

Abstract
Although long believed to be inert, C-peptide has now been shown to have definite biological effects both in vitro and in vivo in diabetic animals and in patients with type 1 diabetes. These effects point to a protective action of C-peptide against the development of diabetic microvascular complications. Underpinning these observations is undisputed evidence of C-peptide binding to a variety of cell types at physiologically relevant concentrations, and the downstream stimulation of multiple cell signaling pathways and gene transcription via the activation of numerous transcription factors. These pathways affect such fundamental cellular processes as re-absorptive and/or secretory phenotype, migration, growth, and survival. Whilst the receptor remains to be identified, experimental data points strongly to the existence of a specific G-protein-coupled receptor for C-peptide. Of the cell types studied so far, kidney tubular cells express the highest number of C-peptide binding sites. Accordingly, C-peptide exerts major effects on the function of these cells, and in the context of diabetic nephropathy appears to antagonise the pathophysiological effects of major disease mediators such as TGFβ1 and TNFα. Therefore, based on its cellular activity profile C-peptide appears well positioned for development as a therapeutic tool to treat microvascular complications in type 1 diabetes.
Keywords: diabetes, C-peptide, receptor, kidney, nephropathy, PPAR, VACM-1, protein kinase, nitric oxide, p38 MAPK, TNF-alpha, TGF-beta1, NF-kappaB, Zn2+
Abbreviations: 3T3 cells - 3-day transfer, inoculum 3 x 105 cells; 3T3-L1 cells - 3T3 cells with fibroblast-like morphology; ATF-1 - activating transcription factor-1; BAEC - bovine aortic endothelial cell; Bcl-2 - B-cell lymphoma 2; COX-2 - cyclooxygenase-2; C-peptide - connecting peptide; CRE - cAMP response element; CREB - CRE-binding protein; EMT - epithelial mesenchymal transformation; ERK - extracellular signal-regulated kinase; EGSLQ - sequence of the human C-peptide carboxy(C)-terminal pentapeptide; EVARQ - sequence of the rat C-peptide carboxy(C)-terminal pentapeptide; GPCR - G-protein-coupled receptor; GTP - guanosine triphosphate; GTPγS - guanosine 5'-3-O-(thio)triphosphate; ICAM-1 - inter-cellular adhesion molecule 1; IRS-1 - insulin receptor substrate 1; JNK - c-Jun N-terminal kinase; LEII cells - lung capillary endothelial cells; MAPK - mitogen-activated protein kinase; Na+/K+-ATPase - sodium, potassium adenosintriphosphatase, also sodium-potassium pump; NO - nitric oxide; eNOS - endothelial nitric oxide synthase; NF-κB - nuclear factor-kappa light-chain enhancer of activated B cells; NPY - neuropeptide Y; OK - opossum kidney; PI-3-kinase - phosphatidylinositide-3-kinase; PKC - protein kinase C; PLC - phospholipase C; PPARγ - peroxisome proliferator-activated receptor; PPRE - peroxisome proliferator response element; PTC - proximal tubular cell; RhoA - ras homolog gene family member A; RNA - ribonucleic acid; T1D - type 1 diabetes; T2D - type 2 diabetes; TGF-β1 - transforming growth factor beta, isoform 1; TNF-α - tumor necrosis factor alpha; TNF-R1 - tumor necrosis factor receptor 1; TNF-R2 - tumor necrosis factor receptor 2; TRAF2 - TNF receptor-associated factor 2; VACM-1 - vasopressin-activated calcium mobilising receptor 1
Introduction
Elsewhere in this issue other authors clearly describe numerous physiological effects of C-peptide in multiple organ systems, and beneficial effects of C-peptide on the development of microvascular complications in type 1 diabetes (T1D). It is now unequivocally established that C-peptide has biological effects, but important questions remain. C-peptide interacts with individual cells to control intracellular processes governing cell functions such as growth, proliferation, death, transport, secretion, and ultimately whole organ function. Appreciation of the mechanisms by which circulating C-peptide interacts with these cells is crucial to the full understanding of a molecule that must surely be regarded as a peptide hormone in its own right. Dissection of the key components of C-peptide signaling pathways from receptors to effector molecules will be necessary to facilitate its full development towards the clinical arena. This paper describes what is known about C-peptide/cell interactions, the intracellular consequences of these interactions, and what can be deduced from this information with respect to the unanswered questions about the C-peptide function.
Activation of cell signaling pathways by C-peptide
It is now well established that application of C-peptide to a variety of cell types results in activation of intracellular signaling pathways. Many of these pathways are relevant to the pathogenesis and/or treatment of microvascular complications.
C-Peptide activation of the Na+/K+-ATPase
The first description of C-peptide signaling functions came from study of the ubiquitous Na+/K+-ATPase. Impaired activity of Na+/K+-ATPase is seen in a variety of cell types in diabetes (Table 1), and contributes to the pathogenesis of diabetic complications [1-4]. The kidney tubule is a rich source of Na+/K+-ATPase. Ohtomo et al. described activation of this enzyme by rat C-peptide in rat kidney tubules at low nanomolar concentrations [5]. This effect was abolished by pertussis toxin, suggesting involvement of a G-protein-coupled receptor (GPCR). The effect also appeared dependent on intracellular Ca2+ concentration. Subsequently, the same investigators studied the ability of many different C-peptide fragments and amino acids to activate Na+/K+-ATPase. They discovered that the C-terminal pentapeptide alone was able to elicit full activity (see also subsection “Structure-function relationships of C-peptide binding and signaling”).
Table 1. Signaling elements influenced by C-peptide according to cell types studied.
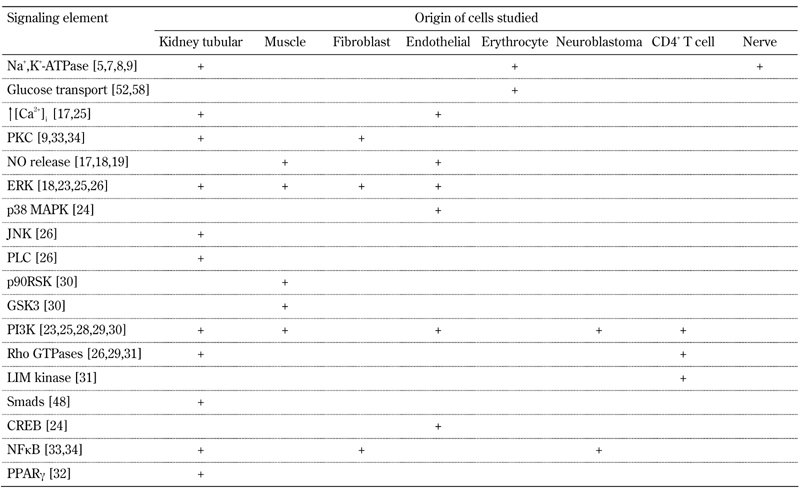
Numbers in brackets refer to the relevant references cited in the text.
Erythrocyte membranes from T1D patients with complete C-peptide deficiency exhibit reduced Na+/K+-ATPase activity resulting in impaired cell deformability and increased blood viscosity [6]. Infusion of C-peptide into such patients increases plasma cGMP and moves erythrocyte membrane Na+/K+-ATPase activity towards normal levels, with a maximal observed effect at achieved plasma C-peptide levels of ~3.5 nM [7]. Analogous improvements in rat nerve Na+/K+-ATPase activity and function have also been observed after exogenous administration of C-peptide [8].
Studies in rat kidney medullary thick ascending limb tubules indicate that C-peptide treatment at physiological concentrations activates Na+/K+-ATPase, with concomitant phosphorylation of the Na+/K+-ATPase α-subunit, and translocation of the Ca2+-dependent protein kinase C (PKC)-α to the membrane [9]. Maestroni et al. studied C-peptide effects on the vasopressin-activated calcium mobilising receptor (VACM-1) [10]. They found that low C-peptide concentrations increased expression of VACM-1 at both RNA and protein levels in human fibroblasts and mesangial cells [10]. Given that vasopressin also stimulates Na+/K+-ATPase activity, enhancement of vasopressin action via upregulation of VACM-1 provides another mechanism for C-peptide action relating to Na+/K+-ATPase.
Consequently, C-peptide at physiological concentrations stimulates Na+/K+-ATPase dependent on intracellular Ca2+ and PKC, and sensitive to pertussis toxin. Replacement of C-peptide in diabetic animals and patients with T1D has a salutary effect on Na+/K+-ATPase activity in a variety of tissues affected by diabetic complications.
The effect of C-peptide on endothelial nitric oxide synthase (eNOS)
Following in vivo administration of C-peptide to patients with T1D, microvascular blood flow to tissues and organs, including muscle, skin, and kidney, is consistently augmented [11], and likely relates to stimulatory effects on nitric oxide (NO) pathways. C-peptide evoked increased glucose utilization in streptozotocin diabetic rats [12]. and increased glucose transport. Metabolism by muscle tissue is also NO-dependent [13, 14]. C-peptide-mediated arteriolar dilatation is similarly reliant on NO [15, 16]. In a bovine aortic endothelial cell (BAEC) model, Wallerath et al. reported that at physiological post-prandial concentrations, C-peptide stimulated NO release following a rise in intracellular Ca2+ (Table 1) [17]. The authors speculated that C-peptide signaled increased cellular Ca2+ influx, and activation of Ca2+-sensitive endothelial NOS (eNOS), thus explaining the vasodilatory effects of C-peptide observed in vivo [17]. Such findings have now been confirmed by other workers who demonstrated upregulation of eNOS gene transcription and NO release dependent on the upstream phosphorylation and activation of extracellular signal-regulated mitogen activated protein kinase (ERK) [18].
In addition to vasodilatation, activation of the NO system by C-peptide may have other consequences. In C-peptide-injected rats, the levels of aortic basal eNOS gene expression and NO production are both increased. At the same time, reduced cell surface levels of the adhesion molecules P-selectin and ICAM-1 on the microvascular endothelium are observed. Consequently, leukocyte/endothelial interactions are attenuated [19]. These observations in non-diabetic rats raise the possibility of an immunomodulatory or anti-inflammatory action of C-peptide, and deserve more study in diabetic animals.
C-peptide mediated stimulation of mitogen-activated protein kinases (MAPKs)
MAPKs are serine threonine-specific kinases that respond to extracellular signals, and link cell-surface receptors, or chemical and physical stresses to fundamental regulatory targets within cells. They control such critical functions as growth, gene expression, survival, and adaptation [20-22]. The MAPK family includes the ERKs 1 and 2, c-Jun N-terminal kinases, p38s, and ERK5.
C-peptide has a clear ability to activate members of the MAPK family. Prompted by the finding of synergy between C-peptide and neuropeptide Y (NPY) signaling in the activation of Na+/K+-ATPase [5], Kitamura and colleagues studied C-peptide effects on MAPK in Swiss 3T3 cells [23]. Both researcher groups used immunoblotting to look for phosphorylated activated ERK1 and ERK2, and an ERK-specific in vitro kinase assay. These workers showed that C-peptide briskly activated ERK in Swiss 3T3 cells detectable at a concentration as low as 1 pM and maximal at 1 nM [23]. This stimulatory effect was also seen with NPY, but not retro-sequenced or D-amino acid human C-peptides, and ERK activation was abolished by pertussis toxin. This C-peptide response was also cell type-specific given that no ERK stimulation was observed in 3T3-L1 cells, L6E9 muscle cells, HepG2 hepatoma cells, NG108.15 neuroblastoma cells, or C6 glioma cells [23].
Activation of ERK is involved in the positive regulation of eNOS gene transcription, suggesting that C-peptide-stimulated eNOS expression may be ERK-dependent. To clarify the potential link between these phenomena studies of transcription factor activation were performed in LEII mouse lung capillary endothelial cells (Table 1) [24]. Here, C-peptide stimulated both p38 and ERK activities, whereas insulin activated only ERK, but not p38 MAPK. In addition, C-peptide activated the cAMP response element (CRE)-binding protein (CREB)/activating transcription factor-1 (ATF-1) in a p38-dependent manner, resulting in the binding of these transcription factors to CRE. It was subsequently confirmed that enhanced eNOS transcription in BAECs following C-peptide treatment was also MAPK-dependent [18]. Differences in C-peptide responses between various cell types clearly exist such that in BAECs ERK activation is required for eNOS gene transcription. Whereas in LEII cells. relevant transcription factor activation follows stimulation of p38.
Renal tubular disease is a prominent feature of diabetic nephropathy, and C-peptide responses in tubular cells have been well studied. In the immortalized opossum kidney (OK) proximal tubular cell line, C-peptide potently activates ERK, maximally at a concentration of 300 pM, and declining thereafter with a bell shaped dose response curve [25]. Furthermore, C-peptide also induced activation of Akt in OK cells. This event was sensitive to wortmannin, and indicative of phosphatidylinositide-3-kinase (PI-3-kinase) activation. The dose response curve for Akt activation revealed a maximal effect at a C-peptide concentration of 5 nM, and remained constant thereafter up to a C-peptide concentration of 100 nM. This effect is quite distinct from that for ERK activation. Moreover, C-peptide evoked Ca2+ influx into OK cells, with a consequent translocation and activation of PKCα [25]. These findings are in agreement with the earlier studies by Tsimaratos et al. that showed PKC-induced activation of kidney Na+/K+-ATPase in C-peptide-exposed tubular segments [9] (see above, section C-Peptide activation of the Na+/K+-ATPase, and Table 1 for a summary of signaling elements in different cell types).
These signaling events had important functional consequences with significant enhancement of proliferation seen in C-peptide-treated cells [25]. All of these events were sensitive to pertussis toxin, providing further evidence of the likely presence of a GPCR for C-peptide. The absence of these growth effects mediated by C-peptide in kidney tubular cells may be of key importance in diabetic nephropathy, where tubular cell loss and tubular atrophy are prominent.
Most recently, C-peptide has been found to promote translocation of the low molecular weight guanosine triphosphate (GTP)-binding protein, RhoA, from the cytoplasm to the membrane of human kidney proximal tubular cells [26]. This effect was completely reliant on the upstream activation of phospholipase C (PLC). In fact, in these cells, the activation of ERK, JNK, and PKC-ε and -δ were each sensitive to PLC inhibition, indicating an obligate dependency on upstream PLC activation by C-peptide. Again, all stimulatory effects of C-peptide were pertussis toxin-sensitive [26].
The signal transduction through which C-peptide activates MAPK can now be described as: i) C-peptide binds to a pertussis toxin-sensitive GPCR, ii) PLC is activated, iii) subsequently increased diacylglycerol and intracellular Ca2+ levels stimulate several PKC isoforms, iv) PKC-dependent activation and translocation of RhoA to plasma membrane occurs, and v) MAPKs are phosphorylated and activated.
C-peptide activation of PI-3-kinase
The PI 3-kinases are a family of enzymes that phosphorylate the hydroxyl group at the third position of the inositol ring of phosphatidyinositol, and regulate a diverse range of cellular functions, including growth, proliferation, survival, differentiation, motility, and intracellular trafficking [27]. Many functions of PI-3-kinases are related to their ability to activate protein kinase B (Akt). The PI-3-kinases are a key component of insulin signaling pathways, and they are of substantial interest in diabetes research [27].
Robust activation of PI-3-kinase by physiological concentrations of C-peptide has now been demonstrated in OK cells [25], Swiss 3T3 fibroblasts [23], SH-SY5Y neuroblastoma cells [28], human CD4+ T cells [29], and L6 myoblasts [30]. In CD4+ lymphocytes, PI-3-kinase is activated via Src kinase, and in turn activates several members of the Rho-GTPase family [29, 31]. Consequently, both Rho kinase and LIM kinase are stimulated, which impact in turn on myosin light chain and cofilin, respectively, to facilitate cell body contraction and migration [31]. As a direct consequence of this signaling, it is now recognized that stimulation of PI-3-kinase by C-peptide acting alone is responsible for: i) enhancement of neuronal and kidney tubular cell proliferation [25, 28], ii) increased T cell migration [29], iii) stimulation of peroxisome proliferator activated receptor-γ (PPARγ) in kidney tubule cells and associated gene transcription [32], and iv) upregulated glycogen synthesis in skeletal muscle cells [30].
Effects of C-peptide on transcription factors
Many of the changes in cell phenotype and function that follow signaling by bioactive molecules are mediated by altered gene and protein expression. It is therefore not surprising that C-peptide regulates activity of multiple cell transcription factors. Kitamura et al. described phosphorylation and activation of CREB, ATF-1 and ATF-2 in LEII cells [24], where 1 nM C-peptide and phorbol ester, used as a positive control, were equipotent. Activated CREB and ATF proteins are transcription factors that bind to specific cAMP response elements (CREs) in DNA, thereby regulating transcription. Gel mobility shift assays clearly showed the binding of CREB to CRE in C-peptide-treated cells although the specific genes subject to regulation were not identified [24].
In neuroblastoma cells, C-peptide treatment enhanced expression and translocation of nuclear factor-κB (NF-κB), and expression of the Bcl-2 protein, a central mediator of NF-κB-controlled anti-apoptotic effects [28]. Modulation of NF-κB activity in Swiss 3T3 by C-peptide has also been demonstrated, where 1 nM C-peptide activates NF-κB-dependent transcription of cyclooxygenase-2 (COX-2) following upon upstream activation of PKC [33]. COX-2, a cytokine-inducible gene, is the rate-limiting enzyme in the conversion of arachidonic acid to prostaglandin, but the potential consequences of its upregulation by C-peptide remain unclear.
We performed detailed studies of transcription factor activation in OK proximal tubular cells (Table 1) [32, 34], and compared C-peptide effects to those of insulin, whilst focusing on PPARγ and NF-κB. PPARγ is a member of the nuclear hormone receptor family, and is the target for the insulin-sensitizing thiazolidinediones, currently used as therapeutic agents in the treatment of type 2 diabetes (T2D) [35]. The expression of PPARγ may also be regulated by insulin [36]. Using transient transfection of a peroxisome proliferator response element, (PPRE)-luciferase reporter construct, we showed that both C-peptide and insulin transactivated PPRE via PPARγ [32]. C-peptide (EC50 4 nM) was more potent than insulin (EC50 10nM) in this regard, but both agents evoked phosphorylation of PPARγ similarly via activation of PI-3-kinase. One consequence of PPARγ activation by C-peptide and insulin was enhanced transcription of the prototypic PPARγ regulated gene, CD36 [32].
Clearly, there is a degree of overlap between insulin and C-peptide signaling with both agents' effects being directed via PI-3-kinase towards PPARγ. However, only the effects of C-peptide were attenuated by pertussis toxin. Therefore, C-peptide must be signaling through a receptor system fundamentally distinct from that of insulin. These data indicate an important novel mechanism, whereby C-peptide and insulin may interact to regulate glycemia, and the expression of PPARγ-regulated genes such as those involved in metabolic control and inflammation.
C-peptide evoked signaling in kidney tubular cells blocks the actions of deleterious mediators in diabetic nephropathy
The identification of signaling pathways regulated by C-peptide in kidney cells has resulted in studies to establish proof of principle that C-peptide may act as a protective agent in diabetic nephropathy. To this end, we investigated whether C-peptide could counteract adverse effects precipitated by the administration of TNF-α or TGF-β1 to kidney proximal tubular cells. TNF-α is recognized as a major player in the development of diabetic nephropathy, and may contribute to tubular cell apoptosis and tubular atrophy prominently observed in diabetic nephropathy [37-40]. TNF-α is a pleiotropic peptide cytokine, capable of eliciting a wide spectrum of cellular responses, including differentiation, proliferation, inflammation, and cell death, via interaction with two members of the TNF receptor family, TNF-R1 and TNF-R2 [39]. TNF-α is predominantly produced by monocytes/macrophages, and also, by T and B-lymphocytes and glomerular mesangial cells [40, 41]. Its binding to TNF-R1 may trigger apoptotic pathways by recruitment of death effector adaptor molecules, with subsequent activation of caspase cascades, and anti-apoptotic pathways by a pathway involving TNF receptor-associated factor 2 (TRAF2) and NF-κB. Integration of these events determines the eventual cellular response to TNF-α stimulation. In particular, NF-κB stimulates transcription of anti-apoptotic factors that modulate the caspase cascade. Thus, NF-κB activity acts as a checkpoint in a cell’s decision to survive or apoptose in response to a given stimulus.
When applied to OK cells, TNF-α markedly reduced viability and induced apoptosis [34]. This was completely prevented by pre-treatment with insulin or C-peptide. Both insulin and C-peptide activated NF-κB simultaneously, but insulin displayed a typical sigmoidal dose with maximal NF-κB activation at an applied concentration of 100 nM. In contrast, C-peptide showed a completely different bell shaped curve of NF-κB stimulation, maximal with a 5 nM applied concentration. Pertussis toxin blocked only the C-peptide effect. In this work, the ability of C-peptide to prevent TNF-α-induced apoptosis appeared to be due to its ability to induce the expression of survival genes such as TRAF2 via NF-κB activation [34].
Overwhelming evidence implicates TGF-β1 is a predominant factor mediating proximal tubular cell (PTC) phenotypic changes and fibrosis in diabetic nephropathy [42, 43]. Production of TGF-β1 by PTC in diabetes is stimulated in part by high glucose and advanced glycation end products [44, 45]. In the proximal tubule, TGF-β1 is a key mediator of epithelial mesenchymal transformation (EMT), and modulates the expression of several epithelial cell recognition and organizational proteins, including cadherins [46], catenins, and the actin cytoskeleton [47]. Blockade of TGF-β1 action is a key therapeutic target in diabetic nephropathy. Application of C-peptide to human proximal tubular cells blocks the typical phenotypic and cytoskeletal changes caused by TGF-β1, as seen at the light microscope level [48]. C-peptide also prevents TGF-β1-induced upregulation of expression of both type I and type II TGF-β1 receptors, and abolishes TGF-β1-mediated phosphorylation and transcriptional activity of Smads -2 and -3 [48]. Where examined, pertussis toxin pre-treatment inhibited the action of C-peptide.
These studies provide new evidence in support of the ability of C-peptide to regulate the expression of beneficial genes in the face of pathophysiological stimuli relevant to the development and pathology of diabetic nephropathy.
Evidence for a C-peptide receptor
To exert hormone-like activity, C-peptide must have a receptor, but this is yet to be identified. The following lines of evidence support the existence of a specific C-peptide receptor.
C-Peptide binding to cells
As a result of studying the binding of radiolabeled C-peptide to a rat islet cell tumor composed predominantly of β-cells, a curvilinear Scatchard plot indicative of specific displaceable C-peptide binding was derived by Flatt et al. [49]. High affinity, specific binding of rhodamine-C-peptide to membranes from human skin fibroblasts, saphenous vein endothelial cells, and renal tubular cells was subsequently demonstrated in sub-fL volumes using sensitive fluorescence correlation microscopy [50]. The maximal numbers of binding sites, 1,000-1,500 per cell, were found on renal tubular cells, and these were 50% occupied by 0.3 nM C-peptide, with full saturation being achieved with 0.9 nM C-peptide. Binding was specific, displaced by excess intact C-peptide (but not by scrambled C-peptide), insulin, or insulin-like growth factors-I or -II [50]. Pertussis toxin pre-treatment also markedly attenuated C-peptide binding, thus implicating a GPCR.
An affinity (Kd) of 0.3nM was in complete agreement with the documented potency of C-peptide in signaling experiments. Furthermore, this affinity suggests that, in the presence of normal pancreatic β-cell function, these receptors should be fully occupied in the face of background circulating C-peptide concentrations. Hence, no further response would necessarily follow exogenous administration, thus explaining absent C-peptide responses after administration to healthy subjects. Although circulating concentrations of C-peptide are known, concentrations of bioactive peptide in the vicinity of physiologically relevant C-peptide receptors, for example in the kidney tubule, have not been studied.
Structure-function relationships of C-peptide binding and signaling
A number of careful studies have evaluated the relative abilities of various C-peptide fragments to recapitulate the activity of the intact molecule using Na+/K+-ATPase activity in rat kidney tubules as a functional readout [5]. Two regions of C-peptide appear to have functional importance in these assays. The rat C-peptide carboxy(C)-terminal pentapeptide, EVARQ, elicited 100% of the activity of intact C-peptide, whereas the remaining portion of the molecule was completely inactive. The corresponding region of human C-peptide (EGSLQ) elicited 75% activity. The C-terminal pentapeptide also has the ability to displace binding of full length C-peptide to cell membranes [50]. Glutamic acid and glutamine residues at positions 1 and 5 of this pentapeptide are generally conserved in mammals. Similar well-defined, functional C-terminal regions are also found in many other hormones, including gastrin and cholecystokinin. The properties of the C-terminal pentapeptide delineated by these studies are typical of those of a peptide ligand agonist acting via a receptor.
Mid-C-peptide sequences only partially recapitulate the activity of the intact molecule. des-(27-31)-C-peptide has no activity, but some non-naturally occurring sequences containing D-amino acid isomers display some bio-activity. Peptides greater than 9 amino acids in length show decreased activity. These findings are reminiscent of those described by Ido et al. (8) who found that some D-amino acid C-peptide enantiomers, and reverse sequence peptides, were able to activate Na+/K+-ATPase, and regulate nerve and vascular function in diabetic rats. This behavior is not compatible with a peptide/receptor interaction. It is more associated with a non-specific peptide/membrane interaction independent of chirality, but dictated by structural features related at least in part to sequence.
The activity balance between these two regions in vivo is likely to be of importance. Most recently, the combination of conserved glutamic acid residues at positions 3, 11, and 27 of C-peptide, together with of helix-promoting residues in the N-terminal segment [51], have been shown to be required for efficient signaling. Taken together, these studies demonstrate that the C-terminal pentapeptide of C-peptide possesses properties typical for a peptide ligand interacting with a specific receptor. Also, it shows that the C-peptide molecule comprises a three-part structure where the N- and C- terminal sections participate in functional interactions, and the mid-region serves as a joining segment [11, 51].
Evidence indicating C-peptide binding to a G-protein coupled receptor
The binding of C-peptide to cell membranes is reduced by pre-treatment with pertussis toxin (see above). Also, the great majority of signaling events, and resulting alterations in cell phenotype mediated by C-peptide, are inhibited by the pre-treatment of cells with pertussis toxin [11, 50]. This toxin ADP-ribosylates and inactivates members of the Gαi and Gαo families of heterotrimeric G-proteins, and suggests that these proteins are involved in C-peptide-activated signaling. In addition, C-peptide stimulates GTPγS binding to Gαi in kidney tubular cells [34]. Such a guanine nucleotide exchange typically occurs on G-protein alpha subunits following agonist ligation of a GPCR. Therefore, the most straightforward explanation of these observations is that C-peptide influences cellular signaling events via a G-protein-coupled receptor involving Gαi.
Insulin-mimetic actions of C-peptide
Some components of C-peptide signaling pathways are shared with those of insulin. It has been suggested that C-peptide may signal through the insulin receptor. In one study, C-peptide activated insulin receptor tyrosine kinase, tyrosine phosphorylation of IRS-1, MAPK, PI-3-kinase, p90 ribosomal S6 kinase, glycogen synthase kinase-3, and glycogen synthesis [30]. However, upstream steps in the signaling pathway were not tested for pertussis toxin sensitivity [30]. In contrast, other workers found no evidence of insulin receptor activation by C-peptide [52]. However, insulin-mediated effects are never sensitive to pertussis toxin, and insulin fails to displace C-peptide binding to cells [50]. Furthermore, using plasmon resonance no interaction of C-peptide with soluble purified insulin receptor is seen [53].
Therefore, a direct insulin receptor-mediated effect of C-peptide seems most unlikely. The balance of evidence points more strongly to C-peptide mediation of its own distinct signaling via a specific GPCR, with possible subsequent transactivation of receptor tyrosine kinases occurring through well established pathways [54, 55].
Current priorities and future perspectives
There can be no doubt that C-peptide is bioactive with multiple functions relevant to the development of complications in T1D. More interesting C-peptide actions are likely to be uncovered in the near future, but identification of the receptor is the really urgent goal. The balance of evidence strongly favors a GPCR, and strenuous efforts should be made to find it. The rewards are potentially huge and the biomedical impact will be substantial.
Although there is a belief that several investigators may have tried to find this, there are no peer reviewed original publications in this area. In a review paper, Luzi et al. reported some unsuccessful attempts to identify a C-peptide receptor using conventional approaches, but methodology was brief and this cannot be regarded as a definitive negative report [56]. Alternative and more intensive approaches may well be successful. Also potentially relevant to this point are recent descriptions of a divalent metal ion dependency of C-peptide activation [57]. We have often observed variable cell responses to C-peptide when C-peptide is applied to cells in defined salt solutions lacking divalent metals. This is particularly evident when measuring changes in intracellular Ca2+. However, studies performed with C-peptide presented in cell culture media containing the requisite metals generally display more robust and easily reproducible responses. Receptor cloning studies dependent on the binding of labeled C-peptide to λ phage expression libraries may need to take these observations into account, as may other affinity chromatographic approaches, as well as studies examining binding of C-peptide to cells and plasma membranes.
It is relatively straightforward to envisage how replacement of C-peptide may elicit biological effects in T1D when the peptide is completely absent. All clinical studies of C-peptide so far have focused exclusively on patients with T1D. However in T2D, elevated C-peptide levels are often found, at least in the earlier stages of the disease, and many affected patients may develop microvascular complications in the face of elevated ambient C-peptide levels. Given the high prevailing C-peptide levels in T2D, and bearing in mind the experimentally derived affinity of C-peptide binding [50], it is likely that any cognate receptor would be fully occupied and quite possibly desensitized with down-regulation of signaling. Very recent data from Meyer et al. suggest that C-peptide resistance may also be present in T2D alongside insulin resistance [58].
The evolution of understanding about C-peptide from inert by-product to bioactive peptide has been fascinating. The physiological effects are solidly underpinned by unequivocal signaling properties, and evidence of activity at the cellular level. The challenge now is to use these observations to guide the development of C-peptide science more firmly towards clinical trials and applications.
Disclosures: The authors report no conflict of interests.
References
- 1.Wald HP, Scherzer P, Rasch R, Popovtzer MM. Renal tubular Na+,K+-ATPase in diabetes mellitus: relationship to metabolic abnormality. Am J Physiol. 1993;265(1 Pt 1):E96–E101. doi: 10.1152/ajpendo.1993.265.1.E96. [DOI] [PubMed] [Google Scholar]
- 2.Raccah D, Lamotte-Jannot MF, Issautier T, Vague P. Effect of experimental diabetes on Na/K-ATPase activity in red blood cells, peripheral nerve and kidney. Diabete Metab. 1994;20(3):271–274. [PubMed] [Google Scholar]
- 3.Raccah D, Lamotte-Jannot MF, Issautier T, Vague P. Erythrocyte Na(+)-K(+)-ATPase activity, metabolic control, and neuropathy in IDDM patients. Diabetes Care. 1996;19:564–568. doi: 10.2337/diacare.19.6.564. [DOI] [PubMed] [Google Scholar]
- 4.Gerbi A, Barbey O, Raccah D, Coste T, Jamme I, Nouvelot A, Ouafik L, Levy S, Vague P, Maixment JM. Alteration of Na, K-ATPase isoenzymes in diabetic cardiomyopathy: effect of dietary supplementation with fish oil (n-3 fatty acids) in rats. Diabetologia. 1997;40:496–505. doi: 10.1007/s001250050707. [DOI] [PubMed] [Google Scholar]
- 5.Ohtomo Y, Aperia A, Sahlgren B, Johansson BL, Wahren J. C-peptide stimulates rat renal tubular Na+, K+-ATPase activity in synergism with neuropeptide Y. Diabetologia. 1996;39:199–205. doi: 10.1007/BF00403963. [DOI] [PubMed] [Google Scholar]
- 6.Dufayet De La Tour D, Raccah D, Jannot MF, Coste T, Rougerie C, Vague P. Erythrocyte Na/K ATPase activity and diabetes: relationship with C-peptide level. Diabetologia. 1998;41:1080–1084. doi: 10.1007/s001250051033. [DOI] [PubMed] [Google Scholar]
- 7.Forst T, Dufayet De La Tour D, Kunt T, Pfutzner A, Goitom K, Pohlmann T, Schneider S, Johansson BL, Wahren J, Lobig M, Engelbach M, Beyer J, Vague P. Effects of proinsulin C-peptide on nitric oxide, microvascular blood flow and erythrocyte Na+, K+-ATPase activity in diabetes mellitus type. Clin Sci. 2000;98:283–290. [PubMed] [Google Scholar]
- 8.Ido Y, Vindigni A, Chang K, Stramm L, Chance R, Heath WF, DiMarchi RD, Di Cera E, Williamson JR. Prevention of vascular and neural dysfunction in diabetic rats by C-peptide. Science. 1997;277:563–566. doi: 10.1126/science.277.5325.563. [DOI] [PubMed] [Google Scholar]
- 9.Tsimaratos M, Roger F, Chabardes D, Mordasini D, Hasler U, Doucet A, Martin PY, Feraille E. C-peptide stimulates Na+, K+-ATPase activity via PKC alpha in rat medullary thick ascending limb. Diabetologia. 2003;46:124–131. doi: 10.1007/s00125-002-0996-1. [DOI] [PubMed] [Google Scholar]
- 10.Maestroni A, Ruggieri D, Dell'Antonio G, Luzi L, Zerbini G. C-peptide increases the expression of vasopressin-activated calcium-mobilizing receptor gene through a G protein-dependent pathway. Eur J Endocrinol. 2005;152:135–141. doi: 10.1530/eje.1.01823. [DOI] [PubMed] [Google Scholar]
- 11.Wahren J, Ekberg K, Johansson J, Henriksson M, Pramanik A, Johansson BL, Rigler R, Jörnvall H. Role of C-peptide in human physiology. Am J Physiol. 2000;278:E759–E768. doi: 10.1152/ajpendo.2000.278.5.E759. [DOI] [PubMed] [Google Scholar]
- 12.Li L, Oshida Y, Kusunoki M, Yamanouchi K, Johansson BL, Wahren J, Sato Y. Rat C peptide I and II stimulate glucose utilization in STZ-induced diabetic rats. Diabetologia. 1999;42:958–964. doi: 10.1007/s001250051254. [DOI] [PubMed] [Google Scholar]
- 13.Etgen GJ, Fryburg DA, Gibbs EM. Nitric oxide stimulates skeletal muscle glucose transport through a calcium/contraction- and phosphatidylininositol-3 kinase-independent pathway. Diabetes. 1997;46:1915–1919. doi: 10.2337/diab.46.11.1915. [DOI] [PubMed] [Google Scholar]
- 14.Young ME, Radda GK, Leighton B. Nitric oxide stimulates glucose transport and metabolism in rat skeletal muscle in vitro. Biochem J. 1997;322:223–228. doi: 10.1042/bj3220223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jensen ME, Messina EJ. C-peptide induces a concentration-dependent dilation of skeletal muscle arterioles only in presence of insulin. Am J Physiol. 1999;276:H1223–H1228. doi: 10.1152/ajpheart.1999.276.4.H1223. [DOI] [PubMed] [Google Scholar]
- 16.Joshua IG, Zhang Q, Falcone JC, Bratcher AP, Rodriguez WE, Tyagi SC. Mechanisms of endothelial dysfunction with development of type 1 diabetes mellitus: role of insulin and C-peptide. J Cell Biochem. 2005;96(6):1149–1156. doi: 10.1002/jcb.20620. [DOI] [PubMed] [Google Scholar]
- 17.Wallerath T, Kunt T, Forst T, Closs EI, Lehmann R, Flohr T, Gabriel M, Schäfer D, Göpfert A, Pfützner A, Beyer J, Förstermann U. Stimulation of endothelial nitric oxide synthase by proinsulin C-peptide. Nitric Oxide. 2003;9:95–102. doi: 10.1016/j.niox.2003.08.004. [DOI] [PubMed] [Google Scholar]
- 18.Kitamura T, Kimura K, Makondo K, Furuya DT, Suzuki M, Yoshida T, Saito M. Proinsulin C-peptide increases nitric oxide production by enhancing mitogen-activated protein-kinase dependent transcription of endothelial nitric oxide synthase in aortic endothelial cells of Wistar rats. Diabetologia. 2003;46(12):1698–1705. doi: 10.1007/s00125-003-1232-3. [DOI] [PubMed] [Google Scholar]
- 19.Scalia R, Coyle K, Levine B, Booth G, Lefer A. C-peptide inhibits leukocyte-enodthelium interaction in the microcirculation during endothelial dysfunction. FASEB J. 2000;14(14):2357–2364. doi: 10.1096/fj.00-0183com. [DOI] [PubMed] [Google Scholar]
- 20.Chang L, Karin M. Mammalian MAP kinase signaling cascades. Nature. 2001;410(6824):37–40. doi: 10.1038/35065000. [DOI] [PubMed] [Google Scholar]
- 21.Chen Z, Gibson TB, Robinson F, Silvestro L, Pearson G, Xu B, Wright A, Vanderbilt C, Cobb MH. MAP kinases. Chem Rev. 2001;101:2449–2476. doi: 10.1021/cr000241p. [DOI] [PubMed] [Google Scholar]
- 22.Turjanski AG, Vaque JP, Gutkin JS. MAP kinases and the control of nuclear events. Oncogene. 2007;26:3240–3253. doi: 10.1038/sj.onc.1210415. [DOI] [PubMed] [Google Scholar]
- 23.Kitamura T, Kimura K, Jung BD, Makondo K, Okamoto S, Canas X, Sakane N, Yoshida T, Saito M. Proinsulin C-peptide rapidly stimulates mitogen-activated protein kinases in Swiss 3T3 fibroblasts: requirement of protein phosphoinositide 3-kinase and pertussi toxin-sensitive G-protein. Biochem J. 2001;355(Pt 1):123–129. doi: 10.1042/0264-6021:3550123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kitamura T, Kimura K, Jung BD, Makondon K, Sakane N, Yoshida T, Saito M. Proinsulin C-peptide activates cAMP response element binding proteins through the p38 mitogen-activated protein kinase pathway in mouse lung capillary endothelial cells. Biochem J. 2002;366(Pt 3):737–744. doi: 10.1042/BJ20020344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Al-Rasheed NM, Meakin F, Royal EL, Lewington AJ, Brown J, Willars GB, Brunskill NJ. Potent activation of multiple signaling pathways by C-peptide in opossum kidney proximal tubular cells. Diabetologia. 2004;47(6):987–997. doi: 10.1007/s00125-004-1404-9. [DOI] [PubMed] [Google Scholar]
- 26.Zhong Z, Davidescu A, Ehren I, Ekberg K, Jornvall H, Wahren J, Chibalin AV. C-peptide stimulates ERK 1/2 and JNK MAP kinases via activation of protein kinase C in human renal tubular cells. Diabetologia. 2005;48:187–197. doi: 10.1007/s00125-004-1602-5. [DOI] [PubMed] [Google Scholar]
- 27.Vanhaesebroeck B, Leevers SJ, Ahmadi K, Timms J, Katso R, Driscoll PC, Woscholski R, Parker PJ, Waterfield MD. Synthesis and function of 3-phosphorylated inositol lipids. Ann Rev Biochem. 2001;70:535–602. doi: 10.1146/annurev.biochem.70.1.535. [DOI] [PubMed] [Google Scholar]
- 28.Li ZG, Zhang W, Sima AA. C-peptide enhances insulin-mediated cell growth and protection against high glucose-induced apoptosis in SH-SY5Y cells. Diabetes Metab Res Rev. 2003;19:375–385. doi: 10.1002/dmrr.389. [DOI] [PubMed] [Google Scholar]
- 29.Walcher D, Aleksic M, Jerg V, Hombach V, Zieske A, Homma S, Strong J, Marx N. C-peptide induces chemotaxis of human CD4-positive cells. Involvement of pertussis toxin–sensitive G-proteins and phosphoinositide 3-kinase. Diabetes. 2004;53:1664–1670. doi: 10.2337/diabetes.53.7.1664. [DOI] [PubMed] [Google Scholar]
- 30.Grunberger G, Qiang X, Li Z, Mathews S, Sbrissa D, Shisheva A, Sima AA. Molecular basis for the insulinometric effects of C peptide. Diabetologia. 2001;44:1247–1257. doi: 10.1007/s001250100632. [DOI] [PubMed] [Google Scholar]
- 31.Aleksic M, Walcher D, Giehl K, Bach H, Grub M, Durst R, Hombach V, Marx N. Signaling processes involved in C-peptide induced chemotaxis of CD4-positive lymphocytes. Cell Mol Life Sci. 2009;66:1974–1984. doi: 10.1007/s00018-009-9057-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Al-Rasheed NM, Chana RS, Baines RJ, Willars GB, Brunskill NJ. Ligand-independent activation of peroxisome proliferator-activated receptor-gamma by insulin and C-peptide in kidney proximal tubular cells: dependent on phosphatidylinositol 3-kinase activity. J Biol Chem. 2004;279:49747–49754. doi: 10.1074/jbc.M408268200. [DOI] [PubMed] [Google Scholar]
- 33.Kitazawa M, Shibata Y, Hashimoto S, Ohizumi Y, Yamakuni T. Proinsulin C-peptide stimulates a PKC/IkappaB/NFkappaB signaling pathway to activate COX-1 gene transcription in Swiss 3T3 fibroblasts. J Biochem. 2006;139:1083–1088. doi: 10.1093/jb/mvj122. [DOI] [PubMed] [Google Scholar]
- 34.Al-Rasheed NM, Willars GB, Brunskill NJ. C-peptide signals via Galpha i to protect against TNF-alpha-mediated apoptosis of opossum kidney proximal tubular cells. J Am Soc Nephrol. 2006;17(4):986–995. doi: 10.1681/ASN.2005080797. [DOI] [PubMed] [Google Scholar]
- 35.Willson TM, Lambert MH, Kliewer SA. Peroxisome proliferator-activated receptor-gamma and metabolic disease. Annu Rev Biochem. 2001;70:341–367. doi: 10.1146/annurev.biochem.70.1.341. [DOI] [PubMed] [Google Scholar]
- 36.Rieusset J, Andreelli F, Auboeuf D, Roques M, Vallier P, Riou JP, Auwerx J, Laville M, Vidal H. Insulin acutely regulates the expression of the peroxisome proliferator-activated receptor-gamma in human adipocytes. Diabetes. 1999;48:699–705. doi: 10.2337/diabetes.48.4.699. [DOI] [PubMed] [Google Scholar]
- 37.Kalantarinia K, Awad AS, Siragy HM. Urinary and renal interstitial concentrations of TNF-alpha increase prior to the rise in albuminuria in diabetic rats. Kidney Int. 2003;64:1208–1213. doi: 10.1046/j.1523-1755.2003.00237.x. [DOI] [PubMed] [Google Scholar]
- 38.Moriwaki Y, Yamamoto T, Shibutani Y, Aoki E, Tustsumi Z, Takahashi S, Okamura H, Koga M, Fukuchi M, Hada T. Elevated level of interleukin-18 and tumor necrosis factor-alpha in serum of patients with type 2 diabetes mellitus: relationship with diabetic nephropathy. Metabolism. 2003;52:605–608. doi: 10.1053/meta.2003.50096. [DOI] [PubMed] [Google Scholar]
- 39.Wajant H, Pfizenmaier K, Scheurich P. Tumor necrosis factor signaling. Cell Death Differ. 2003;10:45–65. doi: 10.1038/sj.cdd.4401189. [DOI] [PubMed] [Google Scholar]
- 40.Baud L, Oudinet JP, Bens M, Noel L, Peraldi MN, Rondeau E, Etienne J, Ardaillou R. Production of tumor necrosis factor by rat mesangial cells in response to bacterial lipopolysaccharide. Kidney Int. 1989;35:1111–1118. doi: 10.1038/ki.1989.98. [DOI] [PubMed] [Google Scholar]
- 41.Hruby ZW, Lowry RP. Spontaneous release of tumor necrosis factor alpha by isolated renal glomeruli and cultured glomerular mesangial cells. Clin Immunol Immunopathol. 1991;59:156–164. doi: 10.1016/0090-1229(91)90089-s. [DOI] [PubMed] [Google Scholar]
- 42.Kanwar YS, Wada J, Sun L, Xie P, Wallner EI, Chen S, Chugh S, Danesh FR. Diabetic nephropathy: mechanisms of renal disease progression. Exp Biol Med. 2008;233:4–11. doi: 10.3181/0705-MR-134. [DOI] [PubMed] [Google Scholar]
- 43.Sharma K, Ziyadeh FN. Hyperglycaemia and diabetic kidney disease. The case for transforming growth factor-beta as a key mediator. Diabetes. 1995;44:1139–1146. doi: 10.2337/diab.44.10.1139. [DOI] [PubMed] [Google Scholar]
- 44.Oldfield MD, Bach LA, Forbes JM, Nikolic-Paterson D, McRobert A, Thallas V, Atkins RC, Osicka T, Jerums G, Cooper ME. Advanced glycation end products cause epithelial-myofibroblast transdifferentiation via the receptor for advanced glycation end products (RAGE) J Clin Invest. 2001;108:1853–1863. doi: 10.1172/JCI11951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Qi W, Chen X, Zhang Y, Waltham M, Schache M, Kelly DJ, Pollock CA. High glucose induces macrophage inflammatory protein-3 alpha in renal proximal tubule cells via a transforming growth factor-beta 1 dependent mechanism. Nephrol Dial Transplant. 2007;11:3147–3153. doi: 10.1093/ndt/gfm365. [DOI] [PubMed] [Google Scholar]
- 46.Tian YC, Fraser D, Attisano L, Philips AO. TGF-beta 1 mediated alterations of renal proximal tubular epithelial cell phenotype. Am J Physiol. 2003;285:F130–F142. doi: 10.1152/ajprenal.00408.2002. [DOI] [PubMed] [Google Scholar]
- 47.Humes HD, Nakamura T, Cieslinski DA, Miller D, Emmons RV, Border WA. Role of proteoglycans and cytoskeleton in the effects of TGF-beta 1 on renal proximal tubule cells. Kidney Int. 1993;43:575–584. doi: 10.1038/ki.1993.85. [DOI] [PubMed] [Google Scholar]
- 48.Hills CE, Al-Rasheed N, Al-Rasheed N, Willars GB, Brunskill NJ. C-peptide reverses TGF-beta1-induced changes in renal proximal tubular cells: implications for treatment of diabetic nephropathy. Am J Physiol. 2009;296:F614–F621. doi: 10.1152/ajprenal.90500.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Flatt PR, Swanston-Flatt SK, Hampton SM, Bailey CJ, Marks V. Specific binding of the C-peptide of proinsulin to cultured B-cells from a transplantable rat islet cell tumor. Biosci Rep. 1986;6:193–199. doi: 10.1007/BF01115006. [DOI] [PubMed] [Google Scholar]
- 50.Rigler R, Pramanik A, Jonasson P, Kratz G, Jansson O, Nygren PA, Stahl S, Ekberg K, Johansson BL, Uhlen S, Uhlen M, Jornvall H, Wahren J. Specific binding of proinsulin C-peptide to human cell membranes. Proc Natl Acad Sci USA. 1999;96:13318–13323. doi: 10.1073/pnas.96.23.13318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Henriksson M, Nordling E, Melles E, Shafqat J, Ståhlberg M, Ekberg K, Persson B, Bergman T, Wahren J, Johansson J, Jörnvall H. Separate functional features of proinsulin C-peptide. Cell Mol Life Sci. 2005;62:1772–1778. doi: 10.1007/s00018-005-5180-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zierath JR, Handberg A, Tally M, Wallberg-Henriksson H. C-peptide stimulates glucose transport in isolated human skeletal muscle independent of insulin receptor and tyrosine kinase activation. Diabetologia. 1996;39:306–313. doi: 10.1007/BF00418346. [DOI] [PubMed] [Google Scholar]
- 53.Henriksson M, Johansson J, Moede T, Leibiger I, Shafqat J, Berggren P, Jörnvall H. Proinsulin C-peptide and insulin: Limited pattern similarities of interest in inter-peptide interactions but no C-peptide effect on insulin and IGF-1 receptor signaling. Cell Mol Life Sci. 2006;63:3055–3060. doi: 10.1007/s00018-006-6426-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gschwind A, Zwick E, Prenzel N, Leserer M, Ullrich A. Cell communication networks: epidermal growth factor receptor transactivation as the paradigm for interreceptor signal transmission. Oncogene. 2001;20:1594–1600. doi: 10.1038/sj.onc.1204192. [DOI] [PubMed] [Google Scholar]
- 55.Rao GN, Delafontaine P, Runge MS. Thrombin Stimulates Phosphorylation of Insulin-like Growth Factor-1 Receptor, Insulin Receptor Substrate-1, and Phospholipase C-1 in Rat Aortic Smooth Muscle Cells. J Biol Chem. 1995;270:27871–27875. doi: 10.1074/jbc.270.46.27871. [DOI] [PubMed] [Google Scholar]
- 56.Luzi L, Zerbini G, Caumo A. C-peptide: a redundant relative of insulin? Diabetologia. 2007;50:500–502. doi: 10.1007/s00125-006-0576-x. [DOI] [PubMed] [Google Scholar]
- 57.Medawala W, McCahill P, Giebink A, Meyer J, Ku CJ, Spence DM. A molecular level understanding of zinc activation of C-peptide and its effects on cellular communication in the bloodstream. Rev Diabet Stud. 2009 doi: 10.1900/RDS.2009.6.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Meyer JA, Froelich JM, Reid GE, Karunarathne WK, Spence DM. Metal-activated C-peptide facilitates glucose clearance and the release of a nitric oxide stimulus via the GLUT1 transporter. Diabetologia. 2008;51:175–182. doi: 10.1007/s00125-007-0853-3. [DOI] [PMC free article] [PubMed] [Google Scholar]