

Abstract
A complementary concept for superarming glycosyl donors through the use of common protecting groups was previously discovered with S-benzoxazolyl (SBox) glycosyl donors. As this strategy can be of benefit to existing oligosaccharide methodologies, it has now been expanded to encompass a wide array of common, stable glycosyl donors. The versatility of this developed technique has been further illustrated in application to a sequential chemoselective oligosaccharide synthesis, wherein a superarmed ethyl thioglycoside was incorporated into the conventional armed-disarmed strategy.
Keywords: Glycosylation, carbohydrates, chemoselectivity
Introduction
Only recently, has the tremendous biological significance and therapeutic potential of carbohydrates and conjugates thereof (glycoproteins, glycolipids, proteoglycans, etc.) begun to emerge.1 However, success in studying these fascinating biomolecules has proven to be directly correlated to the availability of the involved carbohydrates. With the low availability of pure natural isolates, the invention of efficient chemical and enzymatic methods for the synthesis of complex carbohydrates has become increasingly important. This need has led to the development of many excellent new methods for glycoside synthesis,2,3 from which a variety of expeditious strategies for oligosaccharide assembly have surfaced.4,5 Amongst these, the armed-disarmed strategy is of particular interest.6,7 As first introduced by Fraser-Reid and co-workers, this approach allows for the expeditious synthesis of oligosaccharides while necessitating only one type of anomeric leaving group. Thus, the reactivities of the building blocks involved in such chemoselective activations are differentiated by the electronic and/or torsional effects of the protecting groups.8–10 Though initially discovered with O-pentenyl glycosides, the armed-disarmed concept has since been proven with many other classes of glycosyl donors.5
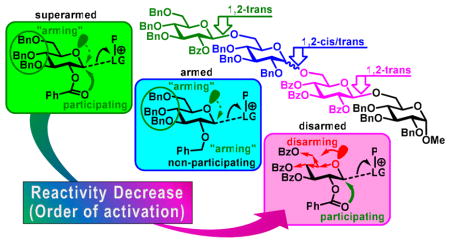
This strategy led to the commonly accepted belief that benzylated derivatives are always significantly more reactive than their benzoylated counterparts, and that the overall glycosyl donor reactivity is in direct correlation with the total number of benzyl substituents.11 However, this presumption was challenged with the discovery of the O-2/O-5 cooperative effect, wherein it was observed that glycosyl donors with mixed protecting group patterns showed unexpected reactivity trends.12 This was the first indication that the reactivity of the glycosyl donor was not limited to the electron withdrawing/donating (or torsional) effects of its protecting groups. Therein, it was proposed that the reactivity of glycosyl donors was also influenced by how well the glycosyl cation, formed upon promoter (P) assisted leaving group (LG) departure, could self-stabilize. As depicted below (Figure 1), in the case of the armed, benzylated glycosyl donor (B), stabilization can be efficiently achieved via the oxacarbenium intermediate, through resonance with the electronically “armed” lone pair electrons of O-5. However, in the case of the per-benzoylated derivative (C), this type of stabilization would be less likely due to the electron-withdrawing substituents at C-4 and C-6. Instead, the acyl substituent at C-2 allows for stabilization via the acyloxonium intermediate. Therefore, the combination of these two opposing effects results in the decreased reactivity of disarmed donor (C).
Figure 1.

Cooperative arming and disarming effects.
It then follows, that a glycosyl donor wherein there is no stabilization/participating group at C-2, and electron-withdrawing groups at the remaining positions, would be even further deactivated, as is the case for superdisarmed glycosyl donor (D).12 Crich and Li additionally investigated this phenomenon for the S-benzoxazolyl (SBox) glycosyl donors of the D-gluco series, finding that in order for this C-2 stabilization to occur, a 1,2-trans anomeric configuration is necessary.13 This also suggests that the lone pair at C-2 must also have access to the developing positive charge as the leaving group departs, and therefore implies that anchimeric assistance is in part, if not fully, responsible for the observed C-2 effect. Building upon this concept, we then determined that glycosyl donors possessing a participating moiety at C-2 and an electronically armed lone pair at O-5 (A), would have exceptionally high reactivity as the both stabilizing effects would be combined.14,15
Results and discussion
This concept of superarming was previously explored with benzoxazolyl 2-O-benzoyl-3,4,6-tri-O-benzyl-1-thio-β-D-glycopyranosides (SBox glycosides) of the D-gluco, D-galacto, and D-manno series,14,15 however it remained unclear whether this principle could be applied to other classes of leaving groups. Previously, the term “super-armed” was coined by Bols and co-workers in their recent publications in reference to conformationally modified glycosyl donors.16–18 Herein, we describe the investigation of stable glycosyl donors of the armed (1) and superarmed (2) series, bearing common leaving groups: O-pentenyl, S-ethyl, S-phenyl, S-tolyl, and S-thiazolinyl (STaz, Figure 2). All test glycosylations were performed with the standard glycosyl acceptor 319 to afford disaccharides 420 and 520 from glycosyl donors of the armed and superarmed series, respectively (Table 1). Additionally, the obtained results were compared to the previously investigated SBox glycosides 1a21 and 2a.14
Figure 2.

Glycosyl donors of the armed and superarmed series.
Table 1.
Comparative activation of armed (1) and superarmed (2) glycosyl donors
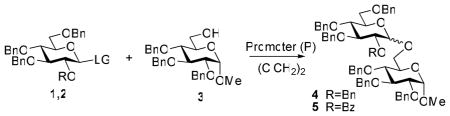 | |||||||
|---|---|---|---|---|---|---|---|
| Entry | D | P | Temp. | Time | CP | Yield | α:β |
| 1 | 1A | DMTST | 0 °C | 2 h | 4 | 92% | 2:1 |
| 2 | 2A | DMTST | 0 °C | 5 min | 5 | 86% | β only |
| 3 | 1A | Cu(OTf)2 | 0 °C | 2 h | 4 | 99% | 2:1 |
| 4 | 2A | Cu(OTf)2 | 0 °C | 20 min | 5 | 96% | β only |
| 5 | 1B | IDCP | −10 °C | 20 h | 4 | 62% | 3:1 |
| 6 | 2B | IDCP | −10 °C | 3 h | 5 | 53% | β only |
| 7 | 1C | IDCP | −10 °C | 96 h | 4 | 82% | 2:1 |
| 8 | 2C | IDCP | −10 °C | 96 h | 5 | 78% | β only |
| 9 | 1C | MeOTf | 0 °C | 18 h | 4 | 95% | 2:1 |
| 10 | 2C | MeOTf | 0 °C | 6 h | 5 | 96% | β only |
| 11 | 1C | DMTST | −20 °C | 1 h | 4 | 90% | 2:1 |
| 12 | 2C | DMTST | −20 °C | 10 min | 5 | 89% | β only |
It should be noted that the key feature of any chemoselective activation lies in choosing suitable reaction conditions that will allow for the reactivity levels of the building blocks to be differentiated. However, establishing a reactivity differentiation becomes increasingly difficult with the highly reactive superarmed and armed glycosyl donors. In our earlier work, we described that the reactivity of armed and superarmed SBox glycosides, 1a and 2a, respectively, can be effectively separated in the presence of dimethyl(methylthio)sulfonium trifluoromethansulfonate22 (DMTST) as promoter (Table 1, Entries 1 and 2).14 Herein, we also determined that an equally successful differentiation can be achieved in the presence of copper(II) trifluoromethanesulfonate (Entries 3 and 4). This result offers further significance, as Cu(OTf)2 is a promoter unique to thioimidoyl leaving groups.
In order to differentiate between armed and superarmed O-pentenyl glycosides, 1b23 and 2b,24 respectively, we chose iodonium(di-γ-collidine)perchlorate25 (IDCP), as this mild promoter has proven effective for chemoselectively activating armed pentenyl glycosides over their disarmed counterparts at rt.6 However, these reaction conditions were found to be too powerful, as both glycosyl donors 1b and 2b were activated in a matter of minutes at rt. Additionally, no significant difference in reactivity was observed when O-pentenyl glycosides were investigated in the presence of NIS (rt), NBS (45 °C), or NIS/TfOH (−20 °C), albeit in a majority of comparative glycosylations, superarmed donor 1b was slightly more reactive (the extended experimental results are available as a part of the SI). Ultimately, the application of IDCP at low temperature (−10 °C) was deemed the most successful in terms of the differential activation of O-pentenyl glycosides. Thus, 20 h was required for the glycosidation of 1b, whereas the glycosidation of 2b was complete in about 3 h (Entries 5 and 6). The yields obtained with both armed and superarmed pentenyl glycosides under these reaction conditions, however, remained modest (62% and 53%, respectively).
Consequently, we investigated S-ethyl glycosides 1c26 and 2c27 in the presence of relatively mild promoters, including IDCP, DMTST, and MeOTf at reduced temperatures (Entries 7–12). While reactions in the presence of IDCP showed insufficient difference in reactivity (Entries 7 and 8), the best differentiation was achieved with MeOTf at 0 °C or DMTST at −20 °C, wherein a three and six-fold increase in reactivity was achieved, respectively (Entries 9–12). Additionally, the yields for both armed and disarmed glycosyl donors were good to excellent across the board. Upon investigating a variety of other known thioglycoside promoters,28–30 we found results with molecular iodine, introduced by Field as a mild promoter for the activation of methyl thioglycosides,31 at −25 °C to be the most impressive. Thus, while activation of the armed glycosyl donor 1c required at least 10 h, the reaction of its superarmed counterpart 2c completed in less than 1 h, displaying at least a ten-fold reactivity increase (Table 2, Entries 1 and 2). Encouraged by these results, iodine was tested as the promoter in subsequent studies with O-pentenyl donors (1b and 2b), as well as common classes of thioglycoside donors,28–30 S-phenyl (1d32 and 2d33), S-tolyl (1e34 and 2e), and STaz glycosides (1f35 and 2f). Although we observed that the superarmed glycosyl donors of both the S-phenyl and S-tolyl series reacted faster (Entries 3–6), no significant differentiation was achieved under these reaction conditions.
Table 2.
Activation of armed (1) and superarmed (2) glycosyl donors in the presence of iodine (3 mol equiv)
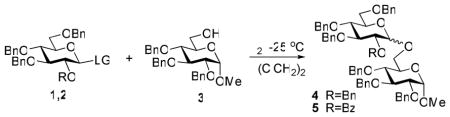 | |||||
|---|---|---|---|---|---|
| Entry | Donor | Time | Product | Yield | Ratio α: β |
| 1 | 1C | 10 h | 4 | 89% | 1:4 |
| 2 | 2C | 1 h | 5 | 93% | β only |
| 3 | 1D | 15 h | 4 | 74% | 1:2 |
| 4 | 2D | 8 h | 5 | 78% | β only |
| 5 | 1E | 15 h | 4 | 63% | 1:2 |
| 6 | 2E | 12 h | 5 | 72% | β only |
| 7 | 1F | 19 h | 4 | 87% | 1:2 |
| 8 | 2F | 19 h | 5 | 85% | β only |
| 9 | 1B | 17 h | 4 | 82% | 1:2.8 |
| 10 | 2B | 17 h | 5 | 84% | β only |
| 11 | 1A | 48 h | 4 | NRa | - |
| 12 | 2A | 20 h | 5 | 95% | β only |
NR – no reaction; although glycosyl donor 1a smoothly reacted at rt in 13 h affording disaccharide 4 in 95% (α:β=1:2)
Furthermore, no reactivity differentiation could be established in the presence of iodine between the armed and superarmed STaz glycosyl donors 1f and 2f, nor between O-pentenyl glycosides 1b and 2b, respectively (Entries 7–10). Although both O-pentenyl and STaz glycosides easily react in accordance with the conventional armed-disarmed activation scheme,6,36 we have found the differentiation between the more closely positioned superarmed and armed glycosides, to be challenging. It is possible, that the protecting group effect is decreased in cases of remote activation,37 typical for both the O-pentenyl and STaz glycosides (alkene7 and nitrogen atom of the thiazoline ring,38 respectively); opposite to that of the direct activation21 cases (anomeric sulfur atom of S-ethyl and SBox glycosyl donors).
Following this rationale, we additionally tested the armed and superarmed SBox donors (1a and 2a, respectively), wherein we found that the reactivity difference between them in the presence of iodine at −25 °C was remarkable (Entries 11 and 12). While no reaction took place with the armed glycosyl donor 1a, the superarmed glycosyl donor 2a reacted very smoothly, and afforded the corresponding disaccharide 5 in 20 h (95%). It should be noted that the armed SBox glycoside 1a can also be activated with iodine, but this was only accomplished at rt. Interestingly, the fact that the SBox glycosides reacted much slower than their S-ethyl counterparts was rather unanticipated, as SBox glycosides are typically much more reactive and can be selectively activated in the presence of alkyl/aryl thioglycosides.21,39
As verification of the promising results obtained with S-ethyl glycosides in the presence of iodine, we set up direct competitive glycosylations. To optimize the reaction conditions for a chemoselective oligosaccharide assembly, the competitive glycosylations were investigated at a variety of reaction temperatures (Table 3). These reactions were carried out by placing equimolar amounts (1.1 equiv each) of the armed 1c and superarmed 2c glycosyl donors in the same reaction vessel with glycosyl acceptor 3 (1 equiv). Upon the addition of iodine (3 equiv), both glycosyl donors competed for the same acceptor to form either disaccharide 4 or 5. The highest product ratio in these competitive glycosylations was also obtained at −25 °C, wherein the 1,2-trans-linked disaccharide 5 (derived from superarmed glycosyl donor 2c) was isolated in 62% yield (Entry 3). Conversely, the disaccharide 4 (derived from armed glycosyl donor 1c) was isolated in only 14% yield (α/β = 1/4).
Table 3.
Competitive activations of S-ethyl glycosyl donors: 1c (armed) vs. 2c (superarmed)
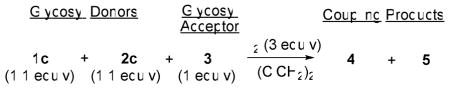 | |||||
|---|---|---|---|---|---|
| Entry | Temp. | Time | Ratio 4:5 a | Yield 4 b (α:β) | Yield 5 b (α:β)b |
| 1 | rt | 1 h | 1:0.8 | 53% (1:1.7) | 45% (β only) |
| 2 | −10 °C | 1 h | 1:2.4 | 22% (1:4) | 51% (β only) |
| 3 | −25 °C | 3 h | 1:4 | 14% (1:4) | 62% (β only) |
| 4 | −35 °C | 24 h | 1:1.5 | 11% (1:5) | 26% (β only) |
– determined by comparing integral intensities of the corresponding signals in the NMR spectra recorded for the crude reaction mixtures;
– isolated yields
Having investigated the superarming and arming effects in direct competitive glycosylations with glycosyl donors of the S-ethyl series, we now turned our attention to a sequential tetrasaccharide synthesis. To begin, the chemoselective activation of the superarmed glycosyl donor 2c over the “armed” glycosyl acceptor 640 was successfully carried out in the presence of iodine, at −25 °C (Scheme 1). Subsequently, the resulting armed disaccharide 7, obtained in 80% yield, was coupled with “disarmed” glycosyl acceptor 8.41 This chemoselective activation was also performed in the presence of iodine, but this time at rt. The resulting trisaccharide 9, obtained in 55% yield (α/β = 1:2.6), was then reacted with glycosyl acceptor 3 in the presence of NIS/TfOH, and the resulting tetrasaccharide 10 was isolated in 72% yield. Overall, this synthesis serves as ultimate proof of the utility of this superarmed approach in chemoselective oligosaccharide synthesis.
Scheme 1.

Sequential activation of differently protected S-ethyl building blocks: synthesis of tetrasaccharide 10.
In conclusion, a promising new concept for superarming glycosyl donors through the use of common protecting groups has now been extended to encompass a range of common glycosyl donors. Although it was initially necessary to fine-tune the reaction conditions and carefully select an adequate (mild) promoter, once established, the reactivity of the superarmed glycosyl donors was able to be exploited. This approach ultimately provides an additional synthetic building block that can be integrated into the conventional chemoselective armed-disarmed strategy. Results obtained herein were both consistent and high yielding, thus, offering this approach as a general method in cases wherein a 1,2-trans linkage must to be introduced prior to other linkages. Additionally, the superarmed glycosyl donors can be easily obtained by conventional synthetic methods. Currently, further application of this superarmed concept toward selective activation and orthogonal approaches to oligosaccharide synthesis is under pursuit in our laboratory.
Experimental section
Preparation of superarmed glycosyl donors 2e and 2f
P-Methylphenyl 2-O-benzoyl-3,4,6-tri-O-benzyl-1-thio-β-D-glucopyranoside (2e)
The title compound was obtained using a modified protocol similar to that previously reported.7,8,14 CH2Cl2 (9.0 mL) and p-toluenethiol (1.6 g, 13.03 mmol) were added to 3,4,6-tri-O-benzyl-1,2-O-(methylorthobenzoate)-α-D-glucopyranoside14 (740 mg, 1.31 mmol) and molecular sieves (3Å, 850 mg), and the resulting mixture was stirred under argon for 45 min. Trimethylsilyl trif-luoromethanesulfonate (6 μL, 0.328 mmol) was added and the reaction mixture was stirred under argon for 16 h at rt. After that, the reaction mixture was neutralized by the addition of triethylamine (~0.1 mL), diluted with CH2Cl2, the solid was filtered-off and was rinsed successively with CH2Cl2. The combined filtrate (~30 mL) was washed with water (10 mL), 20% aq. Na-HCO3 (10 mL), and water (3 × 10 mL). The organic phase was separated, dried, and concentrated in vacuo. The residue was purified by column chromatography on silica gel (ethyl acetate/hexanes gradient elution) to afford the title compound 2e as a white solid in 70% yield. Analytical data for 2e: Rf = 0.54 (ethyl acetate/hexanes, 3/7, v/v); m. p. 134–135 °C (diethyl ether/hexanes); [α]D22 +32.5° (c = 1, CHCl3); 1H-n.m.r: δ, 2.18 (s, 3H, SPhCH3), 3.48 (m, 1H, J5,6a = 1.9 Hz, J5,6b = 3.6 Hz, H-5), 3.62 (dd, 1H, J4,5 = 9.5 Hz, H-4), 3.65–3.78 (m, 3H, H-3, 6a, 6b), 4.42–4.75 (m, 6H, 3 × CH2Ph), 4.61 (d, 1H, J1,2 = 10.0 Hz, H-1), 5.14(dd, 1H, J2,3 = 9.0 Hz, H-2), 6.80–8.00 (m, 24H aromatic) ppm; 13C-n.m.r.: δ, 21.3, 69.1, 72.7, 73.7, 75.2, 75.5, 76.8, 77.2, 77.4, 77.7, 78.0, 79.7, 84.5, 86.4, 127.7 (×3), 127.8 (×3), 127.8 (×3), 128.0, 128.1 (×2), 128.3, 128.4 (×2), 128.5 (×2), 128.7 (×2), 128.8 (×2), 128.9 (×2), 128.9 (×3), 129.1, 130.0 (×2), 130.3 (×2), 130.4, 133.6, 133.9 (×3), 138.1, 138.4, 138.5, 138.7, 165,3 ppm; HR-FAB MS [M+Na]+ calcd for C41H40O6SNa+ 683.2443, found 683.2624.
Thiazolinyl 2-O-benzoyl-3,4,6-tri-O-benzyl-1-thio-β-D-glucopyranoside (2f)
The title compound was obtained using a modified protocol similar to that previously reported.42 3,4,6-tri-O-benzyl-1,2-O-(methylorthobenzoate)-α-D-glucopyranoside14 (200 mg, 0.35 mmol) was mixed with molecular sieves (3Å, 1.2 g) and 2-mercaptothiazoline (420 mg, 3.52 mmol), and dried in vacuo for 15 min. Acetonitrile (2.6 mL) was added and the resulting mixture was stirred under argon for 1 h. Mercuric bromide (13 mg, 0.035 mmol) was added and the resulting reaction mixture was heated at reflux for 16 h. The volatiles have been evaporated under reduced pressure, the residue was diluted with CH2Cl2, the solid was filtered-off and rinsed successively with CH2Cl2. The combined filtrate (30 mL) was washed with 1N aq. NaOH (10 mL) and water (3 × 10 mL). The organic phase was separated, dried, and concentrated in vacuo. The residue was purified by column chromatography on silica gel (ethyl acetate/hexane gradient elution) to afford the title compound 2f as a white solid in 70% yield. Analytical data for 2f: Rf = 0.47 (ethyl acetate/hexane, 4/6, v/v); m. p. 141–143 °C (diethyl ether-hexanes); [α]D22 +81.2° (c = 1, CHCl3); 1H-n.m.r: δ, 3.43 (m, 2H, NCH2), 3.82 (m, 1H, H-5), 3.88–3.98 (m, 2H, H-6a, 6b), 4.03 (dd, 1H, J4,5 = 9.1 Hz, H-4), 4.05 (dd, 1H, J3,4 = 8.8 Hz, H-3), 4.30 (m, 2H, CH2S), 4.75 (dd, 2H, J2 = 11.9 Hz, CH2Ph), 4.85 (dd, 2H, J2 = 10.9 Hz, CH2Ph), 4.87 (dd, 2H, J2 = 11.0 Hz, CH2Ph), 5.55 (dd, 1H, J2,3 = 8.5 Hz, H-2), 5.67 (d, 1H, J1,2 = 10.3 Hz, H-1), 7.22–8.20 (m, 20H, aromatic) ppm; 13C-n.m.r.: δ, 35.6, 64.3, 68.8, 72.4, 73.6, 75.3, 75.5, 77.8, 80.0, 83.1, 84.3, 127.8, 127.9, 128.0, 128.1 (×2), 128.1 (×2), 128.2 (×2), 128.5 (×2), 128.6 (×3), 128.6 (×2), 129.7, 130.1 (×2), 133.5, 137.9, 138.2, 138.3, 163.7, 165.4 ppm; HR-FAB MS [M+Na]+ calcd for C37H37NO6S2Na+ 678.1959, found 678.1977.
Preparation of di- and oligosaccharides (4, 5, 7, 9, and 10)
Method A: Typical DMTST-promoted glycosylation procedure
A mixture containing the glycosyl donor (0.11 mmol), glycosyl acceptor (0.10 mmol), and freshly activated molecular sieves (4Å, 200 mg) in 1,2-dichloroethane (DCE, 1.6 mL) was stirred under argon for1 h. The mixture was chilled to 0 or −20 °C (see Table 1), DMTST22 (0.33 mmol) was added and the reaction mixture was stirred for 5 min – 2 h (see Table 1). Upon completion, the mixture was diluted with CH2Cl2, the solid was filtered-off and rinsed successively with CH2Cl2. The combined filtrate (30 mL) was washed with 20% aq. NaHCO3 (10 mL) and water (3 × 10 mL). The organic phase was separated, dried and concentrated in vacuo. The residue was purified by column chromatography on silica gel (ethyl acetate-hexane gradient elution).
Method B: Typical Cu(OTf)2-promoted glycosylation procedure
A mixture containing the glycosyl donor (0.11 mmol), glycosyl acceptor (0.10 mmol), and freshly activated molecular sieves (4Å, 200 mg) in DCE (1.6 mL) was stirred under argon for 1 h followed by the addition of freshly conditioned Cu(OTf)2 (0.22 mmol). The reaction mixture was stirred for 20 min – 2h at 0 °C (see Table 1) then diluted with CH2Cl2, the solid was filtered-off and rinsed successively with CH2Cl2. The combined filtrate (30 mL) was washed with 20% aq. NaHCO3 (10 mL) and water (3 × 10 mL). The organic phase was separated, dried and concentrated in vacuo. The residue was purified by column chromatography on silica gel (ethyl acetate-hexane gradient elution).
Method C: Typical IDCP-promoted glycosylation procedure
A mixture containing the glycosyl donor (0.11 mmol), glycosyl acceptor (0.10 mmol), and freshly activated molecular sieves (4Å, 200 mg) in DCE (1.6 mL) was stirred under argon for 1 h. The reaction mixture was then chilled to −10 °C, IDCP25 (0.22 mmol) was added and the reaction mixture was stirred for 3–96 h (see Table 1). Upon completion, the mixture was diluted with CH2Cl2, the solid was filtered-off and rinsed successively with CH2Cl2. The combined filtrate (30 mL) was washed with 20% NaHCO3 (10 mL) and water (3 × 10 mL). The organic phase was separated, dried and concentrated in vacuo. The residue was purified by column chromatography on silica gel (ethyl acetate-hexane gradient elution).
Method D: Typical MeOTf-promoted glycosylation procedure
A mixture containing the glyco-syl donor (0.13 mmol), glycosyl acceptor (0.10 mmol), and freshly activated molecular sieves (3Å, 200 mg) in DCE (1.6 mL) was stirred under argon for 1 h. The mixture was chilled to 0 °C, MeOTf (0.39 mmol) was added and the reaction mixture was stirred for 6–18 h (see Table 1). The mixture was then diluted with CH2Cl2, the solid was filtered-off and the residue was rinsed successively with CH2Cl2. The combined filtrate (30 mL) was washed with 20% NaHCO3 (10 mL) and water (3 × 10 mL). The organic phase was separated, dried and concentrated in vacuo. The residue was purified by column chromatography on silica gel (ethyl acetate-hexane gradient elution).
Method E: Typical iodine-promoted glycosylation procedure
A mixture containing the glycosyl donor (0.11 mmol), glycosyl acceptor (0.10 mmol), and freshly activated molecular sieves (3Å, 200 mg) in DCE (1.6 mL) was stirred under argon for 16 h. The mixture was chilled to −25 °C (or as indicated in Table 3 or Scheme 1), iodine (0.33 mmol) was added and the reaction mixture was stirred for 1–48 h (see Table 2). Upon completion, the mixture was quenched with Et3N and diluted with CH2Cl2, the solid was filtered-off and the residue was rinsed successively with CH2Cl2. The combined filtrate (30 mL) was washed with 10% Na2S2O3 (10 mL) and water (3 × 10 mL). The organic phase was separated, dried and concentrated in vacuo. The residue was purified by column chromatography on silica gel (ethyl acetate-toluene gradient elution).
Method F: Typical NIS/TfOH-promoted glycosylation procedure
A mixture containing the glycosyl donor (0.11 mmol), glycosyl acceptor (0.10 mmol), and freshly activated molecular sieves (4Å, 200 mg) in DCE (1.6 mL) was stirred under argon for 16 h. NIS (0.22 mmol) and TfOH (0.022 mmol) were added and the reaction mixture was stirred for 1 h. Upon completion, the mixture was diluted with CH2Cl2, the solid was filtered-off and the residue was rinsed successively with CH2Cl2. The combined filtrate (30 mL) was washed with 10% Na2S2O3 (10 mL) and water (3 × 10 mL). The organic phase was separated, dried and concentrated in vacuo. The residue was purified by column chromatography on silica gel (ethyl acetate-toluene gradient elution).
Methyl O-(2,3,4,6-tetra-O-benzyl-D-glucopyranosyl)-(1→6)-2,3,4-tri-O-benzyl-α-D-glucopyranoside (4)
The spectroscopic and analytical data for the title compound were in good agreement with those reported previously.20
Methyl O-(2-O-benzoyl-3,4,6-tri-O-benzyl-β-D-glucopyranosyl)-(1→6)-2,3,4-tri-O-benzyl-agr;D-glucopyranoside (5)
The spectroscopic and analytical data for the title compound were in good agreement with those reported previously.20
Ethyl O-(2-O-benzoyl-3,4,6-tri-O-benzyl-β-D-glucopyranosyl)-(1→6)-2,3,4-tri-O-benzyl-1-thio-β-D-glucopyranoside (7)
The title compound was obtained as a colorless syrup from 1c26 and 640 by method E in 80% yield. Analytical data for 7: Rf = 0.4 (ethyl acetate/hexanes, 3/7, v/v); [α]D23 +18.4° (c=1, CHCl3); 1H-n.m.r: δ, 1.13 (t, 3H, J = 8.0 Hz, SCH2CH3), 2.51 (m, 2H, SCH2CH3), 3.31 (dd, 1H, J2,3 = 9.0 Hz, H-2), 3.32–3.46 (m, 2H, H-4, 5), 3.48–3.65 (m, 3H, H-3, 5′, 6a), 3.67–3.82 (m, 4H, H-3′, 4′, 6a′, 6b′), 4.10 (d, 1H, J5,6b = J6a,6b = 10.5 Hz, H-6b), 4.32 (d, 1H, J1,2 = 9.7 Hz, H-1), 4.40–4.87 (m, 13H, H-1′, 6 × CH2Ph), 5.31 (br dd, 1H, H-2′), 7.05–8.00 (m, 35H, aromatic) ppm; 13C-n.m.r.: δ, 15.2, 24.7, 29.9, 68.4, 69.2, 74.0, 75.4, 75.7, 75.8, 75.8, 76.0, 77.6, 78.4, 79.1, 82.0, 83.3, 84.9, 86.9, 101.4, 128.0 (×2), 128.1 (×4), 128.2 (×3), 128.2 (×4), 128.4 (×4), 128.4 (×4), 128.6 (×3), 128.8 (×6), 128.8 (×5), 128.8 (×5), 130.2 (×2), 138.2, 138.4 (×2), 138.5, 166.0 ppm; HR-FAB MS [M+Na]+ calcd for C63H66O11SNa+ 1053.4224, found 1053.4238
Ethyl O-(2-O-benzoyl-3,4,6-tri-O-benzyl-β-D-glucopyranosyl)-(1→6)-O-(2,3,4-tri-O-benzyl-D-glucopyranosyl)-(1→6)-2,3,4-tri-O-benzoyl-1-thio-β-D-glucopyranoside (9)
The title compound was obtained as a pale-yellow syrup from 7 and 841 by method E (rt) in 55% yield (α: β = 1:2.6). Analytical data for 9: Rf = 0.3 (ethyl acetate/hexane, 3/7, v/v); 1H-n.m.r. (selected data for β-9): δ, 1.13 (t, 3H, J = 8.0 Hz, SCH2CH3), 2.65 (m, 2H, SCH2CH3), 3.26 (dd, 1H, J2′,3′ = 8.9 Hz, H-2′), 3.30–3.58 (m, 4H, H-3′, 4′, 5′, 6a), 3.62–4.20 (m, 9H, H-3″, 4″, 5, 5″, 6a′, 6a″, 6b, 6b′, 6b″), 4.35 (d, 1H, J1′,2′= 7.7 Hz, H-1′), 4.35–4.95 (m, 14H, H-1, 1″, 12 × CH2Ph), 5.32 (dd, 1H, J2″,3″ = 9.5 Hz, H-2″), 5.38 (dd, 1H, J4,5 = 7.0 Hz, H-4), 5.49 (dd, 1H, J2,3 = 9.7 Hz, H-2), 5.86 (dd, 1H, J3,4 = 9.7 Hz, H-3), 7.05–8.15 (m, 50H aromatic) ppm; 13C-n.m.r.: δ, 15.0 (×2), 24.4, 29.9, 69.0, 70.4, 70.9, 73.7 (×2), 74.1, 74.4, 74.9 (×2), 75.2, 75.2, 75.4, 75.6, 77.7, 78.1, 78.4 (×2), 82.3, 83.0, 83.9, 84.6, 101.4, 103.9, 127.6 (×2), 127.8 (×2), 127.8 (×2), 127.9, 128.0, 128.1 (×4), 128.2 (×2), 128.4, 128.4 (×4), 128.5 (×4), 128.6 (×5), 128.7, 129.0, 129.1, 129.5, 129.9 (×2), 130.0, 130.0 (×2), 130.1, 133.2, 133.4 (×2), 133.6 (×2), 138.1 (×2), 138.2 (×2), 138.3, 138.4 (×2), 138.8 (×4), 165.3 (×2), 165.4 (×2), 165.6 (×3), 166.0 (×2) ppm; HR-FAB MS [M+Na]+ calcd for C90H88O19SNa+ 1527.5538, found 1527.5558.
Methyl O-(2-O-benzoyl-3,4,6-tri-O-benzyl-β-D-glucopyranosyl)-(1→6)-O-(2,3,4-tri-O-benzyl-D-glucopyranosyl)-(1→6)-O-(2,3,4-tri-O-benzoyl-β-D-glucopyranosyl)-(1→6)-2,3,4-tri-O-benzyl-α-D-glucopyranoside (10)
The title compound was obtained as a colorless syrup 3 and 9 by method F in 72% yield. Selected analytical data for β-10: Rf = 0.66 (ethyl acetate/hexane, 2/3, v/v); 1H-n.m.r.: δ, 4.24 (d, 1H, J1″,2″ = 7.8 Hz, H-1″), 4.29 (d, 1H, J1,2 = 3.3 Hz, H-1), 4.44 (d,1H, H-1′), 4.47 (d, 1H, H-1‴) ppm; 13C-n.m.r.: δ, 55.7, 67.7, 68.4, 68.8, 69.8, 70.0, 72.2, 73.3, 73.9, 74.3, 74.8, 74.9, 75.0, 75.4, 75.6, 75.8, 77.6 (×2), 77.9, 78.2, 79.6 (×2), 80.1, 82.2, 83.1, 98.3, 98.5, 101.0, 101.6, 127.4 (×3), 127.8, 128.0 (×3), 128.2 (×6), 128.3 (×4), 128.3 (×4), 128.4, 128.5 (×3), 128.7 (×5), 128.7 (×6), 128.8 (×7), 128.8 (×7), 128.8 (×6), 129.0 (×5), 129.1, 129.2, 129.4 (×3), 129.4, 129.6, 130.0, 130.1 (×3), 130.3, 130.4, 133.5, 133.6, 133.9, 136.2, 138.1, 138.3, 138.4, 138.6, 138.8, 139.3, 165.2, 165.4, 165.8, 166.1 ppm; HR-FAB MS [M+Na]+ calcd for C116H114O25Na+ 1929.7547, found 1929.7583.
Supplementary Material
Acknowledgments
This work was supported by awards from the NIGMS (GM077170) and NSF (CHE-0547566). The authors thank Ms. Mercy Kiiru for experimental assistance and Dr. R. E. K. Winter and Mr. J. P. Kramer (all UM – St. Louis) for HRMS determinations.
Footnotes
Supporting Information Available: Extended experimental data, general experimental procedures and 1H and 13C NMR spectra for all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Varki A, Cummings RD, Esko JD, Freeze HH, Bertozzi CR, Stanley P, Hart GW, Etzler ME, editors. Essentials of Glycobiology. 2. CSH Laboratory Press; New York: 2009. [PubMed] [Google Scholar]
- 2.Demchenko AV. Handbook of Chemical Glycosylation: Advances in Stereoselectivity and Therapeutic Relevance. Wiley-VCH; Weinheim: 2008. [Google Scholar]
- 3.Zhu X, Schmidt RR. Angew Chem Int Ed. 2009;48:1900–1934. doi: 10.1002/anie.200802036. [DOI] [PubMed] [Google Scholar]
- 4.Boons GJ. Tetrahedron. 1996;52:1095–1121. [Google Scholar]
- 5.Smoot JT, Demchenko AV. Adv Carbohydr Chem Biochem. 2009;62:161–250. doi: 10.1016/S0065-2318(09)00005-5. [DOI] [PubMed] [Google Scholar]
- 6.Mootoo DR, Konradsson P, Udodong U, Fraser-Reid B. J Am Chem Soc. 1988;110:5583–5584. [Google Scholar]
- 7.Fraser-Reid B, Udodong UE, Wu ZF, Ottosson H, Merritt JR, Rao CS, Roberts C, Madsen R. Synlett. 1992:927–942. and references therein. [Google Scholar]
- 8.Douglas NL, Ley SV, Lucking U, Warriner SL. J Chem Soc, Perkin Trans 1. 1998:51–65. [Google Scholar]
- 9.Zhang Z, Ollmann IR, Ye XS, Wischnat R, Baasov T, Wong CH. J Am Chem Soc. 1999;121:734–753. [Google Scholar]
- 10.Jensen HH, Nordstrom LU, Bols M. J Am Chem Soc. 2004;126:9205–9213. doi: 10.1021/ja047578j. [DOI] [PubMed] [Google Scholar]
- 11.Paulsen H. Angew Chem Int Edit Engl. 1982;21:155–173. [Google Scholar]
- 12.Kamat MN, Demchenko AV. Org Lett. 2005;7:3215–3218. doi: 10.1021/ol050969y. [DOI] [PubMed] [Google Scholar]
- 13.Crich D, Li M. Org Lett. 2007;9:4115–4118. doi: 10.1021/ol701466u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mydock LK, Demchenko AV. Org Lett. 2008;10:2103–2106. doi: 10.1021/ol800345j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mydock LK, Demchenko AV. Org Lett. 2008;10:2107–2110. doi: 10.1021/ol800648d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pedersen CM, Nordstrom LU, Bols M. J Am Chem Soc. 2007;129:9222–9235. doi: 10.1021/ja071955l. [DOI] [PubMed] [Google Scholar]
- 17.Jensen HH, Pedersen CM, Bols M. Chem Eur J. 2007;13:7576–7582. doi: 10.1002/chem.200700947. [DOI] [PubMed] [Google Scholar]
- 18.Pedersen CM, Marinescu LG, Bols M. Chem Commun. 2008:2465–2467. doi: 10.1039/b801305e. [DOI] [PubMed] [Google Scholar]
- 19.Kuester JM, Dyong I. Justus Liebigs Ann Chem. 1975:2179–2189. [Google Scholar]
- 20.Nguyen HM, Chen YN, Duron SG, Gin DY. J Am Chem Soc. 2001;123:8766–8772. doi: 10.1021/ja015968p. [DOI] [PubMed] [Google Scholar]
- 21.Kamat MN, Rath NP, Demchenko AV. J Org Chem. 2007;72:6938–6946. doi: 10.1021/jo0711844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ravenscroft M, Roberts RMG, Tillett JG. J Chem Soc Perkin Trans 2. 1982:1569–1972. [Google Scholar]
- 23.Ratcliffe AJ, Fraser-Reid B. J Chem Soc, Perkin Trans 1. 1989:1805–1810. [Google Scholar]
- 24.Mach M, Schlueter U, Mathew F, Fraser-Reid B, Hazen KC. Tetrahedron. 2002;58:7345–7354. [Google Scholar]
- 25.Lemieux RU, Morgan AR. Can J Chem. 1965;43:2190–2198. [Google Scholar]
- 26.Andersson F, Fugedi P, Garegg PJ, Nashed M. Tetrahedron Lett. 1986;27:3919–3922. [Google Scholar]
- 27.Ekelof K, Oscarson S. J Org Chem. 1996;61:7711–7718. doi: 10.1021/jo960789p. [DOI] [PubMed] [Google Scholar]
- 28.Codee JDC, Litjens REJN, van den Bos LJ, Overkleeft HS, van der Marel GA. Chem Soc Rev. 2005;34:769–782. doi: 10.1039/b417138c. [DOI] [PubMed] [Google Scholar]
- 29.Garegg PJ. Adv Carbohydr Chem Biochem. 1997;52:179–205. doi: 10.1016/s0065-2318(08)60091-8. [DOI] [PubMed] [Google Scholar]
- 30.Zhong W, Boons G-J. Handbook of Chemical Glycosylation. Wiley-VCH; Weinheim, Germany: 2008. pp. 261–303. [Google Scholar]
- 31.Kartha KPM, Aloui M, Field RA. Tetrahedron Lett. 1996;37:5175–5178. [Google Scholar]
- 32.Pfaeffli PJ, Hixson SH, Anderson L. Carbohydr Res. 1972;23:195–206. [Google Scholar]
- 33.Nicolaou KC, Mitchell HJ, Jain NF, Bando T, Hughes R, Winssinger N, Natarajan S, Koumbis AE. Chem Eur J. 1999;5:2648–2667. [Google Scholar]
- 34.Balavoine G, Berteina S, Gref A, Fischer JC, Lubineau A. J Carbohydr Chem. 1995;14:1217–1236. [Google Scholar]
- 35.Pornsuriyasak P, Demchenko AV. Chem Eur J. 2006;12:6630–6646. doi: 10.1002/chem.200600262. [DOI] [PubMed] [Google Scholar]
- 36.Smoot JT, Pornsuriyasak P, Demchenko AV. Angew Chem Int Ed. 2005;44:7123–7126. doi: 10.1002/anie.200502694. [DOI] [PubMed] [Google Scholar]
- 37.Hanessian S, Bacquet C, Lehong N. Carbohydr Res. 1980;80:c17–c22. [Google Scholar]
- 38.Kaeothip S, Pornsuriyasak P, Rath NP, Demchenko AV. Org Lett. 2009;11:799–802. doi: 10.1021/ol802740b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kamat MN, De Meo C, Demchenko AV. J Org Chem. 2007;72:6947–6955. doi: 10.1021/jo071191s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fei C, Chan TH. Acta Chimica Sinica, Engl Edit. 1989:258–264. [Google Scholar]
- 41.Veeneman GH, van Boom JH. Tetrahedron Lett. 1990;31:275–278. [Google Scholar]
- 42.Beignet J, Tiernan J, Woo CH, Benson MK, Cox LR. J Org Chem. 2004;69:6341–6356. doi: 10.1021/jo049061w. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.