minutes at 50 °C in 6 M urea, 17.8 mM Na-citrate at pH 3.5, 0.9 mM EDTA (Nallagatla et al., 2007).
Reverse cloning of in vitro transcripts
RNA was cloned by a protocol which was similar to a method introduced by Lee and colleagues (Lee and Ambros, 2001) for small RNA cloning: PAGE-purified RNA was polyadenylated using poly(A) polymerase (Epicentre). The cDNA was cloned into the vector pLib (Clontech) using the Creator™ SMART™ cDNA Library Construction Kit (Clontech) according to the manufacturers protocol. Plasmid DNA was prepared from single E. coli colonies and sequenced using M13forward primer. For the alternative approach, RNA molecules were dephosphorylated using calf intestine phosphatase (CIP), phosphorylated with T4 polynucleotide kinase (PNK), purified and converted into cDNA-libraries as described (Hafner et al., 2008) using bar-coded 3′ adapters (Vigneault et al., 2008) and submitted for a one-well 454 sequencing reaction.
Cell culture
Human PBMCs were isolated from whole human blood of healthy, voluntary donors by Ficoll-Hypaque density gradient centrifugation (Biochrom Berlin, Germany). Plasmacytoid dendritic cells (PDC) were positively depleted using magnetically labeled anti-CD304 antibody (Miltenyi Biotec). Untouched monocytes were obtained by negative depletion from PBMCs according to the manufacturers instructions (Human Monocyte Isolation Kit II, Miltenyi Biotec). Viability of all cells was above 95 %, as determined by trypan blue exclusion. If not indicated otherwise, 400,000 cells (PBMCs) or 200,000 cells (monocytes) were cultured in 96-well plates for stimulation experiments. Cells were kept in RPMI 1640 containing 10 % FCS, 1.5 mM L-glutamine, 100 U/ml penicilin and streptomycin 100 μg/ml. All compounds were tested for endotoxin contamination prior to use. Mouse embryonic fibroblasts (MEFs) from MDA-5 -/-, RIG-I -/- and IPS-1 -/- mice were prepared as described (Kato et al., 2006). For transfection 0,2 ug nucleic acid and 0,5 ul Lipofectamine (Invitrogen) were mixed in 50 ul Optimem (Invitrogen), incubated for 20 min and added to the well containing 200000 cells. The studies were approved by the local ethic committee (Ethikkommission der Medizinischen Fakultät Bonn).
Detection of cytokines
The amount of IFN-α production was determined using the IFN-α module set from Bender MedSystems (Vienna, Austria). The ELISA assay was performed according to the manufacturer's protocol. The concentration of cytokines was determined by the standard curve obtained using known amounts of recombinant cytokines.
Flow cytometry
Cell purity was assessed by FACS analysis of cell surface antigens using a FACS LSRII (BD Biosciences, Heidelberg, Germany). Human monocytes were stained with antibody against CD14-FITC or CD14-APC and cell purity was between 83 % and 99 %. Human PDCs were positively labeled with antibody against CD123-PE, HLA-DR-PerCp and negatively for CD11c-APC and a cocktail to lineage markers (FITC). Antibodies were purchased from BD Pharmingen. Data analysis was performed on viable cells using CellQuest (BD Biosciences) and Flowjo (Treestar).
Protein purification and analysis
(His6)-Flag-tagged RIG-I (HF-RIG-I) was transiently overexpressed in 293T cells and lysed in a CHAPS containing lysis buffer (150 mM NaCl, 50 mM Tris/HCl pH7,4, 2 mM MgCl2, 1 mM DTT, 1% CHAPS) including protease inhibitor cocktail (Roche). The lysate was incubated over night at 4°C with anti-FLAG beads (Sigma). Anti-FLAG beads were washed subsequently with lysis buffer and high salt wash buffer (300 mM NaCl, 50 mM Tris/HCl pH7,4, 5 mM MgCl2, 1 mM DTT, 0,1% CHAPS). RIG-I-FLAG was eluted by addtion of FLAG-peptide (300 ug/ml) solution to the beads. Purity of recombinant RIG-I was determined by SDS-PAGE separation and subsequent Coomassie blue stain (Fig. 6H).
ATPase assay
The ATPase assay was performed in assay buffer (50 mM KCl, 55 mM HEPES (pH 7.0) 3 mM MgCl2, 0.5 mM DTT, 0.1 mM ATP). In order to calculate EC50, the RNA was titrated in a range from 6 fM to 4 μM. After 30 min of incubation at 37°C, occurence of ADP was measured using a very sensitive FRET based competitive immunoassay (HTRF® Transcreener™ ADP, Cisbio, Bedford, USA) according to the manufacturers protocol. FRET was measured using an EnVision® Multilabel Reader (PerkinElmer, Waltham, USA). In this assay, inhibition of FRET correlates with the concentration of ADP generated by ATPase activity of RIG-I. ADP concentrations were calculated from an ADP/ATP titration curve according to the manufacturers protocol.
Alpha Screen RIG-I-binding assay
The binding affinity of RNA for (His6)FLAG-tagged RIG-I (HF-RIG-I) was determined as described (Haas et al., 2008; Latz et al., 2007) by an amplified luminescent proximity homogenous assay (AlphaScreen; Perkin Elmer). In this assay purified HF-RIG-I was incubated with increasing concentrations of biotinylated RNA for 1 hour at 37°C in buffer (50 mM Tris/pH7.4, 100 mM NaCl, 0.01% Tween20, 0.1% BSA) and subsequently incubated for 30 min at 25°C with HF-RIG-I-binding Nickel Chelate acceptor beads (Perkin-Elmer) and biotin-RNA-binding Streptavidine donor beads (Perkin Elmer). The donor bead contains the photosensitizer phtalocyanine, which converts ambient oxygen into a ‘singlet’ oxygen after illumination with a 680-nm laser light. During the 4 μs lifetime, the ‘singlet’ oxygen can diffuse up to 200 nm and activate a thioxene derivative on the acceptor bead that is brought into proximity by interaction of the test molecules bound to the beads. The resulting chemiluminescence with subsequent activation of a fluorochrome (contained within the same bead) emitting in the range of 520–620 nm correlates with the number and proximity of associated beads which is inversely correlated with the dissociation constant of donor (biotin-RNA) and acceptor (HF-RIG-I). The assay was performed in wells of 384-well plates (Proxiplate; Perkin-Elmer). Plates were analyzed for emitted fluorescence with a multilabel reader (Envision; Perkin Elmer).
Supplementary Material
Acknowledgments
This study was supported by grants BMBF Biofuture 0311896, SFB 670, SFB 704, KFO115 and KFO 177 to G.H. and grants from German Research Foundation DFG BA3544/1-1 (to W.B.), and by National Institutes of Health grants AI-065483 (to E.L.) and AI-067497 (to K.A.F). This work is part of the theses of Andreas Roth and Cristina Amparo Hagmann at the University of Bonn. We thank Anna Herzner, Christine Schuberth, Klaus Conzelmann, Beate Kümmerer and Christian Drosten for helpful discussion. We thank Agnes Viales at the Genomics Core Laboratory at MSKCC for 454 Sequencing and Robert Sheridan (MSKCC) for bioinformatic processing of the sequencing data.
References
- Bonin M, Oberstrass J, Lukacs N, Ewert K, Oesterschulze E, Kassing R, Nellen W. Determination of preferential binding sites for anti-dsRNA antibodies on double-stranded RNA by scanning force microscopy. Rna. 2000;6:563–570. doi: 10.1017/s1355838200992318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cazenave C, Uhlenbeck OC. RNA template-directed RNA synthesis by T7 RNA polymerase. Proc Natl Acad Sci U S A. 1994;91:6972–6976. doi: 10.1073/pnas.91.15.6972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui S, Eisenacher K, Kirchhofer A, Brzozka K, Lammens A, Lammens K, Fujita T, Conzelmann KK, Krug A, Hopfner KP. The C-terminal regulatory domain is the RNA 5′-triphosphate sensor of RIG-I. Mol Cell. 2008;29:169–179. doi: 10.1016/j.molcel.2007.10.032. [DOI] [PubMed] [Google Scholar]
- Fire A, Xu S, Montgomery MK, Kostas SA, Driver SE, Mello CC. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature. 1998;391:806–811. doi: 10.1038/35888. [DOI] [PubMed] [Google Scholar]
- Gitlin L, Barchet W, Gilfillan S, Cella M, Beutler B, Flavell RA, Diamond MS, Colonna M. Essential role of mda-5 in type I IFN responses to polyriboinosinic:polyribocytidylic acid and encephalomyocarditis picornavirus. Proc Natl Acad Sci U S A. 2006;103:8459–8464. doi: 10.1073/pnas.0603082103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gondai T, Yamaguchi K, Miyano-Kurosaki N, Habu Y, Takaku H. Short-hairpin RNAs synthesized by T7 phage polymerase do not induce interferon. Nucleic Acids Res. 2008;36:e18. doi: 10.1093/nar/gkm1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas T, Metzger J, Schmitz F, Heit A, Muller T, Latz E, Wagner H. The DNA sugar backbone 2′ deoxyribose determines toll-like receptor 9 activation. Immunity. 2008;28:315–323. doi: 10.1016/j.immuni.2008.01.013. [DOI] [PubMed] [Google Scholar]
- Habjan M, Andersson I, Klingstrom J, Schumann M, Martin A, Zimmermann P, Wagner V, Pichlmair A, Schneider U, Muhlberger E, et al. Processing of genome 5′ termini as a strategy of negative-strand RNA viruses to avoid RIG-I-dependent interferon induction. PLoS ONE. 2008;3:e2032. doi: 10.1371/journal.pone.0002032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafner M, Landgraf P, Ludwig J, Rice A, Ojo T, Lin C, Holoch D, Lim C, Tuschl T. Identification of microRNAs and other small regulatory RNAs using cDNA library sequencing. Methods. 2008;44:3–12. doi: 10.1016/j.ymeth.2007.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofacker IL, Stadler PF, Stocsits RR. Conserved RNA secondary structures in viral genomes: a survey. Bioinformatics. 2004;20:1495–1499. doi: 10.1093/bioinformatics/bth108. [DOI] [PubMed] [Google Scholar]
- Hornung V, Ellegast J, Kim S, Brzozka K, Jung A, Kato H, Poeck H, Akira S, Conzelmann KK, Schlee M, et al. 5′-Triphosphate RNA is the ligand for RIG-I. Science. 2006;314:994–997. doi: 10.1126/science.1132505. [DOI] [PubMed] [Google Scholar]
- Kang DC, Gopalkrishnan RV, Wu Q, Jankowsky E, Pyle AM, Fisher PB. mda-5: An interferon-inducible putative RNA helicase with double-stranded RNA-dependent ATPase activity and melanoma growth-suppressive properties. Proc Natl Acad Sci U S A. 2002;99:637–642. doi: 10.1073/pnas.022637199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato H, Takeuchi O, Mikamo-Satoh E, Hirai R, Kawai T, Matsushita K, Hiiragi A, Dermody TS, Fujita T, Akira S. Length-dependent recognition of double-stranded ribonucleic acids by retinoic acid-inducible gene-I and melanoma differentiation-associated gene 5. J Exp Med. 2008;205:1601–1610. doi: 10.1084/jem.20080091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato H, Takeuchi O, Sato S, Yoneyama M, Yamamoto M, Matsui K, Uematsu S, Jung A, Kawai T, Ishii KJ, et al. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature. 2006;441:101–105. doi: 10.1038/nature04734. [DOI] [PubMed] [Google Scholar]
- Kim DH, Longo M, Han Y, Lundberg P, Cantin E, Rossi JJ. Interferon induction by siRNAs and ssRNAs synthesized by phage polymerase. Nat Biotechnol. 2004;22:321–325. doi: 10.1038/nbt940. [DOI] [PubMed] [Google Scholar]
- Latz E, Verma A, Visintin A, Gong M, Sirois CM, Klein DC, Monks BG, McKnight CJ, Lamphier MS, Duprex WP, et al. Ligand-induced conformational changes allosterically activate Toll-like receptor 9. Nat Immunol. 2007;8:772–779. doi: 10.1038/ni1479. [DOI] [PubMed] [Google Scholar]
- Lee RC, Ambros V. An extensive class of small RNAs in Caenorhabditis elegans. Science. 2001;294:862–864. doi: 10.1126/science.1065329. [DOI] [PubMed] [Google Scholar]
- Loo YM, Fornek J, Crochet N, Bajwa G, Perwitasari O, Martinez-Sobrido L, Akira S, Gill MA, Garcia-Sastre A, Katze MG, Gale M., Jr Distinct RIG-I and MDA5 signaling by RNA viruses in innate immunity. J Virol. 2008;82:335–345. doi: 10.1128/JVI.01080-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludwig J, Eckstein F. Rapid and Efficient Synthesis of Nucleoside 5′-0 -(1-Thiotriphosphates), 5′-Triphosphates and 2′,3′-Cyclophosphorothioates Using 2-Chloro-4H- 1,3,2-benzodioxaphosphorin-4-one. J Org Chem. 1989;54:631–635. [Google Scholar]
- Manche L, Green SR, Schmedt C, Mathews MB. Interactions between double-stranded RNA regulators and the protein kinase DAI. Mol Cell Biol. 1992;12:5238–5248. doi: 10.1128/mcb.12.11.5238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marques JT, Devosse T, Wang D, Zamanian-Daryoush M, Serbinowski P, Hartmann R, Fujita T, Behlke MA, Williams BR. A structural basis for discriminating between self and nonself double-stranded RNAs in mammalian cells. Nat Biotechnol. 2006;24:559–565. doi: 10.1038/nbt1205. [DOI] [PubMed] [Google Scholar]
- Nallagatla SR, Hwang J, Toroney R, Zheng X, Cameron CE, Bevilacqua PC. 5′-triphosphate-dependent activation of PKR by RNAs with short stem-loops. Science. 2007;318:1455–1458. doi: 10.1126/science.1147347. [DOI] [PubMed] [Google Scholar]
- Pattnaik AK, Ball LA, LeGrone AW, Wertz GW. Infectious defective interfering particles of VSV from transcripts of a cDNA clone. Cell. 1992;69:1011–1020. doi: 10.1016/0092-8674(92)90619-n. [DOI] [PubMed] [Google Scholar]
- Pichlmair A, Schulz O, Tan CP, Naslund TI, Liljestrom P, Weber F, Reis e Sousa C. RIG-I-mediated antiviral responses to single-stranded RNA bearing 5′-phosphates. Science. 2006;314:997–1001. doi: 10.1126/science.1132998. [DOI] [PubMed] [Google Scholar]
- Plumet S, Herschke F, Bourhis JM, Valentin H, Longhi S, Gerlier D. Cytosolic 5′-triphosphate ended viral leader transcript of measles virus as activator of the RIG I-mediated interferon response. PLoS ONE. 2007;2:e279. doi: 10.1371/journal.pone.0000279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Portela A, Digard P. The influenza virus nucleoprotein: a multifunctional RNA-binding protein pivotal to virus replication. J Gen Virol. 2002;83:723–734. doi: 10.1099/0022-1317-83-4-723. [DOI] [PubMed] [Google Scholar]
- Saito T, Owen DM, Jiang F, Marcotrigiano J, Gale M. Innate immunity induced by composition-dependent RIG-I recognition of hepatitis C virus RNA. Nature. 2008 doi: 10.1038/nature07106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuel CE. Antiviral actions of interferons. Clin Microbiol Rev. 2001;14:778–809. doi: 10.1128/CMR.14.4.778-809.2001. table of contents. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strahle L, Marq JB, Brini A, Hausmann S, Kolakofsky D, Garcin D. Activation of the beta interferon promoter by unnatural Sendai virus infection requires RIG-I and is inhibited by viral C proteins. J Virol. 2007;81:12227–12237. doi: 10.1128/JVI.01300-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahasi K, Yoneyama M, Nishihori T, Hirai R, Kumeta H, Narita R, Gale M, Jr, Inagaki F, Fujita T. Nonself RNA-sensing mechanism of RIG-I helicase and activation of antiviral immune responses. Mol Cell. 2008;29:428–440. doi: 10.1016/j.molcel.2007.11.028. [DOI] [PubMed] [Google Scholar]
- Takeuchi O, Akira S. MDA5/RIG-I and virus recognition. Curr Opin Immunol. 2008;20:17–22. doi: 10.1016/j.coi.2008.01.002. [DOI] [PubMed] [Google Scholar]
- Triana-Alonso FJ, Dabrowski M, Wadzack J, Nierhaus KH. Self-coded 3′-extension of run-off transcripts produces aberrant products during in vitro transcription with T7 RNA polymerase. J Biol Chem. 1995;270:6298–6307. doi: 10.1074/jbc.270.11.6298. [DOI] [PubMed] [Google Scholar]
- Vigneault F, Sismour AM, Church GM. Efficient microRNA capture and bar-coding via enzymatic oligonucleotide adenylation. Nat Methods. 2008;5:777–779. doi: 10.1038/nmeth.1244. [DOI] [PubMed] [Google Scholar]
- Weber F, Wagner V, Rasmussen SB, Hartmann R, Paludan SR. Double-stranded RNA is produced by positive-strand RNA viruses and DNA viruses but not in detectable amounts by negative-strand RNA viruses. J Virol. 2006;80:5059–5064. doi: 10.1128/JVI.80.10.5059-5064.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoneyama M, Kikuchi M, Natsukawa T, Shinobu N, Imaizumi T, Miyagishi M, Taira K, Akira S, Fujita T. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat Immunol. 2004;5:730–737. doi: 10.1038/ni1087. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.