

Abstract
Elevated levels of fluoroquinolone resistance are frequently found among Escherichia coli clinical isolates. This study investigated the antibiotic resistance mechanisms of strain NorE5, derived in vitro by exposing an E. coli clinical isolate, PS5, to two selection steps with increasing concentrations of norfloxacin. In addition to the amino acid substitution in GyrA (S83L) present in PS5, NorE5 has an amino acid change in ParC (S80R). Furthermore, we now find by Western blotting that NorE5 has a multidrug resistance phenotype resulting from the overexpression of the antibiotic resistance efflux pump AcrAB-TolC. Microarray and gene fusion analyses revealed significantly increased expression in NorE5 of soxS, a transcriptional activator of acrAB and tolC. The high soxS activity is attributable to a frameshift mutation that truncates SoxR, rendering it a constitutive transcriptional activator of soxS. Furthermore, microarray and reverse transcription-PCR analyses showed that mdtG (yceE), encoding a putative efflux pump, is overexpressed in the resistant strain. SoxS, MarA, and Rob activated an mdtG::lacZ fusion, and SoxS was shown to bind to the mdtG promoter, showing that mdtG is a member of the marA-soxS-rob regulon. The mdtG marbox sequence is in the backward or class I orientation within the promoter, and its disruption resulted in a loss of inducibility by MarA, SoxS, and Rob. Thus, chromosomal mutations in parC and soxR are responsible for the increased antibiotic resistance of NorE5.
Escherichia coli is the most frequent pathogen isolated from patients with urinary tract infections (UTIs). The prevalence of this microorganism in uncomplicated UTIs is between 71 and 90% throughout the world (3, 18), but the percentage is lower for complicated infections (1, 3). The common therapy for UTIs is ampicillin or trimethoprim-sulfamethoxazole. However, the increasing frequency of resistance to these agents (reaching maximum levels of >40% and >20%, respectively, in countries such as Spain, Portugal, Ireland, and Korea [1, 17, 18]) often necessitates the use of a second antibiotic, such as ciprofloxacin. However, resistance to ciprofloxacin is also increasing in some geographic areas, to >20% (17, 18). Reasonable alternatives for treating uncomplicated infections are nitrofurantoin and fosfomycin, due to the low rates of resistance detected so far (3, 17).
The mechanisms of quinolone resistance in E. coli strains have been studied in detail. They result from both chromosomally encoded mutations and plasmid-mediated quinolone resistance (10). The first mechanism includes mutations of the target genes gyrA and gyrB, which encode DNA gyrase, and parC and parE, encoding topoisomerase IV (29, 35, 41, 43, 44), as well as mutations responsible for a decrease in quinolone permeability, either by increasing the efflux or by decreasing the expression of outer membrane proteins used as entrance channels (5, 7, 9, 34). The second mechanism, the prevalence of which is steadily increasing, relies on the presence of plasmid mediated determinants, such as qnr, qepA, and aac(6′)-Ib-cr (14, 48, 49).
The acquisition of quinolone resistance occurs sequentially. For E. coli strains, the first step usually involves mutations within the quinolone resistance-determining regions (QRDRs) of the target genes and is associated with 32- and 10-fold increases in the MICs of nalidixic acid and ciprofloxacin, respectively (32). The second-step mutations are often within the regulatory loci that control efflux pump expression and usually show a 2- to 8-fold increase in quinolone resistance levels (16). At this step, a multidrug resistance (MDR) phenotype is detected, since efflux pumps have a wide range of exportable substrates, so that cross-resistance with other antibiotics results (7, 10). AcrAB is the main efflux pump described for Enterobacteriaceae and acts in conjunction with TolC (11, 32, 34). Five regulators that play roles in AcrAB expression have been described so far (10, 13). AcrR, the local repressor encoded upstream of acrA (20), and AcrS, located upstream of the acrEF operon (13), repress acrAB. Mutations acquired within the acrR locus have been reported to trigger a truncated/inactivated repressor (45, 46). The three other regulators, SoxS, MarA, and Rob, are members of the AraC/XylS family of transcriptional activators (25). The soxRS region contains two loci divergently expressed; when oxidized, e.g., by treatment with superoxide-generating agents such as paraquat (PQ), SoxR transcriptionally activates soxS expression (2, 8), which in turn activates a wide number of genes, the marA-soxS-rob regulon (25). Constitutive expression of SoxS results from mutations acquired within the C terminus of SoxR, which activate the protein (31). The marRAB operon also encodes a regulator, MarA, that is autorepressed by MarR, the first gene of the operon (6). Mutations within marR result in a loss of repressor activity and allow increased marA expression (6, 33). Exposure to salicylate also reduces MarR repression and increases marA expression (37). Rob is posttranscriptionally activated upon treatment of cells with a bile salt, decanoate or 2,2′-dipyridyl (DIP) (4, 36, 38). However, clinical significance has been associated only with the mutation of AcrR (45, 46) and the overexpression of MarA and SoxS.
SoxS, MarA, and Rob have highly overlapping regulons (25). They bind as monomers to a 20-bp asymmetric sequence with the degenerate consensus sequence AYNGCACNNWNNRYYAAAYN (where N stands for any base, R stands for A or G, W stands for A or T, and Y stands for C or T). This binding site is referred to as the “marbox.” A marbox is present upstream of the promoters of all the regulon genes (such as acrAB, tolC, and marRAB itself, which is autoactivatable) (21). SoxS and MarA bind to the marboxes of different genes and activate them to different extents, such as the fpr promoter, which is more susceptible to activation by SoxS than by MarA (22).
Here we characterize the mechanisms of fluoroquinolone resistance acquired in vitro by a fluoroquinolone-susceptible E. coli clinical isolate upon exposure to increasing concentrations of norfloxacin. A mutation in parC and a novel mutation in soxR appear to be responsible.
MATERIALS AND METHODS
Bacterial strains, plasmids, and growth conditions.
The bacterial strains and plasmids used in this study are listed in Table 1. All cultures were grown in LB broth at 37°C with shaking or on LB plates supplemented with ampicillin (100 μg/ml) or kanamycin (35 μg/ml) when necessary. The indicator 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside (X-Gal) was added to LB plates at a final concentration of 40 μg/ml.
TABLE 1.
Bacteria, plasmids, and phages
| Bacterium, plasmid, or phage | Parent strain | Relevant genotype and/or characteristics | Source or reference |
|---|---|---|---|
| Bacteria | |||
| PS5 | E. coli clinical isolate susceptible to fluoroquinolones | 42 | |
| NorE5 | Fluoroquinolone-resistant mutant selected from PS5 with norfloxacin | 42 | |
| GC4468 | E. coli K-12 (ΔlacsoxRS+) | 12 | |
| DJ901 | GC4468 | ΔsoxRS | 12 |
| JTG936 | GC4468 | soxR(Con) | 12 |
| GC48-F | GC4468 | pRS551 fpr::lacZ | This study |
| P5-F | PS5 | pRS551 fpr::lacZ | This study |
| N5-F | NorE5 | pRS551 fpr::lacZ | This study |
| M4450 | GC4468 | mdtG67::lacZ (−159 to +6) | This study |
| M4452 | GC4468 | mdtG77::lacZ (−98 to +6) | This study |
| M9948 | 834(D3lysS) | pRGM9948 | 21 |
| Plasmids | |||
| pRS551 | pBR322 derivative; Ampr | 40 | |
| pRGM9948 | pET15b carrying the soxS gene; Ampr | 21 | |
| Phage, λRS45 | λimm21; Kanr | 40 |
Cells were treated with 50 μM PQ, 5 mM sodium salicylate (SAL), or 5 mM DIP and were incubated at 37°C with shaking for 1 h where indicated.
Susceptibility testing.
MICs of chloramphenicol, tetracycline, amoxicillin, erythromycin, trimethoprim, and amikacin for strains PS5 and NorE5 were determined by Etest (AB Biodisk, Solna, Sweden) on Mueller-Hinton (MH) plates according to the manufacturer's recommendations.
Microarray analysis.
Total RNA from PS5 and NorE5 was extracted from a mid-exponential-phase culture (optical density at 600 nm [OD600], 0.6) using RNeasy spin columns (Qiagen, Chatsworth, CA). A total of 20 μg of total RNA was labeled with Cy-3-dUTP (RNA from strain PS5) or Cy-5-dUTP (RNA from strain NorE5) in a standard reverse transcriptase (RT) reaction using Superscript II(+) (Gibco BRL, Carlsbad, CA) with 1 μg of random hexamer (Amersham Pharmacia, Piscataway, NJ) primers. After purification through Microcon-30 membranes (Millipore, Billerica, MA), Cy-3- and Cy-5-labeled cDNA samples were mixed with SSC (final concentration, 2.5×; 1× SSC is 0.15 M NaCl plus 0.015 M trisodium citrate [pH 7]), 0.25% sodium dodecyl sulfate (SDS), and 40 μg of E. coli rRNA (Boehringer Mannheim, Ingelheim, Germany) in a final volume of 16 μl and were hybridized with the DNA microarray for 5 h at 65°C. The DNA microarray contained 4,058 open reading frames (ORFs) representing 95% of E. coli ORFs, and hybridization was performed as described in the MGuide (http://cmgm.stanford.edu/pbrown/mguide/index.html). The glass slide was washed and scanned using an Axon Scanner GENPIX 1.0 (Axon Instruments, Foster City, CA) at a resolution of 10 μm per pixel. The resulting 16-bit tagged-image format file (TIFF) images were analyzed using SCANALYZE software. The reproducibility of the technique was assessed in two separate experiments. A normalized relative Cy5/Cy3 ratio of >2 was considered a significant increase in expression, and a normalized relative Cy3/Cy5 ratio of >2 was considered a significant decrease in expression, when observed for both of the two different experiments performed.
RT-PCR.
Fresh overnight cultures of PS5, NorE5, GC4468, DJ901, and JTG936 were diluted 1/100 into 15 ml LB medium and were aerated at 37°C until strains reached OD600 values of 0.5 to 0.6. Three milliliters was then taken and treated with 6 ml of RNAprotect Bacteria reagent (Qiagen, Hilden, Germany). Mixtures were processed according to the manufacturer's instructions. Pellets were resuspended in 200 μl of TE buffer (10 mM Tris-Cl, 1 mM EDTA [pH 8.0]) supplemented with 3 mg/ml lysozyme, vortexed, and incubated at 32°C for 10 min with shaking. The RNA was extracted by using an RNeasy minikit (Qiagen, Hilden, Germany) according to the manufacturer's recommendations. Samples were subsequently treated with DNA-free DNase (Ambion, Austin, TX) according to the manufacturer's recommendations until RNA samples were totally DNA free when checked by PCR using gapA (a housekeeping gene) primers. RT-PCR was performed using the AccessQuick RT-PCR system (Promega, Madison, WI) according to the manufacturer's recommendations with primers gapA.1 (GTATCAACGGTTTTGGCCG) and gapA.2 (AGCTTTAGCAGCACCGGTA) for gapA and primers yceE.RT1 (GCCAGTTCGCGGACATAC) and yceE.RT2 (CTGCGGGCGCTTCTTGGGTTACTT) for mdtG. The retrotranscription process was performed using 500 ng of RNA at 45°C for 45 min, followed by a standard PCR program. Samples were loaded in a GeneGel Excel (GE Healthcare, Uppsala, Sweden) at 600 V, 25 mA, and 15 W for 1.5 h. Gels were stained with a DNA silver-staining kit (GE Healthcare, Uppsala, Sweeden) according to the manufacturer's recommendations. Results were corroborated from two independent mRNA extractions and amplifications.
Sequencing of the soxRS region.
The whole soxRS regions of strains GC4468, PS5, and NorE5 were amplified by PCR using the same primers as previously described (soxRS.F [GGCGAAGCTTCCGCAGGTGTTTATGC] and soxRS.R [CGTCGGGGGAAGCTTTCCTGTGTACC]) (19). The amplified fragments were directly used for sequencing and were compared for the detection of mutations.
Western blotting.
Bacterial strains were grown overnight in 50 ml LB medium and were harvested by centrifugation. The pellet was rinsed twice with 10 mM Tris supplemented with 1% NaCl and was resuspended in 3 ml of the same buffer. Bacterial samples were sonicated on ice on a Vibra-Cell VCX 130 (Sonics) for a total of 3 min (30 s for each cycle of sonication, followed by 30 s of rest) with an amplitude of 50%. Cell debris was removed by centrifugation for 20 min at 4°C and 3,500 rpm, and the supernatant was collected and centrifuged again for 90 min at 4°C and 16,000 rpm. The final pellet was resuspended in 1× phosphate-buffered saline (PBS) (Roche, Mannheim, Germany). Protein quantification was performed using the RC DC protein assay kit (Bio-Rad, Hercules, CA) according to the manufacturer's indications.
Ten micrograms of each protein sample was loaded onto an 8% SDS-polyacrylamide gel electrophoresis (PAGE) gel (Mini Protean II). Transfer from the gel to a nitrocellulose membrane was performed for 2 h at 60 V on ice. The membranes were blocked using 1× PBS containing Tween 20 diluted 1/2,000 (PBS-T) and 5% skim milk for 1 h at room temperature, followed by an overnight incubation at 4°C with the primary antibodies against the AcrB and TolC proteins (Antibody Bcn, Barcelona, Spain) diluted 1/500 in PBS-T. The membranes were washed three times with PBS-T and once with PBS before the secondary antibody, anti-rabbit IgG (GE Healthcare, Buckinghamshire, United Kingdom), diluted 1/2,000 in PBS-T, was added for 1 h of incubation at room temperature. Then the membranes were washed as described above and processed using EZ-ECL (Biological Industries, Kibbutz Beit Haemek, Israel) for chemiluminescence detection of bands in a Fuji LAS-3000 instrument.
DNA manipulations.
Strains GC48-F, P5-F, and N5-F were derived from parental strains GC4468, PS5, and NorE5, respectively, by transformation with plasmid pRS551 (40) harboring the fpr::lacZ transcriptional fusion. The resulting strains were assayed for β-galactosidase activity.
A lacZ transcriptional fusion was constructed with the mdtG promoter (40). Amplification of the promoter was carried out by PCR using chromosomal DNA from strain GC4468 as the template. The mdtG67 fragment was 165 bp long (from −159 to +6, relative to the ATG codon) and was amplified with primers 1756 (AAAAAAGAATTCGGATGCTTCAGAATGGCATCCGGCATTACCACA [the EcoRI site is underlined]) and 1757 (AAAAAAGGATCCTGACATAGCAATCCGCTGTTGGTGCGCCA [the BamHI site is underlined]). A shorter fusion, containing the 104-bp mdtG77 fragment (from −98 to +6), was also constructed, using primers 1767 (AACCAAGAATTCTCTCTGGATTGCGCCCCCTGGAAGT [the EcoRI site is underlined]) and 1757. The amplified DNA fragments were digested with EcoRI and BamHI and were ligated to the similarly cut vector pRS551. Recombinant plasmids were isolated in DH5α cells by selection for ampicillin resistance and were verified by sequence analysis. Recombination between the pRS551 derivatives and λRS45 resulted in lysates bearing the transcriptional fusions. Single λ lysogens of GC4468 were obtained (40) by selection for kanamycin resistance. The mdtG::lacZ fusion lysogens were designated M4450 and M4452, respectively.
β-Galactosidase assays.
Strains M4450 and M4452, as well as strains GC48-F, P5-F, and N5-F, were assayed for β-galactosidase activity, expressed in Miller units, as previously described (26). Bacterial growth to log phase and treatments for 1 h with PQ, SAL, or DIP, where indicated, and at the concentrations mentioned above, were performed as reported previously (38, 39). All assays were carried out twice in duplicate and agreed within 15%.
DNA-binding assays.
SoxS was overexpressed using the soxS gene cloned into a pET15b vector (pRGM9948) by induction of strain M9948 with isopropyl-β-d-thiogalactopyranoside (IPTG) (21). SoxS was then purified to homogeneity, and the histidine tag was removed as previously described (15). Gel mobility experiments were performed three times in 6% acrylamide gels in TAE buffer as previously reported (24). The 20-bp oligonucleotide 5′-AGAGCTTTTATCGCTAAATC-3′ was labeled at its 5′ end using polynucleotide kinase (New England Biolabs, Beverly, MA) according to the manufacturer's recommendations and was annealed with a complementary 20-bp oligonucleotide. Trace quantities (approximately 10 fmol/sample) of the radiolabeled oligonucleotide in 9 μl of buffer containing 1× TAE buffer in 25% glycerol, 50 fmol of nonradioactive dA·dT (20 bp in length), and 0.1 μg serum albumin were mixed with 1-μl samples of purified SoxS that had been diluted in buffer containing 50 mM HEPES (pH 8.0), 0.5 M NaCl, and 25% glycerol to give the final concentrations indicated in Fig. 4. The samples were subjected to electrophoresis at 150 V for 35 min. The amounts of unbound and SoxS-complexed DNA were quantified by analysis on a Molecular Dynamics PhosphorImager.
FIG. 4.
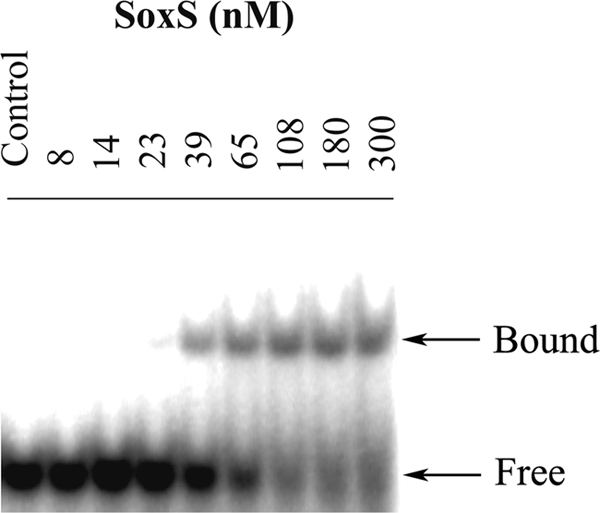
Radioautogram of a 6% acrylamide gel demonstrating the binding of SoxS to the 32P-labeled 20-bp fragment AGAGCTTTTATCGCTAAATC (the marbox of mdtG). The multiple SoxS concentrations (nM), and the unbound DNA (free) and SoxS-complexed DNA (bound), are indicated.
RESULTS
Fluoroquinolone resistance acquisition and MDR phenotype.
Strain NorE5 was selected in vitro from strain PS5, an E. coli clinical isolate, after exposure to increasing concentrations of norfloxacin as previously reported (42). The target gene mutations were described in the previous study (PS5 had an amino acid substitution in GyrA [Ser83Leu], whereas NorE5 acquired an additional change in ParC [Ser80Arg]). In the present study, a broader characterization of the antibiotic susceptibility profile was performed (Table 2). The results showed significant increases in the MICs of nalidixic acid, norfloxacin, and ciprofloxacin, as previously found, and of other classes of antibiotics—chloramphenicol, tetracycline, amoxicillin, erythromycin, and trimethoprim—whereas the levels of susceptibility to amikacin remained unchanged.
TABLE 2.
Mutations detected within the quinolone resistance-determining regions and MIC determinations for the strains
| Strain | Amino acid substitutiona |
MIC (μg/ml)b |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| GyrA | ParC | NOR | CIP | NAL | CHL | TET | AMX | ERY | TMP | AMK | |
| PS5 | S83L | — | 0.5 | 0.5 | 1,024 | 8 | 2 | 48 | 64 | 0.5 | 12 |
| NorE5 | S83L | S80R | 32 | 8 | 2,048 | 64 | 8 | >256 | >256 | 8 | 12 |
S, serine; L, leucine; R, arginine; —, no mutation found.
NOR, norfloxacin; CIP, ciprofloxacin; NAL, nalidixic acid; CHL, chloramphenicol; TET, tetracycline; AMX, amoxicillin; ERY, erythromycin; TMP, trimethoprim; AMK, amikacin. Data for NOR, CIP, NAL, and TET are from reference 38.
Changes in gene expression as determined by microarray and Western blot analyses.
To determine the basis of this MDR, gene expression in strains PS5 and NorE5 was studied using cDNA microarrays. In comparison to PS5, NorE5 overexpressed acrA and acrB approximately 2.5-fold. Since these genes encode two components of the very important AcrAB-TolC drug efflux pump, this finding suggested a likely explanation for the MDR phenotype of NorE5. The ratios of increased expression for each gene, with their known roles in antimicrobial resistance, are listed in Table 3. Protein analysis using antibodies against AcrB corroborated its increased expression in NorE5. Furthermore, overexpression of TolC was also observed in Western blot gels (Fig. 1). AcrAB-TolC does not pump aminoglycosides out of the cell, consistent with the unchanged MIC of amikacin.
TABLE 3.
Altered gene expression in NorE5 as determined by microarrays
| Gene | Product | Fold changea |
||
|---|---|---|---|---|
| Expt 1 | Expt 2 | Expt 3 | ||
| Upregulated | ||||
| soxS | Transcriptional activator of regulator of superoxide response regulon | 8.3 | 6.2 | 10.6 |
| marA | Transcriptional activator of multiple antibiotic resistance | 3.6 | 1.7 | 3.2 |
| acrA | MDR efflux membrane fusion protein | 2.9 | 1.8 | 3.0 |
| acrB | MDR efflux pump | 2.3 | 2.8 | 2.3 |
| mdtG | Predicted drug efflux pump | 2.5 | 1.6 | 2.0 |
| Downregulated (ompF) | Outer membrane porin 1a | 9.1 | 5.9 | 8.3 |
The ratio of expression in strain NorE5 to expression in strain PS5, as determined by microarray analysis, is shown for three independent experiments.
FIG. 1.
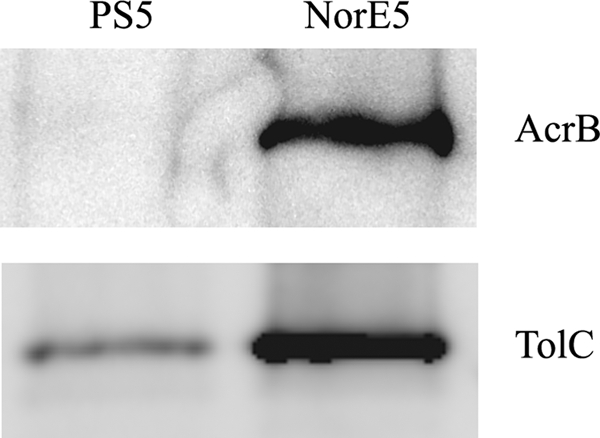
Western blot analysis of PS5 and NorE5 using antibodies against AcrB and TolC.
The microarrays also revealed significantly increased expression of soxS and marA in NorE5, about 8.4- and 2.8-fold, respectively (Table 3). In addition, ompF expression was decreased 7.8-fold, in agreement with the previous study, which showed decreased amounts of OmpF in the NorE5 outer membranes (42). The microarrays also showed that mdtG (locus b1053 of E. coli K-12 NC_000913, also known as mdtG) was upregulated approximately 2-fold in NorE5 (Table 3). This gene has been reported to encode a putative transport protein and to be involved in fosfomycin resistance (30).
A truncated SoxR protein detected in NorE5 is associated with elevated levels of SoxS transcriptional activity.
The significantly increased expression levels of soxS in NorE5 detected by the microarrays could be responsible for the MDR phenotype. We sequenced the full soxRS regions of GC4468, PS5, and NorE5 in order to detect any mutation that may have been acquired during the process of selection with norfloxacin. The results showed two mutations in NorE5 that differ from that in GC4468 and would affect the structure of the SoxR protein. One encodes a Gly74-to-Arg change, which is also present in PS5. The second mutation, an insertion of two adenines at nucleotide position 402, was detected only in NorE5. This second change in soxR encodes a frameshift starting at position Lys134 and generates a stop codon (TAG) 7 codons later. This would remove the last 21 amino acids of the C terminus of SoxR. No other amino acid change within SoxS, and no other mutation within the promoter region, was detected in the two strains in comparison with GC4468.
In order to evaluate in vivo the transcriptional activity of the SoxR protein in GC4468, PS5, and NorE5, each strain was transformed with plasmid pRS551 carrying the SoxS-activatable fpr::lacZ transcriptional fusion. These strains (GC48-F, P5-F, and N5-F) were either left untreated or treated with PQ for 1 h and were then assayed for β-galactosidase activity (Table 4). In the absence of PQ, the fpr::lacZ transcriptional fusion was 8.2- and 5.1-fold more active in N5-F than in GC48-F and P5-F, respectively, indicating constitutive expression. When GC48-F and P5-F cells were treated with PQ, the activity significantly increased over that obtained without treatment, showing that soxS was inducible in these strains, as expected. In contrast, the activity detected in N5-F was almost the same in the absence as in the presence of PQ, indicating that the ability of SoxR to induce SoxS is constitutively elevated in this strain (Table 4). Thus, the overexpression of acrAB and tolC can be attributed to the soxR mutation.
TABLE 4.
Constitutive expression of fpr in N5-F
| Strain | Genetic background | β-Galactosidase activity (Miller units)a |
Fold inductionb | |
|---|---|---|---|---|
| −PQ | + PQ | |||
| GC4468 | 4.8 | 5.8 | 1.2 | |
| GC48-F | fpr::lacZ | 2,149.1 | 19,744.6 | 9.2 |
| PS5 | 5.3 | 6.0 | 1.1 | |
| P5-F | fpr::lacZ | 3,460.8 | 26,360.7 | 7.6 |
| NorE5 | 4.9 | 6.6 | 1.3 | |
| N5-F | fpr::lacZ | 17,548.8 | 29,066.1 | 1.7 |
− PQ, without PQ; + PQ, with the addition of 50 μM PQ.
Calculated as the β-galactosidase activity of the fpr fusion in the presence of PQ divided by the value obtained in the absence of PQ.
Increased expression of mdtG in the soxS-overexpressing strains, NorE5 and JTG936.
The microarray results showed significantly increased expression of the mdtG gene in NorE5. Further analysis by RT-PCR using the RNA extracts from PS5 and NorE5 corroborated this finding; mdtG was clearly overexpressed in NorE5 (Fig. 2A). Similarly, a further RT-PCR analysis of RNA extracts from strain GC4468, strain DJ901 (GC4468 ΔsoxRS), and the constitutively soxR expressing strain JTG936 [GC4468 soxR(Con)] showed that mdtG is also upregulated in the soxR(Con) strain JTG936 (Fig. 2B). This suggests that mdtG is a member of the soxS regulon.
FIG. 2.

RT-PCR analysis to detect the levels of expression of mdtG using RNA extracts from strains PS5 and NorE5 (A) and from GC4468, DJ901, and JTG936 (B). The gapA gene was the internal control used to detect if similar amounts of RNA were added for each strain assay. Lanes 1, PS5; lanes 2, NorE5; lanes 3, GC4468; lanes 4, DJ901; lanes 5, JTG936.
mdtG, a new gene activated by SoxS, MarA, and Rob.
To further characterize the regulation of mdtG expression, the mdtG promoter was cloned to obtain a lacZ transcriptional fusion. Strain M4450, a single-copy-number lysogen containing the whole mdtG promoter (starting 159 bp upstream of the ATG codon), was constructed. This strain was assayed for β-galactosidase activity in the absence and presence of PQ, SAL, and DIP. The results showed clear induction of the promoter in the presence of all three compounds, with maximum activity upon PQ induction (Table 5). Furthermore, a putative marbox sequence was found within the promoter region, in the backward orientation, 7 bp upstream of the −35 signal and 28 bp upstream of the −10 signal for RNA polymerase (RNP) (Fig. 3). This marbox is therefore a rare class I marbox like that found in the acnA promoter (21). A second lacZ fusion to the mdtG promoter, lacking two-thirds of the identified marbox sequence (97 bp upstream of the ATG codon), was made (Fig. 3), and the corresponding lysogen, strain 4452, was obtained. No significant induction of mdtG by PQ, SAL, or DIP was observed (Table 5). In order to confirm the identity of the marbox detected within the mdtG promoter, binding of the SoxS protein to the 20-bp oligonucleotide of the presumed marbox sequence was performed. As shown in Fig. 4, SoxS binds to this sequence with a dissociation constant of ∼100 nM. Thus, mdtG is a new member of the marA-soxS-rob regulon. However, its role in stress response has yet to be determined.
TABLE 5.
Activation of two mdtG promoters measured by mdtG::lacZ expression
| Strain | Promoter length (bp)a | Uninduced β-galactosidase activity (Miller units) | Induction ratiob |
||
|---|---|---|---|---|---|
| + PQ | + SAL | + DIP | |||
| M4450 | 159 | 16.5 | 4.3 | 3.4 | 2.0 |
| M4452 | 97 | 10.5 | 1.1 | 2.2 | 1.3 |
The length of the mdtG promoter sequence fused to the lacZ gene is the number of base pairs immediately upstream of the presumptive initiation codon (ATG).
Calculated as the activity of the promoter in the presence of either PQ, SAL, or DIP divided by the activity for the uninduced control.
FIG. 3.

Sequence of the mdtG promoter showing the relationship of the inferred marbox to the putative −35 and −10 RNP signals, the transcriptional start site (TTS), and the first amino acid of the coding sequence (aa1) (boldface). The backward orientation of the marbox is indicated by the leftward heavy arrow inside the box. The distance between the −10 signal and the marbox is given in the middle of the lightface arrow. The underlined sequences represent the indicated primer binding sites.
DISCUSSION
This study has focused on the mechanisms of fluoroquinolone resistance acquired by an E. coli clinical strain after exposure to two selections with increasing concentrations of norfloxacin in vitro. Two mutations acquired within the target genes were described previously (42). The clinical isolate, PS5, harbored a mutation within GyrA (Ser83L) associated with a nalidixic acid resistance phenotype (MIC, 1,024 μg/ml) and decreased susceptibility to norfloxacin and ciprofloxacin (MICs, 0.5 μg/ml). The norfloxacin-resistant strain selected in vitro, NorE5, acquired a second mutation, within ParC (Ser80Arg), during the two-stage selection process. The MICs of norfloxacin and ciprofloxacin showed 64- and 16-fold increases, reaching 32 μg/ml and 8 μg/ml, respectively. Previous studies established an association between the MIC and the number of target gene mutations. Ciprofloxacin MICs of 1 to 4 μg/ml have been associated with two target gene mutations (one in gyrA and one in parC) (35, 43). Since no other QRDR mutation was found in NorE5, we looked for mutations in other genes that could be responsible for the higher MIC of ciprofloxacin observed for this strain.
A comparative study of gene expression between PS5 and NorE5 was performed using microarrays of cDNA. In agreement with the outer membrane protein profile obtained in the previous study (42), the results revealed significantly decreased expression of ompF in NorE5. The role that reduced expression of porins, such as OmpF and OmpC, plays in conferring fluoroquinolone resistance has been reported previously (5, 7, 9). In terms of efflux pumps, the microarray results showed increased expression of acrAB, >2-fold. Furthermore, Western blotting corroborated this finding for AcrB and extended it to include increased expression of TolC. The AcrAB-TolC pump is the main efflux pump detected in Enterobacteriaceae; its overexpression contributes not only to increasing levels of resistance to quinolones but also to increasing levels of resistance to other, unrelated drugs, such as chloramphenicol, tetracycline, β-lactams, trimethoprim, and erythromycin, but not aminoglycosides (7, 10). Thus, the fluoroquinolone resistance and MDR phenotypes observed in NorE5, representing increases of ≥4- to 16-fold in the MICs of the affected antibiotics, are likely explained by these findings.
Regulatory mechanisms that decrease OmpF expression and increase AcrAB and TolC expression have been elucidated (10). Three members of the AraC/XlyS family of transcriptional activators, SoxS, MarA, and Rob, have been reported to activate those genes containing a marbox in their promoters (25). The marbox is the sequence where these activators bind to interact with RNA polymerase and activate the transcription of the genes of the regulon. The promoters of the acrAB operon, the tolC gene, and the antisense mRNA micF (which blocks ompF translation [28]) contain marboxes (21). The microarray results of this study revealed significantly increased soxS expression, > 8-fold, in NorE5, in addition to slightly increased expression of marA, >2-fold. Since the marRAB operon also contains a marbox in its promoter (23), the elevated SoxS activity could activate marRAB and increase marA expression (15, 27).
To determine the basis of the elevated SoxS expression, the soxRS region was sequenced. The results showed an insertion of two adenines in soxR in NorE5 but not in PS5. This insertion should cause a frameshift at amino acid 134 and transcription termination 7 codons later, leading to a truncated protein. Other mutations within the C-terminal domain of SoxR have been shown to render SoxS constitutively active in the absence of the redox signals that are normally required to activate wild-type SoxR protein and lead to constitutive expression of soxS (19, 31, 47). Constitutive expression of SoxS due to an in-frame internal deletion of SoxR amino acids 136 to 144 (affecting the last 19 amino acids) has been described (31). A second mutation within SoxR, Gly74Arg, found in both PS5 and NorE5, has also been found in several soxS-overexpressing clinical isolates, but accompanied by a second mutation within the same locus, Thr38Ser (19). It seems likely that the Gly74Arg change plays little or no role in the overexpression of SoxS, since the levels of AcrB expression in PS5 were very similar to those in GC4468 by Western blot analysis (data not shown).
The most likely explanation is that the frameshift is responsible for constitutive activity of SoxR leading to constitutive expression of soxS and hence an upregulation of the genes that belong to the regulon. However, due to the facts that soxR and soxS are divergently expressed from the soxRS regulon and the soxS promoter is within the intergenic region (47), the hypothesis that the two nucleotide insertions within soxR may lead to a new promoter responsible for the constitutive expression of soxS was considered. To test this hypothesis, the intergenic region, including the partial sequence of soxR where the nucleotide insertions were detected, was amplified from NorE5 and also from PS5. Both PCR products were digested and cloned into the pRS551 vector in order to assess its putative promoter activity. The corresponding assays of β-galactosidase activity revealed no significant difference in activity between the sequences (data not shown). Thus, the insertions did not create a new soxS promoter.
The constitutivity of the soxS expression was further demonstrated by monitoring the behavior of an fpr::lacZ transcriptional fusion in strains GC4468, PS5, and NorE5. As expected for a regulon promoter, expression of the fpr promoter was 5-fold greater in NorE5 than in the parental strain, PS5 (Table 4). Furthermore, when these cells were treated with PQ to activate SoxR, all three strains showed similar high levels of activity, strongly indicating that the SoxR protein is already in an activated state in NorE5.
This study has also revealed a new member of the marA-soxS-rob regulon, mdtG. mdtG is inducible by PQ, SAL, and DIP, and SoxS binds tightly to the marbox sequence reported within the mdtG promoter. Moreover, disruption of this marbox sequence was accompanied by a loss of inducibility by all three compounds. The MdtG protein, also named YceE, appears to be a member of the major facilitator superfamily of transporters, and it has been reported, when overexpressed, to increase fosfomycin and deoxycholate resistances by 4- and 2-fold, respectively (30). What other roles it may play in antibiotic resistance is not known.
Acknowledgments
We thank B. Demple for kindly providing strains GC4468, DJ901, and JTG936.
This study has been supported by the Spanish Ministry of Health (FIS 05/0068), by 2009 SGR 1256 from the Departament d'Universitats, Recerca i Societat de la Informació de la Generalitat de Catalunya, and by the Ministerio de Sanidad y Consumo, Instituto de Salud Carlos III, Spanish Network for Research in Infectious Diseases (REIPI RE06/0008). This work has also been supported by funding from the European Community (TROCAR contract HEALTH-F3-2008-223031).
Footnotes
Published ahead of print on 14 December 2009.
REFERENCES
- 1.Alos, J. I. 2005. Epidemiology and etiology of urinary tract infections in the community. Antimicrobial susceptibility of the main pathogens and clinical significance of resistance. Enferm. Infecc. Microbiol. Clin. 23(Suppl. 4):3-8. (In Spanish.) [DOI] [PubMed] [Google Scholar]
- 2.Amabile-Cuevas, C. F., and B. Demple. 1991. Molecular characterization of the soxRS genes of Escherichia coli: two genes control a superoxide stress regulon. Nucleic Acids Res. 19:4479-4484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Arslan, H., O. K. Azap, O. Ergonul, and F. Timurkaynak. 2005. Risk factors for ciprofloxacin resistance among Escherichia coli strains isolated from community-acquired urinary tract infections in Turkey. J. Antimicrob. Chemother. 56:914-918. [DOI] [PubMed] [Google Scholar]
- 4.Bennik, M. H., P. J. Pomposiello, D. F. Thorne, and B. Demple. 2000. Defining a rob regulon in Escherichia coli by using transposon mutagenesis. J. Bacteriol. 182:3794-3801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chenia, H. Y., B. Pillay, and D. Pillay. 2006. Analysis of the mechanisms of fluoroquinolone resistance in urinary tract pathogens. J. Antimicrob. Chemother. 58:1274-1278. [DOI] [PubMed] [Google Scholar]
- 6.Cohen, S. P., H. Hachler, and S. B. Levy. 1993. Genetic and functional analysis of the multiple antibiotic resistance (mar) locus in Escherichia coli. J. Bacteriol. 175:1484-1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cohen, S. P., L. M. McMurry, D. C. Hooper, J. S. Wolfson, and S. B. Levy. 1989. Cross-resistance to fluoroquinolones in multiple-antibiotic-resistant (Mar) Escherichia coli selected by tetracycline or chloramphenicol: decreased drug accumulation associated with membrane changes in addition to OmpF reduction. Antimicrob. Agents Chemother. 33:1318-1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ding, H., and B. Demple. 1996. Glutathione-mediated destabilization in vitro of [2Fe-2S] centers in the SoxR regulatory protein. Proc. Natl. Acad. Sci. U. S. A. 93:9449-9453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Everett, M. J., Y. F. Jin, V. Ricci, and L. J. Piddock. 1996. Contributions of individual mechanisms to fluoroquinolone resistance in 36 Escherichia coli strains isolated from humans and animals. Antimicrob. Agents Chemother. 40:2380-2386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fabrega, A., S. Madurga, E. Giralt, and J. Vila. 2009. Mechanism of action of and resistance to quinolones. Microb. Biotechnol. 2:40-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fralick, J. A. 1996. Evidence that TolC is required for functioning of the Mar/AcrAB efflux pump of Escherichia coli. J. Bacteriol. 178:5803-5805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Greenberg, J. T., P. Monach, J. H. Chou, P. D. Josephy, and B. Demple. 1990. Positive control of a global antioxidant defense regulon activated by superoxide-generating agents in Escherichia coli. Proc. Natl. Acad. Sci. U. S. A. 87:6181-6185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hirakawa, H., A. Takumi-Kobayashi, U. Theisen, T. Hirata, K. Nishino, and A. Yamaguchi. 2008. AcrS/EnvR represses expression of the acrAB multidrug efflux genes in Escherichia coli. J. Bacteriol. 190:6276-6279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jacoby, G. A., N. Chow, and K. B. Waites. 2003. Prevalence of plasmid-mediated quinolone resistance. Antimicrob. Agents Chemother. 47:559-562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jair, K. W., R. G. Martin, J. L. Rosner, N. Fujita, A. Ishihama, and R. E. Wolf, Jr. 1995. Purification and regulatory properties of MarA protein, a transcriptional activator of Escherichia coli multiple antibiotic and superoxide resistance promoters. J. Bacteriol. 177:7100-7104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jellen-Ritter, A. S., and W. V. Kern. 2001. Enhanced expression of the multidrug efflux pumps AcrAB and AcrEF associated with insertion element transposition in Escherichia coli mutants selected with a fluoroquinolone. Antimicrob. Agents Chemother. 45:1467-1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kahlmeter, G. 2003. An international survey of the antimicrobial susceptibility of pathogens from uncomplicated urinary tract infections: the ECO.SENS Project. J. Antimicrob. Chemother. 51:69-76. [DOI] [PubMed] [Google Scholar]
- 18.Kim, M. E., U. S. Ha, and Y. H. Cho. 2008. Prevalence of antimicrobial resistance among uropathogens causing acute uncomplicated cystitis in female outpatients in South Korea: a multicentre study in 2006. Int. J. Antimicrob. Agents 31(Suppl. 1):S15-S18. [DOI] [PubMed] [Google Scholar]
- 19.Koutsolioutsou, A., S. Pena-Llopis, and B. Demple. 2005. Constitutive soxR mutations contribute to multiple-antibiotic resistance in clinical Escherichia coli isolates. Antimicrob. Agents Chemother. 49:2746-2752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ma, D., M. Alberti, C. Lynch, H. Nikaido, and J. E. Hearst. 1996. The local repressor AcrR plays a modulating role in the regulation of acrAB genes of Escherichia coli by global stress signals. Mol. Microbiol. 19:101-112. [DOI] [PubMed] [Google Scholar]
- 21.Martin, R. G., W. K. Gillette, S. Rhee, and J. L. Rosner. 1999. Structural requirements for marbox function in transcriptional activation of mar/sox/rob regulon promoters in Escherichia coli: sequence, orientation and spatial relationship to the core promoter. Mol. Microbiol. 34:431-441. [DOI] [PubMed] [Google Scholar]
- 22.Martin, R. G., W. K. Gillette, and J. L. Rosner. 2000. Promoter discrimination by the related transcriptional activators MarA and SoxS: differential regulation by differential binding. Mol. Microbiol. 35:623-634. [DOI] [PubMed] [Google Scholar]
- 23.Martin, R. G., K. W. Jair, R. E. Wolf, Jr., and J. L. Rosner. 1996. Autoactivation of the marRAB multiple antibiotic resistance operon by the MarA transcriptional activator in Escherichia coli. J. Bacteriol. 178:2216-2223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Martin, R. G., and J. L. Rosner. 1995. Binding of purified multiple antibiotic-resistance repressor protein (MarR) to mar operator sequences. Proc. Natl. Acad. Sci. U. S. A. 92:5456-5460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Martin, R. G., and J. L. Rosner. 2002. Genomics of the marA/soxS/rob regulon of Escherichia coli: identification of directly activated promoters by application of molecular genetics and informatics to microarray data. Mol. Microbiol. 44:1611-1624. [DOI] [PubMed] [Google Scholar]
- 26.Miller, J. H. 1972. Experiments in molecular genetics. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 27.Miller, P. F., L. F. Gambino, M. C. Sulavik, and S. J. Gracheck. 1994. Genetic relationship between soxRS and mar loci in promoting multiple antibiotic resistance in Escherichia coli. Antimicrob. Agents Chemother. 38:1773-1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mizuno, T., M. Y. Chou, and M. Inouye. 1984. A unique mechanism regulating gene expression: translational inhibition by a complementary RNA transcript (micRNA). Proc. Natl. Acad. Sci. U. S. A. 81:1966-1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nakamura, S., M. Nakamura, T. Kojima, and H. Yoshida. 1989. gyrA and gyrB mutations in quinolone-resistant strains of Escherichia coli. Antimicrob. Agents Chemother. 33:254-255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nishino, K., and A. Yamaguchi. 2001. Analysis of a complete library of putative drug transporter genes in Escherichia coli. J. Bacteriol. 183:5803-5812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nunoshiba, T., and B. Demple. 1994. A cluster of constitutive mutations affecting the C-terminus of the redox-sensitive SoxR transcriptional activator. Nucleic Acids Res. 22:2958-2962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Oethinger, M., W. V. Kern, A. S. Jellen-Ritter, L. M. McMurry, and S. B. Levy. 2000. Ineffectiveness of topoisomerase mutations in mediating clinically significant fluoroquinolone resistance in Escherichia coli in the absence of the AcrAB efflux pump. Antimicrob. Agents Chemother. 44:10-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Oethinger, M., I. Podglajen, W. V. Kern, and S. B. Levy. 1998. Overexpression of the marA or soxS regulatory gene in clinical topoisomerase mutants of Escherichia coli. Antimicrob. Agents Chemother. 42:2089-2094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Okusu, H., D. Ma, and H. Nikaido. 1996. AcrAB efflux pump plays a major role in the antibiotic resistance phenotype of Escherichia coli multiple-antibiotic-resistance (Mar) mutants. J. Bacteriol. 178:306-308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Qiang, Y. Z., T. Qin, W. Fu, W. P. Cheng, Y. S. Li, and G. Yi. 2002. Use of a rapid mismatch PCR method to detect gyrA and parC mutations in ciprofloxacin-resistant clinical isolates of Escherichia coli. J. Antimicrob. Chemother. 49:549-552. [DOI] [PubMed] [Google Scholar]
- 36.Rosenberg, E. Y., D. Bertenthal, M. L. Nilles, K. P. Bertrand, and H. Nikaido. 2003. Bile salts and fatty acids induce the expression of Escherichia coli AcrAB multidrug efflux pump through their interaction with Rob regulatory protein. Mol. Microbiol. 48:1609-1619. [DOI] [PubMed] [Google Scholar]
- 37.Rosner, J. L., T. J. Chai, and J. Foulds. 1991. Regulation of ompF porin expression by salicylate in Escherichia coli. J. Bacteriol. 173:5631-5638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rosner, J. L., B. Dangi, A. M. Gronenborn, and R. G. Martin. 2002. Posttranscriptional activation of the transcriptional activator Rob by dipyridyl in Escherichia coli. J. Bacteriol. 184:1407-1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rosner, J. L., and J. L. Slonczewski. 1994. Dual regulation of inaA by the multiple antibiotic resistance (mar) and superoxide (soxRS) stress response systems of Escherichia coli. J. Bacteriol. 176:6262-6269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Simons, R. W., F. Houman, and N. Kleckner. 1987. Improved single and multicopy lac-based cloning vectors for protein and operon fusions. Gene 53:85-96. [DOI] [PubMed] [Google Scholar]
- 41.Sorlozano, A., J. Gutierrez, A. Jimenez, L. J. de Dios, and J. L. Martinez. 2007. Contribution of a new mutation in parE to quinolone resistance in extended-spectrum-beta-lactamase-producing Escherichia coli isolates. J. Clin. Microbiol. 45:2740-2742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tavio, M. M., J. Vila, J. Ruiz, J. Ruiz, A. M. Martin-Sanchez, and M. T. Jimenez de Anta. 1999. Mechanisms involved in the development of resistance to fluoroquinolones in Escherichia coli isolates. J. Antimicrob. Chemother. 44:735-742. [DOI] [PubMed] [Google Scholar]
- 43.Vila, J., J. Ruiz, P. Goni, and M. T. De Anta. 1996. Detection of mutations in parC in quinolone-resistant clinical isolates of Escherichia coli. Antimicrob. Agents Chemother. 40:491-493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vila, J., J. Ruiz, F. Marco, A. Barcelo, P. Goni, E. Giralt, and T. Jimenez De Anta. 1994. Association between double mutation in gyrA gene of ciprofloxacin-resistant clinical isolates of Escherichia coli and MICs. Antimicrob. Agents Chemother. 38:2477-2479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang, H., J. L. Dzink-Fox, M. Chen, and S. B. Levy. 2001. Genetic characterization of highly fluoroquinolone-resistant clinical Escherichia coli strains from China: role of acrR mutations. Antimicrob. Agents Chemother. 45:1515-1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Webber, M. A., A. Talukder, and L. J. Piddock. 2005. Contribution of mutation at amino acid 45 of AcrR to acrB expression and ciprofloxacin resistance in clinical and veterinary Escherichia coli isolates. Antimicrob. Agents Chemother. 49:4390-4392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wu, J., and B. Weiss. 1991. Two divergently transcribed genes, soxR and soxS, control a superoxide response regulon of Escherichia coli. J. Bacteriol. 173:2864-2871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yamane, K., J. Wachino, S. Suzuki, and Y. Arakawa. 2008. Plasmid-mediated qepA gene among Escherichia coli clinical isolates from Japan. Antimicrob. Agents Chemother. 52:1564-1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yang, H., H. Chen, Q. Yang, M. Chen, and H. Wang. 2008. High prevalence of plasmid-mediated quinolone resistance genes qnr and aac(6′)-Ib-cr in clinical isolates of Enterobacteriaceae from nine teaching hospitals in China. Antimicrob. Agents Chemother. 52:4268-4273. [DOI] [PMC free article] [PubMed] [Google Scholar]