

Abstract
Calpain-catalyzed proteolysis of αII-spectrin is a regulated event associated with neuronal long-term potentiation, platelet and leukocyte activation, and other processes. Calpain proteolysis is also linked to apoptotic and non-apoptotic cell death following excessive glutamate exposure, hypoxia, HIV-gp120/160 exposure, or toxic injury. The molecular basis for these divergent consequences of calpain action, and their relationship to spectrin proteolysis, is unclear. Calpain preferentially cleaves αII spectrin in vitro in repeat 11 between residues Y1176 and G1177. Unless stimulated by Ca++ and calmodulin (CaM), βII spectrin proteolysis in vitro is much slower. We identify additional unrecognized sites in spectrin targeted by calpain in vitro and in vivo. Bound CaM induces a second αII spectrin cleavage at G1230*S1231. βII spectrin is cleaved at four sites. One cleavage only occurs in the absence of CaM at high enzyme-to-substrate ratios near the βII spectrin COOH-terminus. CaM promotes βII spectrin cleavages at Q1440*S1441, S1447*Q1448, and L1482*A1483. These sites are also cleaved in the absence of CaM in recombinant βII spectrin fusion peptides, indicating that they are probably shielded in the spectrin heterotetramer and become exposed only after CaM binds αII spectrin. Using epitope-specific antibodies prepared to the calpain cleavage sites in both αII and βII spectrin, we find in cultured rat cortical neurons that brief glutamate exposure (a physiologic ligand) rapidly stimulates αII spectrin cleavage only at Y1176*G1177, while βII spectrin remains intact. In cultured SH-SY5Y cells that lack an NMDA receptor, glutamate is without effect. Conversely, when stimulated by calcium influx (via maitotoxin), there is rapid and sequential cleavage of αII and then βII spectrin, coinciding with the onset of non-apoptotic cell death. These results identify: i) novel calpain target sites in both αII and βII spectrin; ii) trans-regulation of proteolytic susceptibility between the spectrin subunits in vivo; and iii) the preferential cleavage of αII spectrin vs. βII spectrin when responsive cells are stimulated by engagement of the NMDA receptor. We postulate that calpain proteolysis of spectrin can activate two physiologically distinct responses: one that enhances skeletal plasticity without destroying the spectrin-actin skeleton, characterized by preservation of βII spectrin; or an alternative response closely correlated with non-apoptotic cell death and characterized by proteolysis of βII spectrin and complete dissolution of the spectrin skeleton.
Keywords: cytoskeleton, proteolysis, necrosis, cell death, glutamate, fodrin
Spectrin is the major component of the cytoskeletal network associated with the plasma membrane of vertebrate cells (for reviews 1, 2, 3). While seven spectrin genes exist, encoding two variants of an alpha spectrin and five beta spectrins, the most common form of this protein is a heterotetramer of αII,βII spectrin. Together with actin and a host of adapter proteins, spectrin controls the distribution of many integral and peripheral membrane proteins, and possibly also the distribution of certain phospholipids. Mutations in spectrin or its associated adapter proteins typically destabilize the membrane, and their absence is often embryonic lethal (4, 5). Calcium-dependent proteolytic modification of αII-spectrin by any of several calpain proteases is linked to platelet activation (6), neutrophil degranulation (7), the onset of long-term potentiation in hippocampal neurons (8–11), dendritic and post synaptic density remodeling (12–14), crystalline lens maturation (15), and receptor-mediated endocytosis (16). Calpain activation occurs in response to fibrinogen binding to the integrin glycoprotein IIb-IIIa in platelets (6, 17) and to NMDA-stimulation of responsive neurons (9, 18–21). Beyond these putative physiologic processes, calpain processing of αII-spectrin also follows hypoxic or ischemic injury (22–34). In the brain, calpain activation is associated with excitatory amino acid toxicity, as occurs experimentally after treatment with kainate or NMDA (18, 19, 35–37), and may be accentuated by the action of HIV-1 gp120/160 (38–40). Calpain-cleavage of spectrin has been observed during apoptosis in monocytic U937 cells (41) and in neurons (42, 43). The molecular basis for these divergent consequences of calpain action, and their relationship to spectrin proteolysis, is unclear.
Previously this laboratory determined that a central region within repeat unit 11 of αII spectrin is hypersensitive to a variety of proteases, including μ-calpain (44), and demonstrated that the preferred site of calpain cleavage is between Y1176*G1177, immediately adjacent to the calmodulin binding domain in this protein (45, 46). Epitope-specific antibodies recognizing the in vitro generated cleavage fragments established that cleavage also occurs at this site in vivo (47), and mutational analysis revealed that the specificity of calpain for this site is determined by complex conformational rather than linear sequence determinants (48). The sensitivity of this site to cleavage is regulated at the substrate level by the phosphorylation of Y1176, a process mediated by src-family kinases and low-molecular-weight phosphotyrosine phosphatase (LMW-PTP) (49, 50).
Calpain also cleaves βII spectrin, but at least in vitro does so efficiently only when CaM is bound to the αII subunit in the αII/βII heterodimer (46), an event that induces a substantial conformational change in sequences associated with the CaM-binding site (51). Other cleavages of βII spectrin by calpain also exist (52, 53), yet beyond the identification in fusion peptides of a susceptible site near the COOH terminus (A2067) of βII spectrin (53), the precise locus of any calpain-cleavage site in βII spectrin or its CaM-dependency is unknown. In vitro studies have established a complex relationship between calpain cleavage of spectrin, Ca++, CaM binding, and the ability of spectrin to oligomerize, bind actin, and bind to membranes (52, 54). These studies suggest that calpain cleavage of αII spectrin converts this molecule into a reversible Ca++ and CaM-regulated actin cross-linking protein, whereas coincident calpain-cleavage of the βII subunit irreversibly disassembles the spectrin-actin cortical membrane skeletal lattice. Thus, two calcium-dependent processes, calmodulin binding and calpain proteolysis, unless suppressed by tyrosine phosphorylation of αII spectrin (49, 50), act synergistically to regulate the proteolysis of spectrin and the organization and integrity of the cortical membrane skeletal lattice.
In the present study, we identify a second calmodulin-regulated site targeted by calpain in αII spectrin, and three novel sites of cleavage in βII spectrin. Using subunit and cleavage specific antibodies to αII and βII spectrin, we find that glutamate stimulation of cortical neurons preferentially induces αII spectrin cleavage as a very early event, while, in non-glutamate responsive SH-SY5Y cells, spectrin is unaffected by glutamate exposure. When calcium is introduced directly into cells by maitotoxin treatment, delayed cell death ensues on a time-scale that correlates with complete calpain-mediated breakdown of both αII and βII spectrin, with αII cleavage again preceding that of βII spectrin. These findings together with the earlier data cited above indicate that αII spectrin cleavage per se is not cell lethal (21), and suggest that as when spectrin is cleaved by caspase 3 during apoptosis (55), the advent of βII subunit cleavage marks an irreversible transition in the cortical cytoskeleton that may contribute to impaired cell viability.
MATERIALS AND METHODS
Protein preparation and expression of recombinant peptides
Fresh calf brain was obtained from a local abattoir and was washed in cold 0.32 M sucrose prior to homogenization. Bovine αII/βII spectrin was purified from demyelinated brain membranes by low ionic strength extraction followed by gel filtration on Sephacryl S-500 HR (56, 57). Recombinant peptides representing various regions of αII and βII spectrin were prepared as glutathione-S-transferase (GST) fusion peptides in E. coli strains CAG-456 and W3110, and purified using glutathione-agarose (48, 58). Purified spectrin and fusion proteins were stored in 40 mM Tris pH 7.5, 50 mM NaCl, 0.5 mM DTT, 0.1mM Pefabloc, 0.1mM Benzamidine. Purified porcine erythrocyte μ-calpain was acquired from Nacalai Tesque, Inc. in Kyoto, Japan. CaM was from Sigma.
Antibodies
PAb RAF-A is a well-documented polyclonal rabbit antibody that recognizes αII-spectrin (46, 59). Alternatively, for some experiments, the monoclonal αII spectrin antibody from Chemicon (#1622) was also used. PAb-10D, is a rabbit polyclonal, was prepared to a recombinant peptide representing residues 1676 to 2204 of human βII spectrin, corresponding to sequence repeat unit 13 to the middle of the COOH-terminal domain III (58). For the PAb’s α-bdp1 and α-bdp2, the synthetic peptides CQQEVY and GMMPRC respectively were each coupled to keyhole limpet hemocyanin (KLH) via the cysteine residue using m-maleimidobenzoyl-N-hydroxysuccinimide ester, and injected into New Zealand White rabbits in complete Freund’s adjuvant following previous protocols (59). For the PAb βII-BDP1, the synthetic peptide CGIEELQ was similarly coupled to maleimide activated KLH (Pierce); IgY was raised in white leghorn chickens and purified from egg yolk by Aves Labs, Inc., Tigand, OR. All cleavage-specific rabbit antibodies were affinity purified against their respective ligands using antigen-coupled Sepharose in which the peptide was linked via its terminal cysteine. Antibodies raised in chickens were affinity purified against peptide coupled by its cysteine to sulfolink coupling gel (Pierce, Inc.) (60).
Calpain Digestion
Unless otherwise specified, 10 μg of purified spectrin was digested by 0.03 μg of μ-calpain in digestion buffer containing 20 mM Tris pH 7.5, 25 mM NaCl, 0.15 mM total CaCl2, and 10 mM DTT, with or without 5 μM CaM (25 μl total volume) at 25ºC. Proteolysis was terminated by the addition of 5x SDS-PAGE solubilizing buffer at 95ºC for 5 minutes (61), or at 37ºC for 15 minutes for samples used for microsequencing. Digestion patterns were analyzed by SDS-PAGE, followed either by Coomassie blue staining or by transfer to Immobilon-P membranes (Millipore) for immunoblot detection or NH2-terminal amino acid sequencing. The apparent sizes of the digestion products were calculated based on migration of the standard BioRad® High Molecular Weight Markers. Purified fusion peptides were similarly treated. In the case of the GST-βII8-CΔ peptide, 70 μg protein was digested with 0.3 μg calpain; digestion was halted by a 10-fold molar excess of EDTA (relative to total calcium). After digestion GST containing fragments were adsorbed with 5 mg of glutathione-agarose in the presence of 0.1 mg/ml BSA by overnight incubation at 4ºC so as to facilitate the isolation of the GST-free COOH-terminal digestion products for sequencing.
Calpain activation in cultured cells
Cerebral cortices were dissected from E16 to E17 day old Sprague-Dawley rat fetuses and hippocampi were dissected from E20–21 day old fetuses, using sterile technique and triturated to suspend the cells. Cells were plated at a density of 6–7 x 105 cells per 35 mm diameter dish. All culture dishes were pre-coated with poly-D-lysine (20 μg/ml; Mr 30,000–70,000). Cultures were grown at 37° C, 5% CO2 in DMEM/F-12 medium supplemented with 30 mM glucose, B-27 serum-free supplement, 40 U penicillin G sodium per ml and 40 μg streptomycin sulfate per ml for 4 to 5 days, after which the medium was changed to Neurobasal Medium supplemented with B27, 0.5 mM glutamine, 10 U penicillin G sodium per ml and 10 μg streptomycin sulfate per ml. Cultures were maintained by replacing half of the medium with fresh medium twice weekly. Tissue culture media and supplements were obtained from Gibco BRL. Experiments were performed on cortical and hippocampal cultures after two to three weeks in vitro. These cultures consisted of greater than 90% neurons. Cells were rinsed once with phosphate buffered saline, and once with Mg++-free Krebs’ Ringer medium (20 mM HEPES, 1 mM Na2HPO4, 128 mM NaCl, 5 mM KCl, 2.7 mM CaCl2, 10 mM glucose, pH 7.2). Then they were incubated in Mg++-free Ringer medium containing 5μM glycine, with or without glutamate (50 –400 μM) or NMDA (50 μM), at 37° C, 5% CO2. In the case of the 24 hour time course, after incubating for 30 minutes with 50 μM NMDA in Mg++-free Krebs’ Ringer medium containing 5 μM glycine, cells were rinsed to remove the NMDA and the Krebs’ Ringer medium was replaced with conditioned supplemented Neurobasal Medium for the remainder of the experiment. 20 μM MK801 (from 10 mM stock in water) and 100 μg/ml Calpeptin (from 50 mg/ml DMSO stock) were applied to cultures prior to the addition of glutamate (from 100 mM stock) or NMDA (from 100 mM stock in 0.01 N NaOH). Controls received equivalent volumes of diluents. At the end of the experimental time period, the buffer was removed and cells were extracted with 150 μl of 1x Laemmli SDS-PAGE sample buffer containing 150 mM NaCl, 2 mM EDTA, 2 mM EGTA, 0.4 mM DTT, 1 mM Pefabloc, 20 μg/ml leupeptin, 1 mM benzamidine, 10 μg/ml aprotinin. Samples were heated to 100° C for 10 minutes..
For maitotoxin experiments, SH-SY5Y cells were grown in MEM/F12 (1:1) with non-essential amino acids which was supplemented with 10% fetal bovine serum, 2mM glutamine, 50 U/ml penicillin G sodium and 50 μg/ml streptomycin sulfate at 37° C, 5% CO2. Experiments were performed in 35mm dishes containing cultures that were 2/3 to 3/4 confluent. The medium was replaced by 2 ml serum-free medium containing 2mM glutamine to which the treatments were added. Calpain inhibitor (100 μg/ml Calpeptin from 50 mg/ml DMSO stock) and caspase inhibitor (100 μM Z-D-DCB from 50 mM stock in DMSO) were added to the medium 15 minutes prior to treatment with 0.03 nM or 0.04 nM Maitotoxin (from 2 μM stock in Ethanol) followed by various times of incubation at 37° C, 5% CO2. Vehicle controls received equivalent volumes of Ethanol and/or DMSO. At the end of the treatment period, the medium containing floating cells was centrifuged @1000 rpm for 2 minutes and rinsed once with PBS. The remaining adherent cells on the dish were rinsed once with PBS and pooled with the medium containing the floating cells prior to centrifugation. The cells remaining on the dish were extracted with 150 μl 1x Laemmli SDS-PAGE sample buffer containing inhibitors, as was used for the primary cultures, and this extract was combined with the floating cell pellet and heated to 100° C for 10 minutes. In parallel experiments, similarly treated SH-SY5Y cells were analyzed for evidence of cell death by flow cytometry. Cells collected as above were resuspended in 300 μl PBS containing 2% goat serum PBS/GS followed by the addition of 300 μl PBS/GS containing 8 μM Ethidium-1 (final concentration of 4 μM). After filtration through a 35 μM nylon mesh filter (BD Falcon) cells were counted by flow cytometry 10 minutes after the addition of Ethidium-1, 30 minutes after termination of the maitoxin treatment.
Other
Protease inhibitors used were Pefabloc SC (Centerchem Inc.), Benzamidine (Sigma), Leupeptin (Sigma), Pepstatin A (Boehringer Mannheim), and Aprotinin (Sigma). NMDA, glutamate, and MK801 were purchased from RBI. Calmodulin (bovine brain) and maitotoxin were from Sigma, calpeptin from Calbiochem, and Ethidium-1 from Molecular Probes. Transfers to Immobilon-P PVDF membrane (Millipore, Inc.) for Western blotting were performed in 25 mM Tris, 192 mM glycine, 10% methanol. Immunoblots were blocked with milk buffer (5% Carnation® Nonfat Dry Milk, 0.1% bovine serum albumin Fraction V (Sigma), 0.02% sodium azide in 1x phosphate buffered saline, pH 7.4) for one hour, incubated in primary antibody diluted in milk buffer for one hour, rinsed twice then washed 1x 15 minutes and 2x 5 minutes in phosphate buffered saline pH 7.4 containing 0.1% Tween 20 (PBS-Tween). After incubation in secondary antibody (goat anti-rabbit horse-radish peroxidase from Pierce, or rabbit anti-chicken horse-radish peroxidase from Accurate Chemical) diluted in PBS-Tween or milk buffer, respectively, for one hour followed by four 5-minute washes, enzymatic activity was detected by chemiluminescence (ECL, Amersham) using Amersham Hyperfilm®, or alternatively was detected colorimetrically with alkaline phosphatase. Estimation of protein was by OD280 and by BCA assay (Pierce). Microsequencing of digested peptides was performed using an Applied Biosystems Protein sequencer, or alternatively by Yale’s Keck Biotechnology Resource Laboratory using similar equipment. Densitometry of gels and fluorgrams were performed on digital images on a Macintosh computer using the public domain NIH Image or J-image programs (developed at the U.S. National Institutes of Health and available on the Internet at ://rsb.info.nih.gov/nih-image/).
RESULTS
Calmodulin enhances the susceptibility of both αII andβII spectrin to μ-calpain
The CaM binding domain in αII spectrin flanks the site of calpain cleavage and undergoes a substantial conformational reordering upon CaM binding (51). Both subunits of spectrin are more susceptible to cleavage when CaM is bound (45, 46). Figures 1 and 2 reveal several additional features of this phenomenon; a summary of all spectrin breakdown products (BDP’s) is presented in Table I.
Figure 1. Calmodulin enhances μ-calpain cleavage of both subunits of αII/βII spectrin.
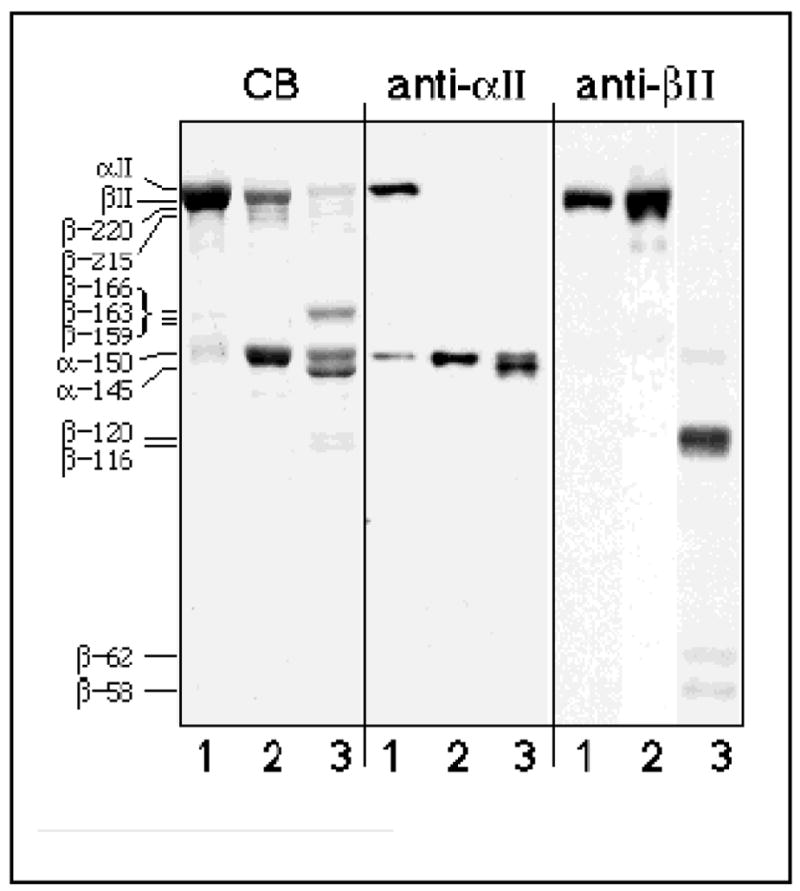
CB) Coomassie Blue-stained 7% SDS-PAGE analysis of purified αII/βII spectrin from bovine brain (lane 1) incubated with μ-calpain (molar E/S =1:18) in the absence (lane 2) or presence (lane 3) of 5 μM calmodulin and 150 μM Ca++ for 15 minutes at 25° C. Note the accentuation of digestion when calmodulin is present. Prominent digestion products are apparent at 166, 150, 145, and 120/116 (fainter bands) kD. Anti-αII) Western blot analysis of the same samples with PAb RAF-A, directed against αII spectrin. Note the additional αII spectrin cleavage product at ≈ 145 kD induced by CaM. Anti-βII) Western blot of same samples examined with PAb 10D, which is directed against βII spectrin, repeat unit 13 (residue 1676) to L2204 (near the COOH-terminus). This antibody does not recognize the prominent βII spectrin BDP’s at ≈ 159–166 kD, but detects the BDP’s at ≈ 116–120 kD and at 62/58 kD derived from the COOH-terminal portions of βII spectrin. Note that there is minimal cleavage of βII spectrin in the absence of CaM. The apparent molecular weights and the subunit of origin of each observed digestion product are indicated.
Figure 2. At high enzyme to substrate ratios, there is limited cleavage of βII spectrin in the absence of CaM.
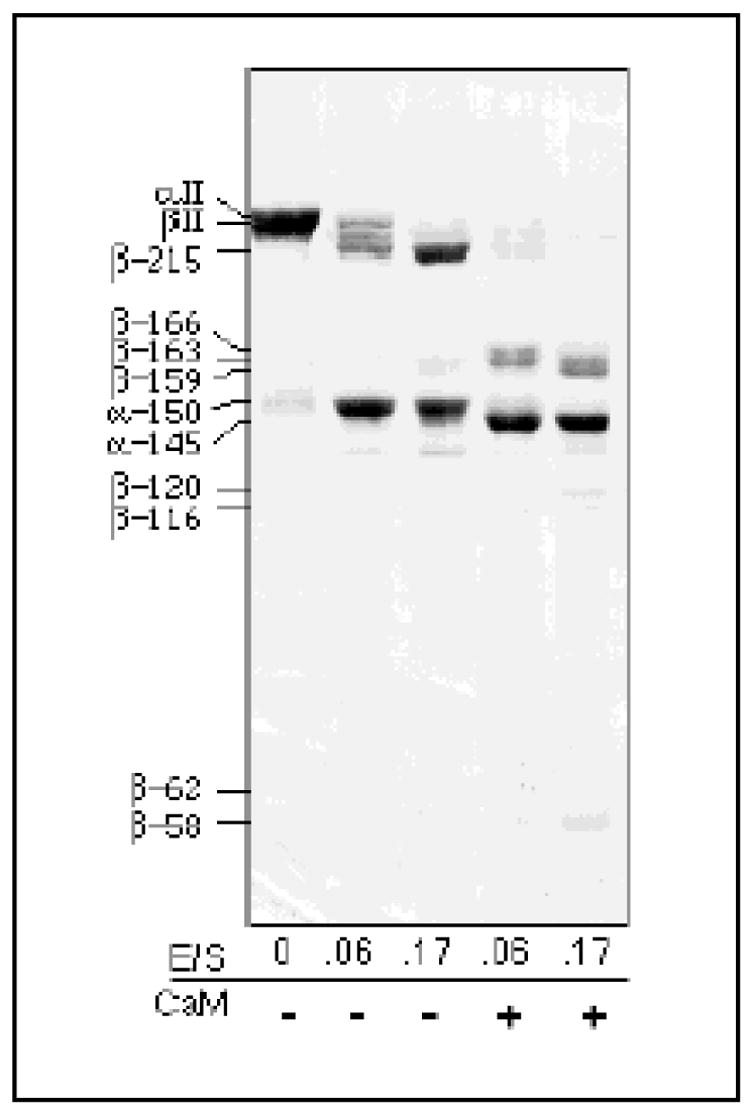
Calpain digestion of αII/βII spectrin (11 μg/lane) was carried out for 45 minutes at 25° C at the molar E/S ratios indicated, with or without CaM and 0.15 mM Ca++. At high E/S ratios, a ≈ 215 kD βII spectrin BDP was generated in the absence of CaM. In the presence of CaM, an additional βII BDP of ≈ 159 kD was generated. These results were independent of Ca++ concentration, within the range of 0.15 mM to 1.5 mM total calcium (data not shown).
Table I.
Major μ-calpain generated spectrin fragments.
| Fragment | NH2-terminal Sequence | Cleavage site | Assumed End | Calc’d MW |
|---|---|---|---|---|
| Native protein | ||||
| αII-BDP150’* | (MDPSGVKVLE) | (start of αII spectrin -assumed) | Y1176 | 135,800 |
| αII-BDP150 | -GMMPRDEXDS | VY1176* G1177MMPRDETD | N2477 | 149,311 |
| αII-BDP145 | -SAHEVQRFHR | QLLG1230* S1231AHEVQRFHR | N2477 | 143,097 |
| αII-BDP145’ | (MDPSGVKVLE) | (start of αII spectrin -assumed) | ND | ≈143,000 |
| βII-BDP220 | (MTTTVATD) | (start of βII spectrin -assumed) | ND | |
| βII-BDP215 | (MTTTVATD) | (start of βII spectrin -assumed) | ND | |
| βII-BDP166 | (MTTTVATD) | (start of βII spectrin -assumed) | Q1440 | 168,286 |
| βII-BDP163 | (MTTTVATD) | (start of βII spectrin -assumed) | ND | |
| βII-BDP159 | (MTTTVATD) | (start of βII spectrin -assumed) | ND | |
| βII-BDP120 | -XQAQALSQEG | EELQ1440* S1441QAQALSQ | K2364 | 106,389 |
| -XEGKSTDEV | QALS1447* Q1448EGKSTDEV | 105,703 | ||
| βII-BDP116 | -MELLXPXN | multiple sequences between | K2364 | 103,424 |
| QTKF1467* M1468ELLEP to | 101,593 | |||
| HNLL1482* A1483SKEIHQFN | ||||
| βII-BDP62 | ND | ND (between 1676 to 2204) | ND | |
| βII-BDP58 | -AXKEIXQFNR | HNLL1482* A1483SKEIHQFN | ND | |
| GST-constructs | ||||
| GST-αII9–12 | (start of αII sequence @E837) | S1267 | 76,331 | |
| 74 kD | -MSPILGYWKI | NH2-terminus of GST | Y1176 | 65,779 |
| 21 kD | -GMMPRDEX | VY1176* G1177MMPRDET | S1267 | 10,570 |
| GST-βII8–CΔ | (start of βII sequence @A1227) | P2189 | 139,676 | |
| 92 kD | -SQAQALSQE | EELQ1440* S1441QAQALSQ | P2189 | 86,591 |
| 92 kD | -QEGKSTDEV | QALS1447* Q1448EGKSTDEV | P2189 | 85,905 |
| 88 kD | -MELXEXL | QTKF1467* M1468ELLEP | P2189 | 83,626 |
| 88 KD | -AXKEIXQFN | HNLL1482* A1483SKEIHQFN | P2189 | 81,795 |
| 70 kD | (MSPILGYWKI)† | NH2-terminus of GST | ND | |
| 47 kD | (MSPILGYWKI)† | NH2-terminus of GST | Q1440 | 53,102 |
| GST-βII16–C | (start of βII sequence@V1908) | K2364 | 80,216 | |
| 45 kD | (MSPILGYWKI)† | NH2-terminus of GST | ND | ND |
| 44 kD | (MSPILGYWKI)† | NH2-terminus of GST | ND | ND |
| GST-βIIN–4 | (no BDP detected) | (start of βII sequence @M1) | S688 | 108,735 |
All digestion’s were carried out in vitro. Sequence numbering based on αIIΣ1 spectrin (GenBank #U83867, ref.80) and βII spectrin (GenBank #M96803, ref. 81). Sequence of GST26 from Schistosomajaponica (82).
Two cleavage products are present in the αII-BDP150 band, representing αII residues 1 to 1176 (calculated MW 135,800) and 1177 to 2477 (45). Only one of these is detected by microsequencing since the NH2-terminus of spectrin is methylated. In separate experiments, we have observed that milli-calpain also cleaves at this site (data not shown).
Not sequenced; end is assumed based on retention of these peptides on glutathione affinity column (see text). ND - not determined.
X- unidentified residue
In the absence of calmodulin, the major cleavage product of spectrin appears at ≈ 150 kD (αII-BDP150). Based on its antibody reactivity, this fragment arises exclusively from αII spectrin, and in the absence of CaM is the only site of αII spectrin cleavage. Previous peptide mapping studies have established that the 150 KD band contains both halves of the αII subunit (45), these are not resolved by SDS-PAGE despite their differences in true MW. NH2-terminal microsequencing of αII-BDP150 confirmed the cleavage between Y1176*G1177 (Table I and 45). Other experiments demonstrated that milli-calpain also cleaves αII spectrin at this site (data not shown). In the absence of CaM, two βII spectrin BDP’s were sub-stoichiometrically generated, yielding βII-BDP220 and βII-BDP215. These appear to be unfavorable cleavages, presumably with a high Kmax, since they are quantitatively significant only at high enzyme/substrate ratios as shown in Figure 2. Thus, to achieve quantitative cleavage of βII spectrin to βII-BDP215 under the conditions used here requires molar ratios of calpain-to-spectrin of 1:6, whereas efficient cleavage of αII spectrin is achieved with at least an order of magnitude less enzyme (44). Microsequencing detected no reliable NH2-terminal sequence information from βII-BDP215.
In earlier studies, we established that CaM bound to the αII-subunit enhances both the rate of αII-BDP150 generation and the rate of βII-subunit cleavage (46). As reported here, CaM also renders several new sites in αII and βII spectrin susceptible to cleavage (Figure 1). In αII spectrin, CaM-binding generates a new susceptibility to proteolysis at G1230*S1231, liberating the CaM binding domain and converting αII-BDP150 to αII-BDP145 (Table I). It also appears that αII-BDP150’, the NH2-terminal half of αII spectrin, undergoes a second cleavage to generate αII-BDP145’, since the entire band at 150 kD disappears coincident with the generation of the band at 145 kD (Figure 2). Since only a single NH2-terminal sequence was detected by microsequencing of the 145 kD band, it is likely that αII-BDP145’ is generated from αII-BDP150’ by additional cleavage near its COOH-terminus.
In βII spectrin, prominent calpain cleavage products appear in the presence of CaM near 166 kD (βII-BDP166, βII-BDP163, and βII-BDP159), and 120 kD (βII-BDP120 and βII-BDP116) (Figures 1,2). While numerous spectrin BDP’s have been observed in earlier studies with CaM, they have not been characterized (44, 46). NH2-terminal sequences were not detected for the βII-BDP166–159 products; these fragments were also not reactive with PAb 10D that is directed against sequence downstream of repeat unit 13 (see methods). Together, these data establish the origin of βII-BDP166–159 from the NH2-terminal portions of βII spectrin. Increasing E/S ratios favored the generation of βII-BDP159 at the expense of βII-BDP166 and βII-BDP163, indicating that once cleavage had occurred, additional sites near the COOH-terminus of βII-BDP166 must become susceptible to calpain (also see results below with the βII-bdp1 antibody). While βII-BDP120 and βII-BDP116 stained poorly by Coomassie blue, they were readily detected by immunoblotting with PAb-10D (Figure 1). These two fragments differed in their NH2-termini. Sequencing of βII-BDP120 yielded cleavage sites of Q1440*S1441 and S1447*Q1448. Sequencing of βII-BDP116 did not yield a discrete sequence, and consisted of an apparent mixture of closely related peptides with NH2-termini derived from a series of cleavages between F1467*M1468 and L1482*A1483.The origins for this variability in the terminus of βII-BDP116 were not determined. Finally, two novel BDP’s were detected at ≈ 62 and ≈ 58 kD (βII-BDP62 and βII-BDP58). These were most apparent after Western blotting with PAb-10D, revealing their origin from the COOH-terminal half of βII spectrin. These were consistently minor products even at high E/S ratios (Figure 2). The NH2-terminus of βII-BDP58 was derived from cleavage between L1482*A1483, similar to one of the cleavages responsible for βII-BDP116. Thus,βII-BDP58 must be derived from βII-BDP116 by an additional COOH-terminal directed cleavage (Table I). The cleavage responsible for the genesis of βII-BDP62 was not determined.
Recombinant spectrin peptides are cleaved by calpain at the native sites
Previous studies established that αII spectrin GST-fusion peptides containing repeat unit 11 and the CaM binding domain are readily cleaved by μ-calpain (48). In the current study, microsequencing of the calpain cleavage products derived from digesting GST-αII9–12 (Figure 3), a fusion peptide encompassing repeat units 9 to 12 (see Fig. 8), established that this peptide is cleaved at a site identical to that of the native αII spectrin heterotetramer (Table I). Calpain did not appreciably digest peptides GST-αII13–18 and GST-αII18–C (data not shown). Calpain digestion of GST-βII8–CΔ, a fusion peptide encompassing βII spectrin repeat units 8 to 17 and a portion of domain III (58), yielded four cleavage fragments at ≈ 92, ≈ 88, ≈ 70, and ≈ 47 kD (Figure 3). Because the ≈ 70 and ≈ 47 kD products are retained on a glutathione affinity column, they must contain GST and thus arise from the NH2-terminal portion of the peptide.
Figure 3. Calpain cleavage of recombinant spectrin peptides.
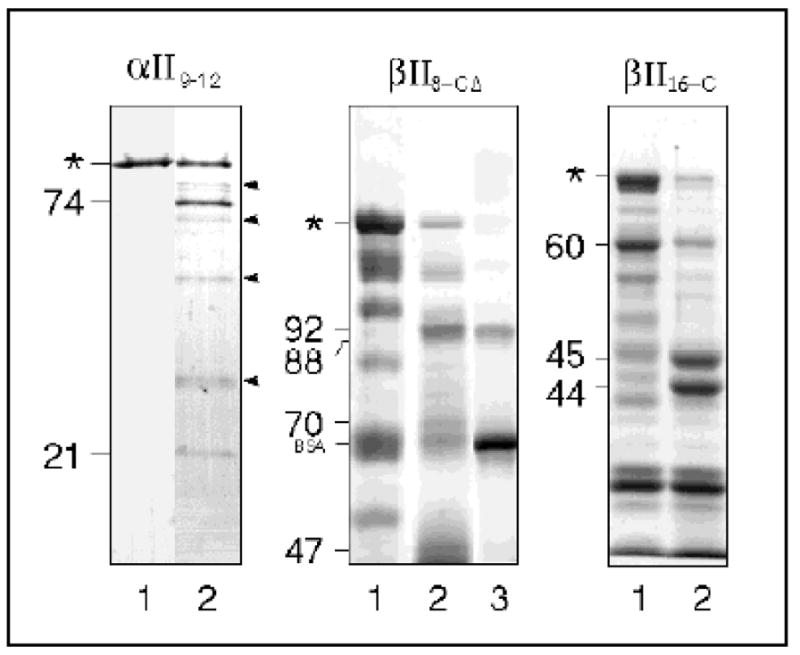
Purified recombinant GST-fusion peptides encompassing the designated regions of αII or βII spectrin were digested with μ-calpain for 15 minutes at 25° C. Lane 1,starting peptide; lane 2, peptide digested with calpain. For βII8–CΔ, the glutathione-S-transferase-containing digestion fragments were removed by incubation with glutathione-agarose beads in the presence of bovine serum albumin, and the remaining material (the GST-free 92 and 88 kD fragments) were analyzed (lane 3). The major detected digestion products for each peptide are indicated (with apparent size in kD), as is the position of bovine serum albumin (BSA). Asterisks identify full-length fusion peptides. Arrowheads point to autolytic fragments of calpain. Coomassie Blue-stained SDS polyacrylamide gels are shown. Duplicate analyses were transferred to Immobilon-P and the NH2-terminal sequence of the major BDP’s determined (Table I).
Figure 8. Summary of the calpain cleavage sites in αII/βII spectrin.
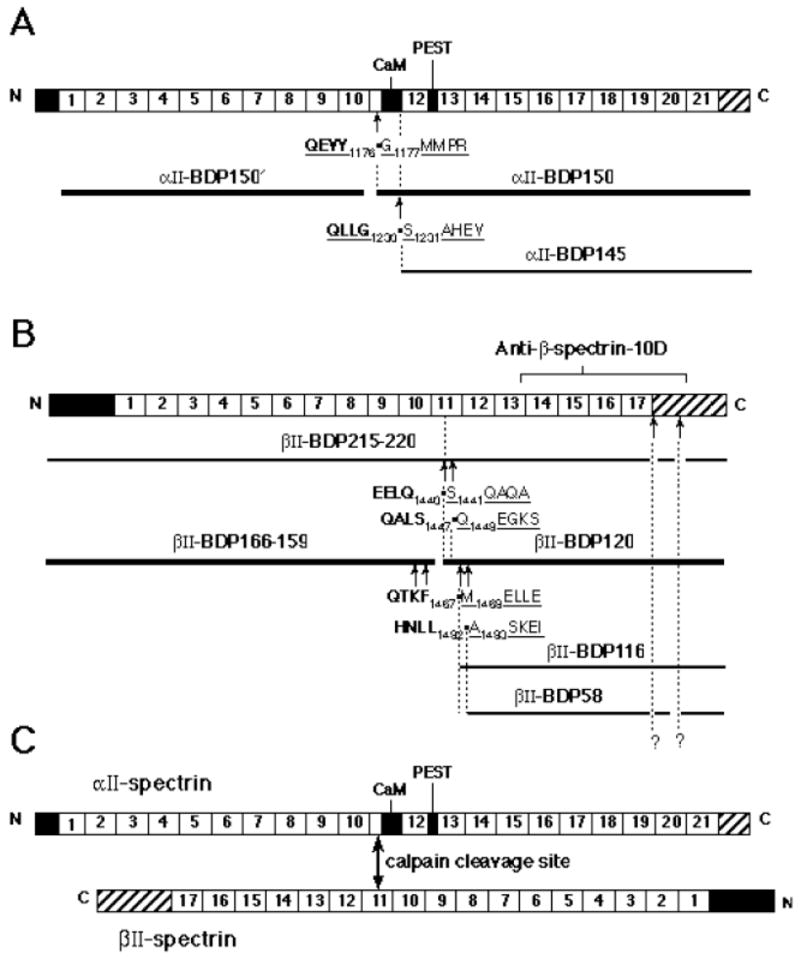
Schematic representation of the structure of αII-spectrin and βII-spectrin illustrates the sites of cleavage and size of the proteolytic fragments. Each subunit consists of three structural domains; domain II contains 21 (α) or 17 (β) structural repeats. Non-homologous sequence domains are present at either end of each subunit. (A) Cartoon of αII spectrin subunit with calpain cleavage sites indicated. The calmodulin binding domain and a region of PEST sequence is indicated; each homologous repeat unit is numbered. (B) Cartoon of βII-spectrin showing the sites of calpain digestion. (C) Schematic illustration of the relationship of the major digestion sites in the αβ-spectrin heterodimer. The structural depiction of the spectrin dimer and the alignment of the two subunits with respect to each other is adapted from Speicher et al. (68). Note the alignment of the major αII and βII calpain cleavage sites in the two subunits.
The prominent non-GST containing band at ≈ 92 kD displayed two NH2-terminal sequences that were out of register by seven residues. These results were identical to the NH2-terminal sequences detected for βII-BDP120 (Table I). The susceptibility of the βII8–CΔ peptide to calpain could not be accounted for by an artifact of its folding, since it had the same α-helical content as intact spectrin ( ≈ 70%) by CD measurement (data not shown), and a similar peptide (βII 8–13) also gave the same digestion pattern (data not shown). The minor non-GST containing 88 kD fragment yielded NH2-terminal sequences identical to those of βII-BDP58 and βII-BDP116. Finally, the calpain cleavage patterns of GST-βIIN–4 and GST-βII16–C were examined to better understand the susceptibility of the ends of βII spectrin to calpain cleavage. No digestion products were detected in the GST-βIIN–4 peptide (data not shown). Calpain generated two prominent digestion products at ≈ 44 and 45 kD from GST-βII16–C. Both of these products were recognized by anti-GST antibody (data not shown), indicating their origin from the NH2- terminal portion of the peptide. While the liberated COOH-terminal fragments could not be identified (Figure 3 and Table I), the fact that this peptide, which begins at V1908, is cleaved by calpain suggests the presence of additional calpain sensitive sites near the COOH-terminus of βII spectrin. Presumably, these are the sites that generate βII-BDP62 and βII-BDP58, or βII-BDP220 and βII-BDP215 from intact βII spectrin (Table I & Figure 8).
Epitope specific antibodies recognize the calpain cleavage products of αII and βII spectrin
With precise information on the sites of calpain cleavage, it is possible to prepare antibodies to the novel epitopes created by calpain cleavage. We have previously demonstrated the utility of such antibodies, prepared on the basis of our earlier determination of the sequences about the calpain cleavage site that generates αII-BDP150 and αII-BDP150’ (45, 47); such antibodies enable the detection of minimal levels of αII-BDP150/150’ in tissues and cells (29, 62). The specificity of two antibodies that detect either the new COOH-terminus or the new NH2-terminus generated by calpain cleavage of αII spectrin is shown in Figure 4. Native αII/βII spectrin was digested with either μ-calpain, or with trypsin, and then analyzed by Western blotting with either α-bdp1 antibody, prepared to the novel COOH-terminal sequence -QQEVY, or α-bdp2 antibody prepared to GMMPR-. Note the absence of reactivity of either antibody with intact spectrin, and the discrimination possible between a 150 kD αII fragment generated by calpain vs. a similar sized fragment generated by trypsin. Antibody β-bdp1 was also prepared to the novel COOH-terminus o the βII-BDP166 peptide. As with α-bdp1 and α-bdp2, β-bdp1 did not react with intact βII spectrin. Interestingly, this antibody reacted only withβII-BDP166, and not with βII-BDP163 or βII-BDP159, confirming the microsequencing studies indicating that the 163 and 159 kD products derived from βII-BDP166 by additional COOH-terminal cleavages.
Figure 4. Antibodies raised to the novel termini generated in spectrin by μ-calpain digestion recognize only the calpain processed αII and βII subunits.
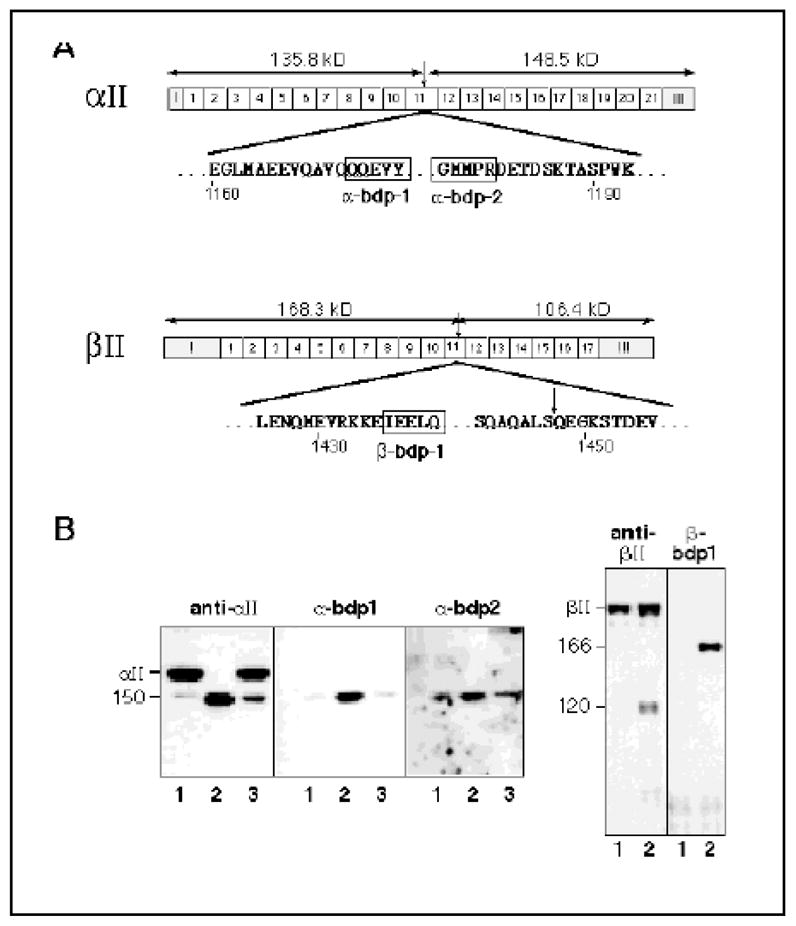
(A) Synthetic peptides representing the novel terminal five residues generated by μ-calpain cleavage of either αII or βII spectrin (boxes) were conjugated to KLH, and used to generate rabbit or chicken PAb antibodies to the novel epitopes created by the cleavages (29, 47). The epitopes used to generate the antibodies α-BDP1, α-BDP2, and β-BDP1, and their loci with respect to the overall structure of spectrin, are depicted. (B) Western blots using either PAb RAF-A (anti-αII), PAb-10D (anti-βII), or the cleavage specific antibodies listed above. Lane 1, intact spectrin (with some endogenous degradation); lane 2, μ-calpain cleaved spectrin; lane 3, trypsin cleaved spectrin. The weak reactivity with the α-BDP1 and α-BDP2 antibodies with the intact spectrin and in trypsin-digested spectrin is due to trace μ-calpain cleaved material contaminating the original preparation. The PAb 10D used to immunoblot βII spectrin here only recognizes the COOH-terminal half of βII spectrin (see text), and thus detects βII-BDP120 but not βII-BDP166, which is recognized by β-BDP1. Note the absence of reactivity of the BDP antibodies to intact αII/βII spectrin.
Glutamate/NMDA and maitotoxin activate distinct pathways of calpain cleavage in cultured cells
To determine how the Ca++ activation of calpain by two distinct pathways affects the pattern of spectrin cleavage in vivo, the response of various cells to either glutamate/NMDA stimulation or calcium loading (via maitotoxin) was evaluated. These results are shown in Figures 5, 6, and 7. In cells containing NMDA receptors, such as rat cortical or hippocampal neurons, glutamate stimulates the opening of gated Ca++ channels, allowing the influx of extracellular Ca++. This process can activate long-term potentiation (reviewed in 63, 64). Figure 5A,B illustrates that brief exposure to glutamate stimulates calpain cleavage of αII but not βII spectrin in cortical neurons, and that only αII-BDP150 (but not αII-BDP145) is generated by such stimulation. This activity is confined to a small pool of the total spectrin in the cell; the absence of aII-BDP145 generation implies that CaM is not active on spectrin during this period. Conversely, glutamate did not enhance spectrin cleavage in SH-SY5Y cells, a neuroblastoma line that lacks NMDA receptors (Figure 5C), demonstrating the indirect effect of glutamate on calpain mediated spectrin cleavage.
Figure 5. Glutamate stimulates calpain cleavage of αII but not βII spectrin in responsive cells.
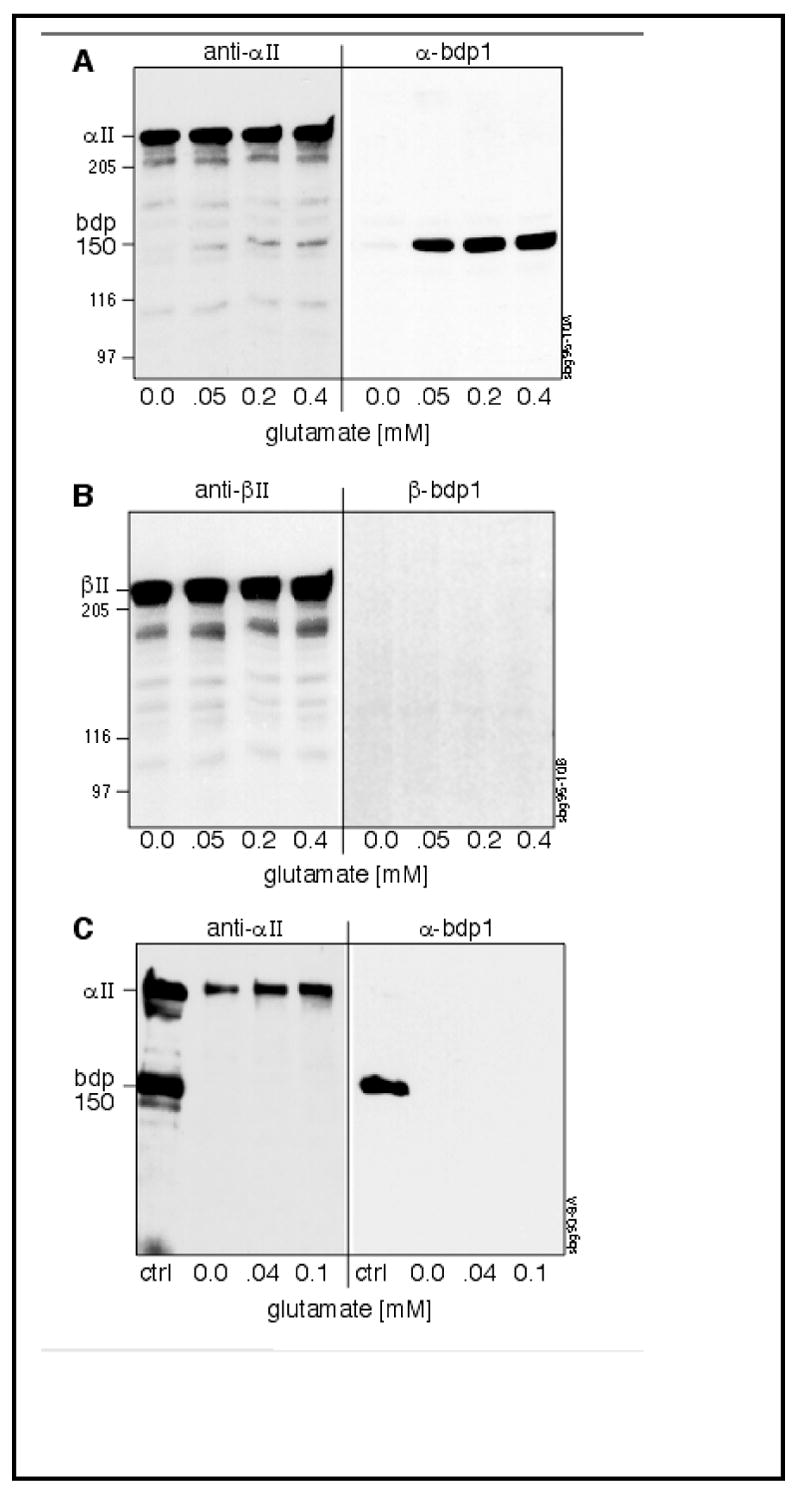
(A, B) Rat cortical neurons, or (C) neuroblastoma SH-SY5Y cells, were incubated for 20 or 45 min. respectively in the presence of increasing concentrations of glutamate. Equivalent numbers of cells at each glutamate concentration were harvested and examined by SDS-PAGE and Western blotting with either Pab RAFA (anti-αII) or α-BDP-1 (A,C) or Pab 10D (anti-βII) /β-BDP-1 (B). Each lane contains 50 μg total cellular protein. A control lane (ctrl) contains purified spectrin (with some endogenous degradation products). The size (kD) of major BDP’s, as well as several MW markers, are shown. Note that with glutamate stimulation of cortical neurons, there is activation of αII-spectrin cleavage by calpain, but no calpain cleavage of βII spectrin. Conversely, in cells that lack significant levels of glutamate receptors (SH-SY5Y), glutamate stimulation does not activate the cleavage of spectrin.
Figure 6. NMDA receptor stimulation selectively activates the calpain-processing of αII spectrin at early time points in neurons.
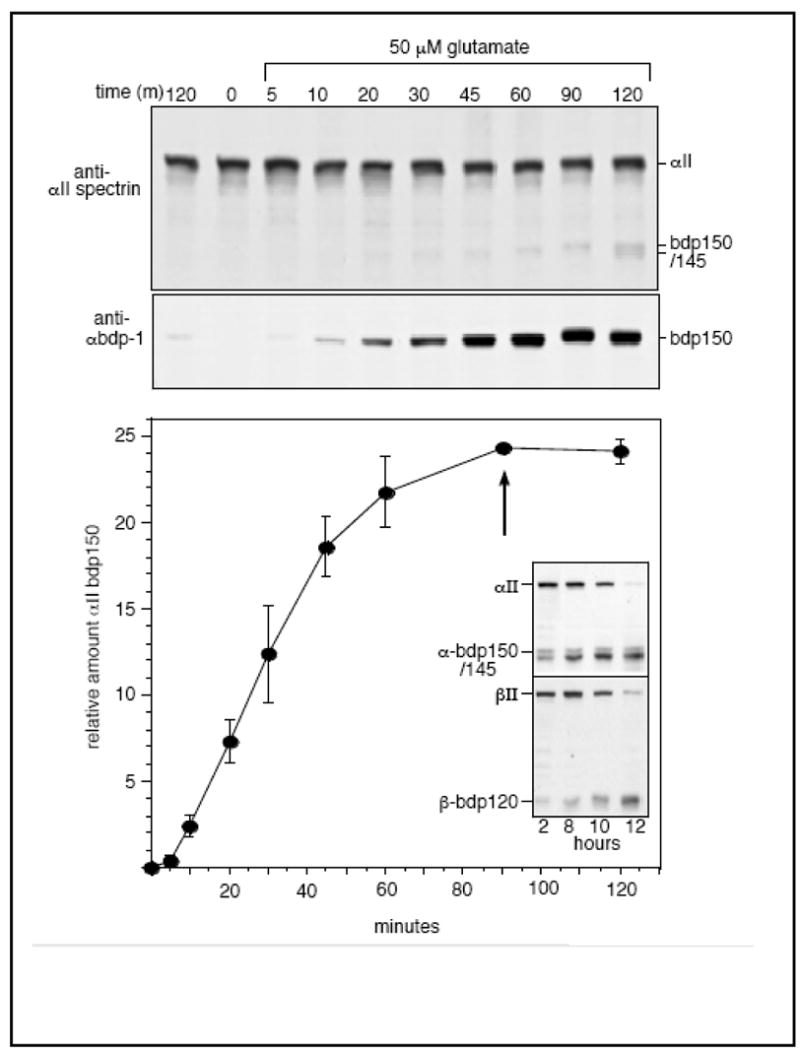
(top)Rat hippocampal cultures were incubated with 50 μM glutamate for the times indicated (0 to 120 minutes). Control cells were incubated for either 0 minutes or for 120 or 60 minutes with vehicle alone or with MK801, a specific blocker of the NMDA receptor. Samples were examined for αII and βII spectrin cleavage by Western blotting. There was no significant βII spectrin cleavage under these conditions (see Fig. 5); MK801 completely blocked all cleavage (not shown). Densitometic analysis of the α-bdp1 blots revealed a biphasic response in αII spectrin cleavage, with a 3–5 min. lag before the onset of proteolysis, and a meta-stable plateau at about 90 min. After this time point (arrow), the α-bdp-145 proteolytic product began to appear, a putative marker of CaM stimulation of αII spectrin cleavage. In parallel experiments with cortical neurons carried out for longer periods after NMDA stimulation (inset), βII spectrin cleavage began to accelerate at about 2 hours, and the calpain-mediated proteolysis of both subunits progressed to completion over a 12 h. period. The graph presents mean values ± SD, N=3.
Figure 7. Maitotoxin treatment of SH-SY5Y cells leads to sequential cleavage of αII/βII spectrin and cell death.
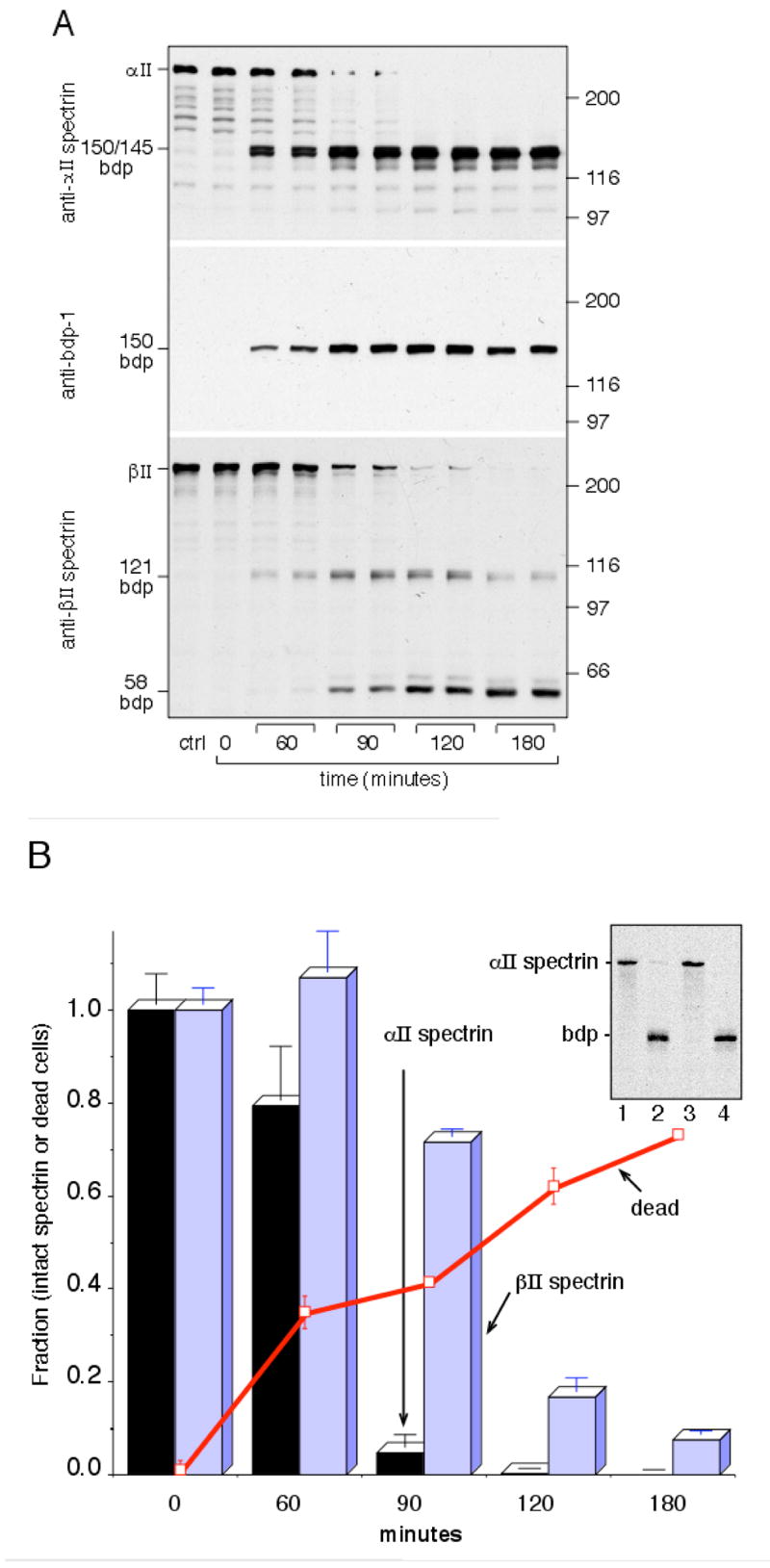
(A) Cultured SH-SY5Y cell were treated with 0.04nM maitotoxin, and the time-course of spectrin breakdown monitored by Western blotting after SDS-PAGE analysis of cell lysates (20 μg/lane) with Pab-RAFA (anti-αII spectrin); α-bdp-1 antibody; and Pab-10D (anti-βII spectrin). Control cells were incubated for three hours without maitotoxin (ctrl). Duplicate experiments are shown for each time point. Note the rapid breakdown of αII spectrin vs. the delayed loss of βII spectrin. The positions of MW markers (÷1000) are depicted on the right; prominent bdp’s are marked on the left. (B) Densitometric evaluation of the extent of spectrin breakdown of the gels shown in (A). The extent of maitotoxin-induced cell death was also evaluated in parallel experiments of maitotoxin treated cells, as measured by flow cytometry after Ethidium-1 staining. Data points are the mean ±SD of three determinations. Background cell death (non-viable cells at t=0) was subtracted from all points. (Inset) Western blot with Pab RAFA of SH-SY5Y cells after 0.03 nM maitotoxin treatment, ± inhibition of calpain or caspase. The maitotoxin induced cleavage of spectrin was entirely inhibited by the calpain inhibitor calpeptin, but not by the caspase inhibitor Z-D-DCB. Lane 1, control SH-SY5Y cells; lane 2, cells after 3 hrs of maitotoxin treatment; lane 3, 3 hrs. maitotoxin + calpeptin; lane 4, 3hrs. maitotoxin + caspase inhibitor.
To better understand the sequential nature of NMDA-receptor stimulation of spectrin cleavage, the processing of spectrin in cultured rat hippocampal neurons was examined at early time points after glutamate or NMDA stimulation (Fig. 6). αII-BDP150’ appeared as early as 4 to 5 minutes after the addition of 50 μM glutamate. The level of cleavage reached a meta-stable plateau after 90 minutes. After this point, the bdp145 product began to appear, a cleavage that is only observed in vitro when CaM is bound to αII spectrin (Fig. 1). As shown in Figure 5A, the amount of cleaved αII spectrin induced in these experiments represents only a very small fraction of the total protein, and cleavage of βII spectrin at these early time-points was not detected (Fig. 5). In control experiments, MK801, a specific NMDA receptor channel blocker, completely blocked the glutamate-induced cleavage of spectrin (not shown), indicating that glutamate was activating calpain processing specifically through the NMDA receptor (NMDA-R), and not via other types of glutamate receptors such as the AMPA or Kainate receptors. Thus, Ca++ entry through the activated NMDA-R channel stimulates within 3–5 minutes calpain proteolysis of a small pool of αII-spectrin, and this process occurs without evidence of either CaM stimulation or βII spectrin cleavage. This apparent plateau in αII-spectrin cleavage without βII-spectrin cleavage is transitory. Prolonged stimulation with NMDA receptor agonists or at levels that rapidly induce neurotoxicity induces the complete cleavage of both αII and βII spectrin by calpain over a 12 hour period (Fig. 6, inset).
Calcium entry into SH-SY5Y cells treated with maitotoxin in the presence of normal culture medium had quite a different effect on spectrin breakdown (Figure 7). Maitotoxin is a marine toxin that opens L-type voltage-sensitive and receptor-operated calcium channels in the plasma membrane. It specifically activates calpain but not caspase and induces non-apoptotic cell death (65, 66). Within 60 minutes of maitotoxin treatment, there is extensive calpain mediated cleavage of αII spectrin, as detected both by Pab-RAFA and by α-bdp1 (Fig. 7A,B). Interestingly, while the generation of the α-bdp145 fragment (a marker of calmodulin action in vitro) is not appreciably delayed, there is delayed onset of proteolysis of βII spectrin. All of this breakdown can be attributed to calpain (vs. caspase), since it is totally inhibited by a calpain inhibitor (Fig. 7A, inset, lane 3) but not by a caspase inhibitor (Fig. 7A, inset, lane 4). These results suggest that a sequential αII/βII cleavage process similar to that we previously identified to be under the control of CaM in vitro (46) is active in vivo. Finally, in parallel experiments, the extent of cellular death was monitored after maitotoxin treatment by the ability of the cells to stain with Ethidium-1 (Fig. 7B). Progressive cell death occurs over the period in which αII and βII spectrin are being rapidly cleaved. While the correlation is not precise, it is noteworthy that while all αII spectrin is proteolyzed by 90 minutes, cell death continues to occur correlating with the onset and continued proteolysis of βII spectrin.
Discussion
This report identifies five novel sites of calpain-catalyzed cleavage in αII and βII-spectrin; establishes that in vivo (as in vitro) the proteolysis of βII spectrin is trans-regulated by CaM binding to the αII subunit; and establishes a correlation between βII spectrin proteolysis and conditions eliciting non-apoptotic cell death. These conclusions are supported by several lines of evidence: i) NH2-terminal microsequencing identifies a second calpain cleavage site in αII spectrin at G1230, immediately downstream of the primary calpain cleavage site at Y1176 and the CaM binding domain of αII-spectrin; ii) this site is only cleaved when CaM is bound; iii) microsequencing identifies a prominent CaM dependent cleavage site in βII spectrin at Q1440, and secondary cleavage sites at S1447 and L1482; iv) additional, less favorable, cleavages have been identified in βII spectrin including a cleavage near the COOH-terminus that may include one (A2067) previously identified (53); and v) recombinant βII spectrin peptides display identical calpain cleavage sites whose susceptibility (in the absence of a paired αII subunit) is CaM independent. These cleavages are summarized in Figure 8. Experiments in cultured cells using subunit or epitope-specific antibodies establish a close correspondence between the proteolytic products observed in vitro and those generated in vivo, and demonstrate that calpain cleavage of αII spectrin can be activated in responsive cells by physiologic receptors without cell death, such as by low-level stimulation of glutamate receptors. Presumably, the early and localized low-level cleavage of just αII spectrin occurs in response to specific signals in susceptible cells and allows for membrane receptor rearrangements. While the correlation of αII spectrin processing and/or calpain activation in response to low level stimulation has been variably noted in several previous studies (9, 18–21, 67), the participation of βII spectrin cleavage in this process has gone unexamined. It is thus interesting that under conditions promoting glutamate toxicity, or actions directly inimical to the cell such as Ca++ loading (e.g. by maitotoxin) (55), we consistently find both very high levels of spectrin proteolysis, as well as cleavage of both the αII and βII subunits.
This study also highlights perhaps the clearest example yet of trans-regulation between the two spectrin subunits. Calmodulin binds exclusively to a single site in αII spectrin. In the presence of bound CaM, new sites in αII spectrin (G1230) and in βII spectrin (Q1440, S1447, and L1482) become susceptible to calpain, and the rate of calpain cleavage of βII spectrin is enhanced many fold. Since recombinant αII spectrin peptides are not readily cleaved at the G1230 bond (also see 48), it follows that CaM must create a new site favorable for cleavage by altering the tertiary conformation of αII spectrin about G1230., a finding consistent with the significant reordering of the 3–D structure of spectrin’s calmodulin binding domain by bound CaM (51). Conversely, isolated βII spectrin recombinant peptides are readily cleaved by calpain at the same site as in CaM-loaded αII/βII spectrin. Based on the best available data for the alignment of the two subunits (68), the preferred sites of cleavage in αII and βII spectrin appear to be adjacent in the heterodimer (Figure 8). Thus, while conformational distortion of βII spectrin by CaM acting through the αII subunit cannot be rigorously excluded, we favor (on the principal of parsimony) the interpretation that calpain access to the susceptible βII cleavage site is blocked by αII spectrin in the intact heterodimer, and that this blockage is relieved by the conformational changes in αII spectrin elicited by CaM (or possibly by the secondary cleavage of αII-BDP150 to αII-BDP145). It is unlikely that the susceptibility of isolated βII peptides derives from their failure to form a native-like structure because they exhibit a level of α-helix consistent with intact spectrin ( ≈ 70% by CD measurement), and unlike other proteases the determinants of calpain-specificity reside not in a linear amino-acid sequence but rather in complex conformational determinants unlikely to be represented in a denatured peptide (48).
The correlation of βII cleavage with cell-lethal manipulation is an interesting one. While glutamate or NMDA exposure of neurons may induce lethality, this is not an obligate effect, and at least in the short-term these compounds may activate long-term potentiation and synaptic remodeling without neuronal death (12, 21, 69). The factors that determine whether glutamate exposed neurons survive or die are complex and incompletely understood, and include changes in nitrous oxide synthetase (70), protein kinase C (71), the level of intracellular Ca++ and other cations (72, 73), and the their interaction with glial cells (74, 75). Our results suggest that the extent of βII spectrin cleavage may be another determining factor. Previously we have established that αII spectrin cleavage by calpain modifies spectrin, such that it becomes a reversible actin cross-linking protein under the control of Ca++ and CaM (54). The native (non-cleaved) protein constitutively cross-links actin and is not modulated by Ca++ or CaM. Thus, by activating the calpain cleavage of αII spectrin, glutamate stimulation of neurons would be expected to convert a relatively fixed cortical spectrin-actin lattice into one dynamically regulated by Ca++ and CaM, presumably facilitating the changes in receptor organization required for long-term potentiation (76) and synaptic plasticity (12, 77). Conversely, cleavage of βII spectrin by calpain in vitro leads to irreversible disassembly of the spectrin-actin complex (54) and the loss of membrane anchoring sites (52), mirroring the in vivo disruption of the cortical spectrin-actin skeleton characteristic of hypoxic cells (29, 78), the early stages of apoptosis (in which both subunits of spectrin are simultaneously cleaved) (55), or the effects of intracellular calcium loading induced by maitotoxin in the experiments reported here. We hypothesize that these associations are more than coincidental, and suggest that the cleavage of βII spectrin, whether by excessive activation of calpain (as in glutamate toxicity or maitotoxin treatment) or by the action of caspase during apoptosis (55), contributes to a degenerative process whereby the spectrin-actin skeleton, membrane integrity, and membrane protein organization are disrupted. It is also interesting to note that while in this model CaM acts as both a reversible regulator of skeletal plasticity, and the activator of βII spectrin cleavage by calpain (with consequential risk to the cell), it also acts at the same time to protect spectrin from cleavage by several executioner caspases (79). Thus, while activating susceptibility to calpain cleavage, CaM may dampen or abrogate certain apoptotic processes. The susceptibility of spectrin to calpain cleavage is also regulated by reversible phosphorylation of Y1176 (49, 50). Thus, the regulation of αII spectrin proteolysis appears to be a point of convergence of several signal transduction pathways. It will be important in future studies to determine the functional consequences of the various regulated cleavages described here, and whether cells can be rendered less susceptible to non-apoptotic cell death by specifically blocking of βII spectrin cleavage.
Supplementary Material
Acknowledgments
The authors thank Drs. Alan Harris, Christian Lombardo, and Mr. Paul Stabach, for providing several reagents and for assistance with the preparation of some constructs and antibodies.
This work supported by NIH grants R01-DK43812 and P01-NS35476 (Ment PI) to JSM and by a NRSA postdoctoral fellowship to SBG.
Abbreviations
- αII–BDP
αII-spectrin breakdown product
- βII–BDP
βII-spectrin breakdown product
- CaM
calmodulin
- DMEM
Dulbecco’s Modified Eagle Medium
- DTT
dithiothreitol
- MDL28170
carboxybenzyl-Val-Phe-H
- MEM
minimum essential medium
- MTX
maitotoxin
- NMDA-R
N-methyl-d-aspartate Receptor
- Pab
polyclonal antibody
- PAGE
polyacrylamide gel electrophoresis
- PBS
phosphate buffered saline
- SDS
sodium dodecyl sulfate
- Z-D-DCB
carbobenzoxy-Asp-CH2OC(O)-2,6-dichlorobenzene
References
- 1.Morrow JS, Rimm DL, Kennedy SP, Cianci CD, Sinard JH, Weed SA. In: Handbook of Physiology. Hoffman J, Jamieson J, editors. Oxford, London: 1997. pp. 485–540. [Google Scholar]
- 2.De Matteis MA, Morrow JS. Spectrin tethers and mesh in the biosynthetic pathway. J Cell Sci. 2000;113:2331–2343. doi: 10.1242/jcs.113.13.2331. [DOI] [PubMed] [Google Scholar]
- 3.Bennett V, Baines AJ. Spectrin and ankyrin-based pathways: metazoan inventions for integrating cells into tissues. Physiol Rev. 2001;81:1353–92. doi: 10.1152/physrev.2001.81.3.1353. [DOI] [PubMed] [Google Scholar]
- 4.Deng H, Lee JK, Goldstein LS, Branton D. Drosophila development requires spectrin network formation. J Cell Biol. 1995;128:71–9. doi: 10.1083/jcb.128.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Peters LL, Birkenmeier CS, Barker JE. Fetal compensation of the hemolytic anemia in mice homozygous for the normoblastosis (nb) mutation. Blood. 1992;80:2122–7. [PubMed] [Google Scholar]
- 6.Fox JE. Transmembrane signaling across the platelet integrin glycoprotein IIb-IIIa. Ann N Y Acad Sci. 1994;714:75–87. doi: 10.1111/j.1749-6632.1994.tb12032.x. [DOI] [PubMed] [Google Scholar]
- 7.Jesaitis AJ, Bokoch GM, Tolley JO, Allen RA. Lateral segregation of neutrophil chemotactic receptors into actin- and fodrin-rich plasma membrane microdomains depleted in guanyl nucleotide regulatory proteins. Journal Cell Biology. 1988;107:921–928. doi: 10.1083/jcb.107.3.921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lynch G, Baudry M. The biochemistry of memory: A new and specific hypothesis. Science. 1984;224:1057–1063. doi: 10.1126/science.6144182. [DOI] [PubMed] [Google Scholar]
- 9.Bednarski E, Vanderklish P, Gall C, Saido TC, Bahr BA, Lynch G. Translational suppression of calpain I reduces NMDA-induced spectrin proteolysis and pathophysiology in cultured hippocampal slices. Brain Res. 1995;694:147–57. doi: 10.1016/0006-8993(95)00851-g. [DOI] [PubMed] [Google Scholar]
- 10.Bahr BA, Kessler M, Rivera S, Vanderklish PW, Hall RA, Mutneja MS, Gall C, Hoffman KB. Stable maintenance of glutamate receptors and other synaptic components in long-term hippocampal slices. Hippocampus. 1995;5:425–39. doi: 10.1002/hipo.450050505. [DOI] [PubMed] [Google Scholar]
- 11.Vanderklish P, Saido TC, Gall C, Arai A, Lynch G. Proteolysis of spectrin by calpain accompanies theta-burst stimulation in cultured hippocampal slices. Brain Res Mol Brain Res. 1995;32:25–35. doi: 10.1016/0169-328x(95)00057-y. [DOI] [PubMed] [Google Scholar]
- 12.Faddis BT, Hasbani MJ, Goldberg MP. Calpain activation contributes to dendritic remodeling after brief excitotoxic injury in vitro. J Neurosci. 1997;17:951–9. doi: 10.1523/JNEUROSCI.17-03-00951.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dosemeci A, Reese TS. Effect of calpain on the composition and structure of postsynaptic densities. Synapse. 1995;20:91–7. doi: 10.1002/syn.890200113. [DOI] [PubMed] [Google Scholar]
- 14.Sheppard AM, Wu JE, Staubli U, Perlmutter LS. Changes in calpain and brain spectrin immunoreactivity accompany sprouting in the deafferented hippocampus. Synapse. 1993;15:239–42. doi: 10.1002/syn.890150309. [DOI] [PubMed] [Google Scholar]
- 15.Lee A, Morrow JS, Fowler VM. Caspase remodeling of the spectrin membrane skeleton during lens development and aging. J Biol Chem. 2001;276:20735–42. doi: 10.1074/jbc.M009723200. [DOI] [PubMed] [Google Scholar]
- 16.Kamal A, Ying Y, Anderson RG. Annexin VI-mediated loss of spectrin during coated pit budding is coupled to delivery of LDL to lysosomes. J Cell Biol. 1998;142:937–47. doi: 10.1083/jcb.142.4.937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fox JE, Taylor RG, Taffarel M, Boyles JK, Goll DE. Evidence that activation of platelet calpain is induced as a consequence of binding of adhesive ligand to the integrin, glycoprotein IIb-IIIa. J Cell Biol. 1993;120:1501–7. doi: 10.1083/jcb.120.6.1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Siman R, Noszek JC. Excitatory amino acids activate calpain I and induce structural protein breakdown in vivo. Neuron. 1988;1:279–287. doi: 10.1016/0896-6273(88)90076-1. [DOI] [PubMed] [Google Scholar]
- 19.del Cerro S, Arai A, Kessler M, Bahr BA, Vanderklish P, Rivera S, Lynch G. Stimulation of NMDA receptors activates calpain in cultured hippocampal slices. Neurosci Lett. 1994;167:149–52. doi: 10.1016/0304-3940(94)91049-9. [DOI] [PubMed] [Google Scholar]
- 20.Manev H, Favaron M, Siman R, Guidotti A, Costa E. Glutamate neurotoxicity is independent of calpain I inhibition in primary cultures of cerebellar granule cells. J Neurochem. 1991;57:1288–95. doi: 10.1111/j.1471-4159.1991.tb08292.x. [DOI] [PubMed] [Google Scholar]
- 21.Di Stasi AM, Gallo V, Ceccarini M, Petrucci TC. Neuronal fodrin proteolysis occurs independently of excitatory amino acid-induced neurotoxicity. Neuron. 1991;6:445–454. doi: 10.1016/0896-6273(91)90252-u. [DOI] [PubMed] [Google Scholar]
- 22.Seubert P, Lee K, Lynch G. Ischemia triggers NMDA receptor-linked cytoskeletal proteolysis in hippocampus. Brain Res. 1989;492:366–70. doi: 10.1016/0006-8993(89)90921-9. [DOI] [PubMed] [Google Scholar]
- 23.Arai A, Vanderklish P, Kessler M, Lee K, Lynch G. A brief period of hypoxia causes proteolysis of cytoskeletal proteins in hippocampal slices. Brain Res. 1991;555:276–80. doi: 10.1016/0006-8993(91)90352-v. [DOI] [PubMed] [Google Scholar]
- 24.Hong SC, Lanzino G, Goto Y, Kang SK, Schottler F, Kassell NF, Lee KS. Calcium-activated proteolysis in rat neocortex induced by transient focal ischemia. Brain Res. 1994;661:43–50. doi: 10.1016/0006-8993(94)91178-9. [DOI] [PubMed] [Google Scholar]
- 25.Lee KS, Frank S, Vanderklish P, Arai A, Lynch G. Inhibition of proteolysis protects hippocampal neurons from ischemia. Proc Natl Acad Sci U S A. 1991;88:7233–7. doi: 10.1073/pnas.88.16.7233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rami A, Krieglstein J. Protective effects of calpain inhibitors against neuronal damage caused by cytotoxic hypoxia in vitro and ischemia in vivo. Brain Res. 1993;609:67–70. doi: 10.1016/0006-8993(93)90856-i. [DOI] [PubMed] [Google Scholar]
- 27.Bartus RT, Hayward NJ, Elliott PJ, Sawyer SD, Baker KL, Dean RL, Akiyama A, Straub JA, Harbeson SL, Li Z, et al. Calpain inhibitor AK295 protects neurons from focal brain ischemia. Effects of postocclusion intra-arterial administration. Stroke. 1994;25:2265–70. doi: 10.1161/01.str.25.11.2265. [DOI] [PubMed] [Google Scholar]
- 28.Doctor RB, Bennett V, Mandel LJ. Degradation of spectrin and ankyrin in the ischemic rat kidney. American Journal of Physiology. 1993;264:C1003–13. doi: 10.1152/ajpcell.1993.264.4.C1003. [DOI] [PubMed] [Google Scholar]
- 29.Glantz SB, Morrow JS. In: Tissue Oxygen Deprivation: Developmental, Molecular and Integrated Function. Haddad GG, Lister G, editors. Marcel Dekker, Inc.; New York: 1996. pp. 153–192. [Google Scholar]
- 30.Gaudio KM, Thulin G, Siegel NJ. Glycolysis is not responsible for the tolerance of immature renal tubules to anoxia. Pediatr Res. 1996;40:457–61. doi: 10.1203/00006450-199609000-00015. [DOI] [PubMed] [Google Scholar]
- 31.Iizuka K, Kawaguchi H, Yasuda H. Calpain is activated during hypoxic myocardial cell injury. Biochem Med Metab Biol. 1991;46:427–31. doi: 10.1016/0885-4505(91)90091-x. [DOI] [PubMed] [Google Scholar]
- 32.Iizuka K, Kawaguchi H, Yasuda H, Kitabatake A. The role of calcium activated neutral protease on myocardial cell injury in hypoxia. Jpn Heart J. 1992;33:707–15. doi: 10.1536/ihj.33.707. [DOI] [PubMed] [Google Scholar]
- 33.Iizuka K, Kawaguchi H, Kitabatake A. Effects of thiol protease inhibitors on fodrin degradation during hypoxia in cultured myocytes. J Mol Cell Cardiol. 1993;25:1101–9. doi: 10.1006/jmcc.1993.1122. [DOI] [PubMed] [Google Scholar]
- 34.Yoshida K, Sorimachi Y, Fujiwara M, Hironaka K. Calpain is implicated in rat myocardial injury after ischemia or reperfusion. Jpn Circ J. 1995;59:40–8. doi: 10.1253/jcj.59.40. [DOI] [PubMed] [Google Scholar]
- 35.Siman R, Noszek JC, Kegerise C. Calpain I activation is specifically related to excitatory amino acid induction of hippocampal damage. J Neurosci. 1989;9:1579–90. doi: 10.1523/JNEUROSCI.09-05-01579.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Seubert P, Larson J, Oliver M, Jung MW, Baudry M, Lynch G. Stimulation of NMDA receptors induces proteolysis of spectrin in hippocampus. Brain Research. 1988;460:189–194. doi: 10.1016/0006-8993(88)91222-x. [DOI] [PubMed] [Google Scholar]
- 37.Bi X, Chang V, Siman R, Tocco G, Baudry M. Regional distribution and time-course of calpain activation following kainate-induced seizure activity in adult rat brain. Brain Res. 1996;726:98–108. doi: 10.1016/0006-8993(95)01360-1. [DOI] [PubMed] [Google Scholar]
- 38.Mucke L, Masliah E, Campbell IL. Transgenic models to assess the neuropathogenic potential of HIV-1 proteins and cytokines. Curr Top Microbiol Immunol. 1995;202:187–205. doi: 10.1007/978-3-642-79657-9_13. [DOI] [PubMed] [Google Scholar]
- 39.Toggas SM, Masliah E, Mucke L. Prevention of HIV-1 gp120-induced neuronal damage in the central nervous system of transgenic mice by the NMDA receptor antagonist memantine. Brain Res. 1996;706:303–7. doi: 10.1016/0006-8993(95)01197-8. [DOI] [PubMed] [Google Scholar]
- 40.Sasaki M, Uchiyama J, Ishikawa H, Matsushita S, Kimura G, Nomoto K, Koga Y. Induction of apoptosis by calmodulin-dependent intracellular Ca2+ elevation in CD4+ cells expressing gp 160 of HIV. Virology. 1996;224:18–24. doi: 10.1006/viro.1996.0502. [DOI] [PubMed] [Google Scholar]
- 41.Vanags DM, Porn-Ares MI, Coppola S, Burgess DH, Orrenius S. Protease involvement in fodrin cleavage and phosphatidylserine exposure in apoptosis. J Biol Chem. 1996;271:31075–85. doi: 10.1074/jbc.271.49.31075. [DOI] [PubMed] [Google Scholar]
- 42.Nath R, Raser KJ, McGinnis K, Nadimpalli R, Stafford D, Wang KK. Effects of ICE-like protease and calpain inhibitors on neuronal apoptosis. Neuroreport. 1996;8:249–55. doi: 10.1097/00001756-199612200-00050. [DOI] [PubMed] [Google Scholar]
- 43.Nath R, Raser KJ, Stafford D, Hajimohammadreza I, Posner A, Allen H, Talanian RV, Yuen P, Gilbertsen RB, Wang KK. Non-erythroid alpha-spectrin breakdown by calpain and interleukin 1 beta-converting-enzyme-like protease(s) in apoptotic cells: contributory roles of both protease families in neuronal apoptosis. Biochem J. 1996;319:683–90. doi: 10.1042/bj3190683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Harris AS, Morrow JS. Proteolytic processing of human brain alpha spectrin (fodrin): identification of a hypersensitive site. J Neurosci. 1988;8:2640–51. doi: 10.1523/JNEUROSCI.08-07-02640.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Harris AS, Croall D, Morrow JS. The calmodulin-binding site in fodrin is near the site of calcium-dependent protease-I cleavage. J Biol Chem. 1988;263:15754–15761. [PubMed] [Google Scholar]
- 46.Harris AS, Croall DE, Morrow JS. Calmodulin regulates fodrin susceptibility to cleavage by calcium-dependent protease I. J Biol Chem. 1989;264:17401–17408. [PubMed] [Google Scholar]
- 47.del Lacoste M-C, Lewis CE, White CL, Morrow JS. Immunocytochemical evidence of accelerated spectrin proteolysis in hippocampus from alzheimer disease patients. Neuroscience. 1992;18:558. (abstract) [Google Scholar]
- 48.Stabach PR, Cianci CD, Glantz SB, Zhang Z, Morrow JS. Site-directed mutagenesis of alpha II spectrin at codon 1175 modulates its mu-calpain susceptibility. Biochemistry. 1997;36:57–65. doi: 10.1021/bi962034i. [DOI] [PubMed] [Google Scholar]
- 49.Nicolas G, Fournier CM, Galand C, Malbert-Colas L, Bournier O, Kroviarski Y, Bourgeois M, Camonis JH, Dhermy D, Grandchamp B, Lecomte MC. Tyrosine phosphorylation regulates alpha II spectrin cleavage by calpain. Mol Cell Biol. 2002;22:3527–36. doi: 10.1128/MCB.22.10.3527-3536.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nedrelow JH, Cianci CD, Morrow JS. c-Src binds alpha II spectrin's Src homology 3 (SH3) domain and blocks calpain susceptibility by phosphorylating Tyr1176. J Biol Chem. 2003;278:7735–41. doi: 10.1074/jbc.M210988200. [DOI] [PubMed] [Google Scholar]
- 51.Simonovic M, Zhang Z, Cianci CD, Steitz TA, Morrow JS. Structure of the calmodulin alpha II-spectrin complex provides insight into the regulation of cell plasticity. J Biol Chem. 2006 doi: 10.1074/jbc.M604613200. in press. [DOI] [PubMed] [Google Scholar]
- 52.Hu RJ, Bennett V. In vitro proteolysis of brain spectrin by calpain I inhibits association of spectrin with ankyrin-independent membrane binding site(s) Journal Biological Chemistry. 1991;266:18200–18205. [PubMed] [Google Scholar]
- 53.Lofvenberg L, Backman L. Calpain-induced proteolysis of beta-spectrins. FEBS Lett. 1999;443:89–92. doi: 10.1016/s0014-5793(98)01697-4. [DOI] [PubMed] [Google Scholar]
- 54.Harris AS, Morrow JS. Calmodulin and calcium-dependent protease I coordinately regulate the interaction of fodrin with actin. Proc Nat Acad Sci (USA) 1990;87:3009–3013. doi: 10.1073/pnas.87.8.3009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang KK, Posmantur R, Nath R, McGinnis K, Whitton M, Talanian RV, Glantz SB, Morrow JS. Simultaneous degradation of alphaII- and betaII-spectrin by caspase 3 (CPP32) in apoptotic cells. J Biol Chem. 1998;273:22490–7. doi: 10.1074/jbc.273.35.22490. [DOI] [PubMed] [Google Scholar]
- 56.Bennett V. Purification of brain analogs of red blood cell membrane skeletal proteins: Ankyrin, protein 4.1 (synapsin), spectrin, and spectrin subunits. Methods in Enzymology. 1986;134:55–69. doi: 10.1016/0076-6879(86)34075-8. [DOI] [PubMed] [Google Scholar]
- 57.Harris AS, Green LAD, Ainger KJ, Morrow JS. Mechanisms of cytoskeletal regulation (1): Functional differences correlate with antigenic dissimilarity in human brain and erythrocyte spectrin. Biochim Biophys Acta. 1985;830:147–158. doi: 10.1016/0167-4838(85)90022-6. [DOI] [PubMed] [Google Scholar]
- 58.Lombardo CR, Weed SA, Kennedy SP, Forget BG, Morrow JS. βII-Spectrin (fodrin) and βIΣ2 -Spectrin (muscle) Contain NH2- & COOH-terminal Membrane Association Domains (MAD1 & MAD2) J Biol Chem. 1994;269:29212–29219. [PubMed] [Google Scholar]
- 59.Harris AS, Anderson JP, Yurchenco PD, Green LAD, Ainger KJ, Morrow JS. Mechanisms of cytoskeletal regulation: Functional and antigenic diversity in human erythrocyte and brain beta spectrin. J Cellular Biochem. 1986;30:51–70. doi: 10.1002/jcb.240300107. [DOI] [PubMed] [Google Scholar]
- 60.Harlow E, Lane D. Antibodies: A laboratory manual. Cold Spring Harbor Laboratory; Cold Spring Harbor: 1988. [Google Scholar]
- 61.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature (London) 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 62.Glantz SB, Friedman JE, Haddad GG, Morrow JS. Calpain cleavage of αII spectrin in primary cortical and hippocampal cultures detected by cleavage specific antibodies. Molecular Biology of the Cell. 1995;6:371a. (abstract) [Google Scholar]
- 63.Miyamoto E, Fukunaga K. A role of Ca2+/calmodulin-dependent protein kinase II in the induction of long-term potentiation in hippocampal CA1 area. Neurosci Res. 1996;24:117–22. doi: 10.1016/0168-0102(95)00991-4. [DOI] [PubMed] [Google Scholar]
- 64.Cain DP. LTP, NMDA, genes and learning. Curr Opin Neurobiol. 1997;7:235–42. doi: 10.1016/s0959-4388(97)80012-8. [DOI] [PubMed] [Google Scholar]
- 65.Wang KKW, Nath R, Raser KJ, Hajimohammadreza I. Maitotoxin induces calpain activation in SH-SY5Y neuroblastoma cells and cerebrocortical cultures. Arch Biochem Biophys. 1996;331:208–14. doi: 10.1006/abbi.1996.0300. [DOI] [PubMed] [Google Scholar]
- 66.Zhao X, Pike BR, Newcomb JK, Wang KK, Posmantur RM, Hayes RL. Maitotoxin induces calpain but not caspase-3 activation and necrotic cell death in primary septo-hippocampal cultures. Neurochem Res. 1999;24:371–82. doi: 10.1023/a:1020933616351. [DOI] [PubMed] [Google Scholar]
- 67.Adamec E, Beermann ML, Nixon RA. Calpain I activation in rat hippocampal neurons in culture is NMDA receptor selective and not essential for excitotoxic cell death. Brain Res Mol Brain Res. 1998;54:35–48. doi: 10.1016/s0169-328x(97)00304-5. [DOI] [PubMed] [Google Scholar]
- 68.Speicher DW, DeSilva TM, Speicher KD, Ursitti JA, Hembach P, Weglarz L. Location of the human red cell spectrin tetramer binding site and detection of a related "closed" hairpin loop dimer using proteolytic footprinting. J Biol Chem. 1993;268:4227–35. [PubMed] [Google Scholar]
- 69.Bear MF, Malenka RC. Synaptic plasticity: LTP and LTD. Curr Opin Neurobiol. 1994;4:389–99. doi: 10.1016/0959-4388(94)90101-5. [DOI] [PubMed] [Google Scholar]
- 70.Wang JY, Chi SI, Wang JY, Hwang CP, Wang JY. Effects of various nitric oxide synthase inhibitors on NMDA-induced neuronal injury in rat cortical neurons. Chin J Physiol. 1996;39:227–33. [PubMed] [Google Scholar]
- 71.Durkin JP, Tremblay R, Chakravarthy B, Mealing G, Morley P, Small D, Song D. Evidence that the early loss of membrane protein kinase C is a necessary step in the excitatory amino acid-induced death of primary cortical neurons. J Neurochem. 1997;68:1400–12. doi: 10.1046/j.1471-4159.1997.68041400.x. [DOI] [PubMed] [Google Scholar]
- 72.Chen QX, Perkins KL, Choi DW, Wong RKS. Secondary activation of a cation conductance is responsible for NMDA toxicity in acutely isolated hippocampal neurons. J Neurosci. 1997;17:4032–6. doi: 10.1523/JNEUROSCI.17-11-04032.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Chidekel AS, Friedman JE, Haddad GG. Anoxia-induced neuronal injury: role of Na+ entry and Na+-dependent transport. Exp Neurol. 1997;146:403–13. doi: 10.1006/exnr.1997.6544. [DOI] [PubMed] [Google Scholar]
- 74.Dugan LL, Bruno VM, Amagasu SM, Giffard RG. Glia modulate the response of murine cortical neurons to excitotoxicity: glia exacerbate AMPA neurotoxicity. J Neurosci. 1995;15:4545–55. doi: 10.1523/JNEUROSCI.15-06-04545.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Auld DS, Robitaille R. Glial cells and neurotransmission: an inclusive view of synaptic function. Neuron. 2003;40:389–400. doi: 10.1016/s0896-6273(03)00607-x. [DOI] [PubMed] [Google Scholar]
- 76.Lynch G, Kessler M, Arai A, Larson J. The nature and causes of hippocampal long-term potentiation. Progress in Brain Research. 1990;83:233–250. doi: 10.1016/s0079-6123(08)61253-4. [DOI] [PubMed] [Google Scholar]
- 77.Park JS, Bateman MC, Goldberg MP. Rapid alterations in dendrite morphology during sublethal hypoxia or glutamate receptor activation. Neurobiol Dis. 1996;3:215–27. doi: 10.1006/nbdi.1996.0022. [DOI] [PubMed] [Google Scholar]
- 78.Molitoris BA, Dahl R, Hosford M. Cellular ATP depletion induces disruption of the spectrin cytoskeletal network. Am J Physiol. 1996;271:F790–8. doi: 10.1152/ajprenal.1996.271.4.F790. [DOI] [PubMed] [Google Scholar]
- 79.Rotter B, Kroviarski Y, Nicolas G, Dhermy D, Lecomte MC. AlphaII-spectrin is an in vitro target for caspase-2, and its cleavage is regulated by calmodulin binding. Biochem J. 2004;378:161–8. doi: 10.1042/BJ20030955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Cianci CD, Morrow JS. Complete cDNA sequence of human fetal brain aII spectrin with a novel alternative splice. GenBank U83867 1997 [Google Scholar]
- 81.Hu R-J, Watanabe M, Bennett V. Characterization of human brain cDNA encoding the general isoform of β-spectrin. Journal Biological Chemistry. 1992;267:18715–18722. [PubMed] [Google Scholar]
- 82.Smith DB, Davern KM, Board PG, Tiu WU, Garcia EG, Mitchell GF. Mr 26,000 antigen of Schistosoma japonicum recognized by resistant WEHI 129/J mice is a parasite glutathione S-transferase [published erratum appears in Proc Natl Acad Sci U S A 1987 Sep;84(18):6541] Proc Natl Acad Sci U S A. 1986;83:8703–7. doi: 10.1073/pnas.83.22.8703. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.