

Summary
Alterations in sensory processing constitute prominent symptoms of Fragile X syndrome; however, little is known about how disrupted synaptic and circuit development in sensory cortex contributes to these deficits. To investigate how the loss of fragile X mental retardation protein (FMRP) impacts the development of cortical synapses, we examined excitatory thalamocortical synapses in somatosensory cortex during the perinatal critical period in Fmr1 knockout mice. FMRP ablation resulted in dysregulation of glutamatergic signaling maturation. The fraction of silent synapses persisting to later developmental times was increased, there was a temporal delay in the window for synaptic plasticity, while other forms of developmental plasticity were not altered in Fmr1 knockout mice. Our results indicate that FMRP is required for the normal developmental progression of synaptic maturation, and loss of this important RNA binding protein impacts the timing of the critical period for layer IV synaptic plasticity.
Keywords: Fragile X syndrome, somatosensory cortex, silent synapse, glutamate receptor, long term potentiation, critical period
Introduction
Fragile X syndrome is the most common cause of inherited male mental retardation and a leading genetic determinant of autism. A tri-nucleotide repeat expansion of the fragile X mental retardation 1 (FMR1) gene results in transcriptional silencing and loss of an RNA-binding protein, fragile X mental retardation protein (FMRP) (Garber et al., 2008; Penagarikano et al., 2007). FMRP is a negative regulator of protein translation and while the precise mechanisms and mRNA targets of FMRP are not yet fully enumerated, loss of this protein results in a severely debilitating and complex neurological phenotype. A central feature of Fragile X syndrome is an alteration in sensory processing that manifests in early infancy and progressively worsens through childhood (Baranek et al., 2008). Similar to the human syndrome, sensory processing deficits are also observed in the mouse model of Fragile X (Fmr1 knockout) (Bakker et al., 1994), including sensory hypersensitivity to auditory stimuli (Chen and Toth, 2001; Yun et al., 2006). In addition, cellular anatomical deficits are observed in the sensory cortex of Fmr1 knockout mice. A preponderance of abnormally long, thin dendritic spines have been reported in pyramidal neurons during early development in the somatosensory cortex (Galvez and Greenough, 2005; Nimchinsky et al., 2001), and aberrant developmental pruning of the layer IV spiny stellate dendrites has been described in Fmr1 knockout mice (Galvez et al., 2003).
Despite the clear alterations in cellular morphology in Fmr1 knockout mice, it is not known whether the anatomical deficits have an impact on the functional development of excitatory glutamatergic synapses in somatosensory cortex. Somatosensory cortex is organized into topographic maps of the body surface. In rodents, discrete barrel patterns in layer IV somatosensory cortex are a representation of vibrissae on the contralateral snout (Woolsey and Van der Loos, 1970) with functional organization such that neuron clusters in each barrel unit preferentially respond to tactile information from a defined principal whisker (Petersen, 2007). Excitatory synapses formed between ascending thalamocortical axons of relay neurons in the ventrobasal (VB) nucleus of the thalamus and layer IV spiny stellate neurons in the primary somatosensory cortex, provide the major sensory input from the periphery to the cortex. Thus, the thalamocortical synaptic connection is the prime mediator of sensory tactile information from the whiskers to the cortex (Feldman and Brecht, 2005; Petersen, 2007). During perinatal development in rodents, activity dependent refinement of thalamocortical synapses leads to a stereotypical maturation of glutamatergic signaling (Barth and Malenka, 2001; Crair and Malenka, 1995). Thalamocortical synapses express long-term potentiation (LTP) and long-term depression (LTD) throughout the critical period for plasticity in layer IV, which approximates the first postnatal week in rodents (Crair and Malenka, 1995; Feldman et al., 1998). Activity-dependent maturation of excitatory thalamocortical synapses during the critical period results in a well characterized and rapid change in the glutamate receptor type (Crair and Malenka, 1995; Daw et al., 2007; Kidd and Isaac, 1999). The AMPA receptor contribution increases relative to the NMDA receptor contribution (Crair and Malenka, 1995), and the proportion of NMDA-only silent synapses is reduced (Isaac et al., 1997). Differences in the biophysical properties of these synaptic receptor types, particularly their divergent deactivation kinetics, shapes the excitatory postsynaptic potential (EPSP), which contributes to a decrease in latency of synaptically evoked spikes and shortens the window for coincidence between separate inputs (Daw et al., 2006). Developmental changes in synaptic glutamate receptors, therefore, contribute to the maturation of cortical circuits and the emergence of temporal coding of sensory signaling in the mature CNS.
Here, we tested whether synaptic development and critical period plasticity were disrupted in Fmr1 knockout animals (Fmr1−/y) by following the development of thalamocortical glutamatergic synapses using electrophysiological recording during the perinatal period. We found that the characteristic functional development of this synapse was profoundly altered. Rather than the decrease in the NMDA/AMPA current ratio that occurs in wildtype animals, loss of FMRP leads to a dramatic increase in this ratio between postnatal day 4 and postnatal day 7, with the largest NMDA/AMPA ratio measured just before closure of the normal critical period. This change in the relative proportion of glutamate receptors was primarily due to an increase in the proportion of NMDA-only silent synapses at the end of the first postnatal week. In parallel with the alterations in the development of synaptic glutamate receptors, the temporally restricted period for LTP expression at thalamocortical synapses was disordered in Fmr1−/y mice. Thalamocortical LTP was most robust at the end of the first postnatal week and LTP persisted beyond closure of the customary defined critical period for layer IV plasticity. Synaptic deficits did not translate into gross anatomical abnormalities; neither the patterning of the somatosensory cortex or spine density of layer IV neurons. Interestingly, the critical period for lesion-induced plasticity of the barrel maps was also unaffected in Fmr1 knockout mice, underlining the specific nature of the delay in critical period for synaptic plasticity. These results highlight previously unknown synaptic deficits, and an important developmental delay, in the circuit underlying sensory information processing in Fmr1 knockout mice.
Results
FMRP expression in layer IV of the somatosensory cortex during perinatal development
Somatosensory cortex undergoes a rapid and dramatic developmental surge in the first two postnatal weeks, during which there is a profusion of synaptogenesis and dendritic rearrangement. The critical period for plasticity in layer IV occurs during the first postnatal week, and is defined by the expression of robust synaptic plasticity and anatomical map plasticity (Daw et al., 2007). Previous work in somatosensory cortex of adult rats demonstrated elevated FMRP expression after whisker stimulation, suggesting that FMRP expression is enhanced by activity-dependent translation of FMRP (Todd and Mack, 2000; Todd et al., 2003). However, the expression pattern of FMRP in developing somatosensory cortex is not known. Using immunohistochemical analysis of sections from layer IV somatosensory cortex, FMRP was detected in both coronal and tangential sections from P4 mice (Figure 1Ai & Aii). Intense staining was observed at P7 through to P14 (Figure 1Bi & Bii), while at later developmental times (P21), the relative intensity of FMRP staining was reduced compared to that at P7 (Figure 1D). Western blot analysis of homogenates from barrel cortex of mice taken at several postnatal ages, confirmed the expression of FMRP in this region during perinatal development (Figure S1A), and the specificity of the antibody was confirmed by the complete loss of staining in Fmr1−/y mice (Figure S1B & C). From these results, it is clear that FMRP is expressed in layer IV during the critical period, with peak expression occurring during the surge in plasticity, synaptogenesis and dendritic rearrangement during the first two postnatal weeks (Daw et al., 2007).
Figure 1. FMRP expression levels parallel the critical period for plasticity in layer IV of the somatosensory cortex.

(Ai, Bi, Ci, Di) Coronal sections containing the primary somatosensory cortex stained with α-FMRP antibody (rAM1) from P4, P7,P14, and P21 wildtype mice. Calibration 250μm (Aii, Bii, Cii, Dii) Tangential sections through layer IV of the PMBSF at each developmental age. The expression of FMRP is highest during the critical period, peaking at P7. Expression in layer IV of older mice is reduced. Calibration 100μm. (Ei-iii) Examples of immunoelectron micrographs from layer IV somatosensory cortex containing labeled asymmetric spines. No FMRP labeling was observed in presynaptic elements at P7. (F) Immunoelectron micrograph of a layer IV neuron from P7 mouse. DAB immunoreactivity is observed in the cytoplasm and primary dendrite. (NU: nucleus. Cyt: cytoplasm)
Although the discrete laminar staining in barrel sections suggests FMRP expression in layer IV neurons, it is possible that FMRP is expressed in thalamic axons and terminals present in layer IV. Indeed, FMRP is found in axons of cultured hippocampal neurons where it could assist in regulating axon pathfinding (Antar et al., 2006). Consequently, immunoelectron microscopy was performed on sections from P7 mice (Figure 1E & F) to determine the subcellular distribution of FMRP in layer IV. DAB immunoreactivity was localized in the cytoplasm and dendritic compartment of neurons in layer IV (Figure 1F). Moreover, immunoreactivity was observed in the postsynaptic region of asymmetric synapses in layer IV (Figure 1Ei-iii), indicating that FMRP is localized close to excitatory synapses in layer IV neurons. Using this subcellular level analysis, labeling of presynaptic structures was not detected at this developmental age (P7). These results demonstrate, for the first time, that FMRP is expressed in layer IV neurons in the somatosensory cortex during the critical period for plasticity.
Development of thalamocortical glutamate receptor signaling is disrupted during the critical period in Fmr1 knockout mice
Thalamocortical afferents to layer IV provide the main sensory input to the cortex via excitatory synaptic connections to stellate neurons and local circuit interneurons. To determine whether thalamocortical synapses develop normally in Fmr1 knockout mice, voltage clamp recordings were made from spiny stellate neurons (Figure 2A) at each postnatal day starting at postnatal day 4. In P4 wildtype mice (Fmr1+/y), the NMDA component of synaptic transmission was large relative to the AMPA component, resulting in an NMDA/AMPA ratio (NA ratio) of 1.5 ± 0.2 (n = 13) (Figure 2B) as previously reported (Crair and Malenka, 1995). In stark contrast, the NA ratio at P4 in Fmr1−/y mice was significantly lower (0.5 ± 0.1, n = 13, p = 0.0003)(Figure 2C). Recordings made on each subsequent postnatal day in Fmr1+/y mice showed a clear progressive decrease in the NA ratio (P5: 0.9 ± 0.2, n = 9; P6: 0.6 ± 0.1, n = 16), until at closure of the critical period for plasticity (P7), the measured NA ratio was 0.5 ± 0.1 (n = 18). Whereas the NA ratio in recordings from Fmr1−/y mice increased between P4 and P7 (P5: 0.8 ± 0.2, n = 10, p > 0.05; P6: 1.1± 0.1, n = 16, p=0.006) with a maximum ratio of 1.5 ± 0.2 (n = 18, p = 0.0007) at the close of the critical period for wildtype animals (Figure 2C). At postnatal days beyond P7, the NA ratio in Fmr1−/y returned back to levels indistinguishable from those observed in wildtype littermates (P10-P14 NA ratio: Fmr1+/y, 0.6 ± 0.1, n = 39; Fmr1−/y, 0.8 ± 0.1, n = 46 p > 0.05 and at >P22 NA ratio: Fmr1+/y, 0.5 ± 0.1, n=9; Fmr1−/y 0.6 ± 0.1, n = 16, p > 0.05).
Figure 2. Critical period maturation of glutamate receptor signaling at thalamocortical synapses is altered in FMRP knockout mice.

(Ai) Cartoon representation of para-coronal thalamocortical slice depicting placement of the recording electrode in the layer IV barrels and the extracellular stimulating electrode in the ventrobasal thalamus (VB) (H: hippocampus; IC: internal capsule; C: caudate). (Aii) Bright field image of the thalamocortical slice and (Aiii) image of the barrels in layer IV. Calibration: 250μm (Aiv) Higher magnification image of a spiny stellate neuron in layer IV (SS: spiny stellate; IN: interneuron. Calibration: 25μm (Bi) Time course of development in NMDA/AMPA ratio at thalamocortical synapses in wildtype mice (Fmr1+/y). Relatively high NMDA/AMPA ratio is observed at P4 when synaptic contribution of NMDA receptors is large. Over the course of the first week NMDA/AMPA ratio declines in wildtype mice and then stabilizes at the close of the critical period (depicted by shaded area). (Bii) Representative EPSC recordings from wildtype Fmr1+/y mice at P4 and P7. AMPA receptor mediated EPSCs were measured as the peak current at −70mV and the NMDA component was measured by depolarizing the cell to +40mV and measuring the mean current over a 2.5ms window, 60ms after the onset of the outward current. At this time point the contribution of the outward AMPA component is negligible and the measured current is mediated solely by NMDA receptors (Marie et al., 2005). Calibration: 20pA, 100ms (Ci) Time course of development of NA ratio in Fmr1−/y mice. NA ratio at P4 is significantly lower than in Fmr1+/y mice (0.5 ± 0.1, n = 13, p = 0.0003). Over the critical period NMDA/AMPA ratio increases to maximal level at P7 (1.5 ± 0.2, n = 18, p = 0.0007) and then reverts to similar levels as wildtype recordings beyond this developmental timepoint. (Cii) Representative EPSC recordings from Fmr1−/y mice at P4 and P7 recorded from spiny stellate neurons voltage clamped at −70mV and +40mV. Calibration: 10pA, 100ms.
Synaptic glutamate receptor expression in Fmr1 knockout mice
The alterations in the relative contribution of NMDA and AMPA receptors at developing thalamocortical synapses in Fmr1−/y mice could represent a change in the complement of glutamate receptors at individual synapses, or an alteration in the proportion of silent synapses containing only NMDA receptors (Isaac et al., 1997), or combined alterations in both these synaptic parameters. Prior studies have reported region specific alterations in AMPA receptor protein expression in Fmr1 knockout mice (Li et al., 2002; Nosyreva and Huber, 2006; Restivo et al., 2005; Schutt et al., 2009); however, it is not known whether AMPA and NMDA receptor expression is altered in the somatosensory cortex during the critical period for plasticity. To directly address this question, sections containing layer IV were isolated and the barrel cortex specifically microdissected to enrich for layer IV neurons. Analysis of protein expression in the synaptoneurosome fraction from tissue during critical period development (P7) demonstrated a selective, significant increase in the expression of NMDA receptor (NR1) protein in synaptic membranes from Fmr1 knockout mice (117 ± 7 %, p = 0.03, n = 3) (Figure 3A). No significant alteration in GluR1 expression was detected. PSD-95, an important synaptic protein whose mRNA interacts with, and is stabilized by, FMRP (Zalfa et al., 2007), appeared to be upregulated although a statistically significant threshold was not reached (114 ± 10% p = 0.06, n = 3) (Figure 3A). Similarly, analysis of whole tissue homogenate only uncovered a significant increase in NR1 protein levels in Fmr1−/y mice at P7 (128 ± 10 %, p = 0.03, n = 3). In contrast, there was no significant difference in NR1, GluR1 or PSD95 protein in synaptic membranes of tissue taken from layer IV of Fmr1−/y or Fmr1+/y after closure of the critical period (P14) (Figure 3B). These results suggest that total NR1 expression in layer IV synapses is enhanced at P7 in Fmr1 knockout mice; nevertheless, it does not discriminate between increases in NMDA receptors at individual synapses, or increases in the number of NMDA-only silent synapses.
Figure 3. Quantal glutamate receptor content is not altered in thalamocortical synapses in Fmr1 knockout mice during the critical period.

(A) Western blot analysis of synaptoneurosome fraction from layer IV S1 tissue during critical period development. Expression of NR1 protein was significantly elevated in the synaptic membrane fraction. (B) Analysis of protein expression in layer IV after closure of the critical period. No significant differences were observed between Fmr1+/y and Fmr1−/y mice (Ci) Cumulative distribution of amplitude of all quantal events measured in Sr2+ from Fmr1+/y (black) and Fmr1−/y mice (grey). (Cii) Mean amplitude of events from all recordings. No difference is observed between Fmr1+/y and Fmr1−/y mice (p>0.05, KS test) (Ciii & iv) Representative traces EPSCs recorded in Ca2+ (upper) and desynchronized events recorded in 6mM Sr2+. Calibration: 10pA, 100ms (Di) Cumulative distribution of quantal NMDA mediated events and (Dii) mean amplitudes from all recordings. No significant difference in the size of quantal NMDA events was observed between Fmr1+/y and Fmr1−/y mice. (Diii & iv) Representative recordings of desynchronized NMDA mediated Sr2+-mEPSCs. Calibration: 5pA, 250ms.
To address any differences in glutamate receptor numbers at developing thalamocortical synapses, evoked AMPA receptor mediated EPSCs were recorded while exchanging the Ca2+-containing extracellular solution with one that contained 6mM strontium (Sr2+). Synaptic release is desynchronized in the presence of Sr2+ and quantal EPSCs (Sr2+-mEPSCs) are observed for several hundreds of milliseconds after stimulation of the inputs (Bannister et al., 2005). Comparison between mice of each genotype during the critical period found no significant difference in AMPA Sr2+-mEPSC amplitudes (Fmr1+/y: 10.2 ± 0.8 pA, n = 24; Fmr1−/y: 9.7 ± 0.5, n = 25 p > 0.05, K-S test) (Figure 3C).
In the same manner, the amplitude of the isolated NMDA receptor mediated component of Sr2+-mEPSCs was measured. Again, recordings were made during critical period development (see methods). No significant differences were found in the quantal NMDA content of individual layer IV thalamocortical synapses during the critical period between Fmr1−/y mice and littermate controls (Fmr1+/y: 6.3 ± 0.2 pA, n = 21; Fmr1−/y: 6.9 ± 0.4, n = 17, p > 0.05, K-S test) (Figure 3D). Therefore, the quantal amplitude of both the AMPA and NMDA components at developing thalamocortical synapses is unaltered during early postnatal development in Fmr1 knockout mice, while there is a significant increase in the expression of synaptic NR1 protein in tissue from layer IV from Fmr1−/y animals.
Silent synapses in Fmr1 knockout mice during the critical period
To determine whether the elevated NA ratio and increase in NR1 protein expression in synaptic membranes was due to an elevation in the abundance of NMDA-only silent synapses, thalamocortical EPSCs were recorded from mice during the critical period (P7), while stimulating just a few inputs to the layer IV neurons using minimal stimulation techniques. Two example experiments are illustrated in Figure 4A & B. Under these conditions, AMPA mediated EPSCs or failures are observed in successive trials when the neuron is voltage clamped at relatively hyperpolarized potentials (−70mV). Depolarizing the neuron to +40mV produces an outward, mixed AMPA, NMDA EPSC; measurement of the outward current 60ms after the onset allows analysis of the largely NMDA-mediated component (Marie et al., 2005). Thus, the fraction of silent synapses can be determined by analysis of the failure rate at −70mV and the failure rate at +40mV (Isaac et al., 1997) (see methods). In Fmr1+/y mice there was no significant difference in the percentage of EPSC failures at −70mV compared to +40mV (failures −70mV: 31 ± 4.7 %; failures at +40mV: 35 ± 6.8 %, n = 8, p > 0.05), similar to previous reports at thalamocortical synapses at the close of the critical period (Isaac et al., 1997) (Figure 4C). In contrast, the number of failures in Fmr1−/y mice at +40 mV was significantly lower than the failures at −70mV, indicating that there was a significant proportion of NMDA-only synapses remaining at the closure of the customary critical period (failures −70mV: 55 ± 7.4 %; failures at +40 mV: 37 ± 7.9 %, n = 7, p < 0.05) (Figure 4D). Representing these failure rates as a failure ratio (percent failures −70mV/percent failures +40mV) allows a direct assessment of the fraction of silent synapses between each genotype where a failure ratio of 1.0 indicates few silent synapses and a larger ratio signifies a greater fraction of NMDA-only synapses. The failure ratio in wildtype mice was close to 1.0 (0.9 ± 0.2) (Figure 4E) suggesting that there were few, if any, silent thalamocortical synapses remaining at P7 in Fmr1+/y mice. In contrast, a relatively high failure ratio was observed in Fmr1−/y mice (1.9 ± 0.4) supporting the conclusion that there are a considerably higher fraction of NMDA-only synapses in these mice (Figure 4E).
Figure 4. Silent synapses are prevalent at the close of the critical period in Fmr1 knockout mice.

(Ai) Representative traces and (Aii) timecourse from a single silent synapse experiment in Fmr1+/y mice. Minimal stimulation was used such that approximately 50% failures were observed when recording with the membrane potential clamped at −70mV. After 100 consecutive trials, the membrane potential was depolarized to +40mV and NMDA EPSCs measured (60ms latency from current onset, see methods). After a further 100 trials, D-APV was applied to block the NMDA component. (Bi & Bii) Representative silent synapse experiment and traces from a Fmr1−/y mouse. Calibration: 20pA, 50ms (C) Percentage of failures for each recording at −70mV and +40mV in Fmr1+/y mice. There is no difference in failure rate of AMPA and NMDA components suggesting that there are few NMDA-only synapses present at P7 in Fmr1+/y mice. (D) Percent failures at −70mV and +40mV in Fmr1−/y mice. A significantly reduced number of failures at +40mV demonstrates that a fraction of NMDA-only synapses remain at P7 in Fmr1−/y mice (p = 0.01). (E) The failure ratio is larger than 1.0 in Fmr1−/y underlining that there is a fraction of silent synapses remaining at P7 in Fmr1 knockout animals.
NMDA receptor stoichiometry switch and spine density of layer IV neurons
In the first week of postnatal development, the NR2B subunit is a prominent constituent of the NMDA receptor complex in layer IV synapses, but it is rapidly downregulated playing a reduced role in synaptic NMDA receptors at the closure of the critical period (Barth and Malenka, 2001). The developmental profile of the NR2B receptor subunit in Fmr1−/y mice was monitored by recording NMDA receptor mediated synaptic currents and determining the relative sensitivity to the NR2B selective antagonist, ifenprodil (3μM) at the beginning, end, and after closure of the critical period. At postnatal day 4, NMDA receptor EPSCs in wildtype mice were inhibited 66 ± 4.4 %, (n = 7), which was not different from the ifenprodil inhibition of synaptic NMDA receptors in Fmr1−/y mice (70 ± 3.6%, n = 10 p > 0.05) (Figure 5A & B). Similarly at P7 and P10, the inhibition of NMDA receptors mice was not significantly different between Fmr1+/y and Fmr1−/y (at P7: Fmr1+/y: 57 ± 5.8 %, n = 11; Fmr1−/y 49 ± 5.7 %, n = 11 p > 0.05; at P10: Fmr1+/y: 31 ± 7.0 %, n = 10; Fmr1−/y 24 ± 5.4 %, n = 11 p > 0.05) (Figure 5A & B). Therefore despite an altered developmental profile of excitatory thalamocortical synapses during the critical period for plasticity, the normal developmental switch in NMDA receptor stoichiometry from NR2B to NR2A is maintained in Fmr1−/y mice.
Figure 5. NMDA receptor stoichiometry and spine density in layer IV stellate neurons.
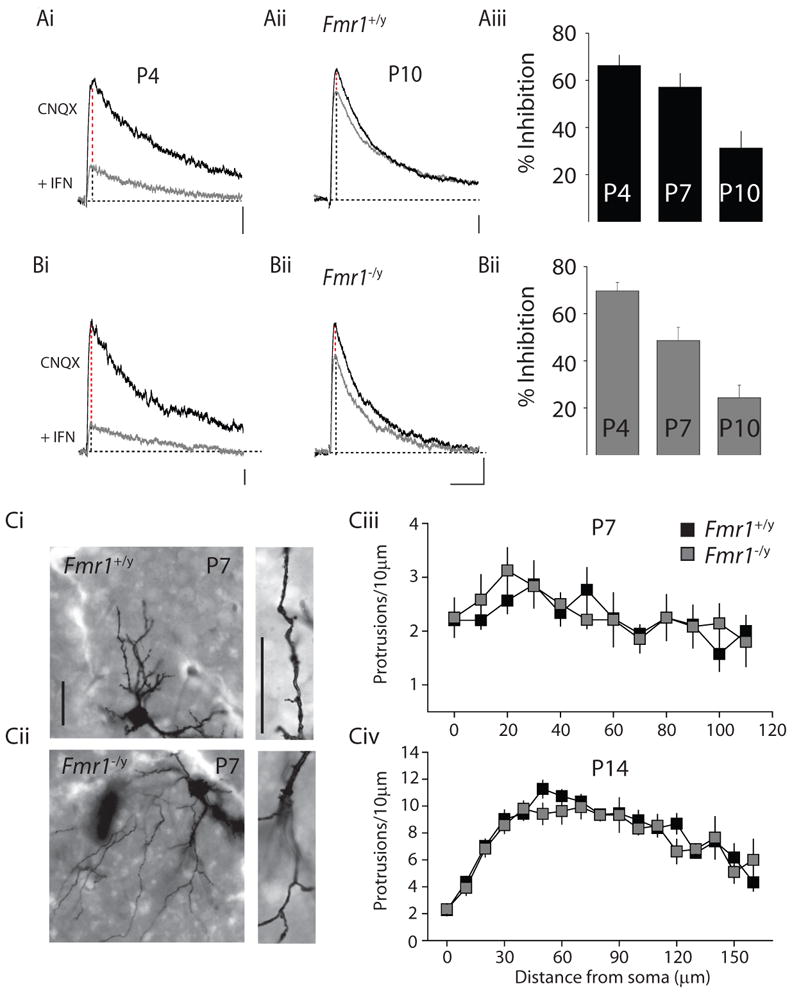
(Ai & Aii) Representative NMDA receptor mediated responses at P4 and P10 before and after application of the NR2B specific antagonist ifenprodil (IFN, 3μM) in Fmr1+/y and (Bi & Bii) Fmr1−/y mice Calibration: 5pA, 200ms (P4) and 5pA, 100ms (P10). (Aiii & Biii) Grouped data from all recordings demonstrating a developmental reduction in the sensitivity of thalamocortical NMDA receptors to ifenprodil between P4 and P10 that is not significantly different between Fmr1+/y and Fmr1−/y mice. (Ci & Cii) Representative pictures of Golgi stained neurons and enlarged dendritic regions from layer IV sections taken from P7 Fmr1 knockout animals and littermate controls. Calibration: 25μm (Ciii) Analysis of spine protrusion density in P7 and (Civ) P14 mice.
Alterations in the development of glutamatergic synapses may be reflected in changes in the density of synaptic connections or spines. A previous study found small but significant increases in the number of spines in layer V somatosensory neurons at P7 (Nimchinsky et al., 2001). In layer IV neurons nascent spines are observed at P7 as dendritic protrusions. Analysis of stellate neuron dendritic protrusions using Golgi stained sections did not uncover any difference in density of putative spines between genotypes at P7 (Figure 5Ci-iii). Moreover, analysis after closure of the critical period at P14 produced similar results, with no observed changes in spine density in Fmr1 knockout mice compared to littermate controls (Figure 5Civ).
Developmental LTP time-window in layer IV is delayed in Fmr1 knockout mice
Long term potentiation at developing thalamocortical synapses is dependent on the activation of NMDA receptors, and is only present during the critical period when topographical rearrangements of somatosensory cortex can be induced by sensory perturbations (Crair and Malenka, 1995). Using perforated patch whole cell recording of spiny stellate neurons at each postnatal day between P3 and P10, LTP was induced using a pairing protocol (Crair and Malenka, 1995; Isaac et al., 1997) in Fmr1+/y and Fmr1−/y mice. At P3-P4 EPSCs potentiated strongly in all our recordings from Fmr1+/y mice (P3: 136 ± 5.1%, n = 6; P4: 138 ± 8.2%, n = 9)(Figure 6Ai & Bi). Using the same induction protocol, essentially no potentiation was observed at the same developmental timepoint in Fmr1−/y mice (P3: 95 ± 5.5%, n = 7, p = 0.0002; P4: 99 ± 7.1%, n = 10, p = 0.003)(Figure 6Aii & Bi). The magnitude of potentiation following pairing declined during subsequent postnatal days in Fmr1+/y mice, as previously reported (Barth and Malenka, 2001; Crair and Malenka, 1995), with only a small potentiation observed in some recordings at postnatal days 6 and 7 (P6: 113 ± 7.0%, n = 7; P7: 104 ± 7.1%, n = 8) (Figure 6Ai & 6Bii). In Fmr1−/y mice, the magnitude of potentiation was larger on each subsequent day after P4, such that the maximal synaptic potentiation in Fmr1−/y mice was obtained at postnatal day 6 and 7 (P6: 141 ± 15%, n = 11; P7: 142 ± 13%, n =12, p = 0.04) (Figure 6Aii & Bii). In Fmr1−/y mice, potentiation was observed following pairing in recordings even beyond the developmental time-point that is considered the closure of the critical period. At P9, potentiation of 118 ± 7.5% (n = 8) was observed in Fmr1−/y whereas no potentiation was evident in Fmr1+/y recordings (98 ± 6.7%, n = 7)(Figure 6A & Biii). These results demonstrate that alterations in NMDA receptors at synapses, due to loss of FMRP, result in correlated changes in the normal progression of plasticity at thalamocortical synapses during the first postnatal week and a delay in the window for synaptic plasticity to later times.
Figure 6. Temporal window for LTP at thalamocortical synapses is shifted in Fmr1 knockout mice.

(Ai) Grouped data from all wildtype recordings at each postnatal day between P3 and P10. Robust potentiation is observed in all recordings at P3 whereas no LTP is observed after closure of the critical period (P7). (Aii) Grouped data from all Fmr1−/y mice thalamocortical LTP recordings. No LTP is observed at P3 whereas increasing amounts of potentiation are observed on subsequent postnatal days with maximal potentiation observed at P6 & P7. In some recordings LTP is observed even beyond closure of the critical period. (Bi-iii) Grouped time-courses of thalamocortical LTP experiments in Fmr1−/y and control mice at P3 & P4, P6 & P7, P9 & P10.
A prior study of developing thalamocortical synapses suggested a differential distribution of synaptic release probabilities between AMPA-containing and NMDA-only synapses (Yanagisawa et al., 2004). Altered presynaptic function may well contribute to the ability of synapses to undergo long term plasticity; therefore as a measure of presynaptic function paired pulse depression of thalamocortical synapses was examined at P4 and P7. Thalamocortical EPSCs demonstrated equivalent degrees of paired pulse depression in Fmr1−/y and Fmr1+/y mice at both developmental ages at interstimulus intervals ranging from 20ms to 500ms, suggesting that presynaptic function is normal in Fmr1 knockout mice (Figure S2).
Somatosensory patterning and lesion induced anatomical map plasticity in Fmr1 knockout mice
The normal patterning of somatosensory cortex is dependent in part on glutamate receptors, and importantly, is disrupted in NMDA receptor knockout mice (Iwasato et al., 2000). To determine whether the shift in the maturation of thalamocortical synaptic glutamate receptors in Fmr1−/y mice affected patterning of barrel cortex, the anatomical segregation of thalamic axons was investigated by examining the barrel patch areas in layer IV of Fmr1−/y mice. In each case, the individual areas of the inner 12 barrels, rows b through d, arcs 1 through 4, were measured and areas compared across genotype. Additionally, the total area occupied by the posterior medial barrel subfield (PMBSF) was measured. There was no significant difference found in the overall cortical territory occupied by the PMBSF in Fmr1−/y mice (Figure 7C), and when the individual barrel areas were normalized to the barrel field area, no consistent difference was seen between Fmr1−/y and Fmr1+/y mice (Figure 7D). Similarly, analysis of barrel areas in adult mice (P60) yielded no differences in arealization of the somatosensory cortex or individual barrel patch areas (Figure S3). These results demonstrate that disruptions in thalamocortical synaptic transmission and plasticity have little impact thalamocortical (TCA) patterning.
Figure 7. Barrel arealization and lesion induced map plasticity in Fmr1 knockout mice.

(Ai & Aii) Representative cytochrome oxidase stained tangential sections through layer IV of the barrel cortex in P7 Fmr1+/y and Fmr1−/y mice. Calibration: 250μm (B) Barrel areas for each individual barrel in rows b-d in wildtype and knockout mice. (C) Total area occupied by the posterior medial barrel subfield (PMBSF) in Fmr1+/y and Fmr1−/y mice (D) Ratio of each individual barrel to the total territory occupied by the PMBSF. (Ei & Eii) Representative cytochrome oxidase stained sections of mice with unilateral c-row whisker lesioning performed at P0. White dashed regions highlight the c-row barrels. (Eiii) Map Plasticity Index of barrel areas for P0 lesioned mice of the ipsilateral and contralateral hemispheres. (Fi & Fii) Representative sections of mice subjected to c-row whisker lesioning at P5. (Fiii) Map Plasticity Index for ipsi- and contralateral barrel areas of P5 lesioned mice.
Lesioning the whiskers in perinatal animals causes a dramatic plasticity of the anatomical structures in the somatosensory cortex. The barrel patch area corresponding to the lost whiskers shrinks, and the surrounding barrel areas invade the deprived whisker associated space (Van der Loos and Woolsey, 1973). The time-window when sensory manipulations can cause anatomical map plasticity overlaps with the critical period for synaptic plasticity in layer IV, which has led to speculation that the two processes may be linked (Crair and Malenka, 1995; Schlaggar et al., 1993). Similarly, a role has been proposed for NMDA receptors in both processes, but for anatomical map plasticity this remains unclear as acute pharmacological block of receptors prevented lesion-induced plasticity, while genetic ablation of NR1 did not (Datwani et al., 2002; Schlaggar et al., 1993). To determine whether the delayed critical period for plasticity was a specific deficits in layer IV, or occurred alongside delays in the critical period for barrel map plasticity in Fmr1 knockout mice, whiskers were lesioned on neonates at P0 and P5. Unilateral lesioning of the c-row whiskers at P0, caused an obvious alteration in the somatotopic patterning in the contralateral S1 region in both Fmr1−/y and Fmr1+/y mice. This was quantified by measuring barrel areas and creating a Map Plasticity Index (MPI) (Lu et al., 2001)(see methods)(Figure 7Ei & Fi). In the control hemisphere (ipsilateral to the lesion), the MPI was close to 1.0, demonstrating that the c-row area was equivalent to the average areas of the b- and d- row barrels (Figure 7Eiii, no lesion). However, c-row whisker lesions caused a severe loss of the corresponding c-row barrels and expansion of the neighboring b- and d- rows in the contralateral (lesion) hemispheres, of both the Fmr1 knockout and wildtype littermate mice, as evidenced by a MPI considerably < 1 (P0 Fmr1+/y: 0.45 ± 0.03, n = 10; P0 Fmr1−/y: 0.47 ± 0.05, n = 6)(Figure 7E).
Whisker lesioning at P0 had an equivalent effect in the Fmr1−/y mice; however it was possible that the window for lesion-induced barrel map plasticity might be extended beyond the close of the critical period at P5. C-row whiskers were lesioned in P5 neonates and somatotopic rearrangement determined by measuring barrel areas as before. In both genotypes, the calculated MPI of c-row barrels after P5 lesioning in both the ipsi- and contralateral hemisphere was close to 1 (P5 Fmr1+/y: 1.1 ± 0.03, n = 8; P5 Fmr1−/y: 0.99 ± 0.04, n = 10) (Figure 7F). Similarly, analyzing plasticity of the c-row, and band d- rows separately (see methods) (Takasaki et al., 2008) at both P0 and P5 did not uncover any differences in either the expansion or reduction of lesioned rows between genotypes (P0 c/total Fmr1+/y: 0.12 ± 0.006, n = 10; P0 c/total Fmr1−/y: 0.12 ± 0.011, n = 6; P5 c/total Fmr1+/y: 0.23 ± 0.005, n = 8; P5 c/total Fmr1−/y: 0.23 ± 0.007, n = 10; P0 b+d/total Fmr1+/y: 0.5 ± 0.007, n = 10; P0 b+d/total Fmr1−/y: 0.5 ± 0.008, n = 6; P5 b+d/total Fmr1+/y: 0.44 ± 0.006, n = 8; P5 b+d/total Fmr1−/y: 0.46 ± 0.006, n = 10). Therefore, there does not appear to be a delay in the critical period for map plasticity, in contrast to the clear temporal shift in synaptic plasticity observed in Fmr1 knockout mice.
Discussion
Altered development of cortical NMDA receptors and synaptic plasticity in Fmr1 knockout mice
Cortical glutamate receptors have been implicated in the development of the barrel map and the refinement of cortical sensory circuits that underlie sensory processing (Schlaggar et al., 1993). Here we demonstrate that early postnatal development of the excitatory connection from the thalamus to layer IV spiny stellate neurons, the thalamocortical synapse, is disrupted in fundamental ways during its critical period in Fmr1 knockout mice. The progressive development of excitatory ionotropic glutamate receptor signaling, that normally occurs over the first postnatal week, is delayed. In wildtype animals, the relative NMDA component of synapses was initially large compared to the AMPA component, resulting in a high NA ratio. Over the course of the first week, this ratio declined as the NMDA receptor component was reduced and the AMPA component increased, agreeing with prior reports (Barth and Malenka, 2001; Crair and Malenka, 1995). In contrast, Fmr1−/y mice have a relatively low NA ratio at the earliest postnatal times, which subsequently increases over the first week, with the highest ratio observed at P7. After this, the NA ratio decreased rapidly, normalizing to wildtype synapse levels at postnatal times beyond closure of the critical period. The altered NA ratio in Fmr1−/y mice is readily explained by the increase in fraction of silent synapses found at the customary closure of the critical period. In wildtype mice at P7, there are essentially no silent synapses present, as previously reported (Isaac et al., 1997); however, at the same developmental timepoint, a fraction of NMDA-only thalamocortical synapses persist in Fmr1−/y mice.
Synaptic plasticity and experience dependent refinement of sensory circuits are inextricably linked, and NMDA receptors play a central role in both processes. Therefore alteration in NMDA receptor signaling and the developmental maturation of silent synapses during the critical period for plasticity will likely have consequences for the mechanisms that regulate development of cortical circuits. Results from LTP experiments performed in Fmr1−/y mice at each postnatal day, starting at P3, demonstrate an alteration in the profile of plasticity that paralleled the changes in NMDA receptors and silent synapses, such that little or no plasticity was found early in development and the most robust LTP was observed at the customary closure of the critical period. LTP continued to be observed in Fmr1−/y mice beyond the developmental timepoint when thalamocortical synaptic potentiation is no longer found in wildtype mice. While critical period shifts have been demonstrated in the visual cortex (Hensch, 2005), this is the first time a temporal delay in the window for developmental synaptic plasticity in layer IV of somatosensory cortex has been observed in a mutant mouse or after manipulations of the sensory whisker system. Therefore, the mouse model of Fragile X could inform us about the mechanisms regulating critical periods in cortical regions (Hensch, 2004).
Prior studies that have attempted to gauge AMPA receptor protein expression in Fmr1 knockout mice have reported either a decrease in specific brain regions or no change in others. In one study GluR1 protein levels were reported to be reduced in the neocortex of Fmr1−/y mice, although no similar alterations in GluR1 were observed in the cerebellum or hippocampus (Li et al., 2002). Other groups also reported a lack of alteration in GluR1 expression in the visual cortex (Restivo et al., 2005) and hippocampus (Nosyreva and Huber, 2006). These data are in agreement with reports that observed no alteration in GluR1 levels in adult Fmr1 knockout mice in various brain regions (Nosyreva and Huber, 2006; Restivo et al., 2005). Likewise, the biochemical and electrophysiological studies of neonatal Fmr1−/y layer IV somatosensory cortex suggest little alteration in AMPA receptor signaling, and the prominent developmental synaptic deficit in Fmr1 knockouts results from a delayed maturation of silent synapses and an associated shift in plasticity.
The erroneous maturation of glutamatergic signaling at thalamocortical synapses during the first postnatal week in Fmr1 knockout mice and the corresponding alterations in the ability to induce LTP, provide insight into the development of sensory synapses. Interestingly, all the alterations in excitatory synapses during early development revert to normalcy in the second postnatal week and beyond. This disruption in functional progression parallels the timecourse of spine morphological abnormalities reported in layer V of Fmr1 knockout mice. The length and density of spines on layer V pyramidal neurons in the PMBSF are significantly altered in Fmr1 knockout mice at P7; however there is little or no alteration in these parameters measured in the 2nd and 4th postnatal weeks (Nimchinsky et al., 2001), although it should be noted that a subsequent study found that spine abnormalities returned in adult mice at 2 months of age (Galvez and Greenough, 2005). We also analyzed dendritic protrusion density (as an indicator of nascent spines) in layer IV neurons in this study. However, no alteration in the density of dendritic protrusions was found at P7 or P14. At these very young ages spines are not fully formed, and there are a large number of shaft synapses present. Therefore the analysis of spine protrusion density does not account for all synaptic contacts, and we cannot say with certainty whether synapse density in layer IV neurons might be altered during the critical period. However the lack of change in spine density in layer IV suggests that the consequences of the loss of FMRP cannot be generalized across cell type and region; moreover, some of the endophenotypes could represent secondary modifications due to alterations in synaptic or circuit development.
This study is also the first to examine expression of FMRP in layer IV somatosensory cortex during perinatal development. FMRP is expressed during the critical period further emphasizing the importance of this protein during development of sensory circuits. Whether these critical period deficits subsequently contribute to alterations in sensory processing in adult animals is not known, but it is possible that synaptic and circuit refinement during these early periods influence other elements in the development of cortical circuits that persist throughout life. Several studies at the molecular and cellular level in the Fmr1 knockout mouse have found delays in normal maturation which return to a normal phenotype later in development. For instance increased spine density during the first postnatal week in layer V pyramidal neurons is not observed in older animals (Nimchinsky et al., 2001), and elevated FMRP during early postnatal development in the hippocampus regulates Map1b (microtubule-associated protein 1B) mRNA during a period of increased synaptogensis in perinatal mice (Lu et al., 2004). A recent study demonstrated that the ascending cortical connection between layer IV spiny stellates and layer 2/3 pyramidal neurons was disrupted in Fmr1 knockout mice (Bureau et al., 2008). This intracortical connection develops after closure of the critical period in layer IV during the second postnatal week. In Fmr1 knockout mice, the strength of this connection is reduced due to a decrease in connection probability between the spiny stellate neurons and the more superficial layer pyramidal neurons (Bureau et al., 2008). The alteration in this ascending pathway is also temporally restricted to early postnatal development; layer IV to II/III connectivity differences between wildtype and knockout mice disappear in the third postnatal week. These prior observations along with the results of the current study are consistent in demonstrating delays in the development of synapses and circuits during cortical critical periods in Fmr1 knockout mice. This theme of delayed maturation at the cellular and synaptic level is particularly compelling as delay in developmental trajectories in motor skills, speech, and social skills are behavioral hallmarks of Fragile X syndrome (Kau et al., 2002).
Synaptic mechanisms implicated in Fragile X syndrome
Fmr1 knockout mice have been an invaluable resource in studying the molecular and cellular aspects of Fragile X syndrome. Loss of FMRP in neurons results in multiple synaptic deficits in the cortex and hippocampus. Synaptic plasticity mechanisms have been a particular focus in defining functional alterations in Fmr1 knockout mice. Several studies have determined that diverse forms of synaptic plasticity expressed in different cortical regions are altered, although it seems that not all synapses are affected. Specific deficits in LTP have been reported in the frontal neocortex (Li et al., 2002; Meredith et al., 2007; Wilson and Cox, 2007), anterior cingulate cortex (Zhao et al., 2005), olfactory cortex (Larson et al., 2005), and in layer V somatosensory cortex (Desai et al., 2006) while LTP in the CA1 of the hippocampus is largely unaltered in Fmr1 knockout mice (Godfraind et al., 1996; Larson et al., 2005; Li et al., 2002)(but see(Lauterborn et al., 2007)). These studies demonstrate that plasticity deficits in some cortical areas persist past early development and could be responsible for alterations in cognitive processes associated with the disease.
Glutamate receptor signaling during the critical period is required for the correct topographic patterning of the somatosensory cortex (Fox et al., 1996). Several manipulations in genes encoding for glutamate receptors and associated regulatory proteins have further demonstrated a role for glutamatergic transmission in the development of the somatosensory cortex. Barrel patterning is disrupted in cortex specific NMDA receptor knockouts (Iwasato et al., 2000) and also when adenylyl cyclase type I (AC1), which influences AMPA receptor trafficking (Lu et al., 2003), is ablated globally (Abdel-Majid et al., 1998) or in the cortex (Iwasato et al., 2008). Moreover, patterning is disrupted in mice that are null for group I metabotropic receptors (mGluR5) (Hannan et al., 2001; She et al., 2009; Wijetunge et al., 2008). The involvement of mGluRs in this process has particular resonance, as there is growing evidence that disruption of normal signaling through group I mGluRs is central to the etiology of Fragile X (Bear, 2005; Bear et al., 2004; Dolen et al., 2007). Indeed there is evidence that the activation of NMDA and metabotropic glutamate receptors in the barrel cortex is linked to increased translation of FMRP (Todd et al., 2003). Thus loss of regulation of FMRP target mRNAs during the critical period, when refinement of synapses is activity-dependent, could underlie the deficits in the normal developmental progression of thalamocortical synapses observed in this study.
The temporal overlap in the critical periods for inducing thalamocortical synaptic plasticity and lesion-induced somatotopic rearrangement of thalamocortical axons, has led to an implied link between these two forms of plasticity. Fmr1 knockout mice had normal somatosensory maps, and plasticity of this map was unaltered. These data are in good agreement with previous findings that neither NMDA receptors (Datwani et al., 2002; Iwasato et al., 2000), or signaling enzymes that regulate synaptic plasticity (Iwasato et al., 2008) are necessary for lesion-induced plasticity. Interestingly, Takasaki and colleagues recently demonstrated a dissociation of the mechanisms which drive retraction of the TCAs from the lesioned whiskers and expansion of TCAs from the adjacent whiskers (Takasaki et al., 2008). Ablation of row c vibrissae in mice lacking the glutamate transporter 1 gene (Glt1) caused a retraction of c-row TCAs, but did not cause the typical expansion of row b and d. Lesions of the infraorbital nerve (which result from whisker follicle cauterization) results in cell death in the thalamus and a lack of barreloid formation, indicating that the shrinkage of the lesioned row area likely results from a loss of TCAs. In contrast the expansion of neighboring row representations almost certainly involves the expansion (or lack of withdrawal) of TCAs into the denervated region. Therefore not surprisingly, it would seem that barrel map plasticity is not simply connected to synaptic plasticity but is also controlled by other mechanisms. Taken together, our data suggest mechanisms independent of NMDA receptors drive lesion-induced plasticity in somatosensory cortex and demonstrate a specific role for FMRP in regulating synaptic plasticity as opposed to a more general role in S1 development.
Glutamate receptors are necessary for barrel map development and thalamocortical synaptic plasticity; however prior studies in glutamate receptor knockout mice models have not revealed shifts in the window for critical period plasticity, as we report in the Fmr1 knockout mouse. Interestingly, BDNF (brain-derived neurotrophic factor) knockout mice demonstrate some similar synaptic phenotypes to Fmr1 knockouts. BDNF, and its receptor TrkB, are expressed in layer IV barrel cortex during early postnatal development, with BDNF peaking at P5 (Lush et al., 2005). At postnatal day 8/9 BDNF knockout mice have a higher N/A ratio at thalamocortical synapses and an elevated fraction of silent synapses (Itami et al., 2000). However, the critical period for lesion induced map plasticity is not altered in BDNF knockout mice as we also found in Fmr1−/y mice (Itami et al 2000). BDNF has been proposed as a negative transcriptional regulator of FMRP mRNA (Castren et al., 2002) and BDNF can rescue hippocampal LTP deficits in Fmr1 knockouts (Lauterborn et al., 2007). Therefore these studies raise the intriguing possibility that BDNF regulation of FMRP could contribute to the development of thalamocortical synapses.
Fragile X individuals display a range of abnormal responses to sensory stimulation due to a heightened sensory response including tactile defensiveness (Baranek et al., 2008) and altered sensorimotor gating (Frankland et al., 2004). Here we demonstrated that the normal development of the principal excitatory sensory input to the primary somatosensory cortices is delayed in Fmr1 knockout mice. The specific alteration in thalamocortical NMDA receptors during the critical period for layer IV plasticity parallels a change in the expression of plasticity and silent synapses over this developmental period, resulting in a shifted time window when plasticity can be induced. The precise timing of critical periods during cortical developmental is essential for the proper organization of synaptic connections and circuits. Thus delayed timing of plasticity windows could contribute to altered refinement of cortical circuits that persist throughout the life of the animal and contribute to sensory processing deficits in Fragile X syndrome.
Experimental Procedures
Electrophysiology
Thalamocortical slices (400 μm) were prepared as described (Agmon and Connors, 1991; Crair and Malenka, 1995) from male postnatal day (P)3-21 Fmr1- ko mice on a congenic C57BL/6 background and their male wildtype littermates. All experiments were performed blind to the genotype of the animal and post hoc genotyping of tail DNA samples was performed following experimentation and analysis. Mice were anesthetized with isoflurane, decapitated, and the brain removed under ice-cold oxygenated sucrose-slicing ACSF containing: 85mM NaCl, 2.5mM KCl, 1.2 mM NaH2PO4, 25mM NaHCO3, 25mM glucose, 75mM sucrose, 0.5mM CaCl2, and 4mM MgCl2, equilibrated with 95% O2/5% CO2. Slices were incubated at 28°C for 30 minutes, while slowly exchanging the sucrose-ACSF in the incubation chamber for oxygenated sodium ACSF solution containing: 125mM NaCl, 2.4mM KCl, 1.2mM NaH2PO4, 25mM NaHCO3, 25mM glucose, 1mM CaCl2, and 2mM MgCl2. After a minimum of 1 hour recovery period, individual slices were transferred to a recording chamber where they were perfused with oxygenated sodium ACSF containing 2mM CaCl2 and 1mM MgCl2.
A tungsten bipolar stimulating electrode was positioned in the VB (see Figure 2A) and whole cell patch clamp recordings were made from spiny stellate neurons in the barrel cortex using a Multiclamp 700B patch clamp amplifier (Axon Instruments). Borosilicate glass electrodes had resistances of 4–5 MΩ when filled with CsF internal solution containing: 95mM CsF, 25mM CsCl, 10mM Cs-HEPES, 10mM Cs-EGTA, 2mM NaCl, 2mM Mg-ATP, 10mM QX-314, 5mM TEA-Cl, 5mM 4-AP, pH adjusted to 7.3 with CsOH. Only stable evoked EPSC recordings that exhibited a constant latency that did not change with increasing stimulation intensity or stimulation rate were accepted as monosynaptic thalamocortical inputs (Feldman et al., 1998). Series resistance was continuously monitored using hyperpolarizing voltage steps generated by pClamp 9 software (Axon Instruments), and recordings were discarded if there was a >15% change during the course of the experiment. All recordings were made in the presence of GABAA receptor antagonists, picrotoxin (50μM) and bicuculline (10 μM). NMDA mediated currents were recorded at +40mV and AMPA receptor mediated currents were measured at a membrane potential of −70mV. In some experiment the NMDA receptor component was individually isolated by including the AMPA/kainate receptor antagonist 6-cyano-7-nitroquinoxaline-2,3-dione (25μM CNQX). Ifenprodil (3μM) was used to block the NR2B containing receptors in some experiments. For the measurement of quantal AMPA receptor EPSCs (Sr2+-EPSCs), the normal extracellular was replaced by ACSF containing 6mM SrCl2 and 2mM MgCl2 and for NMDA receptor mediated Sr2+-EPSCs, the ACSF contained 6mM SrCl2 and 10μM glycine. Comparing the amplitude of Sr2+-mEPSCs at P4 and P7 in Fmr1+/y mice demonstrated that there was no difference in the amplitude of events at these two postnatal times (p = 0.22), therefore recordings were grouped from these two ages during the critical period.
To determine the fraction of silent synapses, whole cell recordings were made from P7 Fmr1+/y and Fmr1−/y mice. EPSCs were elicited using a minimal stimulation technique where stimulation was at first high enough to elicit a reliable EPSC at −70mV. The stimulation intensity was then reduced until the synaptic response had a failure rate around 50% at −70mV and 100 sweeps were recorded. The neuron was depolarized to Then, 100 EPSCs were recorded at +40mV using the same minimal stimulation intensity. To confirm the contribution of the NMDA receptor, D-APV was added to the external solution. The fraction of failures at −70mV was compared to the fraction of failures at +40mV (Isaac et al., 1997; Itami et al., 2000; Liao et al., 1995; Yanagisawa et al., 2004).
For LTP experiments, the perforated patch recording configuration was adopted to eliminate LTP washout. Gramicidin (20μg/ml) was added to a KMeSO4 internal solution: 125mM KMeSO4, 5mM KCl, 5mM NaCl, 11mM HEPES, 1mM MgCl2, 10mM phosphocreatine, 4mM Na-ATP, 0.3mM Na-GTP, pH adjusted to 7.3. Electrodes were front filled with gramicidin-free internal, and then backfilled with gamicidin-containing internal. After formation of a gigaohm seal between the electrode and cell membrane, access resistance was continuously monitored using a hyperpolarizing step. Once stable access was reached (usually ~20mins) synaptic recordings were commenced. LTP was induced by pairing 100 stimuli at 1Hz with postsynaptic depolarization to 0mV.
Data analysis
Properties of EPSCs were analyzed using pClamp 9 (Axon) and Mini Analysis (Synaptosoft) software. Origin 7 (Microcal) was used for statistical analyses. Two sample comparisons were made using the paired Student’s t-test and multiple comparisons were made using a one-way analysis of variance (ANOVA). Nonparametric data was tested for significance using the Kolmogorov-Smirnov test.
FMRP immunohistochemistry and immunoelectron microscopy
Brains from wild type mice at P4, P7, P14 and P21 were used to establish the developmental expression profile of FMRP. Mice were euthanised by a lethal dose of pentobarbital (Euthanol; 20mg/kg; ip) and transcardially perfused with saline followed by 4% paraformaldehyde in 0.1M phosphate buffer. Brains were post-fixed for at least 6hrs and then cryoprotected prior to sectioning on a freezing microtome at 48μm thickness in both the coronal and tangential planes. Tangential sections were obtained from hemispheres that had been flattened by removing the thalamus, entorhinal cortex, hippocampus and striatum, thus leaving only the neocortical sheet. Antigen retrieval was carried out on sections before they were reacted with rAM1 antibody (1:1000 dil; gift from Claudia Bagni). FMRP immunoreactivity was visualized using a Vectastain ABC kit (Vector Laboratories, Burlingame, CA) with diaminobenzidine (DAB) as the chromogen according to previously published methods (Watson et al., 2006).
DAB immunoelectron microscopy was performed on P7 coronal sections through the PMBSF using rAM1 antibody to determine the localization of FMRP at synaptic sites as previously described (Barnett et al., 2006; Watson et al., 2006).
Immunoblotting
Mice were anesthetized with isoflurane, decapitated, and coronal sections containing S1 were prepared as for electrophysiological experiments. Layer IV barrel cortex was microdissected and synaptoneurosome fractions prepared from the resultant tissue (Villasana et al., 2006). Tissue was homogenized in 1 ml of buffer containing 10 mM HEPES, 1 mM EDTA, 2 mM EGTA, 150 mM NaCl, 10 mM Na4P2O7, 0.5 mM DTT, 50 mM NaF, and protease inhibitor cocktail tablets. Small fractions of each homogenate were taken for later analysis; the remainder was filtered through a 100 μm pore nylon filter followed by subsequent filtration through a 5μm PVDF membrane. Filtrates were centrifuged at 4 °C for 10 min at 1,000 RCF, the supernatant was removed, and the pellets, which contained the synaptoneurosomes, were re-suspended in lysis buffer. The protein concentration of each sample was calculated using a BCA Protein Assay Kit. Samples containing 5 or 10 μg of protein from the synaptoneurosome fraction or the whole homogenate, respectively, were mixed in a reducing sample buffer, sonicated for 1 min, and boiled 5 min. Samples were then separated via SDS-PAGE on a 10% polyacrylamide gel and transferred to a PVDF membrane. Membranes were blocked with 5% powdered milk in Tris-buffered saline/0.1% Tween-20 (TBS-T) and incubated with antibodies against GluR1 (1:1000, Chemicon, Temecula, California), NR1 (1:1000, Millipore, Billerica, Massachusetts), PSD95 (1:10,000, Sigma, St. Louis, Missouri) or βactin (1:20,000, Sigma) overnight at 4 °C. Membranes were washed in TBS-T, and then probed with horseradish peroxidase-conjugated antibodies against either rabbit IgG or mouse IgG (1:50,000) for one hour at room temperature. Following a final wash in TBS-T, membranes were incubated with LumiLight Western Blotting Substrate, after which they were allowed to expose a piece of X-ray film. Images were analyzed with ImageJ software (N.I.H., Bethesda, Maryland). Statistical comparisons were performed using a one-tailed t-test.
Histochemistry
Fmr1-ko and wildtype littermates at P7 were anesthetized, perfused, decapitated and the brain rapidly removed. Cortex containing the barrel field was prepared as described (Welker and Woolsey, 1974) and 40–60μm thick sections were made using a vibratome. Sections were stained with a solution containing 0.5mg/ml DAB, 0.18mg cytochrome C, and 40mg/ml sucrose in phosphate buffer at 37 °C for 3–5 hours and dehydrated with an ethanol series (Land and Simons, 1985; Wong-Riley, 1979).
Analysis of barrel cytoarchitecture
Stained barrel fields were imaged with a Nuance camera mounted on a Zeiss Axioskop 50. Quantification of barrel arealization was made by measuring the areas of the individual b-, c, and d- row barrels (ImageJ) and the total area of the PMBSF and comparing values across genotypes.
Whisker lesioning
In order to determine whether experience-dependent barrel map plasticity is altered in Fmr1 knockout mice, c-row whiskers were lesioned on P0 pups and P5 Fmr1−/y and littermate controls. Mice were anesthetized by cooling on ice and the C1-2 whiskers on the left side of the muzzle were cauterized. Pups were warmed before being returned to their mothers until P14 when they were sacrificed and barrel cortex was removed. The same cytochrome oxidase staining protocols were used to visualize the barrels of the lesioned (right) hemisphere and the no-lesion (left) hemisphere. Quantification of the areas of the b-,c-,and d- row barrels was again performed with the experimenter blind to the genotype of the mouse. The Map Plasticity Index (MPI) was calculated by measuring the areas of the lesioned barrels and adjacent barrels (2×(C1+C2)/(B1+B2+D1+D2))(Lu et al., 2001). In addition the reduction in lesioned barrels and expansion of neighbors was analyzed independently a (c/total) and (b+d/total), where (c/total) = (c1+c2)/(a1+a2+b1+b2+c1+c2+d1+d2+e1+e2) and (b+d/total) = (b1+b2+d1+d2)/(a1+a2+b1+b2+c1+c2+d1+d2+e1+e2) (Takasaki et al., 2008).
Supplementary Material
Acknowledgments
We thank Geoffrey Swanson and Gordon Shepherd for comments on the manuscript. The FMRP antibody (rAM1) was kindly made available by Claudia Bagni. This work was supported by grants from the FRAXA Research Foundation, Autism Speaks and the Brain Research Foundation to AC. EGH was funded by the Training Program in Neurobiology of Information Storage NIH/NIMH T32MH067564
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abdel-Majid RM, Leong WL, Schalkwyk LC, Smallman DS, Wong ST, Storm DR, Fine A, Dobson MJ, Guernsey DL, Neumann PE. Loss of adenylyl cyclase I activity disrupts patterning of mouse somatosensory cortex. Nat Genet. 1998;19:289–291. doi: 10.1038/980. [DOI] [PubMed] [Google Scholar]
- Agmon A, Connors BW. Thalamocortical responses of mouse somatosensory (barrel) cortex in vitro. Neuroscience. 1991;41:365–379. doi: 10.1016/0306-4522(91)90333-j. [DOI] [PubMed] [Google Scholar]
- Antar LN, Li C, Zhang H, Carroll RC, Bassell GJ. Local functions for FMRP in axon growth cone motility and activity-dependent regulation of filopodia and spine synapses. Mol Cell Neurosci. 2006;32:37–48. doi: 10.1016/j.mcn.2006.02.001. [DOI] [PubMed] [Google Scholar]
- Bakker C, Verheij C, Willemsen R, van der Helm R, Oerlemans F, Vermey M, Bygrave A, Hoogeveen A, Oostra B, Reyniers E. Fmr1 knockout mice: a model to study fragile X mental retardation. The Dutch-Belgian Fragile X Consortium. Cell. 1994;78:23–33. [PubMed] [Google Scholar]
- Bannister NJ, Benke TA, Mellor J, Scott H, Gurdal E, Crabtree JW, Isaac JT. Developmental changes in AMPA and kainate receptor-mediated quantal transmission at thalamocortical synapses in the barrel cortex. J Neurosci. 2005;25:5259–5271. doi: 10.1523/JNEUROSCI.0827-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baranek GT, Roberts JE, David FJ, Sideris J, Mirrett PL, Hatton DD, Bailey DB., Jr Developmental trajectories and correlates of sensory processing in young boys with fragile X syndrome. Phys Occup Ther Pediatr. 2008;28:79–98. doi: 10.1300/j006v28n01_06. [DOI] [PubMed] [Google Scholar]
- Barnett MW, Watson RF, Vitalis T, Porter K, Komiyama NH, Stoney PN, Gillingwater TH, Grant SG, Kind PC. Synaptic Ras GTPase activating protein regulates pattern formation in the trigeminal system of mice. J Neurosci. 2006;26:1355–1365. doi: 10.1523/JNEUROSCI.3164-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barth AL, Malenka RC. NMDAR EPSC kinetics do not regulate the critical period for LTP at thalamocortical synapses. Nat Neurosci. 2001;4:235–236. doi: 10.1038/85070. [DOI] [PubMed] [Google Scholar]
- Bear MF. Therapeutic implications of the mGluR theory of fragile X mental retardation. Genes Brain Behav. 2005;4:393–398. doi: 10.1111/j.1601-183X.2005.00135.x. [DOI] [PubMed] [Google Scholar]
- Bear MF, Huber KM, Warren ST. The mGluR theory of fragile X mental retardation. Trends Neurosci. 2004;27:370–377. doi: 10.1016/j.tins.2004.04.009. [DOI] [PubMed] [Google Scholar]
- Bureau I, Shepherd GM, Svoboda K. Circuit and plasticity defects in the developing somatosensory cortex of FMR1 knock-out mice. J Neurosci. 2008;28:5178–5188. doi: 10.1523/JNEUROSCI.1076-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castren M, Lampinen KE, Miettinen R, Koponen E, Sipola I, Bakker CE, Oostra BA, Castren E. BDNF regulates the expression of fragile X mental retardation protein mRNA in the hippocampus. Neurobiol Dis. 2002;11:221–229. doi: 10.1006/nbdi.2002.0544. [DOI] [PubMed] [Google Scholar]
- Chen L, Toth M. Fragile X mice develop sensory hyperreactivity to auditory stimuli. Neuroscience. 2001;103:1043–1050. doi: 10.1016/s0306-4522(01)00036-7. [DOI] [PubMed] [Google Scholar]
- Crair MC, Malenka RC. A critical period for long-term potentiation at thalamocortical synapses. Nature. 1995;375:325–328. doi: 10.1038/375325a0. [DOI] [PubMed] [Google Scholar]
- Datwani A, Iwasato T, Itohara S, Erzurumlu RS. Lesion-induced thalamocortical axonal plasticity in the S1 cortex is independent of NMDA receptor function in excitatory cortical neurons. J Neurosci. 2002;22:9171–9175. doi: 10.1523/JNEUROSCI.22-21-09171.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daw MI, Bannister NV, Isaac JT. Rapid, activity-dependent plasticity in timing precision in neonatal barrel cortex. J Neurosci. 2006;26:4178–4187. doi: 10.1523/JNEUROSCI.0150-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daw MI, Scott HL, Isaac JT. Developmental synaptic plasticity at the thalamocortical input to barrel cortex: Mechanisms and roles. Mol Cell Neurosci. 2007 doi: 10.1016/j.mcn.2007.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai NS, Casimiro TM, Gruber SM, Vanderklish PW. Early postnatal plasticity in neocortex of Fmr1 knockout mice. J Neurophysiol. 2006;96:1734–1745. doi: 10.1152/jn.00221.2006. [DOI] [PubMed] [Google Scholar]
- Dolen G, Osterweil E, Rao BS, Smith GB, Auerbach BD, Chattarji S, Bear MF. Correction of fragile X syndrome in mice. Neuron. 2007;56:955–962. doi: 10.1016/j.neuron.2007.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldman DE, Brecht M. Map plasticity in somatosensory cortex. Science. 2005;310:810–815. doi: 10.1126/science.1115807. [DOI] [PubMed] [Google Scholar]
- Feldman DE, Nicoll RA, Malenka RC, Isaac JT. Long-term depression at thalamocortical synapses in developing rat somatosensory cortex. Neuron. 1998;21:347–357. doi: 10.1016/s0896-6273(00)80544-9. [DOI] [PubMed] [Google Scholar]
- Fox K, Schlaggar BL, Glazewski S, O’Leary DD. Glutamate receptor blockade at cortical synapses disrupts development of thalamocortical and columnar organization in somatosensory cortex. Proc Natl Acad Sci U S A. 1996;93:5584–5589. doi: 10.1073/pnas.93.11.5584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frankland PW, Wang Y, Rosner B, Shimizu T, Balleine BW, Dykens EM, Ornitz EM, Silva AJ. Sensorimotor gating abnormalities in young males with fragile X syndrome and Fmr1-knockout mice. Mol Psychiatry. 2004;9:417–425. doi: 10.1038/sj.mp.4001432. [DOI] [PubMed] [Google Scholar]
- Galvez R, Gopal AR, Greenough WT. Somatosensory cortical barrel dendritic abnormalities in a mouse model of the fragile X mental retardation syndrome. Brain Res. 2003;971:83–89. doi: 10.1016/s0006-8993(03)02363-1. [DOI] [PubMed] [Google Scholar]
- Galvez R, Greenough WT. Sequence of abnormal dendritic spine development in primary somatosensory cortex of a mouse model of the fragile X mental retardation syndrome. Am J Med Genet A. 2005;135:155–160. doi: 10.1002/ajmg.a.30709. [DOI] [PubMed] [Google Scholar]
- Garber KB, Visootsak J, Warren ST. Fragile X syndrome. Eur J Hum Genet. 2008;16:666–672. doi: 10.1038/ejhg.2008.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godfraind JM, Reyniers E, De Boulle K, D’Hooge R, De Deyn PP, Bakker CE, Oostra BA, Kooy RF, Willems PJ. Long-term potentiation in the hippocampus of fragile X knockout mice. Am J Med Genet. 1996;64:246–251. doi: 10.1002/(SICI)1096-8628(19960809)64:2<246::AID-AJMG2>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- Hannan AJ, Blakemore C, Katsnelson A, Vitalis T, Huber KM, Bear M, Roder J, Kim D, Shin HS, Kind PC. PLC-beta1, activated via mGluRs, mediates activity-dependent differentiation in cerebral cortex. Nat Neurosci. 2001;4:282–288. doi: 10.1038/85132. [DOI] [PubMed] [Google Scholar]
- Hensch TK. Critical period regulation. Annu Rev Neurosci. 2004;27:549–579. doi: 10.1146/annurev.neuro.27.070203.144327. [DOI] [PubMed] [Google Scholar]
- Hensch TK. Critical period plasticity in local cortical circuits. Nat Rev Neurosci. 2005;6:877–888. doi: 10.1038/nrn1787. [DOI] [PubMed] [Google Scholar]
- Isaac JT, Crair MC, Nicoll RA, Malenka RC. Silent synapses during development of thalamocortical inputs. Neuron. 1997;18:269–280. doi: 10.1016/s0896-6273(00)80267-6. [DOI] [PubMed] [Google Scholar]
- Itami C, Mizuno K, Kohno T, Nakamura S. Brain-derived neurotrophic factor requirement for activity-dependent maturation of glutamatergic synapse in developing mouse somatosensory cortex. Brain Res. 2000;857:141–150. doi: 10.1016/s0006-8993(99)02352-5. [DOI] [PubMed] [Google Scholar]
- Iwasato T, Datwani A, Wolf AM, Nishiyama H, Taguchi Y, Tonegawa S, Knopfel T, Erzurumlu RS, Itohara S. Cortex-restricted disruption of NMDAR1 impairs neuronal patterns in the barrel cortex. Nature. 2000;406:726–731. doi: 10.1038/35021059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwasato T, Inan M, Kanki H, Erzurumlu RS, Itohara S, Crair MC. Cortical adenylyl cyclase 1 is required for thalamocortical synapse maturation and aspects of layer IV barrel development. J Neurosci. 2008;28:5931–5943. doi: 10.1523/JNEUROSCI.0815-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kau AS, Meyer WA, Kaufmann WE. Early development in males with Fragile X syndrome: a review of the literature. Microsc Res Tech. 2002;57:174–178. doi: 10.1002/jemt.10069. [DOI] [PubMed] [Google Scholar]
- Kidd FL, Isaac JT. Developmental and activity-dependent regulation of kainate receptors at thalamocortical synapses. Nature. 1999;400:569–573. doi: 10.1038/23040. [DOI] [PubMed] [Google Scholar]
- Land PW, Simons DJ. Cytochrome oxidase staining in the rat SmI barrel cortex. J Comp Neurol. 1985;238:225–235. doi: 10.1002/cne.902380209. [DOI] [PubMed] [Google Scholar]
- Larson J, Jessen RE, Kim D, Fine AK, du Hoffmann J. Age-dependent and selective impairment of long-term potentiation in the anterior piriform cortex of mice lacking the fragile X mental retardation protein. J Neurosci. 2005;25:9460–9469. doi: 10.1523/JNEUROSCI.2638-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauterborn JC, Rex CS, Kramar E, Chen LY, Pandyarajan V, Lynch G, Gall CM. Brain-derived neurotrophic factor rescues synaptic plasticity in a mouse model of fragile X syndrome. J Neurosci. 2007;27:10685–10694. doi: 10.1523/JNEUROSCI.2624-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Pelletier MR, Perez Velazquez JL, Carlen PL. Reduced cortical synaptic plasticity and GluR1 expression associated with fragile X mental retardation protein deficiency. Mol Cell Neurosci. 2002;19:138–151. doi: 10.1006/mcne.2001.1085. [DOI] [PubMed] [Google Scholar]
- Liao D, Hessler NA, Malinow R. Activation of postsynaptically silent synapses during pairing-induced LTP in CA1 region of hippocampal slice. Nature. 1995;375:400–404. doi: 10.1038/375400a0. [DOI] [PubMed] [Google Scholar]
- Lu HC, Gonzalez E, Crair MC. Barrel cortex critical period plasticity is independent of changes in NMDA receptor subunit composition. Neuron. 2001;32:619–634. doi: 10.1016/s0896-6273(01)00501-3. [DOI] [PubMed] [Google Scholar]
- Lu HC, She WC, Plas DT, Neumann PE, Janz R, Crair MC. Adenylyl cyclase I regulates AMPA receptor trafficking during mouse cortical ‘barrel’ map development. Nat Neurosci. 2003;6:939–947. doi: 10.1038/nn1106. [DOI] [PubMed] [Google Scholar]
- Lu R, Wang H, Liang Z, Ku L, O’Donnell WT, Li W, Warren ST, Feng Y. The fragile X protein controls microtubule-associated protein 1B translation and microtubule stability in brain neuron development. Proc Natl Acad Sci U S A. 2004;101:15201–15206. doi: 10.1073/pnas.0404995101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lush ME, Ma L, Parada LF. TrkB signaling regulates the developmental maturation of the somatosensory cortex. Int J Dev Neurosci. 2005;23:523–536. doi: 10.1016/j.ijdevneu.2005.04.003. [DOI] [PubMed] [Google Scholar]
- Marie H, Morishita W, Yu X, Calakos N, Malenka RC. Generation of silent synapses by acute in vivo expression of CaMKIV and CREB. Neuron. 2005;45:741–752. doi: 10.1016/j.neuron.2005.01.039. [DOI] [PubMed] [Google Scholar]
- Meredith RM, Holmgren CD, Weidum M, Burnashev N, Mansvelder HD. Increased threshold for spike-timing-dependent plasticity is caused by unreliable calcium signaling in mice lacking fragile X gene FMR1. Neuron. 2007;54:627–638. doi: 10.1016/j.neuron.2007.04.028. [DOI] [PubMed] [Google Scholar]
- Nimchinsky EA, Oberlander AM, Svoboda K. Abnormal development of dendritic spines in FMR1 knock-out mice. J Neurosci. 2001;21:5139–5146. doi: 10.1523/JNEUROSCI.21-14-05139.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nosyreva ED, Huber KM. Metabotropic receptor-dependent long-term depression persists in the absence of protein synthesis in the mouse model of fragile X syndrome. J Neurophysiol. 2006;95:3291–3295. doi: 10.1152/jn.01316.2005. [DOI] [PubMed] [Google Scholar]
- Penagarikano O, Mulle JG, Warren ST. The pathophysiology of fragile x syndrome. Annu Rev Genomics Hum Genet. 2007;8:109–129. doi: 10.1146/annurev.genom.8.080706.092249. [DOI] [PubMed] [Google Scholar]
- Petersen CC. The functional organization of the barrel cortex. Neuron. 2007;56:339–355. doi: 10.1016/j.neuron.2007.09.017. [DOI] [PubMed] [Google Scholar]
- Restivo L, Ferrari F, Passino E, Sgobio C, Bock J, Oostra BA, Bagni C, Ammassari-Teule M. Enriched environment promotes behavioral and morphological recovery in a mouse model for the fragile X syndrome. Proc Natl Acad Sci U S A. 2005;102:11557–11562. doi: 10.1073/pnas.0504984102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlaggar BL, Fox K, O’Leary DD. Postsynaptic control of plasticity in developing somatosensory cortex. Nature. 1993;364:623–626. doi: 10.1038/364623a0. [DOI] [PubMed] [Google Scholar]
- Schutt J, Falley K, Richter D, Kreienkamp HJ, Kindler S. Fragile X mental retardation protein regulates the levels of scaffold proteins and glutamate receptors in postsynaptic densities. J Biol Chem. 2009;284:25479–25487. doi: 10.1074/jbc.M109.042663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- She WC, Quairiaux C, Albright MJ, Wang YC, Sanchez DE, Chang PS, Welker E, Lu HC. Roles of mGluR5 in synaptic function and plasticity of the mouse thalamocortical pathway. Eur J Neurosci. 2009;29:1379–1396. doi: 10.1111/j.1460-9568.2009.06696.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takasaki C, Okada R, Mitani A, Fukaya M, Yamasaki M, Fujihara Y, Shirakawa T, Tanaka K, Watanabe M. Glutamate transporters regulate lesion-induced plasticity in the developing somatosensory cortex. J Neurosci. 2008;28:4995–5006. doi: 10.1523/JNEUROSCI.0861-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todd PK, Mack KJ. Sensory stimulation increases cortical expression of the fragile X mental retardation protein in vivo. Brain Res Mol Brain Res. 2000;80:17–25. doi: 10.1016/s0169-328x(00)00098-x. [DOI] [PubMed] [Google Scholar]
- Todd PK, Malter JS, Mack KJ. Whisker stimulation-dependent translation of FMRP in the barrel cortex requires activation of type I metabotropic glutamate receptors. Brain Res Mol Brain Res. 2003;110:267–278. doi: 10.1016/s0169-328x(02)00657-5. [DOI] [PubMed] [Google Scholar]
- Van der Loos H, Woolsey TA. Somatosensory cortex: structural alterations following early injury to sense organs. Science. 1973;179:395–398. doi: 10.1126/science.179.4071.395. [DOI] [PubMed] [Google Scholar]
- Villasana LE, Klann E, Tejada-Simon MV. Rapid isolation of synaptoneurosomes and postsynaptic densities from adult mouse hippocampus. J Neurosci Methods. 2006;158:30–36. doi: 10.1016/j.jneumeth.2006.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson RF, Abdel-Majid RM, Barnett MW, Willis BS, Katsnelson A, Gillingwater TH, McKnight GS, Kind PC, Neumann PE. Involvement of protein kinase A in patterning of the mouse somatosensory cortex. J Neurosci. 2006;26:5393–5401. doi: 10.1523/JNEUROSCI.0750-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welker C, Woolsey TA. Structure of layer IV in the somatosensory neocortex of the rat: description and comparison with the mouse. J Comp Neurol. 1974;158:437–453. doi: 10.1002/cne.901580405. [DOI] [PubMed] [Google Scholar]
- Wijetunge LS, Till SM, Gillingwater TH, Ingham CA, Kind PC. mGluR5 regulates glutamate-dependent development of the mouse somatosensory cortex. J Neurosci. 2008;28:13028–13037. doi: 10.1523/JNEUROSCI.2600-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson BM, Cox CL. Absence of metabotropic glutamate receptor-mediated plasticity in the neocortex of fragile X mice. Proc Natl Acad Sci U S A. 2007;104:2454–2459. doi: 10.1073/pnas.0610875104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong-Riley M. Changes in the visual system of monocularly sutured or enucleated cats demonstrable with cytochrome oxidase histochemistry. Brain Res. 1979;171:11–28. doi: 10.1016/0006-8993(79)90728-5. [DOI] [PubMed] [Google Scholar]
- Woolsey TA, Van der Loos H. The structural organization of layer IV in the somatosensory region (SI) of mouse cerebral cortex. The description of a cortical field composed of discrete cytoarchitectonic units. Brain Res. 1970;17:205–242. doi: 10.1016/0006-8993(70)90079-x. [DOI] [PubMed] [Google Scholar]
- Yanagisawa T, Tsumoto T, Kimura F. Transiently higher release probability during critical period at thalamocortical synapses in the mouse barrel cortex: relevance to differential short-term plasticity of AMPA and NMDA EPSCs and possible involvement of silent synapses. Eur J Neurosci. 2004;20:3006–3018. doi: 10.1111/j.1460-9568.2004.03756.x. [DOI] [PubMed] [Google Scholar]
- Yun SW, Platholi J, Flaherty MS, Fu W, Kottmann AH, Toth M. Fmrp is required for the establishment of the startle response during the critical period of auditory development. Brain Res. 2006;1110:159–165. doi: 10.1016/j.brainres.2006.06.086. [DOI] [PubMed] [Google Scholar]
- Zalfa F, Eleuteri B, Dickson KS, Mercaldo V, De Rubeis S, di Penta A, Tabolacci E, Chiurazzi P, Neri G, Grant SG, Bagni C. A new function for the fragile X mental retardation protein in regulation of PSD-95 mRNA stability. Nat Neurosci. 2007;10:578–587. doi: 10.1038/nn1893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao MG, Toyoda H, Ko SW, Ding HK, Wu LJ, Zhuo M. Deficits in trace fear memory and long-term potentiation in a mouse model for fragile X syndrome. J Neurosci. 2005;25:7385–7392. doi: 10.1523/JNEUROSCI.1520-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.