

Abstract
Oncolytic viruses (OVs) are being used as anticancer agents in preclinical and clinical trials. Propagation of OVs inside infected tumors is critical to their efficacy and is mediated by the productive generation of progeny OVs within infected tumor cells. In turn, this progeny can spread the infection to other tumor cells in successive rounds of oncolysis. Previously, we had found that, in rats, cyclophosphamide (CPA) pretreatment increased infection of brain tumors by an intra-arterially administered herpessimplex virus type 1 OV, because it inhibited activation of complement responses, mediated by innate IgM. We also have previously shown that other pharmacologic inhibitors of complement, such as cobra venom factor (CVF), allowed for increased infection. However, in these studies, further inhibition of complement responses by CVF did not result in additional infection of brain tumor cells or in propagation of OV to surrounding tumor cells. In this study, we sought to determine if CPA did lead to increased infection/propagation from initially infected tumor cells. Unlike our results with CVF, we find that CPA administration does result in a time-dependent increase in infection of tumor cells, suggestive of increased propagation, in both syngeneic and athymic models of brain tumors. This increase was due to increased survival of OV within infected tumors and brain surrounding tumors. CPA’s effect was not due to a direct enhancement of viral replication in tumor cells, rather was associated with its immunosuppressive effects. RT-PCR analysis revealed that CPA administration resulted in impaired mRNA production by peripheral blood mono-nuclear cells (PBMCs) of several cytokines (interferons α/β, interferon γ, TNFα, IL-15, and IL-18) with anti-HSV function. These findings suggest that the CPA-mediated facilitation of OV intraneoplastic propagation is associated with a general decrease of antiviral cytokines mRNAs in PBMCs. These findings not only suggest a potential benefit for the addition of transient immunosuppression in clinical applications of oncolytic HSV therapy, but also suggest that innate immunomodulatory pathways may be amenable to manipulation, in order to increase OV propagation and survival within infected tumors.
Keywords: brain tumors, oncolytic virus, replication-competent virus, immune responses
Introduction
Oncolytic (replication-conditional, replication-compromised, replication-competent, tumor-selective) viruses (OVs) represent genetically mutated strains that target defects in tumor suppressor pathways or activation of oncogenic pathways to facilitate selective replication in and lysis of tumor cells.1–3 Several types of mutants are now being tested in a variety of clinical trials for cancers that have failed standard therapy, with encouraging results in terms of safety.4–6 However, the achievement of therapeutic efficacy might require further manipulation of viral genomes coupled with a better understanding of the processes of viral infection, replication, and propagation within infected tumors and of interactions with host responses against the viral infection.
One of the interesting features of OVs is represented by the intratumoral generation of progeny viruses that can sequentially infect additional tumor cells. Sequential cycles of infection and progeny virus generation would be expected to lead to propagation of the oncolytic effect throughout the tumor mass. In fact, time-dependent comparisons between replication-defective and replication-conditional HSV effects on tumors show that the latter mediate increased trangene expression and viral yields compared to the former.7 However, when compared to in vitro growth curves, the generation of progeny viruses in vivo appears to be relatively suppressed.
The host immune response against an oncolytic viral infection is probably one of the important factors in suppressing virolysis.8 In fact, even in the absence of pre-existing immunity to HSV, pathologic wild-type HSV infection into the central nervous system (CNS) is limited initially by complement as well as by innate cellular responses, consisting of B cells,9 NK cells10 and macrophages.11 These cellular responses are associated with the production of cytokines, such as interferons,12,13 TNFα,11,12 interleukin-15,10,14 and -18.15 Additionally, iNOS production by macrophages, stimulated by IFN-γ, will inhibit viral replication.16,17 Interferons will also activate cellular antiviral responses consisting of oligo-2′5′-adenylate synthase (OAS) and the double-stranded RNA-dependent protein kinase (PKR),18 which lead to shutoff of viral replication. In the presence of pre-existing immunity, such responses are likely to be even more magnified, when the impact of neutralizing antibodies and cytotoxic T cells generally suppresses the occurrence and persistence of active and productive infection.
Although the host immune response has not been studied in great detail in the context of brain tumor infection mediated by a mutant, OV, it is reasonable to hypothesize that it might resemble processes described above. We have previously demonstrated that in athymic rats without pre-existing immunity to HSV, OV infection of human brain tumor xenografts was significantly facilitated by depletion of complement through CVF19 or by inhibition of innate IgM (and IgG)-mediated activation of complement through CPA.20,21 However, complement depletion alone was not sufficient to allow OV propagation in infected tumors.19 Herein, we provide further elucidation of the role of CPA in enhancing propagation of oncolytic HSV within infected tumors. Unlike previous experiments, these were conducted in a syngeneic rat model of glioma, and direct intratumoral inoculation of the OV was carried out to minimize the activation of complement that can occur with intravascular, systemic delivery. We find that CPA significantly enhances OV propagation in tumors as well as increased survival of progeny virions within the neoplasm. We find that one of the most rapid effects (within 60 h) of CPA is the impaired production of several cytokines with known antiviral activity by peripheral blood mononuclear cells (PBMCs). Some of these mRNAs belong to an inflammatory pathway, previously implicated in inhibiting HSV spread in the nervous system.11,16,17 This suggests a possible mechanism for the drug’s observed action on OV intratumoral propagation and survival and the possibility for manipulation of specific pathways in the host immune responses that will facilitate OV therapy of brain tumors.
Results
Generation of a syngeneic rat glioma cell line, permissive for oncolytic HSV infection
One difficulty in studying infection and propagation of an oncolytic HSV in syngeneic models results from the relative nonpermissivity of rodent tumor lines to HSV1 infection, presumably due to lack of the HSV1 viral entry receptors (HveA and HveC). We thus genetically modified rat D74 (also known as RG2)22 and rat 9L glioma cells, which are relatively impervious to HSV infection, by retroviral transduction with a cDNA encoding the herpesvirus receptor HveC or HveA.23 Several clones were selected, which had acquired increased infectability to HSV. With one clone for each cell line (D74/HveC and 9L/HveC cells), 100% of the cells show cytopathic affect 48 h after they were infected with hrR3 at moi of 0.1 (data not shown). HveC receptor expression seemed to provide more efficient infection than expression of HveA (data not shown). Further, D74/HveC cells and 9L/HveC retained tumorigenicity in the brains of immunocompetent Fischer rats (data not shown). There was little, if any, effect of HveC expression in tumor growth in the brain. The relative immunogenicity (in terms of providing a long-term protective, vaccination effect upon subsequent rechallenges with the same tumor cell) of D74/HevC and 9L/HveC glioma celll lines compared to parental cells has not been determined.
CPA facilitates viral-mediated transgene expression in brain tumors infected with oncolytic HSV
We had previously observed that CPA enhanced transgene expression mediated by hrR3, delivered intra-arterially within human U87dEGFR brain tumor xenografts in athymic rats.20 We sought to determine if this observation remained valid when: (1) an immunocompetent rat model was used and (2) intraneoplastic administration of OV with systemic administration of CPA was employed. While the relevance of the former is self-evident, that of the latter derives from the finding that a very small number of OVs (estimated at approximately 50) will have infected an intracerebral tumor upon intra-arterial administration of 109 PFUs.21 One might expect that immunosuppressive effects of CPA may be more relevant with a small viral load, particularly in animals without pre-existing immunity to the OV. Instead, intratumoral administration should allow the delivery of much higher concentrations of OV within a tumor and thus, one might hypothesize that CPA effects may not be as significant.
Therefore, a lacZ-expressing oncolytic HSV (hrR3) was inoculated directly into the tumors at day 7 after tumor cell implantation, when tumors had reached a diameter of approximately 4 mm. CPA was administered systemically (i.p.) either the same (simultaneous) or 2 days before (pretreatment) OV injection. After 3 days, the anatomic extent of lacZ transgene expression was much higher in animals that had been treated with CPA compared to controls (Figure 1a). In fact, animals pretreated with CPA showed evidence of increased anatomic distribution of lacZ trangene expression compared to the simultaneously treated animals. This anatomic distribution was quantitated to demonstrate that rats, pretreated with CPA, expressed significantly higher areas of lacZ expression within tumors compared to rats treated simultaneously with CPA or to those treated with saline (Figure 1b) (P <0.05, Student’s t-test). In fact, this area of transgene distribution was also higher in the ‘simultaneous CPA’ group compared to the control group. These results showed that CPA increased viral-mediated transgene expression (as measured by lacZ trangene expression) in OV-infected tumors, and suggested that it enhanced OV propagation within infected tumors.
Figure 1.
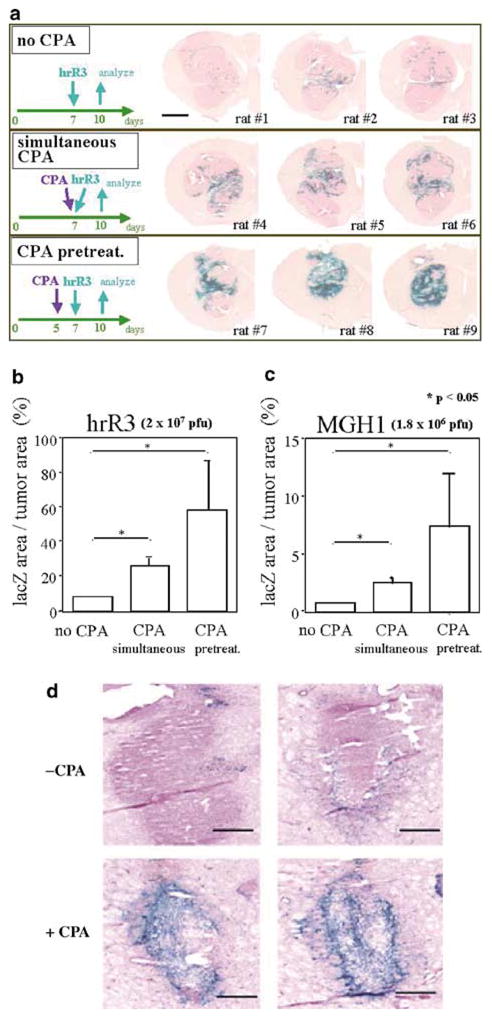
CPA increases the anatomical area of intratumoral infection by oncolytic HSV. In panel a, Fischer 344 rats from each experimental group (n=3), harboring 7-day old D74/HveC brain tumors, were killed 3 days after the injection of hrR3. Brains were then stained for expression of β-galactosidase. CPA treatment (either administered simultaneously or in a pretreatment fashion) markedly increased the anatomic area of β-galactosidase expression. The scale bar represents 2 mm. In panels b and c, the percentage of lacZ-expressing area of tumor was quantified using computer-assisted analysis. In panel d, Fisher 344 rats harboring rat 9L/HveC glioma tumors were injected with hrR3. Vehicle (upper two microphotographs) or CPA (lower two microphotographs) was administered i.p. the same day of hrR3 injection. After 3 days, brain tumors were harvested and stained for lacZ transgene expression. The scale bar represents 400 μm. In panels a, b, and d, 2 × 107 PFUs of hrR3 (ICP6-defective, KOS strain) were injected while in panel c, 1.8 × 106 PFUs of MGH-1 (ICP6 and ICP34.5-defective, F strain) were inoculated. For panels b and c, three tumor sections (each from the anterior, middle, and posterior one-third of the tumor) were selected for computer-assisted analyses of lacZ gene expression. The bars represent the mean percentage of lacZ-expressing area per tumor. Error bars represent the s.d. *P <0.05.
To ensure that these findings were not peculiar to hrR3, experiments were repeated with MGH1, an oncolytic OV derived from a different HSV strain (F versus KOS).24 CPA enhanced lacZ gene expression within MGH1-infected tumors both qualitatively and quantitatively (Figure 1b). The differences in the percentages of lacZ transduction were due to the fact that less MGH1 was injected compared to hrR3 (1.8 × 106 PFU for MGH1 versus 2 × 107 PFU for hrR3), and that MGH1 burst size in cells is less than that of hrR3. Therefore, CPA treatment significantly increased viral-mediated transgene expression in injected tumors independent of HSV mutant strain used.
In addition, to ensure that these findings were not peculiar to the employed cell line, we repeated them using a 9L/HveC glioma model established in animal brains. Figure 1d shows that there was enhanced anatomic expression of the lacZ transgene after hrR3 inoculation when animals were treated with CPA. This was in agreement with previously published findings, in which CPA enhanced the anatomical distribution of lacZ transgene expression in athymic rats with intracranial human U87ΔEGFR20 and human Gli36ΔEGFR gliomas.21
We next sought to determine if the increase in lacZ transgene expression corresponded to an increase in HSV capsid antigen within infected tumors. Therefore, immunohistochemical staining for HSV capsid protein was carried out. Tumors from CPA-treated rats (Panel b – Figure 2), particularly CPA-pretreated rats (panel c – Figure 2), demonstrated that the majority of the brain tumor was replete with HSV capsid antigen, while in saline-treated animals only a minority of the tumor was (panel a – Figure 2). The area of tumor in which HSV1 capsid immunoreactivity was observed corresponded grossly to that in which lacZ transgene expression was seen (compare Figure 2 to 1a). Taken together with the previous data, this showed that CPA enhanced the area of tumor infected by the OV.
Figure 2.
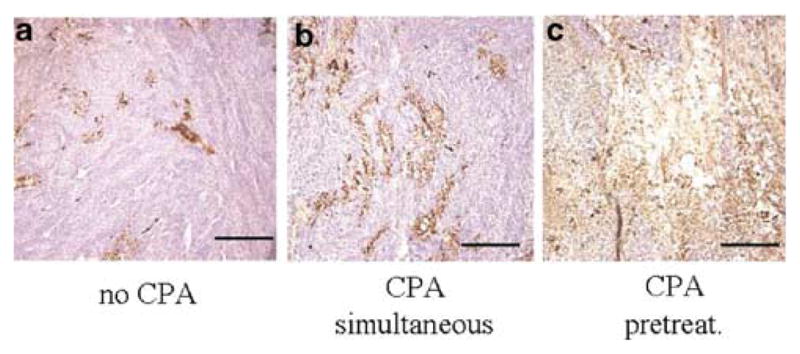
Immunohistochemical detection of HSV capsid protein. In parallel to the experiment described in Figure 1, brain sections were also immunocytochemically processed for the expression of HSV1 capsid antigens. In panel a, a representative tumor section from animals treated only with hrR3 is shown with brown precipitates indicative of HSV capsid antigen expression. In panel b, a representative section is shown from tumors harvested from animals treated with hrR3 and CPA on the same day. In panel c, a representative section is shown from tumors harvested from animals treated with hrR3 and pretreated with CPA (2 days before). The scale bar represents 100 μm.
CPA pretreatment promotes survival of OV within tumors
The previous finding suggested that CPA facilitated OV replication within tumors. We sought to find evidence for this by performing tumor explant assays. In these assays, plaque-forming infectious virus is recovered from whole brain tumor lysates. At 10 h after infection, approximately 106 PFUs were recovered from both CPA- and control-treated tumors (data not shown). However, 36 h after OV injection in nonimmunosuppressed animals, there was a decrease in OV titers that measured approximately 2 logarithmic units in value, when compared to titers measured at 10 h (P <0.01, Student’s t-test). At 60 h, this titer remained relatively stable (Figure 3a). In contrast, brains from CPA-pretreated animals did not show evidence for a significant drop in titers at the tested time points and, both at the 36 and 60 htime points, there were 2 log units higher amounts of OV recovered from CPA-treated compared to control brains. Therefore, CPA promoted the survival of OV within infected tumors and, without CPA, a rapid and significant loss of OV was observed in infected tumors.
Figure 3.
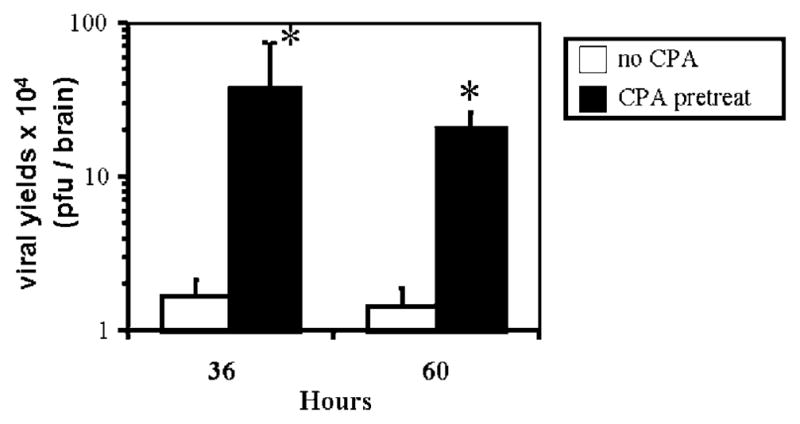
CPA treatment improves the survival of OV within infected brain tumors. Titers of hrR3 were assayed in D74/HveC syngeneic brain tumors implanted in Fisher 344 rats that were either untreated or pretreated with CPA for 2 days before direct OV injection. Time points represent time after injection of OV. After recovering the OV from rat brains as described in Materials and methods, it was titrated on Vero cell monolayers. *P <0.01.
Survival studies
We asked if the observed effect of CPA on increasing OV survival within tumors and thus increasing oncolytic effects resulted in a significant prolongation of animal survival. Four different treatment groups (n=6–8 rats per each group) were employed, consisting of (1) no treatment, (2) intraperitoneal injection of CPA only, (3) intratumoral injection of hrR3 only and (4) both intraperitoneal injection of CPA and intratumoral injection of hrR3. Rats in group 1 had succumbed to death from intracranial tumors within 15 days of tumor cell implantation, because of the highly aggressive phenotype of the D74HveC tumor cells (Wakimoto and Chiocca, unpublished observations). Rats in groups 2 and 3 survived significantly longer than those in group 1 (Figure 4) (P <0.01, Wilcoxon test). This was expected for group 2, because CPA is not only an immunosuppressant, but also possesses anticancer effects on its own. However, survival of rats within group 4 was significantly prolonged compared to groups 2 and 3 (P <0.01, Wilcoxon test). This result demonstrated that addition of CPA resulted in increasing the OV effect against a highly aggressive rat brain tumor.
Figure 4.
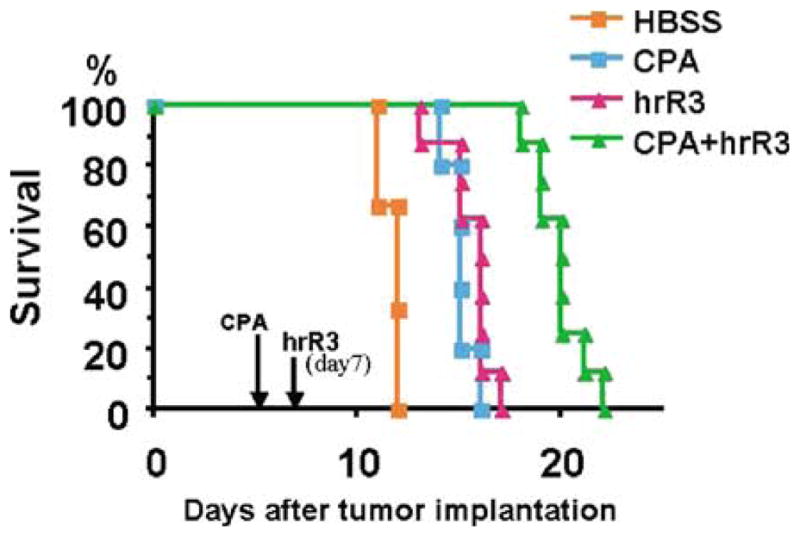
Long-term survival analysis of immunocompetent rats harboring rat malignant glioma. Fischer 344 Rats harboring D74/HveC glioma were treated intraperitoneally with either saline or CPA, 5 days after tumor cell implantation. After 2 days, animals were anesthetized and either hrR3 or HBSS was stereotactically injected into the tumor mass. All animals were observed daily to assess the survival. The treatment with CPA alone or hrR3 alone produced a significant increase in survival over mock treatment (P< 0.01, Wilcoxon test). The prolongation of survival was also significant with CPA + hrR3 combined treatment compared to the treatment with CPA alone or hrR3 alone (P <0.01, Wilcoxon test).
The effect of CPA is not related to enhancement of OV replication in infected tumor cells
Enhancement of viral replication in cells treated with DNA-damaging radiation has been reported,25 and a number of chemotherapeutic agents, such as CPA, function by damaging DNA. This led us to seek whether this could provide an explanation for the observed results. CPA is an inactive prodrug that is metabolized to its active metabolite(s), first 4-hydroxyCPA and then phosphoramide mustard.26 The latter is the active metabolite that damages DNA and is responsible for CPA’s anticancer and immunosuppressive effects. We thus measured effects of the active CPA metabolite on OV replication in cultured D74/Hve C cells. While exposure of cells to saline led to a widespread destruction by the OV, exposure to low or high doses of 4-hydroperoxyCPA (a pharmacologically stable form of 4-hydroxyCPA, which spontaneously converts into PM) inhibited OV-mediated expression of its lacZ transgene as well as widespread cytopathic effects (Figure 5a). The same findings were also obtained with human U87dEGFR glioma cells (data not shown). If the prodrug CPA was added instead of the active metabolite, there was no effect on cell viability or viral yields (Figure 5b). These results suggested that a direct effect of CPA or its activated metabolite on glioma cells was not likely responsible for the observed facilitation of OV survival within the infected tumors.
Figure 5.
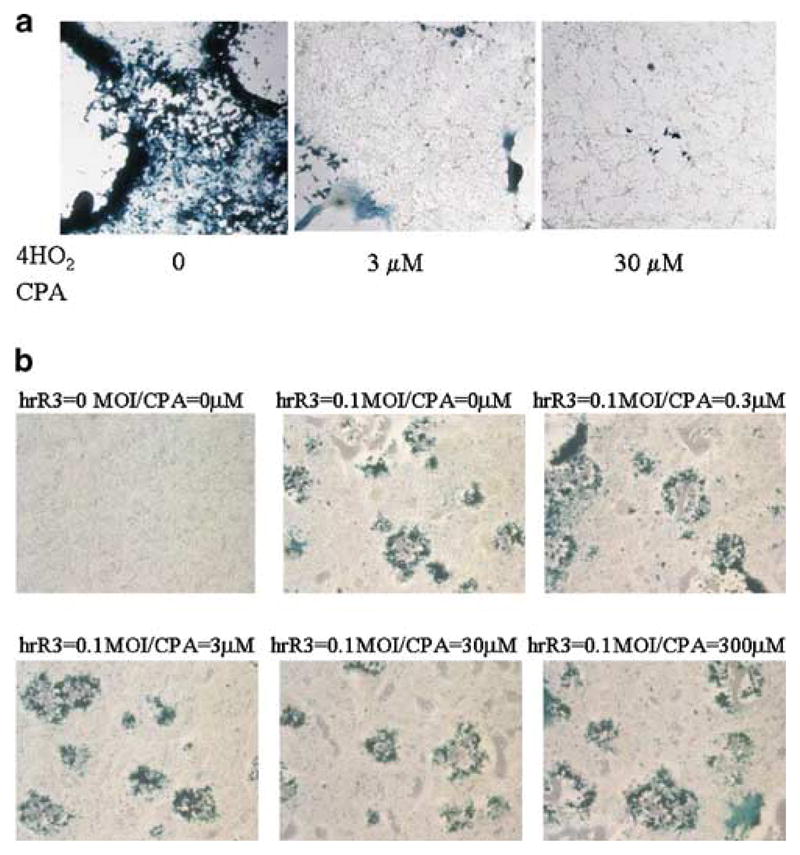
The active metabolite of CPA inhibits OV propagation in rat glioma cells in vitro. In panel a, three concentrations of 4-hydroperoxyCPA (0, 3, or 30 μM, as indicated) were added to D74/HveC cells in culture as cells were being infected with hrR3 (moi=0.01). After 30 h, cells were fixed in 0.5% glutaraldehyde and stained with X-gal to visualize the formation of plaques. The concentration of 30 μM was selected as the maximum value, because slightly higher concentrations resulted into apparent toxicity to cells in vitro (data not shown). In panel b, hrR3 (moi of 0.1) or vehicle was added onto cells in the presence of increasing concentrations of CPA (from 0 to 300 μM). Staining for lacZ expression was performed as described above.
The CPA effect is replicated in athymic rats
The previous findings rendered it unlikely that the observed facilitation of viral survival/replication was due to a direct effect of CPA or its metabolites on infected tumor cell DNA. We sought to elucidate if the observed effect of CPA depended on the presence of an intact immune system. Although we had previously shown that CPA’s facilitatory effects were maintained in athymic rats when the OV was delivered via the circulation,19–21 we asked if the CPA effect was maintained when the OV was delivered intratumorally into the brains of athymic rats (nu/nu, ie NIH rnu strain). Rat D74/HveC glioma tumors were established in athymic rat brains and then OV was inoculated into the tumor in animals that were pretreated or that were left untreated with CPA. Figure 6 shows that the percentage of D74/HveC tumor that showed lacZ transgene expression was significantly larger in the CPA-treated versus - untreated rats. This differential was comparable to that measured in immunocompetent rats (see Figure 1b), thus eliminating a role for T cells derived from thymus (but not for extrathymic-derived T cells) in the observed CPA responses.
Figure 6.
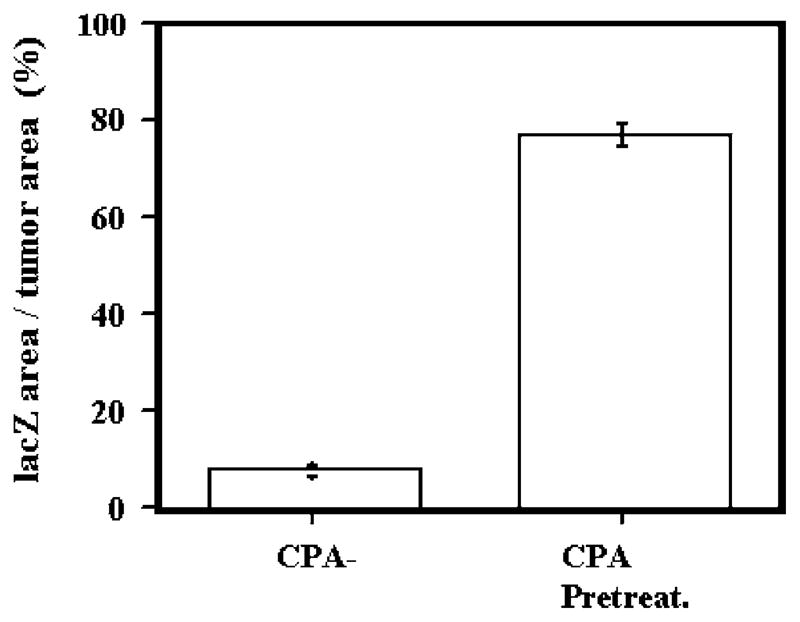
CPA treatment also increases the anatomic area of OV-mediated transgene expression in athymic rats. D74/HveC glioma-harboring athymic rats were injected with hrR3 intratumorally after CPA or saline treatment. After 3 days, brain tumors were harvested and lacZ gene expression was quantitated by staining with X-gal and then by computer-assisted measurements of percentage of lacZ-expressing tumor area.
CPA suppresses the antiviral cytokine response mediated by PBMCs in response to OV infection of brain tumors
CPA is known to produce myelosuppression and, indeed, we did measure a rapid drop of the PBMC count. At 24 h, PBMC count was a little more than half of baseline and by 48 h it was a third of the baseline (Figure 7a). This suppression of PBMC counts lasted for 1 week after a single dose of 80 mg/kg before gradually recovering to basal levels (data not shown).
Figure 7.
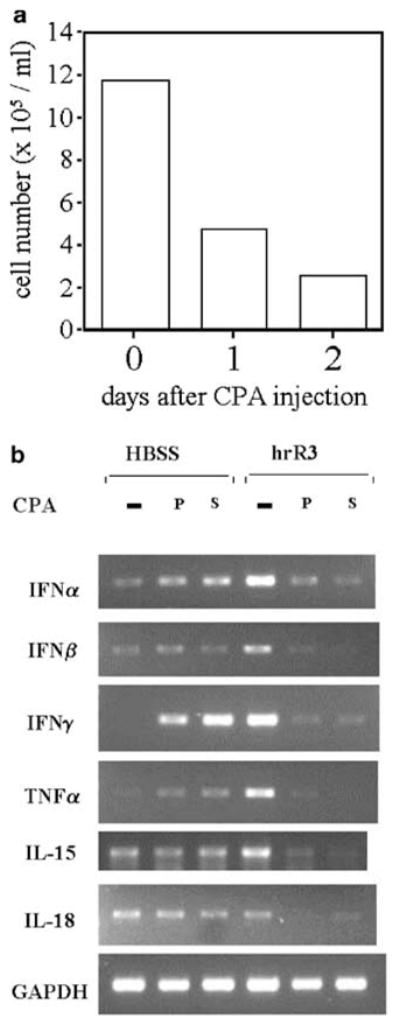
PBMCs and antiviral cytokine gene expression is decreased by CPA treatment. In panel a, the number of PBMCs isolated from Fisher 344 rats with D74/HveC brain tumors is shown 0, 24, and 48 h after treatment with CPA. In panel b, Fisher 344 rats with D74/HveC brain tumors were treated with saline (row labeled ‘−’), were treated with CPA for 2 days (row labeled ‘pre’), or were treated with CPA the same day (row labeled ‘simul.’) that brain tumors were injected with hrR3 or HBSS. At 12 h after OV injection, total RNA was prepared from PBMC and used for RT-PCR analysis to investigate the expression of a variety of cytokines. Equal numbers of PBMCs from each group were used to prepare RNAs.
The innate immune system provides the first line of defense against viral infections. We had previously shown that complement activation limited infection of tumors, when the OV was delivered intravascularly, but complement depletion did not facilitate further propagation of the OV inside the tumor that had already been infected.21 PBMCs (monocytes/macrophages, NK cells, and γ/δ TCR + T lymphocytes) provide another line of cellular innate defenses and react to virus infections by expressing several cytokines (IFNα, β, and γ, TNFα, IL15 and IL18) with antiviral activity.10–17 We thus analyzed expression of these cytokines’ mRNAs by PBMCs, 12 h after OV injection in control rats and in rats that had been pretreated with CPA. As expected under normal circumstances (ie saline-treated rats), IFNα, IFNβ, IFNγ, TNFα, and IL-15 mRNA production by PBMCs was significantly induced by the intraneoplastic injection of oncolytic HSV, while production of IL-18 mRNA was unchanged (compare the −CPA lanes for HBSS versus hrR3 groups in Figure 7b). However, animals belonging to the simultaneous or pretreated CPA group exhibited suppressed mRNA levels of all tested cytokines within 12 h of OV injection into tumors. Such a rapid decrease in mRNA production was even more significant in the light of the finding that CPA treatment alone in the HBSS-treated animals appeared to induce production of some cytokine mRNAs, particularly that for IFNγ (compare the P or S lanes to the −CPA lane in the HBSS group). This CPA-mediated induction of cytokines in PMBCs has been previously reported2,25,27 and it relates to the effect of a single dose of CPA causing a Th1 commitment by CD4 + T cells and production of the cytokine by tumor-infiltrating macrophages. For these experiments, the same number of PBMCs from HBSS-treated and CPA-treated groups were used to prepare RNA, and thus observed changes are indicative of changes in mRNA levels for the same number of PBMCs in the HBSS-treated versus CPA-treated groups. These results thus indicated that CPA impaired the innate PBMC response to oncolytic HSV – a response that consisted of antiviral cytokine mRNA production.
CPA does not increase systemic distribution of intratumorallyadministered OV
Suppression of PBMC production of antiviral cytokines might suggest that systemic dissemination of intratumorally administered OV could occur. To address this, we performed an extremely sensitive Southern/PCR analysis to detect the presence of viral genome in a variety of organs from CPA-untreated and pretreated rats. As shown in Figure 8, amplification of HSV glycoprotein C sequences was detected in tumors from CPA-treated and control rats. In addition, in CPA-treated rats, HSV gC sequences were observed in brain that had been dissected from adjacent tumor, but no sequences were assayed in control rats. However, there was no evidence for HSV genomes in trigeminal ganglia, liver, spleen, or rat plasma even with radioactive probing of PCR products. This result indicated that CPA treatment, while decreasing PBMC counts and antiviral cytokine production, did not result in systemic dissemination of intratumorally administered OV.
Figure 8.
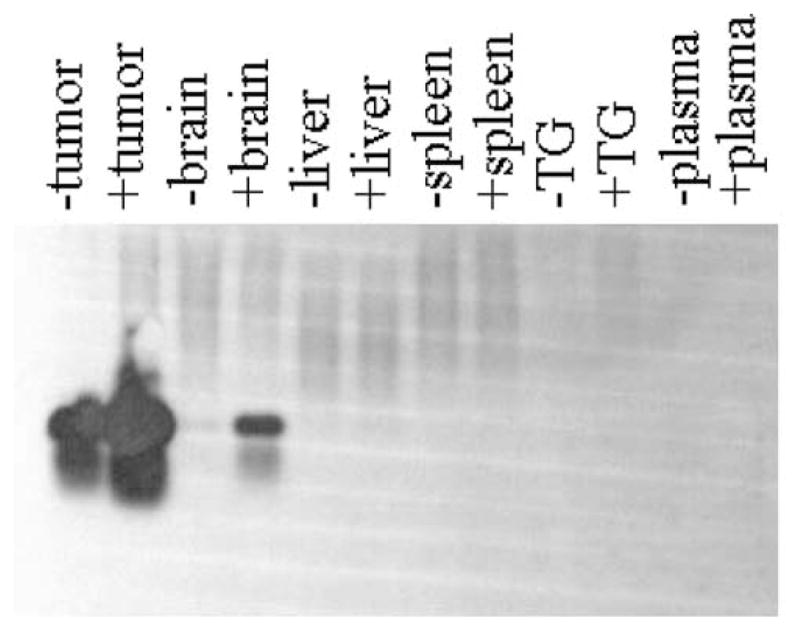
PCR analysis of HSV genomes in rat organs after intratumoral administration of hrR3. Fischer 344 rats harboring D74/HveC brain tumors that were treated with saline (lanes labeled ‘−’) or with CPA (lanes labeled ‘+’) were then treated with intratumoral injection of hrR3. After 12 h, animals were euthanized and the indicated organs were harvested. DNA was prepared as described in Materials and methods and was then analyzed by PCR using primers specific to the sequence for glycoprotein C. Southern analysis of the PCR products was performed using a glycoprotein C DNA fragment as a probe. TG, trigeminal ganglia.
Discussion
The objective of the current study was to further our understanding on mechanisms that facilitate the observed increase in OV anticancer action after immunosuppression with CPA. We were able to show that (1) CPA treatment increased the area of tumor infected by an OV and survival of animals harboring brain tumors, (2) this was associated with an increase in OV genomes within tumor and surrounding brain, but not in other organs or plasma, (3) CPA treatment allowed for increased survival of replicative OV within infected tumors and prevented the rapid decrease in OV concentration, observed within infected tumors, (4) this outcome was not likely due to a direct enhancement of OV replication within an infected tumor cell by the prodrug’s metabolites, (5) this effect was associated with a relatively rapid suppression of antiviral cytokine mRNA production by PBMCs. These results thus argue that CPA may prevent the rapid and innate antiviral response of the host that is associated with the observed decrease in OV within infected tumors. This innate antiviral response may be one of the factors responsible for limiting viral oncolysis during the initial phases of OV infection and propagation.
In our previous studies, we had shown that, in athymic rat models, OV administration through the vasculature of the animal resulted in complement activation, even in the absence of neutralizing immunity.19,20 Interestingly, the pathways of complement activation differed according to species used: while rats and humans appeared to activate the complement cascade through virion binding by innate IgM and IgG (‘classical’ pathway), mice (but also rats) activated complement through the lectin or mannose-binding protein pathway.21 Inhibition of complement activation using CVF (to deplete the C3 component) or CPA (to deplete B cells responsible for innate IgM production, thus limiting binding to OV) resulted in significant enhancement of initial tumor infection by intravascular OV. In fact, although administration of 109 PFUs of OV could be accomplished by carotid artery injection, only 0–4 PFUs of OV could be initially found in infected brain tumors without inhibiting complement, while approximately 30–50 PFUs were measured when complement inhibition was applied. Curiously, complement depletion alone (for example with CVF), while leading to an increase in initial tumor infection by intravascular OV, did not result in subsequent increases in subsequent rounds of tumor infection by progeny viruses (‘propagation’). In fact, we found that, only upon addition of CPA, did increases in propagation occur. This suggested that the effects of CPA and CVF must have differed: in addition to complement inhibition, and unlike CVF, CPA must have also acted on additional pathways that limited OV propagation within infected tumors. In fact, herein we found that CPA’s action was associated with a rapid decrease in PBMC counts, and even residual PBMCs were suppressed in expression of antiviral cytokine genes. Therefore, we propose a model in which a state of relative tumor nonpermissivity for OV propagation, possibly induced by PBMCs within the vasculature of the tumor and/or by γ/δ T cells, NK cells, and macrophages found within the tumor, can be modified by CPA by decreasing the concentration of such PBMCs and suppressing their production of several antiviral cytokines. It is interesting to note how these cytokines and their mediators have been linked in their ability to modulate HSV replication in the nervous system. IFNγ, produced primarily by extrathymic γ/δ TCR + T cells, can synergize with TNFα, produced primarily by macrophages, in inhibiting HSV replication in the nervous system.11 It has been shown to stimulate nervous system production of iNOS, an inhibitor of HSV replication.11,16 IFNγ can also activate OAS, leading to cellular inhibition of viral replication.
Our results also seem to suggest that pretreatment with CPA appeared to provide increased viral replication in the tumor compared to treatment administered the same day of OV injection. This would be consistent with an immunosuppressive effect by the prodrug’s metabolites, which in 24–48 h produced a decrease in PBMCs (see Figure 7a) and decreased cytokine mRNA production by existing PBMCs (see Figure 7b). How this time frame of CPA action might translate to a human therapeutic situation would depend on dosing and schedules commonly used in humans to achieve immunosuppression and will require some careful dose-escalation or changes in schedule studies to fully determine.
In this report, we employed immunocompetent models of syngeneic rat brain tumors essentially replicating the same findings that were observed in athymic models of human brain tumor xenografts.19–21 We explored initially the expression of several cytokines known to possess antiviral action. Interferons α/β are perhaps the most powerful antiviral cytokines and endow cells with a state of relative nonpermissivity for productive infection and replication by HSV, by inducing OAS and PKR.28,29 Interestingly, PKR activation has been linked to activation of the proinflammatory NF-KB pathway as well as to iNOS production in response to influenza virus.30–32 The role of interferon γ in limiting HSV infection has been debated, with some authors advocating a relatively minor role,29 while others finding that it is an important antiviral mediator.12,33 It is produced by activated macrophages and extrathymic, γδ-TCR + T cells in response to viral infections.11 IFNγ can directly inhibit HSV replication as well as stimulate production of iNOS, whose generation of nitric oxide has been shown to be a powerful inhibitor of HSV replication.16,17 Interleukin-15 and -18 represent interesting cytokines. In humans, IL-15 is produced by activated PBMCs and it is responsible for the activation of NK cells against HSV1.10,14 IL18 is produced by activated macrophages and it induces IFNγ production by mouse helper T cells and NK cells.15 Similarly, TNFα is an important initial media of the inflammatory response and inhibition of its expression would appear to be a necessary step for efficient viral infection and replication.11 In addition to the cytokines that we tested, there may be several other mediators of antiviral responses whose expression is likely to be modulated by CPA. In fact, in analyses of gene expression profiles between CPA-treated and control PBMCs, we have found that the expression of several mediators of acute antiviral responses is rapidly downregulated (Fulci and Chiocca, unpublished results). In addition, experiments are in progress to determine the effects of CPA on virus-mediated induction of cytokine expression within the brain tumor itself. It will be interesting to try to define if there is one particular set of antiviral responses that is responsible for limiting OV survival in tumors, because pharmacologic means could be then employed to inhibit these in a fashion that is more selective than the pleiotropic immunosuppressive actions mediated by CPA. In the experiments presented in this study, we sought to measure acute responses in PBMCs that occurred in CPA-treated or saline-treated animals, 12 h after direct intratumoral injection of the OV. We reasoned that such acute responses were better measured by assaying mRNA expression rather than protein level/function, which would be assayable at later time points. It should not be too surprising that PBMCs are affected by intratumoral OV since these tumors are well vascularized, and thus PBMCs circulating through the brain tumor are likely to be exposed to administered virus.
CPA has been used experimentally in the study of wild-type HSV pathogenesis and, in fact, it is routinely employed to enhance CNS ‘spread’ of wild-type HSV1 from systemic sites.9,34 This has always been attributed to its myelosuppressive effects without attempts to further define in more detail the molecular basis for this capacity. Interestingly, CPA has been associated with a decrease in iNOS production in alveolar macrophages in response to mycoplasma.35 For the first time, this report associates OV survival and propagation within tumors with suppressed antiviral cytokine responses in response to CPA. Additional analyses though will be needed to determine which cytokines may be more relevant in our models. The alternative explanation to our findings is that the primary mechanism of CPA on the observed increase in virus-mediated oncolysis is not immunosuppressive, rather it relates to its anticancer action. CPA’s active metabolite, phosphoramide mustard will alkylate DNA strands producing DNA strand breaks during mitosis that lead to tumor cell apoptosis. One thus would predict that such an effect should be observed in vitro as well. However, our in vitro experiments clearly show that addition of the activated form of CPA (4-hydroperox-yCPA) leads to an effect that is opposite to that observed in vivo. We thus believe that these data argue for an initial immunosuppressive mechanism of augmentation of viral oncolysis rather than a mechanism in which CPA-mediated cellular apoptosis enhances viral replication. It should be noted that the in vivo long-term survival data shown in Figure 4 show an effect of CPA treatment on animal survival. This effect is likely due to the prodrug’s anticancer effect, but this type of assay measures long-term effects (over several days) and not the short-term effects shown in Figures 1, 2, 3, and 5–8. Our data argue that the initial effect of CPA may be to cause immunosuppression that allows for increased viral oncolysis. This is then followed by CPA’s anticancer effects (even in vitro such effects can take several days for detection36), which can provide increased survival of animals with brain tumors and further combine with viral oncolysis to cause additive effects.
Although there was a significant increase in oncolytic effect and a significant prolongation of animal survival when treated with the CPA + OV combination, we failed to detect long-term cures. This may be due to at least three factors: (1) The generated D74/HveC glioma line is very aggressive, producing rapid animal death within 2 weeks of implantation. Most other glioma lines usually produce animal deaths after 4 weeks of implantation. (2) Treatment was instituted at day 7 after implantation, when animals already showed clinical signs of disease (decreased grooming behavior) and when, by histology, tumors had already reached diameters of at least one mm. (3) In spite of HveC expression, infection of rat glioma cells is still relatively inefficient compared to infection of human glioma cells, accounting for the better survival rates observed with the latter models. (4) D74 glioma cells have been shown to be poorly immunogenic compared to other glioma cell lines (F98 or 9L) and thus additional anticancer effects mediated by cell line immunogenicity were not operative.22
In conclusion, transient immunosuppression with CPA mediates improved survival and propagation of OV within infected brain tumors. This is associated with alterations in the expression of a multitude of cytokines involved in antiviral action within PBMCs. Further studies should elucidate the relevance of different inflammatory pathways in limiting OV action.
Materials and methods
OV, cell culture
hrR3 is a genetically engineered HSV mutant, derived from HSV-1 KOS, with a disruption of the UL39 gene by insertion of the Escherichia coli lacZ gene under the control of the ICP6 promoter.37–39 MGH1 is another genetically engineered HSV mutant, derived from HSV-1 F strain, with deletion of both γ34.5 loci in addition to defective UL39 by the insertion of lacZ gene.24 Viral stocks were generated in African green monkey kidney cell culture (Vero), purified by ultracentrifugation at 20 000 rpm for 3 h through a cushion of 25% sucrose in phosphate-buffered saline (PBS). Titers were determined by plaque assays. Vero cells and D74 cells (undifferentiated rat glial tumor cells) were purchased from the American Type Culture Collection and grown at 37°C in an atmosphere containing 5% CO2 in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% heat-inactivated fetal calf serum, 100 U/ml of penicillin, and 100 μg/ml of streptomycin (complete media). Since D74 cells22 are very resistant to HSV-1 infection, we generated an HSV-infectable cell line (D74/HveC) by transducing D74 with a recombinant retrovirus RxIR-ESbsr (purchased from RIKEN DNA bank, Tsukuba, Japan), carrying a cDNA encoding human herpesvirus entry mediator C (nectin 1) (kindly provided by Dr Patricia G Spear, Northwestern University, IL),23 to facilitate viral infection. These cells were grown in complete medium with 7.5 μg/ml Blasticidin S (Calbiochem, La Jolla, CA, USA).
Animal studies
Fischer rats were purchased from Charles River laboratory (Wilmington, MA, USA) and athymic rats (nu/nu) were purchased from the National Cancer Institute. Animals were used for in vivo study at the age of 8–10 weeks. All animal experiments were performed according to the guidelines of the Subcommittee on Research Animal Care of Massachusetts General Hospital. A rat brain tumor model was created as described.19,20 Briefly, anesthesized rats were fixed in a stereotactic apparatus and a burr hole was drilled at 2.5 mm lateral to the bregma. Rat glioma D74/HveC cells (2 × 105 cells in 2 μl HBSS) were implanted into the right frontal lobe at 4 mm depth from brain surface. Some rats received i.p. injection of CPA (Bristol-Myers Squibb, Princeton, NJ, USA) at a dose of 80 mg/kg at the time points indicated in figure legends. At 7 days after tumor cell implantation, rats were anesthesized and OV (2 × 107 PFU for hrR3 or 1.8 × 106 PFU for MGH1) suspended in 5 μl HBSS was stereotactically inoculated at the same coordinates. At 3 days after virus injection, animals were killled by intracardiac perfusion with 4% paraformaldehide followed by removal of the brains. Following overnight fixation, the samples were transferred to 30% sucrose solution in PBS. Histological sections were obtained by cryostat cutting to a thickness of 20 μm throughout the tumors and stained by histochemistry using X-gal (5-bromo-4-chloro-3-indolyl-β-D-galactopyranoside) as the substrate as described,40 followed by counterstaining with neutral red. The anatomical extent of Lac-Z-expressing tumor area was calculated with the aid of computer using an image analysis software, ImagePro plus. In a survival study, animals were observed daily and euthanized when found moribund.
Immunohistochemistry
The HSV major capsid protein was detected as follows: paraformaldehyde-fixed frozen sections with 10 μm thickness were reacted with 0.5%. hydrogen peroxide in methanol for 10 min followed by 3 min incubations in 90 and 70% ethanol. After washing in PBS, the sections were subjected to microwave treatment in Na-citrate buffer for 15 min. After washing in PBS, sections were blocked with 10% normal horse serum in PBS with 1% BSA for 30 min at room temperature. A polyclonal anti-HSV1 capsid antibody (Fitzgerald, Concord, MA, USA) was applied at 1:75 dilution as the primary antibody to the sections, which were then incubated overnight at 4°C. Biotin-conjugated horse anti-mouse antibody was used as the secondary antibody. After washing, the sections were incubated with Vectastain ABC reagent (Vector, Burlingame, CA, USA) for 30 min. After extensive washing, diaminobenzidine (DAKO, Carpinteria, CA, USA) was applied as a chromogen. Counterstain was done with hematoxylin.
Virus yield assay
Titers of infectious viral particles recovered from virus-inoculated rat brains were measured as follows. Freshly removed cerebrums in 2 ml of DMEM were homogenized using a teflon homogenizer, followed by three cycles of freezing–thawing. After the final thaw, the lysate was sonicated for 20 s to ensure the release of the virus. Cellular debris was then pelleted by centrifugation. Virus-containing supernatant was collected prior to titration on Vero cells by plaque formation.
In vitro CPA metabolite assays
To study in vitro the effect of action of CPA on viral replication, 4-hydroperoxyCPA (4-HO2-CPA, kindly provided by Dr S M Ludeman at Duke University, NC, USA) or CPA was utilized. In solution, 4-HO2-CPA is spontaneously converted to 4-hydroxyCPA, which is the first active metabolite formed in the metabolism of CPA. D74/HveC cells were seeded at 1 × 105 cells/well onto 24-well plate. The following day, the medium was replaced with a medium that contained serial dilutions of freshly prepared 4-HO2-CPA, CPA, or vehicle. After 16 h, the medium was replaced with the medium containing the same concentrations of freshly prepared 4-HO2-CPA as well as hrR3, which was calculated to provide infection at various moi’s. After 30 h, the cells were fixed in 0.5% glutaraldehyde and stained with X-gal for demonstration of plaques.
RT-PCR
Reverse transcription-PCR was performed to assay cytokine mRNA expression. PBMC were isolated from heparinized rat blood by centrifugation over Lymphocyte mammal (Cederlane, Ontario, Canada). Fischer 344 rats harboring D74/HveC brain tumors with or without systemic CPA treatment were injected intratumorally with hrR3 or HBSS. After 12 h, PBMCs were isolated from heparinized rat blood by centrifugation over Lymphocyte mammal (Cederlane, Ontario, Canada). Total RNA from equal numbers of PBMCs from each experimental group was isolated by using Trizol reagent (GibcoBRL, Bethesda, MD, USA). One microgram of RNA was reverse-transcribed by using Omniscript reverse transcriptase kit (Qiagen, Valencia, CA, USA) with oligo-(dT)15 as a primer for 1 h at 37°C. PCR was performed on 2 μl of each cDNA sample using hotstar-Taq DNA polymerase (Qiagen). The PCR conditions used were 45 s denaturation at 94°C, 45 s annealing at 57°C, and 35 s extension at 72°C, and amplification was done for 30 cycles followed by additional 10 min extension. These conditions were used for all reactions. The specific primer pairs used were as follows. IFN-α, 5′-ACACTCCTGGCACAAATGAG-3′ (sense) and 5′-GTGATTCTGTGGAAGTATTCC-3′ (antisense); IFN-β, 5′-ATCGACTACAAGCAGCTCCAG-3′ (sense) and 5′-TCCTGAAGACTTCTGCTCGGA-3′ (antisense); IFN-γ, 5′-ACACTCATTGAAAGCCTAGAA-3′ (sense) and 5′-TGATGAGTTCATTGACAGCTT-3′ (antisense); IL-15, 5′-TAGGATCCATAATGAAAATTTTGAAACCATATAT-3′ (sense) and 5′-TAGAATTCTCAGGACGTGTTGATGAA-CAT-3′ (antisense); IL-18, 5′-CAGAA-GAAGGCTCTTGTGTCAA-3′ (sense) and 5′-CTTTGATGTAAGTTAGTAAGAGTG-3′(antisense);TNFα, 5′-CGCTCTTCTGTCTACTGAAC-3′ (sense) and 5′-AGG-TACATGGGCTCATACCA-3′ (antisense); GAPDH, 5′-CACTCAGAAGACTGTGGATG-3′ (sense) and 5′-GTCATTGAGAGCAATGCCAG-3′ (antisense). Amplified products (10 μl) were separated on 2.0% agarose.
PCR for viral genome
DNA was isolated from tumors, surrounding brain tissues, trigeminal ganglias, and livers of the rats using a commercially available kit (Qiagen). DNA was also isolated from serum using viral RNA easy kits from Qiagen.
Statistical analysis
Student’s t-test or Wilcoxon’s test was used for the statistical analyses as described in the figure legends. P-values of less than 0.05 were considered significant.
Acknowledgments
We thank Y Saeki and other members of the EAC laboratory for their input in the described experiments. We acknowledge J Basilion and M Pasternack for useful discussions. We dedicate this work to the memory of Dr Keiro Ikeda (MGH and Keio University). This work was supported by a NIH research Grant (CA 69246), the Berkowitz-Knott Fund for Brain Tumor Research, and a grant from the Uehara Memorial Foundation (Tokyo, Japan).
References
- 1.Chiocca EA, Smith ER. Oncolytic viruses as novel anticancer agents: turning one scourge against another (In Process Citation) Expert Opin Investig Drugs. 2000;9:311–327. doi: 10.1517/13543784.9.2.311. (MEDLINE record in process) [DOI] [PubMed] [Google Scholar]
- 2.Kirn D, Martuza RL, Zwiebel J. Replication-selective virotherapy for cancer: biological principles, risk management and future directions. Nat Med. 2001;7:781–787. doi: 10.1038/89901. [DOI] [PubMed] [Google Scholar]
- 3.Chiocca EA. Oncolytic viruses. Nat Rev Cancer. 2002;2:938–950. doi: 10.1038/nrc948. [DOI] [PubMed] [Google Scholar]
- 4.Markert JM, et al. Conditionally replicating herpes simplex virus mutant, G207 for the treatment of malignant glioma: results of a phase I trial (see comments) Gene Therapy. 2000;7:867–874. doi: 10.1038/sj.gt.3301205. [DOI] [PubMed] [Google Scholar]
- 5.Rampling R, et al. Toxicity evaluation of replication-competent herpes simplex virus (ICP 34.5 null mutant 1716) in patients with recurrent malignant glioma. Gene Therapy. 2000;7:859–866. doi: 10.1038/sj.gt.3301184. [DOI] [PubMed] [Google Scholar]
- 6.Khuri FR, et al. A controlled trial of intratumoral ONYX-015, a selectively-replicating adenovirus, in combination with cisplatin and 5-fluorouracil in patients with recurrent head and neck cancer (see comments) Nat Med. 2000;6:879–885. doi: 10.1038/78638. [DOI] [PubMed] [Google Scholar]
- 7.Ichikawa T, Chiocca EA. Comparative analyses of transgene delivery and expression in tumors inoculated with a replication-conditional or -defective viral vector. Cancer Res. 2001;61:5336–5339. [PubMed] [Google Scholar]
- 8.Wakimoto H, Johnson PR, Knipe DM, Chiocca EA. Effects of innate immunity on herpes simplex virus and its ability to kill tumor cells. Gene Therapy. 2003;10:983–990. doi: 10.1038/sj.gt.3302038. [DOI] [PubMed] [Google Scholar]
- 9.Mitchell BM, Stevens JG. Neuroinvasive properties of herpes simplex virus type 1 glycoprotein variants are controlled by the immune response. J Immunol. 1996;156:246–255. [PubMed] [Google Scholar]
- 10.Fawaz LM, Sharif-Askari E, Menezes J. Up-regulation of NK cytotoxic activity via IL-15 induction by different viruses: a comparative study. J Immunol. 1999;163:4473–4480. [PubMed] [Google Scholar]
- 11.Kodukula P, et al. Macrophage control of herpes simplex virus type 1 replication in the peripheral nervous system. J Immunol. 1999;162:2895–2905. [PubMed] [Google Scholar]
- 12.Feduchi E, Alonso MA, Carrasco L. Human gamma interferon and tumor necrosis factor exert a synergistic blockade on the replication of herpes simplex virus. J Virol. 1989;63:1354–1359. doi: 10.1128/jvi.63.3.1354-1359.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Goodbourn S, Didcock L, Randall RE. Interferons: cell signalling, immune modulation, antiviral response and virus countermeasures. J Gen Virol. 2000;81:2341–2364. doi: 10.1099/0022-1317-81-10-2341. [DOI] [PubMed] [Google Scholar]
- 14.Ahmad A, Sharif-Askari E, Fawaz L, Menezes J. Innate immune response of the human host to exposure with herpes simplex virus type 1: in vitro control of the virus infection by enhanced natural killer activity via interleukin-15 induction. J Virol. 2000;74:7196–7203. doi: 10.1128/jvi.74.16.7196-7203.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fujioka N, et al. Interleukin-18 protects mice against acute herpes simplex virus type 1 infection (viruses: a comparative study) J Virol. 1999;73:2401–2409. doi: 10.1128/jvi.73.3.2401-2409.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Karupiah G, et al. Inhibition of viral replication by interferon-gamma-induced nitric oxide synthase. Science. 1993;261:1445–1448. doi: 10.1126/science.7690156. [DOI] [PubMed] [Google Scholar]
- 17.Croen KD. Evidence for antiviral effect of nitric oxide. Inhibition of herpes simplex virus type 1 replication. J Clin Invest. 1993;91:2446–2452. doi: 10.1172/JCI116479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Leib DA, et al. Specific phenotypic restoration of an attenuated virus by knockout of a host resistance gene. Proc Natl Acad Sci USA. 2000;97:6097–6101. doi: 10.1073/pnas.100415697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ikeda K, et al. Complement depletion facilitates the infection of multiple brain tumors by an intravascular, replication-conditional herpes simplex virus mutant. J Virol. 2000;74:4765–4775. doi: 10.1128/jvi.74.10.4765-4775.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ikeda K, et al. Oncolytic virus therapy of multiple tumors in the brain requires suppression of innate and elicited antiviral responses. Nat Med. 1999;5:881–887. doi: 10.1038/11320. [DOI] [PubMed] [Google Scholar]
- 21.Wakimoto H, et al. The complement response against an oncolytic virus is species-specific in its activation pathways. Mol Ther. 2002;5:275–282. doi: 10.1006/mthe.2002.0547. [DOI] [PubMed] [Google Scholar]
- 22.Tzeng JJ, Barth RF, Orosz CG, James SM. Phenotype and functional activity of tumor-infiltrating lymphocytes isolated from immunogenic and nonimmunogenic rat brain tumors. Cancer Res. 1991;51:2373–2378. [PubMed] [Google Scholar]
- 23.Geraghty RJ, et al. Entry of alpha herpesviruses mediated by poliovirus receptor-related protein 1 and poliovirus receptor. Science. 1998;280:1618–1620. doi: 10.1126/science.280.5369.1618. [DOI] [PubMed] [Google Scholar]
- 24.Kramm CM, et al. Therapeutic efficiency and safety of a second-generation replication-conditional HSV1 vector for brain tumor gene therapy. Hum Gene Ther. 1997;8:2057–2068. doi: 10.1089/hum.1997.8.17-2057. [DOI] [PubMed] [Google Scholar]
- 25.Advani SJ, et al. Enhancement of replication of genetically engineered herpes simplex viruses by ionizing radiation: a new paradigm for destruction of therapeutically intractable tumors. Gene Therapy. 1998;5:160–165. doi: 10.1038/sj.gt.3300546. [DOI] [PubMed] [Google Scholar]
- 26.Clarke L, Waxman DJ. Oxidative metabolism of cyclophosphamide: identification of the hepatic monooxygenase catalysts of drug activation. Cancer Res. 1989;49:2344–2350. [PubMed] [Google Scholar]
- 27.Matar P, Rozados VR, Gervasoni SI, Scharovsky GO. Th2/Th1 switch induced by a single low dose of cyclophosphamide in a rat metastatic lymphoma model. Cancer Immunol Immunother. 2002;50:588–596. doi: 10.1007/s00262-001-0237-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Leib DA. Counteraction of interferon-induced antiviral responses by herpes simplex viruses. Curr Top Microbiol Immunol. 2002;269:171–185. doi: 10.1007/978-3-642-59421-2_11. [DOI] [PubMed] [Google Scholar]
- 29.Leib DA, et al. Interferons regulate the phenotype of wild-type and mutant herpes simplex viruses in vivo. J Exp Med. 1999;189:663–672. doi: 10.1084/jem.189.4.663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zamanian-Daryoush M, Mogensen TH, DiDonato JA, Williams BR. NF-kappaB activation by double-stranded-RNA-activated protein kinase (PKR) is mediated through NF-kappaB-inducing kinase and IkappaB kinase. Mol Cell Biol. 2000;20:1278–1290. doi: 10.1128/mcb.20.4.1278-1290.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Deb A, et al. RNA-dependent protein kinase PKR is required for activation of NF-kappa B by IFN-gamma in a STAT1-independent pathway. J Immunol. 2001;166:6170–6180. doi: 10.4049/jimmunol.166.10.6170. [DOI] [PubMed] [Google Scholar]
- 32.Uetani K, et al. Central role of double-stranded RNA-activated protein kinase in microbial induction of nitric oxide synthase. J Immunol. 2000;165:988–996. doi: 10.4049/jimmunol.165.2.988. [DOI] [PubMed] [Google Scholar]
- 33.Geiger KD, et al. Interferon-gamma protects against herpes simplex virus type 1-mediated neuronal death. Virology. 1997;238:189–197. doi: 10.1006/viro.1997.8841. [DOI] [PubMed] [Google Scholar]
- 34.Halford WP, Schaffer PA. Optimized viral dose and transient immunosuppression enable herpes simplex virus ICP0-null mutants to establish wild-type levels of latency in vivo. J Virol. 2000;74:5957–5967. doi: 10.1128/jvi.74.13.5957-5967.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hickman-Davis JM, Lindsey JR, Matalon S. Cyclophosphamide decreases nitrotyrosine formation and inhibits nitric oxide production by alveolar macrophages in mycoplasmosis. Infect Immun. 2001;69:6401–6410. doi: 10.1128/IAI.69.10.6401-6410.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wei MX, et al. Diffusible cytotoxic metabolites contribute to the in vitro bystander effect associated with the cyclophosphamide/cytochrome P450 2B1 cancer gene therapy paradigm. Clin Cancer Res. 1995;1:1171–1177. [PubMed] [Google Scholar]
- 37.Goldstein DJ, Weller SK. Herpes simplex virus type 1-induced ribonucleotide reductase activity is dispensable for virus growth and DNA synthesis: isolation and characterization of an ICP6 lacZ insertion mutant. J Virol. 1988;62:196–205. doi: 10.1128/jvi.62.1.196-205.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Goldstein DJ, Weller SK. Factor(s) present in herpes simplex virus type 1-infected cells can compensate for the loss of the large subunit of the viral ribonucleotide reductase: characterization of an ICP6 deletion mutant. Virology. 1988;166:41–51. doi: 10.1016/0042-6822(88)90144-4. [DOI] [PubMed] [Google Scholar]
- 39.Goldstein DJ, Weller SK. An ICP6::lacZ insertional mutagen is used to demonstrate that the UL52 gene of herpes simplex virus type 1 is required for virus growth and DNA synthesis. J Virol. 1988;62:2970–2977. doi: 10.1128/jvi.62.8.2970-2977.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Boviatsis EJ, et al. Gene transfer into experimental brain tumors mediated by adenovirus, herpes simplex virus, and retrovirus vectors. Hum Gene Ther. 1994;5:183–191. doi: 10.1089/hum.1994.5.2-183. [DOI] [PubMed] [Google Scholar]