

Abstract
Sustained activation of Akt kinase acts as a focal regulator to increase cell growth and survival, which cause tumorigenesis including breast cancer. Statins, potent inhibitors of 3-hydroxy-3-methylglutaryl coenzyme A reductase, display anticancer activity. The molecular mechanisms by which statins block cancer cell growth are poorly understood. We demonstrate that in the tumors derived from MDA-MB-231 human breast cancer cell xenografts, simvastatin significantly inhibited phosphorylation of Akt with concomitant attenuation of expression of the anti-apoptotic protein BclXL. In many cancer cells, BclXL is a target of NFκB. Simvastatin inhibited the DNA binding and transcriptional activities of NF κ B resulting in marked reduction in transcription of BclXL. Signals transmitted by anti-neoplastic mechanism implanted in the cancer cells serve to obstruct the initial outgrowth of tumors. One such mechanism represents the action of the tumor suppressor protein PTEN, which negatively regulates Akt kinase activity. We provide the first evidence for significantly increased levels of PTEN in the tumors of simvastatin-administered mice. Importantly, simvastatin markedly prevented binding of NFκB to the two canonical recognition elements, NFRE-1 and NFRE-2 present in the PTEN promoter. Contrary to the transcriptional suppression of BclXL, simvastatin significantly increased the transcription of PTEN. Furthermore, expression of NFκ B p65 subunit inhibited transcription of PTEN, resulting in reduced protein expression, which leads to enhanced phosphorylation of Akt. Taken together, our data present a novel bifaceted mechanism where simvastatin acts on a nodal transcription factor NFκ B, which attenuates the expression of anti-apoptotic BclXL and simultaneously derepresses the expression of anti-proliferative/proapoptotic tumor suppressor PTEN to prevent breast cancer cell growth.
Keywords: Statin, Breast tumor, BclXL, Akt kinase
1. Introduction
Statins are a class of drugs that prevent cardiovascular and cerebrovascular diseases [1, 2]. They act as specific inhibitors of 3-hydroxy-3-methylglutaryl Coenzyme A reductase (HMG CoA reductase), the rate limiting enzyme in the mevalonate cascade, resulting in the attenuation of synthesis of cholesterol and isoprenoid compounds such as farnesyl pyrophosphate and geranylgeranyl pyrophosphate [3]. The hydrophobic isoprenoids covalently bind to the various proteins including the G-protein members Ras, Rho, Rac, Rap and Rab [4]. This modification regulates the plasma membrane localization of these small G-proteins, thereby has significant effects on cell growth and survival.
In the last few years, use of statins gained interest for cancer prevention. Data using rodent models showed positive results in myeloid leukemia and in colon, prostate, mammary, lung, melanoma and glioma tumorigenesis [5–16]. Contrasting results exist where statins showed no efficacy in mammary carcinogenesis induced by methylnitrosourea in rats [17]. Initial evaluation of a randomized controlled trial showed an increased risk of breast cancer in patients using statin [18]. Additionally a recent British case control study showed no significant association of reduction in colon, skin and breast cancer risks [19]. However, a separate study revealed a significant 20% reduction in overall cancer risk in patients with statin use [20]. Recently a large study conducted with statin-using veterans showed a significant 50–60% reduction in advanced prostate cancer risk [21, 22]. Use of statin was reported to be associated with significant reduction in cancer risk of colon, lung, pancreas and esophagus along with melanoma and B and T cell lymphoma [23–25]. Another study where women used statin more than 4 years showed significant 74% reduced risk of breast cancer [26]. Moreover, a recent multicenter study with women using statin indicated significantly decreased risk of breast cancer as compared to the nonusers [27]. The literature thus favors a beneficial effect from statin use in various cancers including breast cancer.
The exact mode of action of antiproliferative and proapoptotic effects of statins on cancer cells is not clearly understood. The effect in blocking mevalonate pathway may play an important role as it regulates the production of geranylgeranyl and farnesyl pyrophosphates, which modify the Rho and Ras GTPases [28]. Rho proteins are involved in proliferative and invasive potential of cancer cells including breast cancer cells [24, 29–33]. Therefore, inhibition of geranylgeranylation of RhoA may represent a potent mechanism for statin-induced attenuation of breast cancer cell growth. Additionally, statin regulates proliferation and apoptosis of the tumor cells by modulating MAPK and CDK2, which in turn reduce the expression of p21 and p27 cyclin kinase inhibitors [34–36].
Activation of phosphatidylinositol (PI) 3 kinase and its downstream target Akt kinase has been implicated in tumorigenesis and anti-apoptosis of cancer cells [37]. The tumor suppressor protein PTEN (phosphatase and tensin homolog deleted in chromosome 10) acts as negative regulator of PI 3 kinase signaling. The lipid phosphatase activity of PTEN maintains the dynamic levels of the second messenger PI 3,4,5-trisphosphate (PIP3), the product of PI 3 kinase [38]. Mutation/deletion of PTEN has been reported in many cancers including breast tumor [38–41]. Inactivation of PTEN thus increases the levels of PIP3, which activates Akt kinase. To test the molecular mechanism of breast cancer cell growth inhibition by statin, we considered the involvement of Akt/PTEN axis as a mediator. Using the MDA-MB-231 human breast cancer cell xenograft model in mice, we identified that simvastatin significantly inhibited phosphorylation of Akt in the breast tumor tissues. In vitro, we demonstrate simvastatin-mediated attenuation of NFκB activation results in reduction in the anti-apoptotic BclXL protein expression. Additionally, we show an increase in PTEN protein expression in the breast tumors of mouse treated with simvastatin and in the simvastatin-treated MDA-MB-231 breast cancer cells in vitro. Finally, our results demonstrate a derepression mechanism involving NFκB to upregulate PTEN levels in breast tumor cells in response to simvastatin.
2. Materials and Methods
2.1. Cell culture
The MDA-MB-231 human breast cancer cells purchased from American Type Culture Collection were grown in Dulbecco’s modified Eaglemedium with penicillin, streptomycin and 10% fetal bovine serum in 5% CO2 incubator at 37°C.
2.2. Antibodies and Reagents
Antibodies against phospho-Akt and Akt were purchased from Cell Signaling. PTEN, Erk1/2, p65 NFκB subunit and BclXL antibodies were obtained Santa Cruz Biotechnology. Nuclear extract isolation kit and dual luciferase assay kit were purchased from Pierce and Promega, respectively. Double stranded consensus NFκB DNA element (5′ AGTTGAGGGGACTTTCCCAGGC-3′) was obtained from Santa Cruz Biotechnology. The NFκB recognition elements 1 (5′-TGGGGGAAGGGGGAATCTCTAGGCAAAGG-3′) and 2 (CAAGGGGGGAGGGTATTCCCCTTGCAGGGA) from PTEN promoter were synthesized in the core facility at the University of Texas Health Science Center at San Antonio [42]. The reporter plasmid with BclXL promoter driving luciferase cDNA (BclXL-Luc) was a kind gift from Dr. G. Nunez, University of Michigan. pCMV-p65 and PTEN promoter-driven luciferase reporter plasmid have been described previously [43].
2.3. Animal Study
The nude mice were obtained from the NIH animal facility. They were used according to the guide of use of small laboratory animals. The animal protocols were approved by the Institutional Animal Care and Use Committee, The University of Texas of Health Science Center at San Antonio. The mice were kept in normal lab chow diet. Mice were injected with 5 mg/kg body weight of simvastatin every day for 7 days. Control mice were given phosphate buffer saline (PBS). MDA-MB-231 cells were trypsinized, counted and 1×106 cells in phosphate buffered saline were injected in each mammary fat pad of these mice. At three weeks, the mice were sacrificed and the tumors were collected.
2.4. Preparation of cell and tumor lysates
Excised tumors were homogenized using radioimmunoprecipitation assay (RIPA) buffer (20 mM Tris-HCl, pH 7.5, 150 mM NaCl, 5 mM EDTA, 1 mM phenylmethylsulfonile fluoride, 0.05% aprotinin and 1% Nonidet P-40). Homogenates were centrifuged at 4°C at 12,000xg for 30 minutes. Similarly, the MDA-MB-231 cell monolayer was washed with PBS and the cells were lysed in RIPA buffer followed by centrifugation at 4°C. Cleared supernatant was collected for immunoblotting.
Immunoblotting
Protein concentration was determined in the tumor lysates using BioRad reagent as described previously [44, 45]. Equal amounts of proteins were separated by SDS polyacrylamide gel electrophoresis. The immunoblotting of these separated proteins was performed using required antibodies as described [44–47].
2.5. Electrophoretic mobility shift assay
Nuclear extracts were prepared from MDA-MB-231 breast cancer cells using a kit according to the protocol provided by the vendor [45, 47]. Double stranded oligonucleotides representing NFκB elements were labeled at the 5′ end using γ32P-ATP and T4 polynucleotide kinase. EMSA was performed using 10 μg nuclear extracts. The protein-DNA complex was separated by 5% non-denaturing polyacrylamide gel electrophoresis. The gel was dried on filter paper followed by autoradiography. p65 antibody was used to determine the presence of NFκB in the DNA-protein complex in the supershift assays as described previously [45, 47]. Erk1/2 antibody was used as control in this assay.
2.6. Transient transfection and luciferase activity
Semiconfluent MDA-MB-231 breast cancer cells were transfected with the reporter plasmids using Fugene HD as described [47]. Twenty-four hours posttransfection the cells were incubated with 10 μM simvastatin for 24 hours as indicated. The cells were harvested and lysates were used to perform luciferase assay using the kit. The data are presented as mean of luciferase activity per microgram protein as arbitrary units ± SE of triplicate measurements as described previously [43–45, 47].
2.7. Statistical analysis of data
Statistical significance of the data was determined using analysis of variance followed by Student-Newman-Keuls analysis as described previously [43–45, 47]. Significance levels was considered as p value < 0.05.
3. Results
3.1. Simvastatin inhibits Akt activation in breast tumor
The beneficial effect of statins in reducing tumor growth using rodent models has been extensively studied [5–16]. To examine the mechanism of action of simvastatin, we investigated its effect on Akt kinase. Akt contributes to increased survival and resistance to apoptosis of many cancers including breast tumor cells [48–50]. A group of mice treated with simvastatin for one week was inoculated with the MDA-MB-231 breast cancer cells in the mammary fat pads. As expected the tumor growth was significantly inhibited in the mice treated with simvastatin as compared to the tumors in the control mice (Supplementary Fig. S1A and S1B). Similarly, simvastatin markedly blocked the proliferation of MDA-MB-231 cells in vitro (Supplementary Fig. S2).
We have reported previously that in MDA-MB-231 breast cancer cells, a growth factor receptor tyrosine kinase-mediated signal transduction pathway is upregulated, which increases phosphorylation of Akt, resulting in its activation [43, 46]. To investigate the state of Akt activation, MDA-MB-231 cells were incubated with simvastatin in vitro. Immunoblotting was performed with the cell lysates using a specific antibody that recognizes the phosphorylated (activated) form of Akt. Simvastatin inhibited phosphorylation of Akt without any effect on the levels of Akt (Fig. 1A). To examine the effect of simvastatin in vivo, immunoblotting of the tumor lysates from the control and simvastatin-treated mice was performed using phospho-Akt antibody. Akt phosphorylation was significantly reduced in the tumor samples isolated from animals treated with simvastatin (Fig. 1B, compare lanes 4–6 with lanes 1–3; Fig. 1C). These results demonstrate an inhibitory effect of statin on Akt activation in the breast tumors generated in an animal model.
Figure 1.
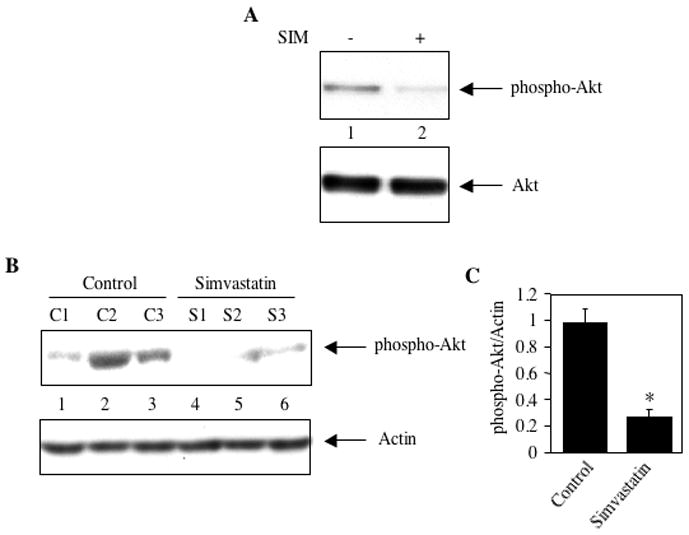
Simvastatin inhibits phosphorylation of Akt. (A) Effect of simvastatin on phosphorylation of Akt in MDA-MB-231 breast cancer cells. Lysates of cells incubated with 10 μM simvastatin (SIM) for 24 hours were immunoblotted with phospho-Akt (Ser-473) and Akt antibodies respectively (B) Simvastatin prevents phosphorylation of Akt in the MDA-MB-231 xenografts. Lysates of tumors from independent control and independent simvastatin-treated mice were immunoblotted with phospho-Akt and actin antibodies as indicated. (C) Quantification of phosphorylated Akt in the panel B. Mean ± SE of three independent animals is shown. *p < 0.01 vs, control.
3.2. Simvastatin inhibits expression of BclXL
Increased expression of mitochondrial anti-apoptotic proteins contribute to augmented survival of cancer cells including breast cancer. We have previously reported increased constitutive levels of anti-apoptotic BclXL protein in MDA-MB-231 cells [43]. Abundant expression of BclXL was detected in the tumor lysates of mice inoculated with MDA-MB-231 cells (Fig. 2A, lanes 1–3). We tested the effect of simvastatin on the expression of BclXL. Levels of BclXL were significantly reduced in the tumor lysates prepared from mice treated with simvastatin (Fig. 2A, compare lanes 4–6 with lanes 1–3; Fig. 2B). Furthermore, incubation of MDA-MB-231 cells with simvastatin in vitro inhibited the expression of BclXL (Fig. 2C, compare lane 2 with lane 1).
Figure 2.
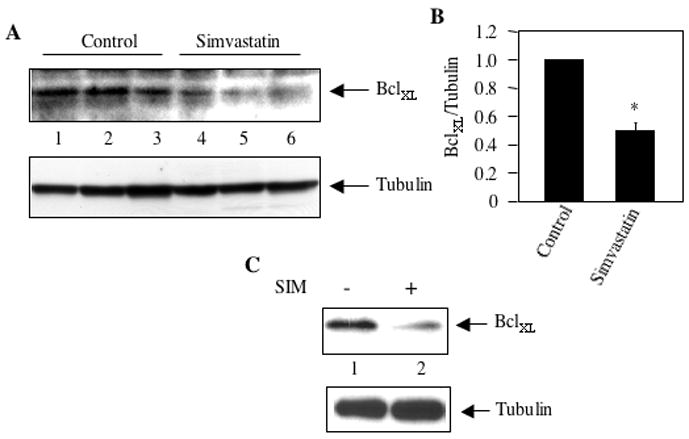
Effect of simvastatin on anti-apoptotic protein BclXL expression in mammary cancer. (A) Tumor lysates from independent control and simvastatin-treated mice were immunoblotted with antibodies against BclXL (top panel) and tubulin (bottom panel). (B) Quantification of BclXL expression in panel A. Mean ± SE of three independent animals is shown. *p < 0.01 vs. control. (C) Lysates of MDA-MB-231 breast cancer cells incubated with 10 μM simvastatin for 24 hours were immunoblotted with BclXL and tubulin antibodies, respectively.
We have shown above that simvastatin inhibits Akt activation in MDA-MB-231 breast cancer cells (Fig. 1A). Akt regulates survival of cancer cells using a myriad of transcription factors including NFκB [51–53]. In MDA-MB-231 human breast cancer cells, NFκB is constitutively active [54]. We tested the effect of simvastatin on activation of NFκB using EMSA with nuclear extracts from MDA-MB-231 cells. Double stranded oligonucleotide representing a consensus NFκB recognition sequence was used as probe. As expected, DNA-protein complex was detected with nuclear extract prepared from MDA-MB-231 cells (Fig. 3A, lane 1). Incubation of nuclear extracts with cold NFκB recognition sequence inhibited the DNA-protein complex formation, demonstrating specificity of the interaction (Fig. 3A, compare lanes 2 and 3 with lane 1). Use of AP2 DNA element as cold competitor oligonucleotide did not have any effect on the formation of DNA protein complex using NFκB sequence as probe (Fig. 3A, compare lane 4 with lane 1). To identify the presence of NFκB in this protein-DNA complex, we performed EMSA in the presence of antibody against p65 NFκB subunit. Incubation of the nuclear extracts with p65 antibody showed supershifted DNA-protein complex as compared to the control (Fig. 3B, compare lane 2 with lane 1, arrow indicates the supershifted protein-DNA complex). Erk1/2 antibody was used as IgG control (Fig. 3B, compare lane 2 with lane 3). Next, we determined the effect of simvastatin on NFκB-DNA complex formation. Nuclear extracts isolated from simvastatin-treated MDA-MB-231 cells were used in EMSA. Simvastatin prevented protein-DNA complex formation (Fig. 3C, compare lane 2 with lane 1). To test whether this inhibition of DNA binding results in transcriptional attenuation, we used a reporter plasmid in which luciferase cDNA is driven by three copies of NFκB binding DNA element. The reporter construct was transfected into MDA-MB-231 cells. Incubation of transiently transfected cells with simvastatin significantly inhibited NFκB-dependent transcription of the reporter gene (Fig. 3D).
Figure 3.
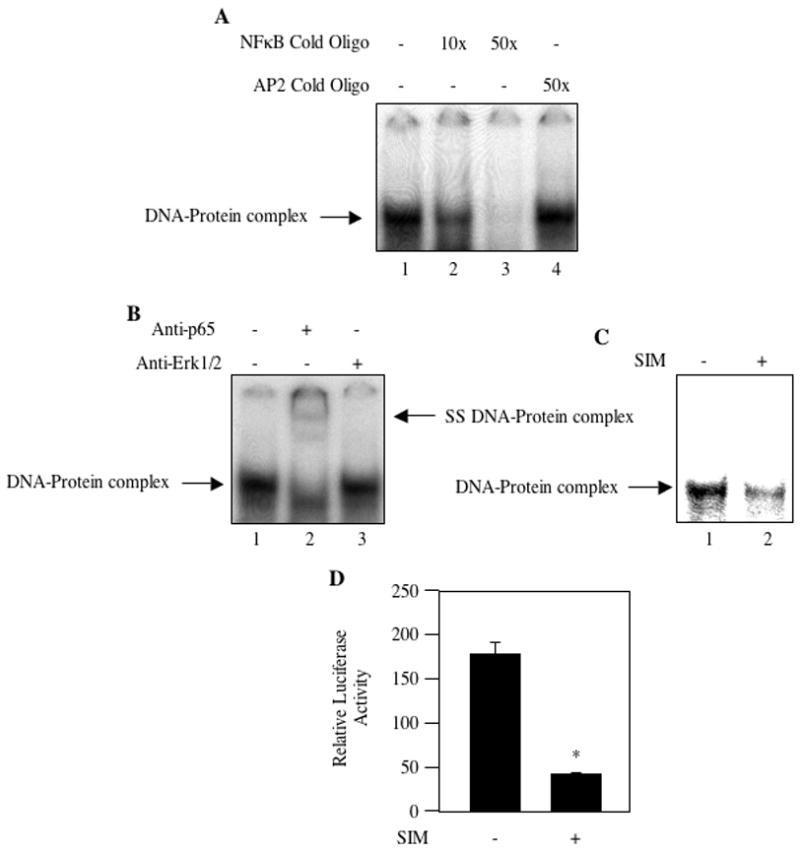
Simvastatin inhibits NFκB activation. (A) Nuclear extracts from MDA-MB-231 breast cancer cells were used in EMSA with 32P-labeled consensus NFκB DNA sequence as described in the Materials and Methods. Cold double stranded oligonucleotide representing NFκB element (lanes 2 and 3) and AP2 transcription factor binding element (lane 4) were used for competition. (B) Nuclear extracts from MDA-MB-231 cells were incubated with anti-p65 and anti-Erk1/2 antibodies prior to incubation with 32P-labeled double stranded NFκB DNA element as described in the Materials and Methods. (C) MDA-MB-231 cells were incubated with 10 μM simvastatin. Equal amounts of nuclear extracts were used in EMSA with double-stranded 32P-labeled NFκB DNA element as described in the Materials and Methods. Arrows in the left margin show the protein-DNA complex. Right arrow in panel B indicates the supershifted DNA-protein comlex in lane 2. (D) Effect of simvastatin on NFκB transcriptional activity. The reporter plasmid 3xNFκB-Luc was transfected into MDA-MB-231 breast cancer cells. Transiently transfected cells were incubated with 10 μM simvastatin (SIM) as indicated. Luciferase activity was determined in the cell lysates as described in the Materials and Methods. Mean ± SE of triplicate measurements is shown. *p < 0.01 vs. control.
Activation of NFκB induces expression of many anti-apoptotic genes including BclXL [55, 56]. We have shown above that simvastatin inhibits expression of BclXL in MDA-MB-231 breast cancer cells in vitro and in xenograft model (Fig. 2). We tested the effect of simvastatin on transcription of BclXL using a reporter construct in which BclXL promoter drives the luciferase cDNA (BclXL-Luc) [43]. Incubation of BclXL-Luc-transfected MDA-MB-231 cells with simvastatin showed reduced transcription of BclXL in the reporter assay (Fig. 4A). To confirm the involvement of NFκB, we tested the effect of p65 on transcription of BclXL. MDA-MB-231 cells were cotransfected with p65 and BclXL-Luc. Expression of p65 significantly elevated transcription of BclXL (Fig. 4B). Additionally, transfection of p65 increased the expression of BclXL protein level (Fig. 4C, compare lane 2 with lane 1). These results indicate that simvastatin targets NFκB to regulate the expression of BclXL.
Figure 4.
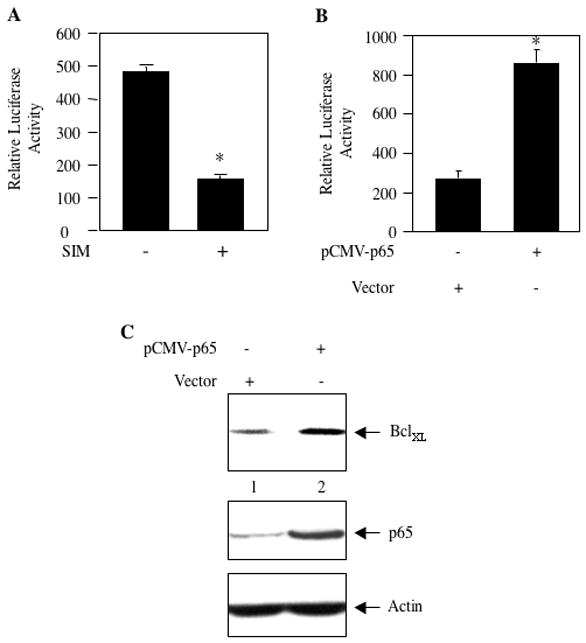
Simvastain inhibits transcription of BclXL. (A) MDA-MB-231 cells were transfected with BclXL-Luc reporter plasmid. Transiently transfected cells were incubated with 10 μM simvastatin (SIM). (B) MDA-MB-231 cells were transfected with BclXL-Luc along with vector or pCMV-p65 expression plasmid as indicated. Luciferase activity was determined in the cell lysates as described in the Materials and Methods. Mean ± SE of triplicate measurements is shown. *p < 0.001 vs. control in panel A. *p < 0.001 vs vector-transfected in panel B. (C) MDA-MB-231 cells were transfected with pCMV-p65 or vector plasmid. Lysates of transfected cells were immunoblotted with antibodies recognizing BclXL, p65 and actin as indicated.
3.3. Simvastatin increases expression of PTEN in xenograft tumor model
Akt activity is upregulated in many cancers [57]. Among various mechanisms mutation/deletion or malfunctioning of the tumor suppressor protein PTEN activates Akt by constitutively increasing the levels of PI 3 kinase product PIP3 [38, 58, 59]. We have shown that simvastatin inhibits Akt activation in both MDA-MB-231 breast cancer cells in vitro and in the mice tumors derived from these cells (Fig. 1). As a mechanism, we tested the involvement of PTEN, which is expressed at low levels in MDA-MB-231 breast cancer cells (Fig. 5A, lane 1). Incubation of these cells with simvastatin increased the expression of PTEN (Fig. 5A, compare lane 2 with lane 1). Exogenous expression of PTEN using a plasmid vector significantly inhibited proliferation of MDA-MB-231 breast cancer cells (Supplementary Fig S3). Next, we examined effect of simvastatin on expression of PTEN in the tumors produced by MDA-MB-231 cells. The tumor lysates from the control mice showed significantly low levels of PTEN protein (Fig. 5B, lanes 1–4). Simvastatin treatment significantly increased the expression of PTEN in the tumors of mice (Fig. 5B, compare lanes 5–8 with lanes 1–4 and Fig. 5C). These results indicate that simvastatin may utilize increased PTEN levels in the mouse breast tumors to reduce activation of Akt.
Figure 5.
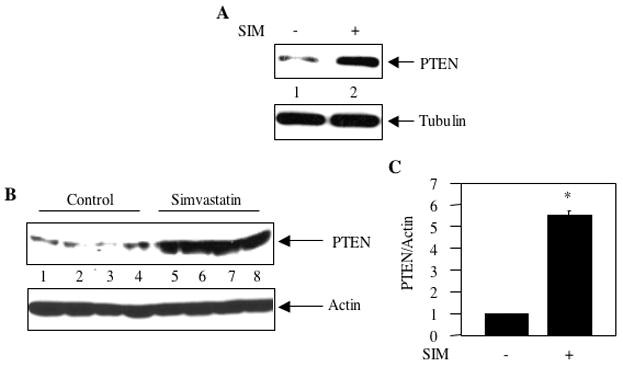
Effect of simvastatin on expression of PTEN. (A) MDA-MB-231 breast cancer cells were incubated with 10 μM simvastatin for 24 hours. The cell lysates were immunoblotted with anti-PTEN and anti-tubulin antibodies as indicated. (B) Tumor lysates from independent control and independent simvastatin-treated mice were immunoblotted with PTEN and actin antibodies respectively. (C) Quantification of PTEN expression in panel B. Mean ± SE of four independent animals is shown. *p < 0.01 vs. control.
3.4. Simvastatin inhibits binding of NFκB to NFRE-1 and NFRE-2 in the PTEN promoter
Expression of PTEN is known to be regulated by a transcriptional mechanism [38]. MDA-MB-231 breast cancer cells possess constitutively active NFκB (Fig. 3) [54]. Two NFκB recognizing DNA elements (NFRE-1 at −1565 bp and NFRE-2 at −1441 bp from ATG start codon) have been identified in the PTEN promoter (Fig. 6A) [42, 60]. We tested binding of NFκB to these elements using nuclear extracts prepared from MDA-MB-231 breast cancer cells. Similar to the results obtained above with consensus NFκB DNA element (Fig. 3A), both NFRE-1 and NFRE-2 formed protein-DNA complex (Fig. 6B and 6C, lanes 1). Use of cold oligonucleotides showed inhibition of protein-DNA complex formation (Figs. 6B and 6C; compare lanes 2 and 3 with lane 1). Cold oligonucleotide recognizing AP2 transcription factor did not block DNA binding (Figs. 6B and 6C; lane 4). These results indicate specificity of NFκB binding to elements present in the PTEN promoter. To examine the identity of NFκB in this protein-DNA complex, we performed supershift analysis using MDA-MB-231 nuclear extracts in the presence of antibody recognizing p65 subunit of NFκB. Fig. 6D shows supershift of the DNA protein complex using NFRE-1 probe as compared to the control (compare lane 2 with lane 1, indicated by asterisk). The modest supershift may be due to less affinity of the antibody-NFκB complex for labeled probe. Erk1/2 antibody was used as a separate IgG control (Fig. 6D, lane 3). Similar supershift was evident with NFRE-2 probe and using p65 antibody, relative to the protein-DNA complex formed in the absence or in the presence of Erk1/2 antibody (Figs. 6E, compare lane 2 with lane 1 and 3, indicated by asterisk). These results indicate the presence of NFκB in the NFRE-1- and NFRE-2-protein complex. Next, we examined the effect of simvastatin on NFκB binding to both these elements using EMSA. Simvastatin inhibited DNA-protein complex formation with the NFRE-1 and NFRE-2 probes, respectively (Figs. 6F and 6G, compare lane 2 with lane 1). These results demonstrate that simvastatin blocks the binding of NFκB with the two DNA elements present in the PTEN promoter.
Figure 6.
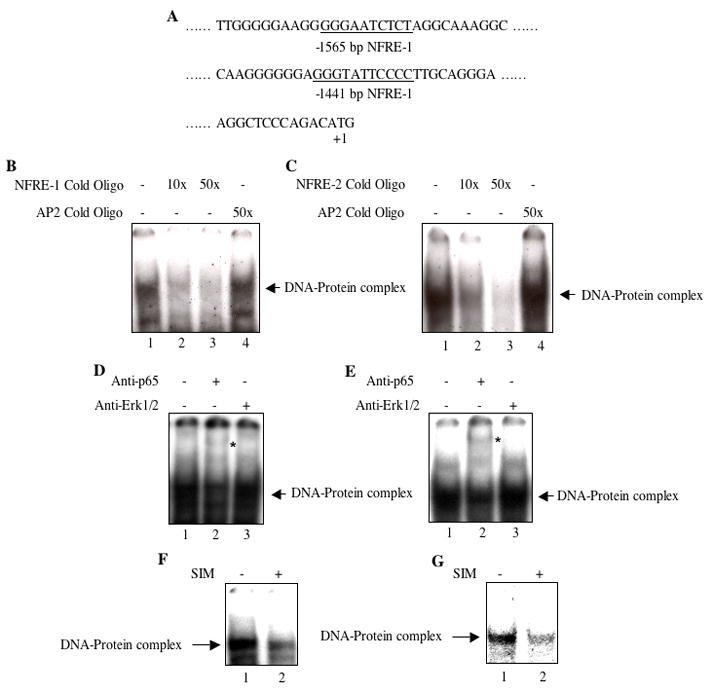
Simvastatin inhibits NFκB binding to NFRE-1 and NFRE-2 present in the PTEN promoter. (A) Partial DNA sequence showing the positions of NFRE-1 and NFRE-2 in the 5′ flanking sequence of PTEN gene (underlined). (B and C) Nuclear extracts from MDA-MB-231 breast cancer cells were used in EMSA with 32P-labeled double stranded NFRE-1 (panel B) and NFRE-2 (panel C) as described in the Materials and Methods. Cold double stranded oligonucleotide representing NFRE-1 and NFRE-2 (lanes 2 and 3 in panels B and C) and AP2 transcription factor binding element (B and C, lane 4) were used for competition. (D and E) Nuclear extracts from MDA-MB-231 cells were incubated with anti-p65 and anti-Erk1/2 antibodies prior to incubation with 32P-labeled double stranded NFRE-1 (panel D) and NFRE-2 (panel E) in EMSA as indicated. (F and G) MDA-MB-231 cells were incubated with 10 μM simvastatin (SIM). Equal amounts of nuclear extracts were used in EMSA with double-stranded 32P-labeled NFRE-1 (panel F) and NFRE-2 (panel G) as described in the Materials and Methods. Arrows indicate the protein-DNA complexes. Asterisks in panels D and E show the supershifted DNA-protein complex in lane 2.
3.5. Simvastatin induces derepression of PTEN via NFκB
Recent reports showed increased expression of PTEN by NFκB [61–63]. However, in lung, colon and cervical cancer cells, NFκB inhibits expression of PTEN [42, 60, 64]. We have shown above that PTEN protein levels are low in MDA-MB-231 breast cancer cells. We examined transcription of PTEN by transient transfection assays using a luciferase reporter plasmid that contained a PTEN genomic fragment including 1,978 bp 5′ from the ATG initiation site (PTEN-Luc) [43, 47, 61–63, 65]. Incubation of PTEN-Luc-transfected MDA-MB-231 cells with simvastatin showed significant increase in PTEN transcription (Fig. 7A). We have shown above that simvastatin inhibits NFκB activation. To test the involvement of NFκB in PTEN transcription, we cotransfected p65 subunit of NFκB along with PTEN-Luc reporter plasmid into MDA-MB-231 cells. p65 significantly inhibited transcription of PTEN (Fig. 7B). Additionally expression of p65 reduced the levels of PTEN protein (Fig. 7C, compare lane 2 with lane 1). This reduction in PTEN was associated with increased phosphorylation of Akt (Fig. 7C, compare lane 2 with lane 1). These results suggest that simvastatin may inhibit the transcriptional suppressor activity of NFκB, resulting in derepression to increase expression of PTEN. To test this hypothesis, we examined the effect of simvastatin on p65-mediated transcriptional repression of PTEN. PTEN-Luc reporter plasmid was cotransfected with p65. The transiently transfected cells were incubated with simvastatin. Simvastatin reversed the repression of PTEN transcription induced by p65 (Fig. 7D). These data demonstrate that simvastatin targets NFκB to inhibit its transcriptional repression activity that downregulates PTEN expression in the human breast cancer cells and confers a growth advantage. Thus simvastatin induces derepression of PTEN to increase its expression, leading to inhibition of MDA-MB-231 breast cancer cell proliferation.
Figure 7.
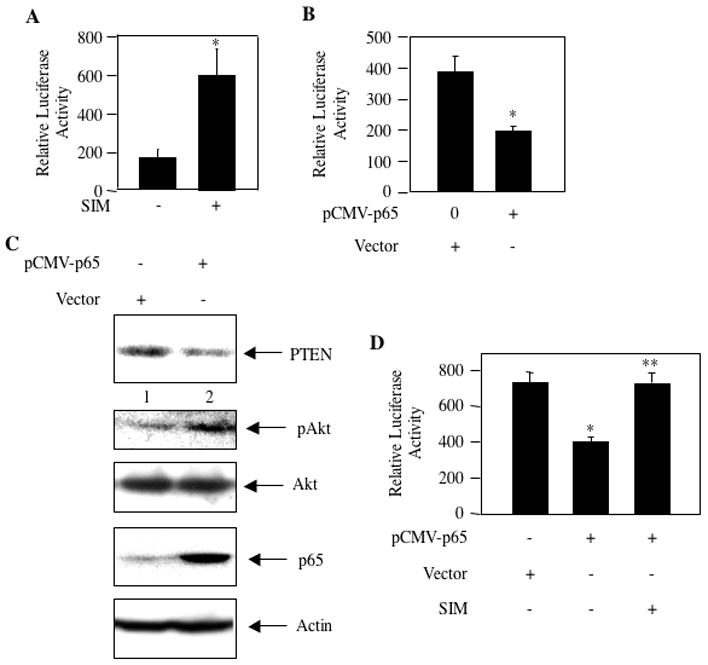
Simvastatin increases transcription of PTEN by NFκB. PTEN-Luc reporter plasmid (panel A) and PTEN-Luc plus pCMV-p65 (panels B and D) as indicated were transfected into MDA-MB-231 cells. In panel A and D, the transiently transfected cells were incubated with 10 μM simvastatin (SIM) as indicated. Cell lysates were used to determine luciferase activity as described in the Materials and Methods. Mean ± SE of triplicate measurements is shown. *p < 0.01 vs. control in panel A. In panel B, *p < 0.05 vs. control. In panel D, *p < 0.01 vs control; **p < 0.01 vs pCMV-p65 alone. (Panel C) MDA-MB-231 cells were transfected with vector or pCMV-p65 expression plasmids as indicated. Equal amounts of cell lysates were immunoblotted with PTEN, phospho-Akt, Akt, p65 and actin antibodies as indicated.
4. Discussion
Recent studies reveal significantly reduced breast cancer risk among women who used a statin [26, 27]. Studies with experimental models show growth inhibitory effect of statins in many cancers [5–16]. This study has explored the effect of simvastatin on key signal transduction pathways and has identified novel points of connection among Akt, NFκB, BclXL and PTEN (Fig. 8). These data reveal that simvastatin inhibits activation of the survival kinase Akt in the tumor tissues of mice developed by the MDA-MB-231 human breast cancer cell xenograft. Significant attenuation of the anti-apoptotic BclXL protein expression by simvastatin in the same mouse model is shown. This defect in BclXL expression is mediated by simvastatin-induced inhibition of NFκB transcriptional activity. Additionally, we demonstrate increased levels of PTEN in simvastatin-treated tumor bearing animals, thus providing a mechanism for growth inhibitory effect of the statin. Increased expression of PTEN correlated with simvastatin-induced reduction in transcriptional repression activity of NFκB in the MDA-MB-231 breast cancer cells. These results provide the first evidence for inhibition of tumor cell growth by simvastatin, which increases the expression of PTEN by relieving NFκB-mediated transcriptional repression.
Figure 8.
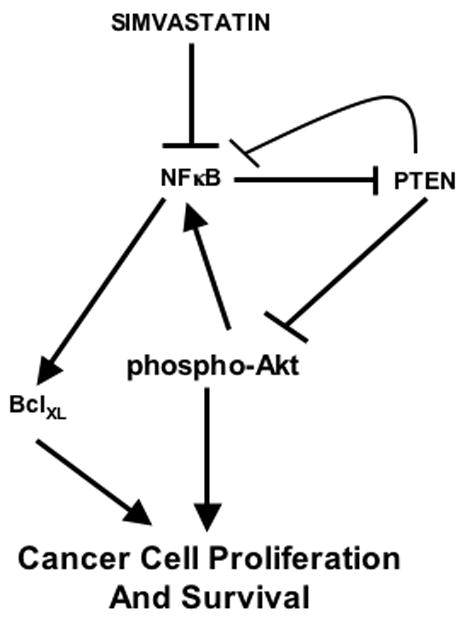
Schematic diagram showing summary of our results. Simvastatin blocks transcriptional activation and transcriptional repression of NFκB to decrease and increase expression of anti-apoptotic BclXL and proapoptotic/antiproliferative PTEN, respectively.
A complex interplay among hormones, growth factors and their receptors such as IGF and members of EGF receptors contribute to the proliferation and survival of breast cancer cells [66]. Constitutive Akt activation is reported in in situ breast tumor and invasive breast cancer. Also, Akt confers chemoresistance and radioresistance to breast cancer [38, 39, 49]. Marked increase in Akt phosphorylation was found in approximately 30–40% breast cancer specimens [67]. Elevated Akt activity correlated with increased malignancy of breast cancer and poor prognosis [67–70]. In the MDA-MB-231 xenograft breast tumor model, we find increased constitutive levels of phosphorylated Akt indicative of activation of this kinase (Fig. 1B and 1C). Our data show that simvastatin significantly reduced the Akt kinase activity in the breast tumor (Fig. 1B and 1C).
Statins have been shown to activate Akt kinase activity in cells in culture and in animal models of endothelium angiogenesis [71–73]. Contrary to these results, recent studies indicate that statins influence Akt activity in many cancer cells including breast cancer cells in culture [21, 74–76]. In a recent study, atorvastatin blocked nuclear localization of phosphorylated Akt [77]. However, phosphorylated Akt was detected in the cytosol indicating a role of the statin in nuclear translocation of Akt without having any effect on its phosphorylation. However, this inhibition of Akt signaling was attributed to statin-mediated activation of mTOR kinase, which inhibits Akt by a negative feedback loop utilizing IRS1/2 [77, 78]. In contrast to these results, statins have been shown to inhibit mTOR activity [79]. In a more recent study, it was shown that statin blocked phosphorylation of Akt with concomitant inhibition of Akt expression [80]. We found inhibition of Akt phosphorylation (Fig. 1A). However, we were unable to see any change in Akt expression in response to simvastatin (Fig. 1A). Many cytostatic drugs activate Akt in tumor cells [81]. Statins have been shown to sensitize tumor cells for cytostatic drugs [75, 82–84]. A recent study demonstrated that although statin inhibited Akt, p53 deficiency was required for sensitivity of the cancer cells to cytostatic drugs [77]. These results suggest that attenuation of tumor cells growth by decreased Akt activity may occur in only p53 null tumor cells. Note that the MDA-MB-231 cells used in the present study possess wild type 53. Thus our observation showing statin-mediated inhibition of Akt and the subsequent tumor cell growth may not involve p53.
Akt kinase acts as a regulator of mammary tumorigenesis. It is associated with increased tumor cell survival and resistance to apoptosis [49]. Hyperactive Akt phosphorylates Bad, leading to increased cell survival by modulation of anti-apoptotic proteins in the mitochondrial membrane [85, 86]. In fact the ratio of antiapoptotic protein such as BclXL to the proapoptotic proteins acts as a rheostat to determine the susceptibility of the cancer cells to apoptosis [87, 88]. Thus the presence of constitutively high levels of anti-apoptotic proteins in the tumor cells pose resistance to apoptosis. We detected elevated levels of BclXL in the MDA-MB-231 breast cancer cells (Fig. 2C). Simvastatin inhibited the expression of BclXL (Fig. 2C). These results are in contrast with the report where no change in BclXL levels was observed in statin-treated cells [80]. Furthermore, we for the first time demonstrate significantly lower expression of BclXL in the tumors of MDA-MB-231 xenografts in the mice treated with simvastatin (Fig. 2A and 2B).
Activated Akt regulates tumor cell proliferation by inactivating a plethora of negative growth regulatory proteins such as p21, p27, tuberin and PRAS40 [89–94]. Akt also attenuates expression of proapoptotic proteins by negatively regulating the transcription factors FoxO and p53 [95–97]. Furthermore, Akt positively regulates the transcriptional activity of NFκB, which target many anti-apoptotic genes including BclXL [53, 55, 56, 98, 99]. Proliferative/anti-apoptotic signals in the tumor cells induces Akt-mediated phosphorylation/activation of IKK complex, which phosphorylates the IκB protein complexed with the NFκB heterodimer p65/p50 [53]. Phosphorylated IκB undergoes degradation, resulting in translocation of NFκB heterodimer to the nucleus to bind to the cognate DNA elements for induction of anti-apoptotic gene expression. MDA-MB-231 cells contain constitutively active NFκB in the nucleus [54]. We confirmed this observation by demonstrating the formation of NFκB-DNA complex using EMSA (Fig. 3A and 3B). Simvastatin prevented this NFκB-DNA complex formation that resulted in inhibition of NFκB-dependent transcription (Fig. 3C and 3D). Furthermore, simvastatin significantly attenuated the transcription of BclXL, which is regulated by NFκB (Fig. 4). These results provide a mechanism how statin may intercept the expression of BclXL to prevent tumor growth in the mice.
One significant mechanism of increased Akt activity in many cancers including breast cancer results from deletion/mutation of the lipid phosphatase PTEN [38–41]. Germ line mutations of PTEN gene cause hamartoma syndromes such as Cowden disease; cells bearing such mutations are characterized by increased incidence of early malignant transformation [38]. Also, women with PTEN mutations suffer from bilateral hypertrophy of the virginal breasts harboring malignant tumors [41]. Recently, it has been shown that humans with BRCA1 mutations possess microdeletion in the PTEN gene [59]. A significant contribution of this lipid phosphatase was established with the PTEN heterozygous mice, which developed tumors in several organs including breast [100–102]. Furthermore, homozygous deletion of PTEN in the mammary gland leads to formation of breast cancer similar to that developed by the activation of Wnt signaling involving inactivation of GSK3β and activation of β-catenin [103]. In fact, the wild type allele of PTEN was lost in the Wnt-1-induced mammary tumor in the PTEN heterozygous background [104]. Expression of PTEN was undetectable in both luminal and myoepithelial cells of these tumors while stromal cells within and outside the tumor expressed PTEN [105]. Also, PTEN negative cells showed significant expression of cyokeratin 6 and Sca-1, indicating that loss of PTEN occurred in common progenitor of luminal and myoepithelial tumor cells [105]. These results indicated a role of PTEN in cancer stem cell generation and maintenance. More recently Korkaya et al reported that down regulation of PTEN in normal mammary epithelial cells resulted in increased Akt phosphorylation concomitant with formation of mammospheres with increased aldehyde dehydrogenase positive mammary stem cells. These mammospheres when inoculated in the humanized NOD/SCID mice produced atypical hyperplasia believed to be the precursor for the development of DCIS (ductal carcinoma in situ) in humans [106]. Similar results were obtained when PTEN was inhibited in the side population of human breast cancer cells, which produced tumorsphere in vitro and resulted in significantly increased tumorigenicity in NOD/SCID mice [106]. Treatment of these tumor-bearing mice with an Akt inhibitor blocked aldehyde dehydrogenase producing cells concomitant with attenuated tumor growth in the xenograft model. These results conclusively demonstrate that Akt activation by dysregulated PTEN function results in generation of breast cancer stem cells, which contribute to chemo- and radio-resistance [107, 108].
Genetic mutation of PTEN in breast cancer is rare and amounts to only 5% of the cases [39, 40]. However, alternative mechanisms of functional PTEN inactivation in the cancer cells with wild type allele have been identified. For example, increased expression of the E3 ubiquitin ligase NEDD4-1 downregulates PTEN in many cancers resulting in increased Akt phosphorylation [109, 110]. Also, reduced immunoreactive wild type PTEN was observed in breast cancer specimens where expression of a redox sensitive protein DJ-1 was increased [58]. More recently a PTEN interacting protein, P-REX2a, has been identified [111]. P-REX2a inhibits PTEN phosphatase activity. In a cohort of breast cancer patients with wild type PTEN, positive correlation between increased levels of P-REX2a mRNA and activating PIK3CA mutation was established. These results indicate that agents that will upregulate active PTEN protein may represent beneficial drugs for breast cancer patients. Our results demonstrate reduced levels of PTEN protein in the MDA-MB-231 breast cancer cell xenograft. Treatment of these mice with simvastatin significantly increased the expression of immunoreactive PTEN in the tumors (Fig. 5). These results for the first time indicate an in vivo role of simvastatin on PTEN protein levels in a model of breast cancer.
Apart from protein stability and protein-protein interaction, expression of PTEN has been shown to be regulated at the level of transcription. For example, in the PTEN promoter, recognition sequences for the transcription factors Egr1, p53, USF, NFκB, PPARγ, Myc and Hif1α are identified, which positively regulate transcription of this lipid phosphatase [47, 61–63, 65, 112–114]. Our results show significant increase in PTEN transcription in response to simvastatin in MDA-MB-231 cells (Fig. 7). Recently simvastatin-induced activation of PPARγ has been shown to correlate with transcription of PTEN [47, 115]. Therefore, the transcriptional upregulation observed in our study could result from activation of PPARγ in response to simvastatin. However, two PPARγ DNA elements present in the PTEN 5′ flanking sequence were mapped to −13,327 and −15,376 bp relative to the transcription start site [47, 113]. Note that in our study, we have used a shorter PTEN promoter, which spans −1978 bp upstream of translation initiation site and thus does not contain the PPARγ DNA elements [43, 47, 61–63, 65]. These results indicate that increased transcription of PTEN in response to simvastatin may not involve PPARγ in our study.
NFκB acts as a transcriptional activator for many antiapoptotic genes including BclXL [55, 56] (Fig. 4). Although NFκB increases the transcription of PTEN, it also acts as a repressor similar to the transcriptional repressor Snail1 and HES1 to inhibit expression of PTEN in many cancer cells [42, 61–64, 112, 116]. Two independent NFκB recognition sequences, NFRE-1 and NFRE-2, have been identified in the PTEN promoter [42]. MDA-MB-231 breast cancer cells contain constitutively active nuclear NFκB [54]. In line with these results, we detected binding of NFκB to the consensus DNA element (Fig. 3). Furthermore, in MDA-MB-231 cells, we show constitutive binding of NFκB to the NFRE-1 and NFRE-2 present in the PTEN promoter (Fig. 6). Simvastatin inhibited binding of NFκB to both these elements (Fig. 6). Additionally, expression of NFκB prevented the transcription of PTEN resulting in reduced protein expression (Fig. 7B and 7C). Furthermore, simvastatin attenuated the p65-dependent transcriptional repression of PTEN (Fig. 7D). These results indicate that simvastatin-mediated inhibition of NFκB activity induces PTEN expression.
Existing breast cancer therapies pose limitations to achieve long-term patient survival due to tumor relapse as a result of chemoresistant mammary cancer cells. Abundant evidence indicates that PTEN/Akt pathway is utilized by breast cancer cells to acquire growth advantage and chemo- and radio-resistances. Our results present the first demonstration of inhibition of transcriptional repressor activity of NFκB in response to simvastatin, which leads to upregulation of PTEN expression, resulting in inhibition of Akt phosphorylation. Thus our results establish a novel mechanism for statin action where it acts as a double-edged sword to repress antiapoptotic BclXL expression and derepress the expression of proapoptotic/antiproliferative PTEN by targeting NFκB.
Supplementary Material
Acknowledgments
The authors thank Patricia St Clair for excellent technical assistance. We thank Brent Wagner, M.D., for critically reading the manuscript. This work was supported by NIH RO1 AR52425 and VA Research Service Merit Review grants (NGC). GGC is recipient of Senior Research Career Scientist Award from the Department of Veterans Affairs. GGC is supported by NIH RO1 DK50190, VA Research Service Merit Review and Juvenile Diabetes Research Foundation 1-2008-185 grants.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Halcox JP, Deanfield JE. Circulation. 2004;109(21 Suppl 1):II42–48. doi: 10.1161/01.CIR.0000129500.29229.92. [DOI] [PubMed] [Google Scholar]
- 2.Jain MK, Ridker PM. Nat Rev Drug Discov. 2005;4(12):977–987. doi: 10.1038/nrd1901. [DOI] [PubMed] [Google Scholar]
- 3.Goldstein JL, Brown MS. Nature. 1990;343(6257):425–430. doi: 10.1038/343425a0. [DOI] [PubMed] [Google Scholar]
- 4.Jackson SM, Ericsson J, Edwards PA. Subcell Biochem. 1997;28:1–21. doi: 10.1007/978-1-4615-5901-6_1. [DOI] [PubMed] [Google Scholar]
- 5.Agarwal B, Rao CV, Bhendwal S, Ramey WR, Shirin H, Reddy BS, Holt PR. Gastroenterology. 1999;117(4):838–847. doi: 10.1016/s0016-5085(99)70342-2. [DOI] [PubMed] [Google Scholar]
- 6.Alonso DF, Farina HG, Skilton G, Gabri MR, De Lorenzo MS, Gomez DE. Breast Cancer Res Treat. 1998;50(1):83–93. doi: 10.1023/a:1006058409974. [DOI] [PubMed] [Google Scholar]
- 7.Clutterbuck RD, Millar BC, Powles RL, Newman A, Catovsky D, Jarman M, Millar JL. Br J Haematol. 1998;102(2):522–527. doi: 10.1046/j.1365-2141.1998.00783.x. [DOI] [PubMed] [Google Scholar]
- 8.Hawk MA, Cesen KT, Siglin JC, Stoner GD, Ruch RJ. Cancer Lett. 1996;109(1–2):217–222. doi: 10.1016/s0304-3835(96)04465-5. [DOI] [PubMed] [Google Scholar]
- 9.He L, Mo H, Hadisusilo S, Qureshi AA, Elson CE. J Nutr. 1997;127(5):668–674. doi: 10.1093/jn/127.5.668. [DOI] [PubMed] [Google Scholar]
- 10.Inano H, Suzuki K, Onoda M, Wakabayashi K. Carcinogenesis. 1997;18(9):1723–1727. doi: 10.1093/carcin/18.9.1723. [DOI] [PubMed] [Google Scholar]
- 11.Kikuchi T, Nagata Y, Abe T. J Neurooncol. 1997;34(3):233–239. doi: 10.1023/a:1005753523949. [DOI] [PubMed] [Google Scholar]
- 12.Narisawa T, Fukaura Y, Tanida N, Hasebe M, Ito M, Aizawa R. Tohoku J Exp Med. 1996;180(2):131–138. doi: 10.1620/tjem.180.131. [DOI] [PubMed] [Google Scholar]
- 13.Narisawa T, Morotomi M, Fukaura Y, Hasebe M, Ito M, Aizawa R. Jpn J Cancer Res. 1996;87(8):798–804. doi: 10.1111/j.1349-7006.1996.tb02103.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shibata MA, Kavanaugh C, Shibata E, Abe H, Nguyen P, Otsuki Y, Trepel JB, Green JE. Carcinogenesis. 2003;24(3):453–459. doi: 10.1093/carcin/24.3.453. [DOI] [PubMed] [Google Scholar]
- 15.Swamy MV, Cooma I, Reddy BS, Rao CV. Int J Oncol. 2002;20(4):753–759. [PubMed] [Google Scholar]
- 16.Weis M, Heeschen C, Glassford AJ, Cooke JP. Circulation. 2002;105(6):739–745. doi: 10.1161/hc0602.103393. [DOI] [PubMed] [Google Scholar]
- 17.Lubet RA, Boring D, Steele VE, Ruppert JM, Juliana MM, Grubbs CJ. Cancer Prev Res (Phila Pa) 2009;2(2):161–167. doi: 10.1158/1940-6207.CAPR-08-0134. [DOI] [PubMed] [Google Scholar]
- 18.Sacks FM, Pfeffer MA, Moye LA, Rouleau JL, Rutherford JD, Cole TG, Brown L, Warnica JW, Arnold JM, Wun CC, Davis BR, Braunwald E. N Engl J Med. 1996;335(14):1001–1009. doi: 10.1056/NEJM199610033351401. [DOI] [PubMed] [Google Scholar]
- 19.Kaye JA, Jick H. Br J Cancer. 2004;90(3):635–637. doi: 10.1038/sj.bjc.6601566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Graaf MR, Beiderbeck AB, Egberts AC, Richel DJ, Guchelaar HJ. J Clin Oncol. 2004;22(12):2388–2394. doi: 10.1200/JCO.2004.02.027. [DOI] [PubMed] [Google Scholar]
- 21.Graaf MR, Richel DJ, van Noorden CJ, Guchelaar HJ. Cancer Treat Rev. 2004;30(7):609–641. doi: 10.1016/j.ctrv.2004.06.010. [DOI] [PubMed] [Google Scholar]
- 22.Shannon J, Tewoderos S, Garzotto M, Beer TM, Derenick R, Palma A, Farris PE. Am J Epidemiol. 2005;162(4):318–325. doi: 10.1093/aje/kwi203. [DOI] [PubMed] [Google Scholar]
- 23.Dellavalle RP, Nicholas MK, Schilling LM. Am J Ther. 2003;10(3):203–210. doi: 10.1097/00045391-200305000-00007. [DOI] [PubMed] [Google Scholar]
- 24.Demierre MF, Higgins PD, Gruber SB, Hawk E, Lippman SM. Nat Rev Cancer. 2005;5(12):930–942. doi: 10.1038/nrc1751. [DOI] [PubMed] [Google Scholar]
- 25.Poynter JN, Gruber SB, Higgins PD, Almog R, Bonner JD, Rennert HS, Low M, Greenson JK, Rennert G. N Engl J Med. 2005;352(21):2184–2192. doi: 10.1056/NEJMoa043792. [DOI] [PubMed] [Google Scholar]
- 26.Beck P, Wysowski DK, Downey W, Butler-Jones D. J Clin Epidemiol. 2003;56(3):280–285. doi: 10.1016/s0895-4356(02)00614-5. [DOI] [PubMed] [Google Scholar]
- 27.Cauley JA, Zmuda JM, Lui LY, Hillier TA, Ness RB, Stone KL, Cummings SR, Bauer DC. J Womens Health (Larchmt) 2003;12(8):749–756. doi: 10.1089/154099903322447710. [DOI] [PubMed] [Google Scholar]
- 28.Denoyelle C, Vasse M, Korner M, Mishal Z, Ganne F, Vannier JP, Soria J, Soria C. Carcinogenesis. 2001;22(8):1139–1148. doi: 10.1093/carcin/22.8.1139. [DOI] [PubMed] [Google Scholar]
- 29.Kusama T, Mukai M, Iwasaki T, Tatsuta M, Matsumoto Y, Akedo H, Nakamura H. Cancer Res. 2001;61(12):4885–4891. [PubMed] [Google Scholar]
- 30.Malliri A, Collard JG. Curr Opin Cell Biol. 2003;15(5):583–589. doi: 10.1016/s0955-0674(03)00098-x. [DOI] [PubMed] [Google Scholar]
- 31.Mazieres J, Tillement V, Allal C, Clanet C, Bobin L, Chen Z, Sebti SM, Favre G, Pradines A. Exp Cell Res. 2005;304(2):354–364. doi: 10.1016/j.yexcr.2004.10.019. [DOI] [PubMed] [Google Scholar]
- 32.Simpson KJ, Dugan AS, Mercurio AM. Cancer Res. 2004;64(23):8694–8701. doi: 10.1158/0008-5472.CAN-04-2247. [DOI] [PubMed] [Google Scholar]
- 33.Zugaza JL, Caloca MJ, Bustelo XR. Oncogene. 2004;23(34):5823–5833. doi: 10.1038/sj.onc.1207768. [DOI] [PubMed] [Google Scholar]
- 34.Rao S, Porter DC, Chen X, Herliczek T, Lowe M, Keyomarsi K. Proc Natl Acad Sci U S A. 1999;96(14):7797–7802. doi: 10.1073/pnas.96.14.7797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ukomadu C, Dutta A. J Biol Chem. 2003;278(44):43586–43594. doi: 10.1074/jbc.M307194200. [DOI] [PubMed] [Google Scholar]
- 36.Wu J, Wong WW, Khosravi F, Minden MD, Penn LZ. Cancer Res. 2004;64(18):6461–6468. doi: 10.1158/0008-5472.CAN-04-0866. [DOI] [PubMed] [Google Scholar]
- 37.Vivanco I, Sawyers CL. Nat Rev Cancer. 2002;2(7):489–501. doi: 10.1038/nrc839. [DOI] [PubMed] [Google Scholar]
- 38.Cully M, You H, Levine AJ, Mak TW. Nat Rev Cancer. 2006;6(3):184–192. doi: 10.1038/nrc1819. [DOI] [PubMed] [Google Scholar]
- 39.Cantley LC, Neel BG. Proc Natl Acad Sci U S A. 1999;96(8):4240–4245. doi: 10.1073/pnas.96.8.4240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li J, Yen C, Liaw D, Podsypanina K, Bose S, Wang SI, Puc J, Miliaresis C, Rodgers L, McCombie R, Bigner SH, Giovanella BC, Ittmann M, Tycko B, Hibshoosh H, Wigler MH, Parsons R. Science. 1997;275(5308):1943–1947. doi: 10.1126/science.275.5308.1943. [DOI] [PubMed] [Google Scholar]
- 41.Stiles B, Groszer M, Wang S, Jiao J, Wu H. Dev Biol. 2004;273(2):175–184. doi: 10.1016/j.ydbio.2004.06.008. [DOI] [PubMed] [Google Scholar]
- 42.Vasudevan KM, Gurumurthy S, Rangnekar VM. Mol Cell Biol. 2004;24(3):1007–1021. doi: 10.1128/MCB.24.3.1007-1021.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ghosh-Choudhury T, Mandal CC, Woodruff K, St Clair P, Fernandes G, Choudhury GG, Ghosh-Choudhury N. Breast Cancer Res Treat. 2008 doi: 10.1007/s10549-008-0227-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ghosh-Choudhury N, Mandal CC, Choudhury GG. J Biol Chem. 2007;282(7):4983–4993. doi: 10.1074/jbc.M606706200. [DOI] [PubMed] [Google Scholar]
- 45.Ghosh-Choudhury N, Singha PK, Woodruff K, St Clair P, Bsoul S, Werner SL, Choudhury GG. J Biol Chem. 2006;281(29):20160–20170. doi: 10.1074/jbc.M511071200. [DOI] [PubMed] [Google Scholar]
- 46.Ghosh-Choudhury N, Woodruff K, Qi W, Celeste A, Abboud SL, Ghosh Choudhury G. Biochem Biophys Res Commun. 2000;272(3):705–711. doi: 10.1006/bbrc.2000.2844. [DOI] [PubMed] [Google Scholar]
- 47.Mahimainathan L, Ghosh-Choudhury N, Venkatesan B, Das F, Mandal CC, Dey N, Habib SL, Kasinath BS, Abboud HE, Ghosh Choudhury G. J Biol Chem. 2009 doi: 10.1074/jbc.M109.028860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Larue L, Bellacosa A. Oncogene. 2005;24(50):7443–7454. doi: 10.1038/sj.onc.1209091. [DOI] [PubMed] [Google Scholar]
- 49.Manning BD, Cantley LC. Cell. 2007;129(7):1261–1274. doi: 10.1016/j.cell.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Testa JR, Tsichlis PN. Oncogene. 2005;24(50):7391–7393. doi: 10.1038/sj.onc.1209100. [DOI] [PubMed] [Google Scholar]
- 51.Kane LP, Shapiro VS, Stokoe D, Weiss A. Curr Biol. 1999;9(11):601–604. doi: 10.1016/s0960-9822(99)80265-6. [DOI] [PubMed] [Google Scholar]
- 52.Madrid LV, Mayo MW, Reuther JY, Baldwin AS., Jr J Biol Chem. 2001;276(22):18934–18940. doi: 10.1074/jbc.M101103200. [DOI] [PubMed] [Google Scholar]
- 53.Ozes ON, Mayo LD, Gustin JA, Pfeffer SR, Pfeffer LM, Donner DB. Nature. 1999;401(6748):82–85. doi: 10.1038/43466. [DOI] [PubMed] [Google Scholar]
- 54.Nakshatri H, Bhat-Nakshatri P, Martin DA, Goulet RJ, Jr, Sledge GW., Jr Mol Cell Biol. 1997;17(7):3629–3639. doi: 10.1128/mcb.17.7.3629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ahn KS, Sethi G, Krishnan K, Aggarwal BB. J Biol Chem. 2007;282(1):809–820. doi: 10.1074/jbc.M610028200. [DOI] [PubMed] [Google Scholar]
- 56.Konishi T, Sasaki S, Watanabe T, Kitayama J, Nagawa H. Oncogene. 2006;25(22):3160–3169. doi: 10.1038/sj.onc.1209342. [DOI] [PubMed] [Google Scholar]
- 57.Baker SJ. Cell. 2007;128(1):25–28. doi: 10.1016/j.cell.2006.12.023. [DOI] [PubMed] [Google Scholar]
- 58.Kim RH, Peters M, Jang Y, Shi W, Pintilie M, Fletcher GC, DeLuca C, Liepa J, Zhou L, Snow B, Binari RC, Manoukian AS, Bray MR, Liu FF, Tsao MS, Mak TW. Cancer Cell. 2005;7(3):263–273. doi: 10.1016/j.ccr.2005.02.010. [DOI] [PubMed] [Google Scholar]
- 59.Saal LH, Gruvberger-Saal SK, Persson C, Lovgren K, Jumppanen M, Staaf J, Jonsson G, Pires MM, Maurer M, Holm K, Koujak S, Subramaniyam S, Vallon-Christersson J, Olsson H, Su T, Memeo L, Ludwig T, Ethier SP, Krogh M, Szabolcs M, Murty VV, Isola J, Hibshoosh H, Parsons R, Borg A. Nat Genet. 2008;40(1):102–107. doi: 10.1038/ng.2007.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kim S, Domon-Dell C, Kang J, Chung DH, Freund JN, Evers BM. J Biol Chem. 2004;279(6):4285–4291. doi: 10.1074/jbc.M308383200. [DOI] [PubMed] [Google Scholar]
- 61.Chandrasekar B, Boylston WH, Venkatachalam K, Webster NJ, Prabhu SD, Valente AJ. J Biol Chem. 2008;283(36):24889–24898. doi: 10.1074/jbc.M804236200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chandrasekar B, Valente AJ, Freeman GL, Mahimainathan L, Mummidi S. Biochem Biophys Res Commun. 2006;339(3):956–963. doi: 10.1016/j.bbrc.2005.11.100. [DOI] [PubMed] [Google Scholar]
- 63.Zabalgoitia M, Colston JT, Reddy SV, Holt JW, Regan RF, Stec DE, Rimoldi JM, Valente AJ, Chandrasekar B. Free Radic Biol Med. 2008;44(3):284–298. doi: 10.1016/j.freeradbiomed.2007.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Xia D, Srinivas H, Ahn YH, Sethi G, Sheng X, Yung WK, Xia Q, Chiao PJ, Kim H, Brown PH, Wistuba, Aggarwal BB, Kurie JM. J Biol Chem. 2007;282(6):3507–3519. doi: 10.1074/jbc.M610141200. [DOI] [PubMed] [Google Scholar]
- 65.Virolle T, Adamson ED, Baron V, Birle D, Mercola D, Mustelin T, de Belle I. Nat Cell Biol. 2001;3(12):1124–1128. doi: 10.1038/ncb1201-1124. [DOI] [PubMed] [Google Scholar]
- 66.Kurokawa H, Arteaga CL. Clin Cancer Res. 2001;7(12 Suppl):4436s–4442s. discussion 4411s–4412s. [PubMed] [Google Scholar]
- 67.Tokunaga E, Kimura Y, Mashino K, Oki E, Kataoka A, Ohno S, Morita M, Kakeji Y, Baba H, Maehara Y. Breast Cancer. 2006;13(2):137–144. doi: 10.2325/jbcs.13.137. [DOI] [PubMed] [Google Scholar]
- 68.Kirkegaard T, Witton CJ, McGlynn LM, Tovey SM, Dunne B, Lyon A, Bartlett JM. J Pathol. 2005;207(2):139–146. doi: 10.1002/path.1829. [DOI] [PubMed] [Google Scholar]
- 69.Sahoo S, Brickley DR, Kocherginsky M, Conzen SD. Eur J Cancer. 2005;41(17):2754–2759. doi: 10.1016/j.ejca.2005.07.018. [DOI] [PubMed] [Google Scholar]
- 70.Zhou X, Tan M, Stone Hawthorne V, Klos KS, Lan KH, Yang Y, Yang W, Smith TL, Shi D, Yu D. Clin Cancer Res. 2004;10(20):6779–6788. doi: 10.1158/1078-0432.CCR-04-0112. [DOI] [PubMed] [Google Scholar]
- 71.Kureishi Y, Luo Z, Shiojima I, Bialik A, Fulton D, Lefer DJ, Sessa WC, Walsh K. Nat Med. 2000;6(9):1004–1010. doi: 10.1038/79510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Skaletz-Rorowski A, Lutchman M, Kureishi Y, Lefer DJ, Faust JR, Walsh K. Cardiovasc Res. 2003;57(1):253–264. doi: 10.1016/s0008-6363(02)00618-1. [DOI] [PubMed] [Google Scholar]
- 73.Skaletz-Rorowski A, Walsh K. Curr Opin Lipidol. 2003;14(6):599–603. doi: 10.1097/00041433-200312000-00008. [DOI] [PubMed] [Google Scholar]
- 74.Denoyelle C, Albanese P, Uzan G, Hong L, Vannier JP, Soria J, Soria C. Cell Signal. 2003;15(3):327–338. doi: 10.1016/s0898-6568(02)00124-9. [DOI] [PubMed] [Google Scholar]
- 75.Khanzada UK, Pardo OE, Meier C, Downward J, Seckl MJ, Arcaro A. Oncogene. 2006;25(6):877–887. doi: 10.1038/sj.onc.1209117. [DOI] [PubMed] [Google Scholar]
- 76.Peres C, Yart A, Perret B, Salles JP, Raynal P. FEBS Lett. 2003;534(1–3):164–168. doi: 10.1016/s0014-5793(02)03832-2. [DOI] [PubMed] [Google Scholar]
- 77.Roudier E, Mistafa O, Stenius U. Mol Cancer Ther. 2006;5(11):2706–2715. doi: 10.1158/1535-7163.MCT-06-0352. [DOI] [PubMed] [Google Scholar]
- 78.Paajarvi G, Roudier E, Crisby M, Hogberg J, Stenius U. Faseb J. 2005;19(3):476–478. doi: 10.1096/fj.04-2745fje. [DOI] [PubMed] [Google Scholar]
- 79.Woodard J, Sassano A, Hay N, Platanias LC. Clin Cancer Res. 2008;14(14):4640–4649. doi: 10.1158/1078-0432.CCR-07-5232. [DOI] [PubMed] [Google Scholar]
- 80.Aberg M, Wickstrom M, Siegbahn A. Thromb Res. 2008;122(2):191–202. doi: 10.1016/j.thromres.2007.09.017. [DOI] [PubMed] [Google Scholar]
- 81.West KA, Castillo SS, Dennis PA. Drug Resist Updat. 2002;5(6):234–248. doi: 10.1016/s1368-7646(02)00120-6. [DOI] [PubMed] [Google Scholar]
- 82.Holstein SA, Hohl RJ. Mol Cancer Ther. 2001;1(2):141–149. [PubMed] [Google Scholar]
- 83.Kozar K, Kaminski R, Legat M, Kopec M, Nowis D, Skierski JS, Koronkiewicz M, Jakobisiak M, Golab J. Int J Oncol. 2004;24(5):1149–1157. [PubMed] [Google Scholar]
- 84.Wang W, Collie-Duguid E, Cassidy J. FEBS Lett. 2002;531(3):415–420. doi: 10.1016/s0014-5793(02)03575-5. [DOI] [PubMed] [Google Scholar]
- 85.Datta SR, Dudek H, Tao X, Masters S, Fu H, Gotoh Y, Greenberg ME. Cell. 1997;91(2):231–241. doi: 10.1016/s0092-8674(00)80405-5. [DOI] [PubMed] [Google Scholar]
- 86.Harada H, Andersen JS, Mann M, Terada N, Korsmeyer SJ. Proc Natl Acad Sci U S A. 2001;98(17):9666–9670. doi: 10.1073/pnas.171301998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Danial NN, Korsmeyer SJ. Cell. 2004;116(2):205–219. doi: 10.1016/s0092-8674(04)00046-7. [DOI] [PubMed] [Google Scholar]
- 88.Spierings D, McStay G, Saleh M, Bender C, Chipuk J, Maurer U, Green DR. Science. 2005;310(5745):66–67. doi: 10.1126/science.1117105. [DOI] [PubMed] [Google Scholar]
- 89.Kwiatkowski DJ, Manning BD. Hum Mol Genet. 2005;14(Spec No 2):R251–258. doi: 10.1093/hmg/ddi260. [DOI] [PubMed] [Google Scholar]
- 90.Liang J, Zubovitz J, Petrocelli T, Kotchetkov R, Connor MK, Han K, Lee JH, Ciarallo S, Catzavelos C, Beniston R, Franssen E, Slingerland JM. Nat Med. 2002;8(10):1153–1160. doi: 10.1038/nm761. [DOI] [PubMed] [Google Scholar]
- 91.Sancak Y, Thoreen CC, Peterson TR, Lindquist RA, Kang SA, Spooner E, Carr SA, Sabatini DM. Mol Cell. 2007;25(6):903–915. doi: 10.1016/j.molcel.2007.03.003. [DOI] [PubMed] [Google Scholar]
- 92.Shin I, Yakes FM, Rojo F, Shin NY, Bakin AV, Baselga J, Arteaga CL. Nat Med. 2002;8(10):1145–1152. doi: 10.1038/nm759. [DOI] [PubMed] [Google Scholar]
- 93.Viglietto G, Motti ML, Bruni P, Melillo RM, D’Alessio A, Califano D, Vinci F, Chiappetta G, Tsichlis P, Bellacosa A, Fusco A, Santoro M. Nat Med. 2002;8(10):1136–1144. doi: 10.1038/nm762. [DOI] [PubMed] [Google Scholar]
- 94.Zhou BP, Liao Y, Xia W, Spohn B, Lee MH, Hung MC. Nat Cell Biol. 2001;3(3):245–252. doi: 10.1038/35060032. [DOI] [PubMed] [Google Scholar]
- 95.Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS, Anderson MJ, Arden KC, Blenis J, Greenberg ME. Cell. 1999;96(6):857–868. doi: 10.1016/s0092-8674(00)80595-4. [DOI] [PubMed] [Google Scholar]
- 96.Mayo LD, Donner DB. Proc Natl Acad Sci U S A. 2001;98(20):11598–11603. doi: 10.1073/pnas.181181198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Zhou BP, Liao Y, Xia W, Zou Y, Spohn B, Hung MC. Nat Cell Biol. 2001;3(11):973–982. doi: 10.1038/ncb1101-973. [DOI] [PubMed] [Google Scholar]
- 98.Angileri FF, Aguennouz M, Conti A, La Torre D, Cardali S, Crupi R, Tomasello C, Germano A, Vita G, Tomasello F. Cancer. 2008 doi: 10.1002/cncr.23407. [DOI] [PubMed] [Google Scholar]
- 99.Takase O, Minto AW, Puri TS, Cunningham PN, Jacob A, Hayashi M, Quigg RJ. Kidney Int. 2008;73(5):567–577. doi: 10.1038/sj.ki.5002563. [DOI] [PubMed] [Google Scholar]
- 100.Di Cristofano A, Pesce B, Cordon-Cardo C, Pandolfi PP. Nat Genet. 1998;19(4):348–355. doi: 10.1038/1235. [DOI] [PubMed] [Google Scholar]
- 101.Podsypanina K, Ellenson LH, Nemes A, Gu J, Tamura M, Yamada KM, Cordon-Cardo C, Catoretti G, Fisher PE, Parsons R. Proc Natl Acad Sci U S A. 1999;96(4):1563–1568. doi: 10.1073/pnas.96.4.1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Stambolic V, Tsao MS, Macpherson D, Suzuki A, Chapman WB, Mak TW. Cancer Res. 2000;60(13):3605–3611. [PubMed] [Google Scholar]
- 103.Li G, Robinson GW, Lesche R, Martinez-Diaz H, Jiang Z, Rozengurt N, Wagner KU, Wu DC, Lane TF, Liu X, Hennighausen L, Wu H. Development. 2002;129(17):4159–4170. doi: 10.1242/dev.129.17.4159. [DOI] [PubMed] [Google Scholar]
- 104.Li Y, Podsypanina K, Liu X, Crane A, Tan LK, Parsons R, Varmus HE. BMC Mol Biol. 2001;2:2. doi: 10.1186/1471-2199-2-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Li Y, Welm B, Podsypanina K, Huang S, Chamorro M, Zhang X, Rowlands T, Egeblad M, Cowin P, Werb Z, Tan LK, Rosen JM, Varmus HE. Proc Natl Acad Sci U S A. 2003;100(26):15853–15858. doi: 10.1073/pnas.2136825100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Korkaya H, Paulson A, Charafe-Jauffret E, Ginestier C, Brown M, Dutcher J, Clouthier SG, Wicha MS. PLoS Biol. 2009;7(6):e1000121. doi: 10.1371/journal.pbio.1000121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Berns K, Horlings HM, Hennessy BT, Madiredjo M, Hijmans EM, Beelen K, Linn SC, Gonzalez-Angulo AM, Stemke-Hale K, Hauptmann M, Beijersbergen RL, Mills GB, van de Vijver MJ, Bernards R. Cancer Cell. 2007;12(4):395–402. doi: 10.1016/j.ccr.2007.08.030. [DOI] [PubMed] [Google Scholar]
- 108.Shafee N, Smith CR, Wei S, Kim Y, Mills GB, Hortobagyi GN, Stanbridge EJ, Lee EY. Cancer Res. 2008;68(9):3243–3250. doi: 10.1158/0008-5472.CAN-07-5480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Trotman LC, Wang X, Alimonti A, Chen Z, Teruya-Feldstein J, Yang H, Pavletich NP, Carver BS, Cordon-Cardo C, Erdjument-Bromage H, Tempst P, Chi SG, Kim HJ, Misteli T, Jiang X, Pandolfi PP. Cell. 2007;128(1):141–156. doi: 10.1016/j.cell.2006.11.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Wang X, Trotman LC, Koppie T, Alimonti A, Chen Z, Gao Z, Wang J, Erdjument-Bromage H, Tempst P, Cordon-Cardo C, Pandolfi PP, Jiang X. Cell. 2007;128(1):129–139. doi: 10.1016/j.cell.2006.11.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Fine B, Hodakoski C, Koujak S, Su T, Saal LH, Maurer M, Hopkins B, Keniry M, Sulis ML, Mense S, Hibshoosh H, Parsons R. Science. 2009;325(5945):1261–1265. doi: 10.1126/science.1173569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Palomero T, Sulis ML, Cortina M, Real PJ, Barnes K, Ciofani M, Caparros E, Buteau J, Brown K, Perkins SL, Bhagat G, Agarwal AM, Basso G, Castillo M, Nagase S, Cordon-Cardo C, Parsons R, Zuniga-Pflucker JC, Dominguez M, Ferrando AA. Nat Med. 2007;13(10):1203–1210. doi: 10.1038/nm1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Patel L, Pass I, Coxon P, Downes CP, Smith SA, Macphee CH. Curr Biol. 2001;11(10):764–768. doi: 10.1016/s0960-9822(01)00225-1. [DOI] [PubMed] [Google Scholar]
- 114.Stambolic V, MacPherson D, Sas D, Lin Y, Snow B, Jang Y, Benchimol S, Mak TW. Mol Cell. 2001;8(2):317–325. doi: 10.1016/s1097-2765(01)00323-9. [DOI] [PubMed] [Google Scholar]
- 115.Teresi RE, Shaiu CW, Chen CS, Chatterjee VK, Waite KA, Eng C. Int J Cancer. 2006;118(10):2390–2398. doi: 10.1002/ijc.21799. [DOI] [PubMed] [Google Scholar]
- 116.Escriva M, Peiro S, Herranz N, Villagrasa P, Dave N, Montserrat-Sentis B, Murray SA, Franci C, Gridley T, Virtanen I, Garcia de Herreros A. Mol Cell Biol. 2008;28(5):1528–1540. doi: 10.1128/MCB.02061-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.