

Abstract
Diabetic encephalopathy is a recently recognized complication in type 1 diabetes. In this review, we summarize a series of experimental results obtained longitudinally in the spontaneously type 1 diabetic BB/Wor-rat, and bringing out the beneficial effects of C-peptide replacement. It is increasingly clear that lack of insulin and C-peptide, and perturbations of their signaling cascades in type 1 diabetes are detrimental to the regulation of neurotrophic factors and their receptors. Other consequences of such deficits and perturbations are innate inflammatory responses with effects on synaptogenesis, neurite degeneration, and early behavioral abnormalities. Replacement of C-peptide, which does not effect hyperglycemia, has beneficial effects on a variety of pro-apoptotic stressors, oxidative stressors, and finally on apoptosis. Eventually, this cascade of events leads to neuronal loss and decreased densities of white matter myelinating cells, with more profound deficits in behavioral and cognitive function. Such changes are likely to underlie gray and white matter atrophy in type 1 diabetes, and are significantly prevented by full C-peptide replacement. Present data demonstrate that C-peptide replacement has beneficial effects on numerous sequential and partly interrelated pathogenetic mechanisms, resulting in prevention of neuronal and oligodendroglial cell loss, with significant prevention of neurobehavioral and cognitive functions.
Keywords: type 1 diabetes, C-peptide, encephalopathy, BB/Wor-rat, inflammation, hyperglycemia, neuronal loss, hippocampus, synaptic connectivity, cerebral atrophy, intracerebral insulin signaling, gray matter density
Abbreviations: 8-OHdG - 8-Hydroxy-2'-deoxyguanosine; ADP - adenosine diphosphate; AGE - advanced glycation end products; AMP - adenosine monophosphate; cAMP - cyclic adenosine monophosphate; AMPA - α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid; Bax - Bcl-2-associated X protein (promotes apoptosis); BB/Wor rat - BioBreeding/Worcester rat (animal model of spontaneous autoimmune diabetes); Bcl-2 - B-cell lymphoma 2; BDNF - brain-derived neurotrophic factor; Ca2+- bivalent ionic calcium; CA1 - Cornu Ammonis area 1 (also known as Ammon's horn); CNS - central nervous system; C-peptide - connecting peptide; CREB - cyclic AMP response element-binding protein; DNA - desoxyribonucleic acid; DPN - diabetic peripheral neuropathy; EEG - Electroencephalogram; Fas - transmembrane protein of the TNF family that induces apoptosis (also known as CD95L); FDG - 18F-fluorodeoxyglucose; GFAP - glial fibrillary acidic protein; GluR - postsynaptic glutamate receptor; IGF - insulin-like growth factor; IL - interleukin; IQ - intelligence quotient; MRI - magnetic resonance imaging; Na+/K+-ATPase - sodium, potassium adenosintriphosphatase, also sodium-potassium pump; NGF - nerve growth factor; NGFR-p75 - nerve growth factor receptor p75; NGF-TrA - nerve growth factor tyrosine kinase A; NF-κB - nuclear factor-kappa light-chain enhancer of activated B cells; PARP - poly (ADP-ribose) polymerase; p-Akt - phosphorylated akt; p-GSK-3β - glycogen synthase kinase-3 beta phosphorylated on serine 9 (also known as phosphorylated stress kinase); RAGE - receptor for advanced glycation end products; rMRG1c - regional metabolic rate of glucose 1c; STZ - streptozotocin; TNFα - tumor necrose factor alpha; Trk - tropomyosin receptor kinase (also known as tyrosine kinase); TrkA - tyrosine kinase A (highly affine to the NGF receptor); TUNEL - terminal deoxynucleotidyl transferase (Tdt)- mediated dUTP-biotin nick end labeling
Introduction
Diabetes is an increasingly common metabolic disorder with well known serious secondary complications affecting kidney, retina, peripheral nerve and vasculature. Diabetic peripheral neuropathy (DPN) was long considered the only complication involving the nervous system. Whereas, the central nervous system (CNS) was believed to be relatively spared from the direct effects of diabetes. In the last decade, it has become clear that diabetes may be both primarily and secondarily responsible for CNS complications, with adverse functional and cognitive effects. Primary diabetic encephalopathy is caused by direct metabolic perturbations due to hyperglycemia, insulin deficiency, or hyperinsulinemia. Secondary diabetic encephalopathy occurs as a result of micro- and macrovascular disorders, or severe and repeated episodes of hypoglycemia [1-3].
Primary diabetic encephalopathy affects both type 1 and type 2 diabetes. However, as in the DPN occurring in the two types of diabetes, the underlying mechanisms seem to differ, and the cognitive outcomes are different in the two types of diabetes [2, 4].
In type 2 diabetes, several factors predispose to cognitive impairment such as hyperglycemia, insulin resistance, hyperinsulinemia, and age alone [2, 4, 5]. Additional components of the commonly associated metabolic syndrome, such as elevated cholesterol levels and hypertension, are likely to provide significant contributing factors [2, 4, 5]. Several studies have demonstrated an association between type 2 diabetes and Alzheimer's disease [6-11]. Such an association has also been corroborated in an animal model of type 2 diabetes [12].
Several neurobehavioral studies in children with type 1 diabetes have demonstrated deficits in attention, processing speed, executive function, intelligence, and memory. Such deficits translate into lower IQs, worse school performances, and greater likelihood to fail grades [13-17]. In one study, early onset of diabetes correlated with lower IQ performance, and lower full scale IQ. In contrast, a positive history of hypoglycemic episodes correlated with lower verbal IQ [18]. It has been recognized repeatedly that early onset of diabetes leads to worse neuropsychological outcomes [14, 15, 19]. Also, males are more vulnerable than females [15, 20]. In contrast to earlier beliefs, most studies do not seem to associate cognitive dysfunctions with episodes of hypoglycemia due to intensive insulin treatment [21-23].
Neurostructural deficits in type 1 diabetic patients
Recently, neuroimaging studies of type 1 diabetes patients, have reported increased glucose levels in frontal white matter and cortex. These findings were associated with increased concentrations of myoinositol, and decreased levels of N-acetylaspartate [24]. Structural studies of patients with onset of diabetes before the age of six have revealed a high incidence of mesial temporal lobe sclerosis, which was not associated with a history of hypoglycemia [25]. Similarly, volumetric MRI assessments of patients with type 1 diabetes for 12 years showed significantly decreased gray matter volumes in thalami, hippocampal regions, insular cortex, and decreased white matter volumes in parahippocampus, temporal lobe, and in the middle frontal areas [18]. Voxel-based morphometry of a type 1 population with 15-25 years duration of diabetes demonstrated decreased gray matter densities of thalamus, superior and middle temporal gyri, and middle frontal gyri [26]. Based on these recent studies, it appears that cerebral atrophy does occur, and that limbic temporal structures and cortices are preferentially involved.
Only few neuropathological reports have described structural abnormalities. A recent study of two young patients, who succumbed to the complications of ketoacidosis, reported severe neuronal loss in hippocampus and frontal cortex [27]. This was associated with white matter atrophy in the frontal and temporal regions, and was consistent with neuroimaging data. Such pathologies are likely to correlate with deficits in various cognitive domains, such as memory, information processing speed, executive function, attention and motor speed. Interestingly, deficits in such cognitive functions are associated with impaired functional connectivity, a measure of functional interactions between brain regions [28] and loss of fast β-frequency bands on quantitative EEG examinations [29]. These changes did not correlate with previous episodes of hypoglycemia.
Therefore, it is becoming accepted that type 1 diabetes leads to deficits in various cognitive domains associated with structural gray matter deficits, particularly in limbic structures and white matter atrophy. Such abnormalities appear to be more prevalent in patients with early onset of disease.
Experimental studies in animal models
Early studies in streptozotocin-induced diabetes in rats demonstrated neuronal loss in frontal cortex and white matter deficits [30]. Functional studies using the Morris water maze paradigm have demonstrated neurobehavioral deficits [31], which were prevented with insulin treatment from onset of diabetes [32]. Deficits in water maze learning were associated with impaired hippocampal long-term potentiation as a measure of synaptic plasticity [31]. This was also prevented by insulin treatment [32]. However, intervention with insulin to normalize hyperglycemic levels did not reverse water maze learning deficits, but it had a partial effect on long-term potentiation [32]. These data suggest that both pre- and post-synaptic activities are affected in hippocampus [33].
Recent data obtained from streptozotocin-induced diabetes in Swiss Webster wild-type mice show cerebral atrophy associated with cognitive decline over time [34]. However, in this model the cerebral atrophy was not associated with neuronal loss, but was ascribed to atrophy of the white matter. One factor in this study associated with white matter atrophy was upregulation of the receptor for advanced glycation end products (RAGE) [35]. This occurred together with downregulation of insulin signaling intermediaries, transcription factor cyclic adenosine monophosphate (cAMP), response element binding protein (CREB), and synaptophysin. Interestingly, administration of intranasal insulin to this type 1 diabetic murine model, slowed the development of neurobehavioral deficits, ameliorated white matter atrophy, and corrected insulin signaling intermediaries [35]. These results are consistent with, and strongly support, our previous data suggesting that insulin deficiency per se may play a pivotal role in the development of cognitive dysfunction in type 1 diabetes [1, 36].
In this review, we describe the sequential and longitudinal findings of cognitive deficits, metabolic, molecular, and structural abnormalities occurring in the encephalopathy in the BB/Wor-rat. This is a robust model that closely simulates human type 1 diabetes, which occurs spontaneously and secondary to an immune-mediated β-cell loss, with complete insulin and C-peptide deficiency [37]. Also, the model has been used widely to explore mechanisms underlying other type 1 diabetic complications [38]. For information regarding the metabolic profile of this model, the reader is referred to the accompanying paper on DPN in this issue [39].
The effect of C-peptide on early functional and neurotrophic deficits
Changes in cerebral-evoked potentials reflected by somatosensory, visual, and auditory evoked potentials, occur in both STZ-induced diabetic rats and in the BB/Wor-rat. These changes are followed by progressive degenerative changes in the dorsal columns of the spinal cord and the optic nerve [3, 38, 40, 41]. Such changes can be modified by insulin treatment [3].
Various neurobehavioral tests can be applied to rodents to examine different spheres of cognition. One paradigm that we have used for testing the BB/Wor-rat is the radial arm maze [42]. This consists of eight radial arms, of which three are baited with food and associated with visual cues. The animals are trained with three consecutive trials per day, over a period of one week. The set-up has an integrated video camera to record latencies, type I and type II errors. Latency is the time it takes to retrieve all baits. Type I error is entrance into an arm without a bait (reference memory), and type II error is recorded when entering an arm from which the bait was already collected (working memory) (Figure 1). This paradigm has been closely related to hippocampal activity, long-term potentiation, and mossy fiber pathology [42-44]. Testing of 3-month diabetic BB/Wor-rats using this paradigm showed significantly increased frequencies of type I and type II errors, whereas the latencies were unaltered. Diabetic rats replaced with full substitution of C-peptide from onset of diabetes showed normal frequencies of type I and type II errors at the same duration of diabetes (Figure 1). This demonstrated that deficits were preventable in both reference and working memories for spatial orientation abilities.
Figure 1. Results from behavioral testing of 2-mo control, diabetic, and C-peptide replaced diabetic BB/Wor-rats using the radial arm paradigm.
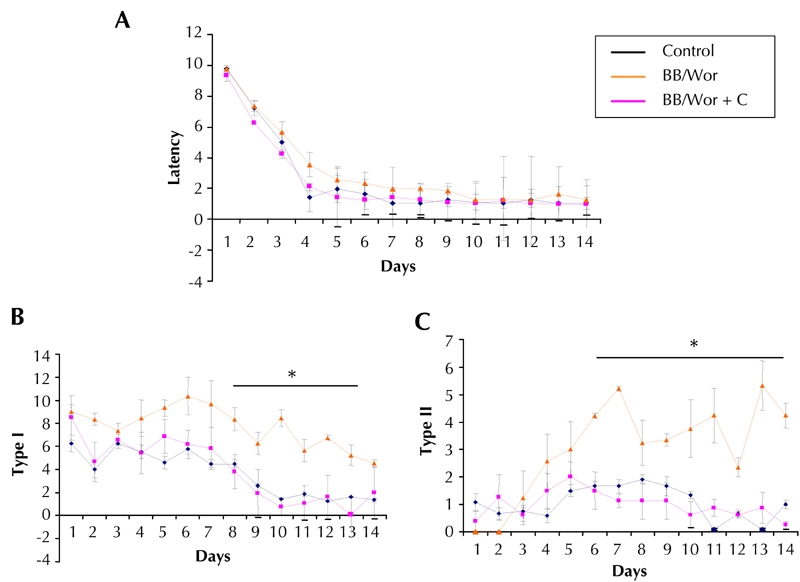
A: Total latencies were not altered in diabetic or C-peptide-treated diabetic rats as compared to age-matched control animals. Type I (B) type II (C) errors were significantly more common in non-treated diabetic rats, signifying impaired reference and working memory, respectively. These error types were not recorded in C-peptide-treated diabetic rats (B and C).
As previously shown, the findings suggest that correction of impaired insulin function alleviates early behavioral abnormalities, and long-term potentiation [32]. The behavioral abnormalities were preceded by impaired insulin signaling activities [36], and a 3-fold increase in glucose uptake in hippocampi of 2-month diabetic rats (Figure 2). At the same duration of diabetes, both the expression of the insulin-receptor, IGF-1-receptor, and NGF and NGF-TrA receptor were significantly suppressed in diabetic BB/Wor-rats (Figure 3). On the other hand, IGF-I expression was only modestly suppressed, whereas that of IGF-II was severely decreased to about 30% of that of control rats (Figure 3). These abnormalities were significantly, if not fully, prevented in C-peptide replaced rats (Figure 3). Simultaneously, insulin signal intermediaries such as p-Akt and p-GSK-3β were significantly decreased and increased respectively. These abnormalities were significantly prevented in C-peptide-replaced diabetic rats. Thus, C-peptide replacement corrects the insulin signaling cascade in type 1 diabetic hippocampus.
Figure 2. Micro-PET data from three 3-month diabetic BB/Wor-rats (A) and age-matched control rats (B).
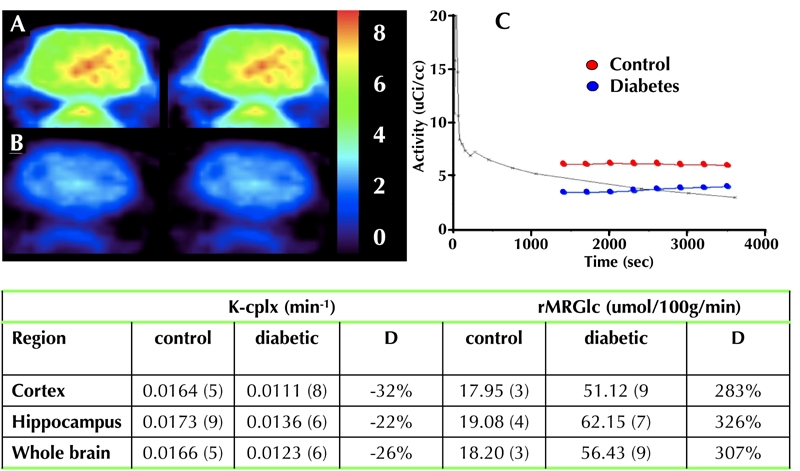
The uptake rate constant of the FDA tracer (K-cplx) was decreased by 22-32% in diabetic rats as compared to control rats (C and Table). Despite this, the metabolic rate of glucose (rMRGlc) was increased approximately 3-fold in cerebral cortex, hippocampus, and whole brain (Table).
Figure 3. Neurotrophic profiles in hippocami of 2-month diabetic and C-peptide treated rats (IR, IGF-R, IGF-1 and IGF-2).
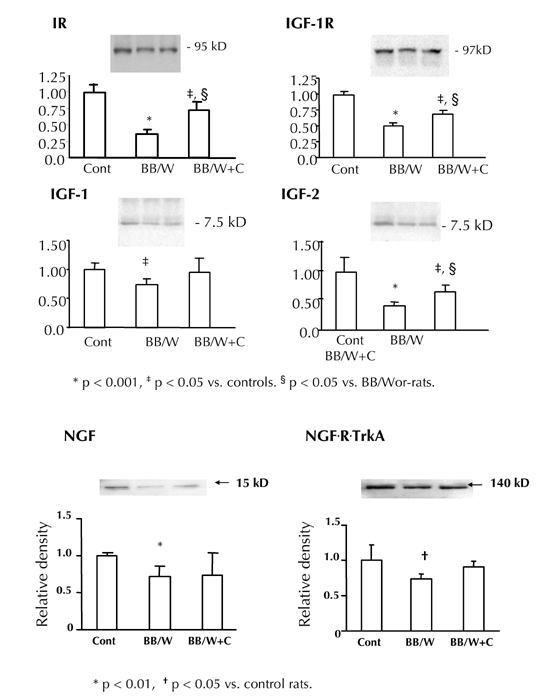
Note the significant suppression of the expression of these factors and receptors in diabetic rats, abnormalities that were significantly prevented by C-peptide replacement. Also, NGF and its receptor Trk A showed decreased expression in hippocampi of 8-month diabetic BB/Wor-rat with modest prevention in C-peptide treated rats. Equal loading of lanes was evaluated by staining of membranes with Ponceau reagent.
Both insulin and NGF provide potent neurotrophic support in hippocampus [45, 46]. Insulin is closely involved in neurotransmitter synthesis including acetylcholine and glutamate [45], and NGF secretion exerts a protective effect of cholinergic neurons [46, 47]. Interestingly, IGF-II expression was markedly suppressed in diabetic rats, which was largely prevented following C-peptide supplementation. On the other hand, IGF-I expression was only mildly decreased. It is known that endogenous IGF-I inhibits hippocampal acetylcholine release, whereas IGF-II has the opposite effect [48, 49].
Although not examined in the BB/Wor-rat, brain-derived neurotrophic factor (BDNF) is known to modulate pre- and post-synaptic transmission and placticity [50]. The early perturbations of trophic factor activities were followed by presynaptic degeneration in hippocampi of 4-month diabetic rats. This was demonstrated by decreased synaptophysin expression, and decreased numbers of presynaptic terminals in strata oriens of Ammon's horn area 1 (CA1), a region rich of pyramidal cells. The degenerative changes of presynaptic terminal neurites, and expression of synaptophysin, were fully prevented by C-peptide replacement (Figure 4). Therefore, it is highly likely that the perturbations of various neurotrophic factors, including insulin itself, are involved in neurite degeneration, and abnormalities in hippocampal plasticity. This is evidenced by suppressed long-term potentiation [31, 32], and spatial learning defects, as shown here (Figure 1). Importantly, full replacement of C-peptide, which exerts insulin-like effects [51], prevents this series of early degenerative events.
Figure 4.
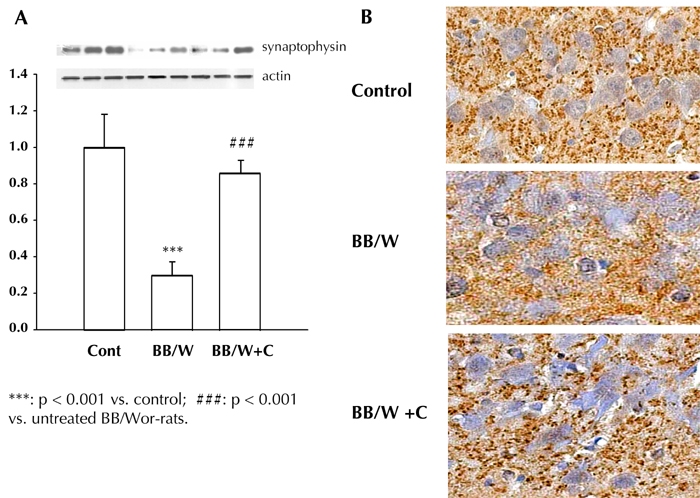
A: The expression of hippocampal presynaptic synaptophysin was significantly decreased in 4-month diabetic rats, and was prevented by C-peptide replacement from onset of diabetes. B: This corresponded to a decreased density of synaptophysin stained synapses in the CA1 region of diabetic rat hippocampi, suggesting presynaptic degeneration. The normal density of presynaptic buttons was preserved in C-peptide treated rats.
Glucose metabolism, prevention of RAGE, and innate inflammatory responses by C-peptide
The diabetic BB/Wor-rats were maintained at high glycemic levels, but free of ketonuria. Blood glucose levels were not influenced by C-peptide treatment [36, 39]. MicroPet imaging of diabetic BB/Wor-rats revealed an approximate 30% decrease in the uptake rate constant of 18F-fluorodeoxyglucose (FDG) into hippocampus and cerebral cortex (Figure 2). However, the regional metabolic rate of glucose (rMRG1c) showed a three-fold increase in whole brain, as well as in hippocampus and cerebral cortex (Figure 2). These data are consistent with those reported in humans [24]. Increased cerebral glucose was associated with an increased expression of RAGE [49]. In the hippocampus, RAGE was mainly colocalized with glial fibrillary acidic protein (GFAP)-positive proliferating astrocytes. To a lesser extent it was localized with hippocampal pyramidal cells and white matter oligodendrocytes [52]. These findings are similar to those reported in studies of brains of human type 1 diabetes subjects [27], and in a STZ-induced diabetic mouse model [34]. RAGE is a multi-ligand receptor for AGE, formed as a consequence of elevated glucose or oxidative stress. It is also a receptor for β-amyloid, and even prion proteins [53]. RAGE activation stimulates NF-κB, which is upregulated in the hippocampus of BB/Wor-rats. It should be mentioned that increased expression of NF-κB also occurs as a consequence of impaired insulin signaling via phosphorylation of IκB [53-55].
Interestingly and unexpectedly, C-peptide replacement prevented the upregulations of RAGE and NF-κB in diabetic hippocampi [52]. NF-κB is also known to upregulate RAGE itself, thereby providing a self-perpetuating loop. These data suggest that upregulation of RAGE and its response to C-peptide supplementation may largely be due to an impaired insulin signaling mechanism [51, 54, 57] rather than a downstream effect of hyperglycemia. RAGE was localized to activated astrocytes, which together with activated microglia play central roles in the activation of inflammatory mediators. Upregulation of RAGE and NF-κB, was accompanied by upregulation of TNF-α, IL-1β, IL-2, and IL-6. Whereas, the anti-inflammatory interleukin IL-10 was downregulated [52, 58]. Consequent to the normalization of RAGE and NF-κB after C-peptide replacement, TNF-α as well as the pro- and anti-inflammatory interleukins were normalized in the hippocampus [52, 58]. As discussed , this cascade of partly interactive and self-perpetuating activities of inflammatory factors 1. affect the already compromised insulin-signaling cascade [35], 2. enhance oxidative stress, and 3. promote apoptotic stress.
The effect of C-peptide on late cognitive function and structural deficits
As mentioned above, deficits in reference and working memory occur relatively early after onset of diabetes. Longitudinal testing in the Morris water maze system [59] revealed normal performances in diabetic rats of 4-month duration of diabetes. Significant deficits in latencies were evident only after 6-month of diabetes (Figure 5). At 8-month duration of diabetes, C-peptide replacement from onset showed significant preventative effects on Morris water maze latencies [1, 36] (Figure 5). Multiple cognitive components are involved in this task such as problem solving, formation of internal representation of the environment, and storage and retrieval of relevant information [1, 59]. The data indicate that progressive learning and memory deficiencies occur in a duration-related fashion, and are significantly prevented by C-peptide. Such changes are indicative of altered synaptic plasticity and cognitive deficits. They can be associated with depression of long-term potentiation, and enhanced long-term depression [31, 32, 60].
Figure 5. Latencies in the Morris water maze set up (right upper corner) in control and diabetic BB/Wor-rats at 2 (A); 4 (B); 6 (C), and 8 months (D) of diabetes.
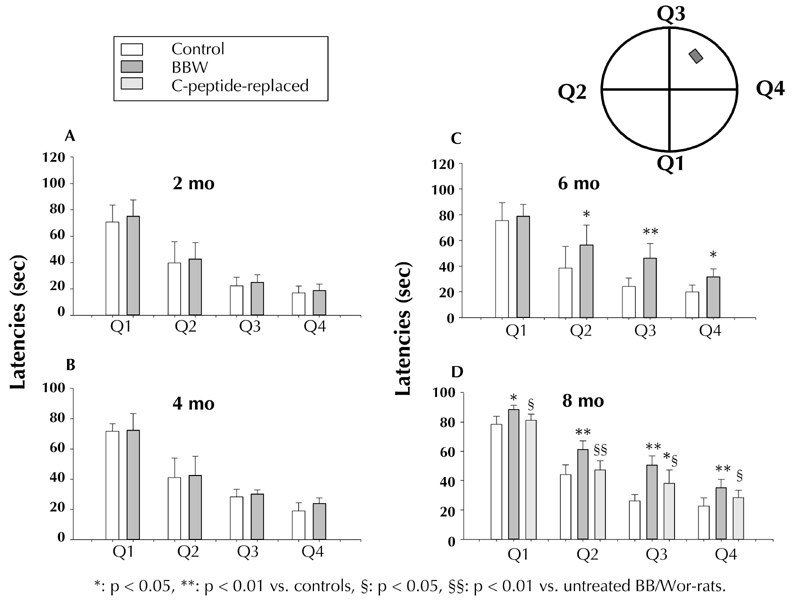
Q1-Q4 signify the four quadrants of the water maze. Note, significantly prolonged latencies are seen only after 6 months of diabetes (C). Diabetic rats treated with C-peptide from onset of diabetes showed significant prevention in the latencies at 8 months (D), suggesting prevention of spatial memory and long-term memory deficits.
Seven-month diabetic rats showed increased expression of the postsynaptic glutamate receptor 2 (GluR2) and 4 (GluR4) subunits (Figure 6), of which GluR2 is the most prevalent of the subunits in the forebrain and hippocampus [61, 62]. The GluR2 subunit controls Ca2+ permeability of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor channels, and plays a crucial role in several forms of long-term synaptic plasticity [63]. Increased presence of GluR2 decreases, or abolishes, Ca2+ permeability [64]. C-peptide replacement fully prevented the change in GluR2, and partially prevented that of the GluR4 subunit (Figure 7). Future studies have to explore whether these changes relate to changes in long-term depression. At this stage of the encephalopathy, hippocampal pre-synaptic synaptophysin remained significantly suppressed in both hippocampus and frontal cortex, and was significantly, but not fully, prevented by C-peptide (data not shown). These findings will inevitably have profound effects on our understanding of the intrinsic hippocampal neuronal networking. Therefore, it is apparent that numerous factors emanating from impaired insulin-signaling, and possibly hyperglycemia, lead to sequential and progressive behavioral deficits, perturbations in expression of neurotrophic factors, and alterations in synaptic connectivity.
Figure 6.
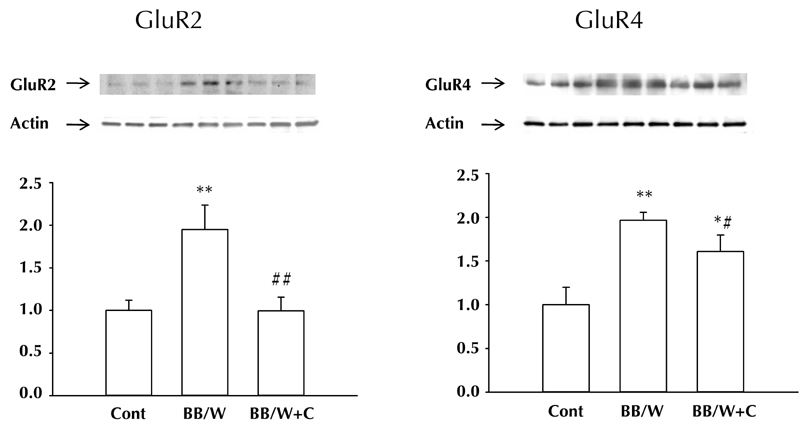
Expression of post-synaptic excitatory GluR2 and GluR4 in hippocampus were both significantly increased in 7-month diabetic BB/Wor-rats. C-peptide substitution from diabetes onset fully prevented the more abundant GluR2 subunit, whereas the effect was less striking with respect to the less abundant GluR4.
Figure 7. White matter changes in control, diabetic, and C-peptide replaced diabetic rats.
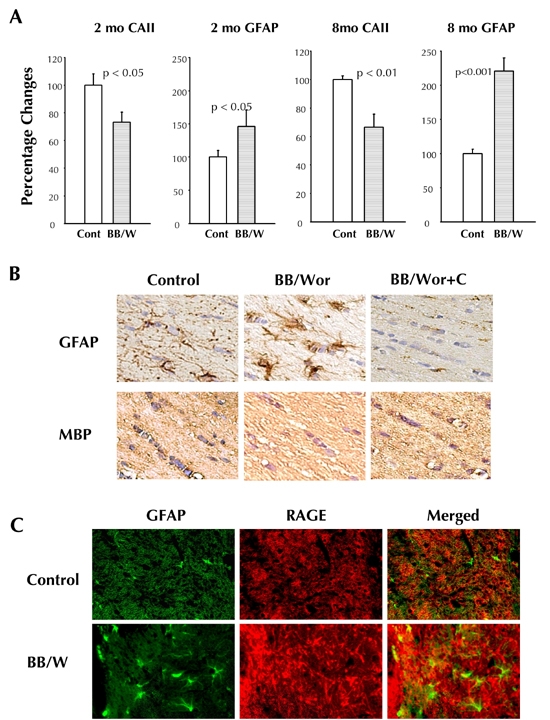
A: Already in 2-month diabetic rats, there is a significant decrease in the number of oligodendroglial cells (identified by the oligo marker CAII) in temporal white matter, which is associated with increased proliferation of astrocytes. These differnces are sustained and increased in 8-month diabetic rats. B: Immunocytochemical identification of astrocytes (GFAP), which are increased in 4-month diabetic rats, which simultaneously show decreased stainability for the oligo marker myelin basic protein (MBP). There is close to normal stainability of astrocytes and oligodendroglia cells in C-peptide treated animals. C: RAGE is markedly upregulated in the white matter of 8-month diabetic rats, and colocalizes with GFAP-positive astrocytes.
Both insulin and C-peptide display strong anti-apoptotic effects. In previous studies [1, 36, 54, 65], we have shown that insulinopenic diabetes in the BB/Wor-rat is associated with upregulation of a number of pro-apoptotic factors such as NGFR-p75, Fas, Bax, PARP, oxidative stress-induced DNA damage, caspase 12, and active caspase 3 expression [36, 65, 66]. Activation of such stressors was accompanied by increased TUNEL staining of hippocampal pyramidal cell neurons, increased DNA laddering of hippocampus, and decreased density of pyramidal cell neurons, particularly in the CA1 region [36]. C-peptide replacement for the duration of 8 months of diabetes, significantly prevented pro-apoptotic factors (except for Fas), partially prevented hippocampal pyramidal cell loss, and normalized associated behavioral deficits, as reflected by the latencies of the Morris water maze [36] (Figure 5).
These findings underline the pivotal role of impaired insulin signaling activities in the development of type 1 diabetic encephalopathy, which are normalized by C-peptide substitution [36, 51]. Recently, this concept was supported by the beneficial effects of direct intranasal insulin delivery to the CNS, which was observed to influence cerebral atrophy, cognitive deficits, and intracerebral insulin signaling intermediaries in STZ-diabetic mice [35]. Neither intranasal insulin administration, nor C-peptide replacement, alter systemic hyperglycemia. These results may shed light on mechanisms underlying hippocampal and cortical neuron loss in human type 1 diabetes [27].
White matter changes in the BB/Wor-rat
Preliminary studies have revealed early deficits in temporal white matter consisting of increased RAGE expression, and pro-inflammatory TNF-α and IL-6. RAGE colocalized with GFAP-positive proliferating astrocytes (Figure 7). These changes were associated with increased expression of Bax, cleaved poly-ADP-ribose polymerase (PARP), and increased expression of active caspase 3 in white matter. As an indicator of oxidative DNA damage, there was increased stainability of 8-OHdG in white matter oligodendrocytes. Quantitative analyses of myelinating oligodendroglia cells and white matter astrocytes, demonstrated a significant decrease in oligodendroglia cell densities in 2-month diabetic rats. A progressive increase was seen in the density of astrocytes from 2- to 8-month duration of diabetes (Figure 7). These changes were significantly prevented by C-peptide replacement, and interestingly, partly reversed by C-peptide treatment between 4 and 7 months of diabetes. These preliminary data suggest that factors similar to those operative in gray matter structures also affect the myelinating white matter. Furthermore, it appears that the white matter deficits, such as loss of myelinating oligodendroglia, occur earlier than the neuronal deficits described in hippocampus [34, 35]. One may speculate that this apparent difference in susceptibility to the metabolic insults caused by insulin and C-peptide deficiencies, may relate to the later occurring maturation of white matter structures, both in rats and humans [67].
Summary and conclusions
It is now established that type 1 diabetes may result in intellectual and cognitive deficits. In this regard, the timing of diabetes onset is critical, since it appears that a developing brain is more vulnerable to the deleterious effects of diabetes than a developed brain [68, 69]. Given that the type 1 diabetic population is globally increasing, and the age of diabetes onset is becoming progressively younger, patients will be more frequently affected at ages when the brain is still developing [70-72].
As outlined in this review, it is apparent that the abnormalities underlying type 1 diabetic encephalopathy are complex, and not well understood. It is clear though that hyperglycemia and its consequences, like non-enzymatic glycation and oxidative stress, are playing important pathogenetic roles, as in other complications. However, recently it is being increasingly recognized that impaired insulin action plays an equally important role, and that C-peptide replacement may be important in correcting the defects caused by insulin deficiency. The potential mechanisms by which C-peptide exerts its insulin-like effects are detailed in the accompanying review on the effect of C-peptide on diabetic polyneuropathy. Insulin replacement alone is not likely to fully normalize these defects. Therefore, type 1 diabetes appears to be a defect in the interaction between two failing hormones, insulin and C-peptide [73].
As demonstrated in this review, diabetic encephalopathy evolves with duration of insulin and C-peptide deficiencies. A conceptual construct of the progressive and interrelated abnormalities underlying the development of encephalopathy in type 1 diabetes, as we understand it at the present time, is illustrated in Figure 8. A major initiating event appears to be impaired insulin signaling, with consequences for the expression of other neurotrophic factors. Such defects are likely to impact on neurotransmitter synthesis, expression of neuroskeletal proteins, and to enhance activation of innate inflammatory factors. The latter may be further enhanced by hyperglycemia per se with upregulation of RAGE. Interestingly though, whilst full replacement of C-peptide does not correct hyperglycemia, it prevents, and in some instances corrects, these early metabolic perturbations. Additionally, it has beneficial effects on trophic factor deficits and perturbations of pre-synaptic connectivity, leading to correction of early behavioral deficits in reference and working memory.
Figure 8. Conceptual depiction of the temporal inter-relationships between pathogenetic mechanisms emanating from hyperglycemia and insulin deficiency in type 1 diabetic encephalopathy.
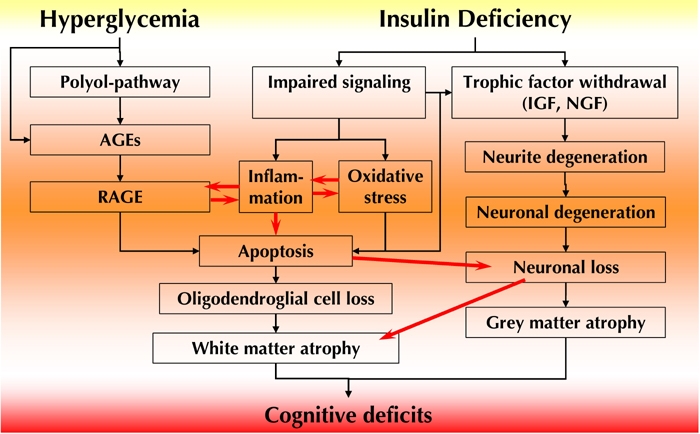
Hyperglycemia leads to activation of the polyol pathway and AGEs, with upregulation of RAGE and activation of innate inflammatory factors. Inflammation and RAGE activation are also induced by increased expression of NF-κB secondary to impaired insulin signaling activity. Suppressed insulin signaling leads to decreased expression of IGF, NFG, and their respective receptors, with consequent neurite degeneration, and loss of presynaptic connections. This results in early (radial-arm paradigm) neuro behavioral deficits. Innate inflammatory activities and suppressed insulin signaling result in oxidative and apoptotic stresses. This results in apoptosis and cell loss of both neurons and oligodendroglial cells, with consequent gray and white matter atrophy and cognitive deficits. As outlined in this review, full substitution of C-peptide has significant beneficial effects on several of these pathogenetic components, resulting in substantial prevention of behavioral and cognitive deficits.
Upregulation of RAGE and induction of inflammatory interleukins have downstream effects on oxidative and apoptotic stressors. Somewhat unexpectedly, C-peptide substitution largely corrected the inflammatory responses and subsequent oxidative and apoptotic DNA damage. The latter benefits are likely (at least in part) due to the normalization of the insulin-signaling cascade by C-peptide. Both insulin and C-peptide have documented anti-apoptotic effects. Oxidative and apoptotic activities eventually result in neuronal cell loss in gray matter structures, such as the hippocampus, and early loss of myelinating oligodendroglia in the white matter. Inevitably, these changes lead to disturbances in brain interconnectivity and disruption of regional circuitry systems, such as the hippocampus, with consequences for storage and retrieval of information, and long-term memory.
In summary, C-peptide replacement from onset of type 1 diabetes prevents the neuronal consequences of the disease. The case of early onset diabetes is particularly important, because the developing brain is more vulnerable to damage by diabetes effects than the matured brain. The data we obtained from the representative model of type 1 diabetes strongly suggest that replacement of C-peptide in type 1 diabetes will have marked beneficial effects on type 1 diabetic encephalopathy.
Disclosures: The authors report no conflict of interests.
References
- 1.Sima AA, Kamiya H, Li ZG. Insulin, C-peptide hyperglycemia and central nervous system complications in diabetes. Europ J Pharmacol. 2004;490:187–197. doi: 10.1016/j.ejphar.2004.02.056. [DOI] [PubMed] [Google Scholar]
- 2.Biessels GJ, Deary TJ, Ryan CM. Cognition and diabetes: a lifespan perspective. Lancet Neurol. 2008;7:184–190. doi: 10.1016/S1474-4422(08)70021-8. [DOI] [PubMed] [Google Scholar]
- 3.Biessels GJ. Diabetic encephalopathy. In: Veves A, Malik RA, editors. Diabetic Neuropathy - Clinical Management. Humana Press; Totowa, NJ: 2007. pp. 187–205. [Google Scholar]
- 4.Sima AA. Pathobiology of diabetic encephalopathy. In: Biessels GJ, Luchsinger JA, editors. Humana Press; Totowa, NJ: 2009. pp. 405–427. [Google Scholar]
- 5.Zhang W, Sima AA. The role glucose, cholesterol and insulin have in amyloidogenesis in human neuroblastoma cells. Toronto, Canada. Proc XIX in Meeting of Neurodiab; 2009. p. 21. Abstract. [Google Scholar]
- 6.Ott A, Stolk RP, van Harskamp F, Pos HA, Hofman A, Breteler MM. Diabetes mellitus and risk of dementia: the Rotterdam study. Neurology. 1999;58:1937–1941. doi: 10.1212/wnl.53.9.1937. [DOI] [PubMed] [Google Scholar]
- 7.Arvanitakis Z, Wilson RS, Bievas JL, Evans DA, Bennett DA. Alzheimer's disease and decline in cognitive function. Arch Neurol. 2004;61:661–666. doi: 10.1001/archneur.61.5.661. [DOI] [PubMed] [Google Scholar]
- 8.Peila R, Rodriquez BL, Lanner LJ. Type 2 diabetes, APOE gene and risk for dementia and related pathologies: The Honolulu-Asia Aging Study. Diabetes. 2002;51:1256–1262. doi: 10.2337/diabetes.51.4.1256. [DOI] [PubMed] [Google Scholar]
- 9.Yoshitake T, Kiyohara Y, Kato I, Ohmura T, Inamoto H, Nakayama K, Ohmori S, Nomiyama K, Kawano H, Ueda K. Incidence and risk factors of vascular dementia and Alzheimer's disease in a defined elderly Japanese population. The Hisayama Study. Neurology. 1995;445:1161–1168. doi: 10.1212/wnl.45.6.1161. [DOI] [PubMed] [Google Scholar]
- 10.Leibson CL, Rocca WA, Hanson VA, Cha R, Kokmere E, O'Brien PC, Palumbo PJ. The risk of dementia among persons with diabetes mellitus: a population based cohort study. Ann NY Acad Sci. 1997;26:422–427. doi: 10.1111/j.1749-6632.1997.tb48496.x. [DOI] [PubMed] [Google Scholar]
- 11.Sima AA, Li ZG. Diabetes and Alzheimer's disease - is there a connection? Rev Diabet Stud. 2007;4:161–168. doi: 10.1900/RDS.2006.3.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li ZG, Zhang W, Sima AA. Alzheimer-like changes in rat models of spontaneous diabetes. Diabetes. 2007;56:1817–1824. doi: 10.2337/db07-0171. [DOI] [PubMed] [Google Scholar]
- 13.Desrocher M, Rovet J. Neurocognitive correlates of type 1 diabetes mellitus in childhood. Child Neuropsychol. 2004;10:36–52. doi: 10.1076/chin.10.1.36.26241. [DOI] [PubMed] [Google Scholar]
- 14.Ryan C, Vega A, Drash A. Cognitive deficits in adolescents who developed diabetes early in life. Pediatrics. 1985;75:921–927. [PubMed] [Google Scholar]
- 15.Schoenle EJ, Schoenle D, Molinari L, Largo RH. Impaired intellectual development in children with type 1 diabetes: association with HbA(1c), age at diagnosis and sex. Diabetologia. 2002;45:108–114. doi: 10.1007/s125-002-8250-6. [DOI] [PubMed] [Google Scholar]
- 16.Northam EA, Anderson PJ, Jacobs R, Hughes M, Warne GL, Werther GA. Neuropsychological profiles of children with type 1 diabetes 6 years after disease onset. Diabetes Care. 2001;24:1541–1546. doi: 10.2337/diacare.24.9.1541. [DOI] [PubMed] [Google Scholar]
- 17.Dahlquist G, Kallen B. School performance in children with type 1 diabetes - a population-based register study. Diabetologia. 2007;50:957–964. doi: 10.1007/s00125-007-0615-2. [DOI] [PubMed] [Google Scholar]
- 18.Northam EA, Rankins D, Lin A, Wellard RM, Pell GS, Finch SJ, Werther GA, Cameron FJ. Central nervous system function in youth with type 1 diabetes 12 years after disease onset. Diabetes Care. 2009;32:445–450. doi: 10.2337/dc08-1657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ryan CM. Why is cognitive dysfunction associated with the development of diabetes early in life? The diathesis hypothesis. Pediatr Diabetes. 2006;7:289–297. doi: 10.1111/j.1399-5448.2006.00206.x. [DOI] [PubMed] [Google Scholar]
- 20.Fox MA, Chen RS, Holmes CS. Gender differences in memory and learning in children with insulin-dependent diabetes mellitus (IDDM) over a 4-year follow-up interval. J Pediatr Psychol. 2003;28:569–578. doi: 10.1093/jpepsy/jsg047. [DOI] [PubMed] [Google Scholar]
- 21.Kramer L, Fasching P, Madl C, Schneider B, Damjancic P, Waldhäusl W, Irsigler K, Grimm G. Previous episodes of hypoglycemic coma are not associated with permanent cognitive brain dysfunction in IDDM patients on intensive insulin treatment. Diabetes. 1998;47:1909–1914. doi: 10.2337/diabetes.47.12.1909. [DOI] [PubMed] [Google Scholar]
- 22.The Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications (DCCT/EDIC) Study Research Group. Long-term effect of diabetes and its treatment on cognitive function. N Engl J Med. 2007;356:1842–1852. doi: 10.1056/NEJMoa066397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Autin EJ, Deary IJ. Effects of repeated hypoglycemia on cognitive function: a psychometrically validated reanalysis of the diabetes control and complications trial data. Diabetes Care. 1999;22:1273–1277. doi: 10.2337/diacare.22.8.1273. [DOI] [PubMed] [Google Scholar]
- 24.Heikkila O, Lundbom N, Timonen M, Groop PH, Heikkinen S, Makimattila S. Hyperglycemia is associated with changes in the regional concentrations of glucose and myo-inositol within the brain. Diabetologia. 2009;52:534–540. doi: 10.1007/s00125-008-1242-2. [DOI] [PubMed] [Google Scholar]
- 25.Ho MS, Weller NJ, Ives FJ, Carne CL, Murray K, Vanden Driesen RI, Nguyen TP, Robins PD, Buisara M, Davis EA, Jones TW. Prevalence of structural central nervous system abnormalities in early-onset type 1 diabetes mellitus. J Pediatr. 2008;153(3):385–390. doi: 10.1016/j.jpeds.2008.03.005. [DOI] [PubMed] [Google Scholar]
- 26.Musen G, Lyoo IK, Sparks CR, Weinger K, Hwang J, Ryan CM, Jimerson DC, Henneu J, Renshaw PF, Jacobson AM. Effects of type 1 diabetes on gray matter density as measured by voxel-based morphometry. Diabetes. 2006;55:326–333. doi: 10.2337/diabetes.55.02.06.db05-0520. [DOI] [PubMed] [Google Scholar]
- 27.Hoffman WH, Artlett CM, Zhang W, Kreipke CW, Passmore GG, Rafols J, Sima AA. Receptor for advanced glycation end products and neuronal deficit in the fatal brain edema of diabetic ketoacidosis. Brain Research. 2008;1238:154–162. doi: 10.1016/j.brainres.2008.08.041. [DOI] [PubMed] [Google Scholar]
- 28.van Duinkerkeu E, Klein M, Schoonenboom NS, Hoogma RP, Moll AC, Snock FJ, Stain CJ, Diamant M. Functional brain connectivity and neurocognitive functioning in patients with longstanding type 1 diabetes with and without microvascular complications. Diabetes. 2009;58:2335–2343. doi: 10.2337/db09-0425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brismar T, Hyllenmark L, Ekberg K, Johansson BL. Loss of temporal lobe beta power in young adults with type 1 diabetes mellitus. Neuro Report. 2002;13:2469–2473. doi: 10.1097/00001756-200212200-00019. [DOI] [PubMed] [Google Scholar]
- 30.Jakobsen J, Sidenius P, Gundersen HJ, Osterby R. Quantitative changes of cerebral neocortical structure in insulin treated long-term streptozotocin-induced diabetes in rats. Diabetes. 1987;36:597–601. doi: 10.2337/diab.36.5.597. [DOI] [PubMed] [Google Scholar]
- 31.Biessels GJ, Kamal A, Ramakers GM, Urban IJ, Spruijt BM, Erkeleus DL, Gispen WH. Place learning and hippocampal synaptic plasticity in streptozotocin-induced diabetic rats. Diabetes. 1996;45:1259–1266. doi: 10.2337/diab.45.9.1259. [DOI] [PubMed] [Google Scholar]
- 32.Biessels GJ, Kamal A, Urban IJ, Spruijt BM, Erkeleus DW, Gispen WH. Water maze learning and hippocampal synaptic plasticity in streptozotocin-diabetic rats: effect of insulin treatment. Brain Res. 1998;800:125–135. doi: 10.1016/s0006-8993(98)00510-1. [DOI] [PubMed] [Google Scholar]
- 33.Kamal A, Biessels GJ, Gispen WH, Ramakers GM. Synaptic transmission changes in the pyramidal cells of the hippocampus in streptozotocin-induced diabetes mellitus in rats. Brain Res. 2006;1073:276–280. doi: 10.1016/j.brainres.2005.12.070. [DOI] [PubMed] [Google Scholar]
- 34.Toth C, Schmidt AM, Tuor UI, Francis G, Foniok T, Brusse V, Kaur J, Yan SF, Martinez JA, Barber PA, Buchan A, Zochodne DW. Diabetes, leucoencephalopathy and RAGE. Neurobiol Dis. 2006;23:445–461. doi: 10.1016/j.nbd.2006.03.015. [DOI] [PubMed] [Google Scholar]
- 35.Francis GJ, Martinez JA, Liu WQ, Xu K, Ayer A, Fine J, Tuor UI, Glazner G, Hanson LR, Frey WH 2nd, Toth C. Intranasal insulin prevents cognitive decline, cerebral atrophy and white matter changes in murine type 1 diabetic encephalopathy. Brain. 2008;131(Pt 12):3311–3334. doi: 10.1093/brain/awn288. [DOI] [PubMed] [Google Scholar]
- 36.Sima AA, Li ZG. The effect of C-peptide on cognitive dysfunction and hippocampal apoptosis in type 1 diabetes. Diabetes. 2005;54:1497–1505. doi: 10.2337/diabetes.54.5.1497. [DOI] [PubMed] [Google Scholar]
- 37.Mordes JP, Bortell R, Groen H, Guburski D, Rossini AA, Greiner DL. Autoimmune diabetes mellitus in the BB-rat. In: Sima AA, Shafrir E, editors. Animal Models of Diabetes. Harwood Academic Publ.; Amsterdam: 2001. pp. 1–42. [Google Scholar]
- 38.Sima AA (ed) Harwood Academic Publ.; Amsterdam: 2000. Chronic complications in diabetes. Animal Models of Diabetes. [Google Scholar]
- 39.Kamija H, Zhang W, Sima AA. The beneficial effects of C-peptide on diabetic polyneuropathy. Rev Diabet Stud. 2009 doi: 10.1900/RDS.2009.6.187. This issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sima AA, Yagihashi S. Central-peripheral distal axonopathy in the spontaneously diabetic BB- rat: Ultrastructural and morphometric findings. Diabetes Res Clin Prac. 1986;1(5):289–298. doi: 10.1016/s0168-8227(86)80037-7. [DOI] [PubMed] [Google Scholar]
- 41.Kamijo M, Cherian PV, Sima AA. The preventive effect of aldose reductase inhibition on diabetic optic neuropathy in the BB/W-rat. Diabetologia. 1993;36:893–898. doi: 10.1007/BF02374469. [DOI] [PubMed] [Google Scholar]
- 42.Crusio WE, Schwegler H. Learning spatial orientation tasks in the radial-maze and structural variation in the hippocampus in inbred mice. Behav Brain Funct. 2005;1(1):3. doi: 10.1186/1744-9081-1-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wiener SI, Paul CA, Eichenbaum H. Spatial and behavioral correlates to hippocampal neuronal activity. J Neurosci. 1989;9:2737–2763. doi: 10.1523/JNEUROSCI.09-08-02737.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kreipke CW, Morgan R, Kallakuri S, Rafols JA. Behavioral pre-conditioning enhances angiogenesis and cognitive outcome after brain trauma. Neurol Res. 2007;29:388–394. doi: 10.1179/016164107X204710. [DOI] [PubMed] [Google Scholar]
- 45.Balakrishnan S, Mathew J, Paulose CS. Cholinergic and glutamergic receptor functional regulation in long-term, low dose somatotropine and insulin treatment to aging rats: rejuvenation of brain function. Mol Cell Endocrinol. 2010;314:23–30. doi: 10.1016/j.mce.2009.07.028. [DOI] [PubMed] [Google Scholar]
- 46.Corner JM, Franks KM, Titterness AK, Russel K, Merrill DA, Christie BR, Sejnowski TJ, Tuszynski MH. NGF is essential for hippocampal plasticity and learning. J Neurosci. 2009;35:10883–10889. doi: 10.1523/JNEUROSCI.2594-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Moon E, Her Y, Lee JB, Park JH, Lee EH, Kim SH, Oh MS, Jang CG, Kim SY. The multi-herbal medicine Gongjin-dan enhances memory and learning tasks via NGF regulation. Neurosci Lett. 2009;466(3):114–119. doi: 10.1016/j.neulet.2009.09.033. [DOI] [PubMed] [Google Scholar]
- 48.Seto D, Zheng WH, McNicoll A, Collier B, Quirion R, Kar S. Insulin-like growth factor-I inhibits endogenous acetylcholine release from rat hippocampal formation: possible involvement of GABA in mediating the effects. Neuroscience. 2002;115(2):603–612. doi: 10.1016/s0306-4522(02)00450-5. [DOI] [PubMed] [Google Scholar]
- 49.Kar S, Seto D, Dore S, Hanisch U, Quirion R. Insulin-like growth factors-1 and -2 differentially regulate endogenenous acetylcholine release from the rat hippocampal formation. Proc Natl Acad Sci USA. 1997;94:14054–14059. doi: 10.1073/pnas.94.25.14054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Madara JC, Levine ES. Presynaptic and postsynaptic NMDA receptors mediate distinct effects of brain-derived neurotrophic factor on synaptic transmission. J Neurophysiol. 2008;100:3175–3184. doi: 10.1152/jn.90880.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Grunberger G, Qiang X, Li ZG, Mathews ST, Sbriessa D, Shisheva A, Sima AA. Molecular basis for the insulinomimetic effects of C-peptide. Diabetologia. 2001;44:1247–1257. doi: 10.1007/s001250100632. [DOI] [PubMed] [Google Scholar]
- 52.Sima AA, Zhang W, Kreipke CW, Rafols JA, Hoffman WH. Inflammation in diabetic encephalopathy is prevented by C-peptide. Rev Diabet Stud. 2009;6:37–42. doi: 10.1900/RDS.2009.6.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ramasamy R, Vanucci SJ, Yau SD, Herold K, Yau SF, Schmidt AM. Advanced glycation end products and RAGE: a common thread in aging, diabetes, neurodegeneration and inflammation. Glycobiology. 2004;15:16R–28R. doi: 10.1093/glycob/cwi053. [DOI] [PubMed] [Google Scholar]
- 54.Li ZG, Zhang W, Sima AA. C-peptide enhances insulin-mediated cell growth and protection against high glucose induced apoptosis in SH-SY5Y cells. Diabetes Metab Res Rev. 2003;19:375–385. doi: 10.1002/dmrr.389. [DOI] [PubMed] [Google Scholar]
- 55.Hayden MS, Shosh S. Signaling to NF-kappaB. Genes Dev. 2004;18:2195–2224. doi: 10.1101/gad.1228704. [DOI] [PubMed] [Google Scholar]
- 56.Cifarelli V, Luppi P, Tse HM, He J, Piganelli J, Trucco M. Human proinsulin C-peptide reduces high-glucose induced proliferation and NF-kappaB activation in vascular smooth muscle cells. Atherosclerosis. 2008;301:248–257. doi: 10.1016/j.atherosclerosis.2007.12.060. [DOI] [PubMed] [Google Scholar]
- 57.Grunberger G, Sima AA. The C-peptide signaling. Exp Diab Res. 2004;5:25–36. doi: 10.1080/15438600490424497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sima AA, Zhang W, Kreipke CW. Immune responses in diabetic encephalopathy and the effects of C-peptide. Vienna, Austria. C-peptide Satellite Symposium, EASD; 2009. Abstract. [Google Scholar]
- 59.Morris R. Development of a water maze procedure for studying spatial learning in the rat. J Neurosci. 1984;11:47–60. doi: 10.1016/0165-0270(84)90007-4. [DOI] [PubMed] [Google Scholar]
- 60.Kamal A, Biessels GJ, Duis SE, Grispen WH. Learning and hippocampal plasticity in streptozotocin-diabetic rats: interaction of diabetes and aging. Diabetologia. 2000;43:500–506. doi: 10.1007/s001250051335. [DOI] [PubMed] [Google Scholar]
- 61.Medvedev NI, Rodriquez-Arellano JJ, Popov VI, Davies HA, Tigaret CM, Schoepfer R, Stewart MG. The glutamate receptor 2 subunits controls post-synaptic density complexity and spine shape in the dentate gyrus. Eur J Neurosci. 2008;27:315–325. doi: 10.1111/j.1460-9568.2007.06005.x. [DOI] [PubMed] [Google Scholar]
- 62.Tanaka H, Grooms SY, Bennett MV, Zukin RS. The AMPAR subunit GLUR2: still front and center stage. Brain Res. 2000;886:190–207. doi: 10.1016/s0006-8993(00)02951-6. [DOI] [PubMed] [Google Scholar]
- 63.Isaac JT, Asky M, McBain CJ. The role of the GluR2 subunit in AMPA receptor function and synaptic plasticity. Neuron. 2007;54:859–871. doi: 10.1016/j.neuron.2007.06.001. [DOI] [PubMed] [Google Scholar]
- 64.McCormack SG, Stornetta RL, Zhu JJ. Synaptic AMPA receptor exchange maintains bidirectional plasticity. Neuron. 2006;50:76–88. doi: 10.1016/j.neuron.2006.02.027. [DOI] [PubMed] [Google Scholar]
- 65.Li ZG, Zhang W, Sima AA. The role of impaired insulin/IGF action in primary diabetic encephalopathy. Brain Res. 2005;1037:12–24. doi: 10.1016/j.brainres.2004.11.063. [DOI] [PubMed] [Google Scholar]
- 66.Li ZG, Sima AA. C-peptide and central nervous system complications in diabetes. Exp Diab Res. 2004;5:79–90. doi: 10.1080/15438600490424550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Donaldson HH. A comparison of the albino rat with respect to the growth of the brain and the spinal cord. J Comp Neurol. 1903;33:345–393. [Google Scholar]
- 68.Dobbing J, Sands J. Vulnerability of developing brain: IX The effect of nutritional growth retardation on the timing of brain growth spurt. Biol Neonate. 1971;19:363–378. doi: 10.1159/000240430. [DOI] [PubMed] [Google Scholar]
- 69.Winick M, Noble A. Quantitative changes in DNA, RNA and protein during prenatal and post natal growth in the rat. Develop Biol. 1965;12:451–466. doi: 10.1016/0012-1606(65)90009-6. [DOI] [PubMed] [Google Scholar]
- 70.Ehehalt S, Blumenstock G, Willasch AM, Hub R, Ranke MB, Neu A DIARY-Study Group Baden-Württemberg. Continuous rise in incidence of childhood type 1 diabetes in Germany. Diabet Med. 2008;25:755–757. doi: 10.1111/j.1464-5491.2008.02450.x. [DOI] [PubMed] [Google Scholar]
- 71.Harjutsalo V, Sjöberg L, Tuomiletho J. Time trends in the incidence of type 1 diabetes in Finnish children: a cohort study. Lancet. 2008;371:1777–1782. doi: 10.1016/S0140-6736(08)60765-5. [DOI] [PubMed] [Google Scholar]
- 72.Kumar P, Krishna P, Reddy SC, Gurappa M, Aravind SR, Munichoodappa C. Incidence of type 1 diabetes mellitus and associated complications among children and young adults: results from Karnataka Diabetes Registry 1995-2008. J Int Med Assoc. 2008;106:708–711. [PubMed] [Google Scholar]
- 73.Sima AA, Kamiya H. Is C-peptide replacement the missing link for successful treatment of neurological complications in type 1 diabetes? Current Drug Targets. 2008;9:37–46. doi: 10.2174/138945008783431745. [DOI] [PubMed] [Google Scholar]