

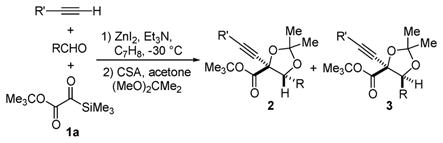
Multicomponent coupling reactions can effect sequential bond constructions and rapidly increase molecular complexity in a single synthetic operation. In this context, acylsilanes1 have found broad utility as reagents that act as a center of reactivity for both nucleophiles and electrophiles by virtue of a [1,2]-Brook rearrangement.2 Metallocyanide3,4 and metallophosphite5,6 nucleophiles reveal latent (silyloxy)carbanions of acylsilanes following a nucleophilic addition/[1,2]-Brook rearrangement sequence (Scheme 1). Because silyl migration is facilitated by the stabilization of the nascent anion by the nitrile or phosphonate groups, one might conjecture that extant electron-withdrawing functionality in the acylsilane moiety may also promote silyl migration. For this reason silylglyoxylates7 (1), a class of acylsilanes that possess an embedded anion stabilizing group, captured our attention. Herein we report three-component coupling reactions of silylglyoxylates (1), terminal alkynes, and aldehydes to furnish glycolate aldols that bear two contiguous stereocenters (eq 1, Table 1).
Scheme 1.

Table 1.
Reaction Scope with 1aa
| ||||||
|---|---|---|---|---|---|---|
| entry | R′ | R | yield (%)b | 2:3c | ||
| 1 | Ph | Ph | 77 | 78:22 | ||
| 2 | n-C6H13 | Ph | 77 | 74:26 | ||
| 3 | BnOCH2 | Ph | 60 | 72:28 | ||
| 4 | PhthNCH2 | Ph | 48 | 71:29 | ||
| 5 | Me3Si | Ph | 81 | 76:24 | ||
| 6 | Me3Si | 2-(Me)C6H4 | 65 | 80:20 | ||
| 7 | Me3Si | 4-(MeO)C6H4 | 73 | 80:20 | ||
| 8 | Me3Si | 4-(Cl)C6H4 | 76 | 78:22 | ||
| 9 | Me3Si | 2-furyl | 66 | 47:53 | ||
| 10 | Me3Si | (E)-PhCHd=CH | 77 | 53:47 | ||
Reaction carried out using 1.0 equiv of 1a, 3.0 equiv of alkyne, 1.5 equiv of PhCHO, 3.0 equiv of ZnI2, and 3.3 equiv of Et3N for 24–48 h.
Average of two isolated yields. All reported yields are for two steps.
Determined by 1H NMR analysis of the crude reaction. See Supporting Information for determination of relative stereochemistry.
Kuwajima8 and Reich9 have shown that alkynylation of aliphatic or vinylic acylsilanes initiates [1,2]-Brook rearrangement to furnish propargyl anions that can engage a variety of electrophiles (H+, DMF, 1° iodides, and disulfides) to give substituted silyloxyallenes. A constraint in this chemistry that is common to multicomponent couplings is reagent incompatibility: the high reactivity of the alkali and alkaline earth acetylides employed by Kuwajima and Reich necessitate a stepwise introduction of the acylsilane and terminating electrophiles. We were interested in testing the feasibility of a single-pot process in which the metal acetylide was generated in the presence of both electrophilic components. This would be viable only if the metal acetylide was a highly discriminating nucleophile; in situ metal acetylide formation with a Lewis acid and amine base as described by Carreira and Yamaguchi was the projected method of choice.10,11
An evaluation of zinc salts confirmed the underlying premise and revealed ZnI2 as the optimal Lewis acid to promote the desired three-component coupling.12 The silylglyoxylate structure was initially optimized for conversion and diastereoselectivity by variation of the ester functionality.13 A screen of trimethylsilylglyoxylates indicated that tert-butyl trimethylsilylglyoxylate (1a) furnished the desired adducts in the highest levels of conversion and diastereocontrol.
Employing tert-butyl trimethylsilylglyoxylate (1a), we explored the scope of the reaction. The initial silyl ether adducts were found to be susceptible to O–O silyl migration during the course of the reaction and therefore were isolated as their derived acetonides. A variety of alkynes (alkyl, aryl, trimethylsilyl, and heteroatom-substituted) were tolerated, affording the intended adducts (2/3) in moderate to good yield (48–81%) and approximately 75:25 diastereoselectivity when benzaldehyde was the coupling partner (entries 1–5, Table 1). Substituted aromatic aldehydes also exhibited good reactivity and diastereoselectivity (75:25 to 80:20; entries 6–8, Table 1); disappointingly, aliphatic aldehydes were not suitable substrates. Cinnamaldehyde and 2-furaldehyde also provided the desired adducts in good yield, albeit with no diastereoselectivity (53:47; entries 9 and 10, Table 1).
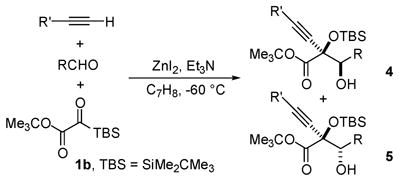
A second round of modifications to the silylglyoxylate structure was undertaken to further improve the diastereocontrol.13 Systematic variation of the silyl group revealed that by using tert-butyl tert-butyldimethylsilylglyoxylate (1b), a marked increase in diastereoselectivity could be achieved (cf. Table 2, entries 1–3 with Table 1, entries 1, 2, and 6). The use of 1b allows for the isolation of silyl-protected adducts (4/5) as differentially protected glycols. Furthermore, deprotection of (4/5) with TBAF (1.1 equiv) followed by a single recrystallization from hexanes afforded the diol in diastereomerically pure form (entry 2).
Table 2.
Reaction Scope with 1ba
| ||||||
|---|---|---|---|---|---|---|
| entry | R′ | R | yield (%) | 4:5b | ||
| 1 | Ph | Ph | 73 | 83:17 | ||
| 2 | n-C6H13 | Ph | 73 (53) | 90:10 (>99:1) | ||
| 3 | Me3Si | 2-(Me)C6H4 | 70c | 91:9 | ||
| 4 | n-C6H13 | 4-(MeO)C6H4 | 72c | 90:10 | ||
| 5 | n-C6H13 | 2-(Me)C6H4 | 68c | 92:8 | ||
1.0 equiv of 1b, 4.0 equiv of alkyne, 1.5 equiv of PhCHO, 4.0 equiv of ZnI2, and 4.4 equiv of Et3N for 48 h.
Determined by 1H NMR analysis of the crude reaction.
5.0 equiv of alkyne, 5.0 equiv of ZnI2, and 5.5 equiv of NEt3 used. Parenthetical yield and dr refers to the diol, revealed after TBAF deprotection and recrystallization (two-step yield).
Although the zinc acetylide appears to react in a chemoselective manner with the silylglyoxylate, we also considered the possibility of initial reversible alkynylation of the aldehyde. To test this hypothesis, a suspension of phenylacetylene, benzaldehyde, Et3N, and ZnI2 in toluene was allowed to stir for 48 h at −30 °C, giving rise to an undefined intermediate A (presumably a zinc alkoxide, Scheme 2). Aqueous ammonium chloride was added, and propargyl alcohol 6 was isolated in 99% yield. If intermediate A was instead treated with 1a, after 24 h propargyl alcohol 6 was obtained 77% yield, whereas 2a and 3a were formed in ≤8% yield (employing the aforementioned deprotection/ketalization protocol).14 Because the propargyl alcohol is formed in high yield and only trace amounts of 2a/3a were observed, we are inclined to believe that zinc acetylide addition to the silylglyoxylate occurs as the result of a kinetic preference.
Scheme 2.

Mechanistic Study
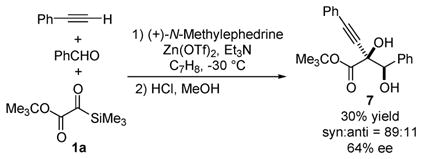
Initial experiments suggest that asymmetric induction may be realized in this tandem process. Using Carreira’s conditions (Zn-(OTf)2/(+)-N-methylephedrine),11 diol 7 was isolated after HCl/MeOH workup in 30% yield, 89:11 dr, and 64% ee (eq 3).
 |
(3) |
Not only was there a marked increase in diastereoselectivity relative to the ZnI2 system (75:25 dr; cf. entry 1, Table 1), but it has been demonstrated, using unoptimized conditions, that an enantioselective version of the title reaction is possible.15
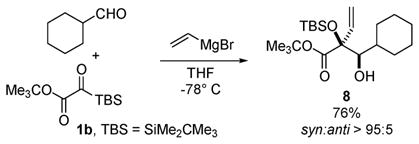
We have extended this three-component coupling concept to include aliphatic aldehydes (eq 4). Treatment of a solution of cyclohexanecarboxaldehyde and 1b in THF at −78 °C with vinyl Grignard results in the formation of glycol 8 in 76% yield and complete diastereoselectivity.
 |
(4) |
In summary, a tandem alkynylation/Brook rearrangement/aldol reaction of silylglyoxylates has been reported. This mode of reactivity is divergent from that of the Kuwajima and Reich systems, affording glycolate aldols rather than silyloxyallene aldols. The reaction exhibits exquisite chemoselectivity at every potential branch point in the reaction sequence. The identity of the silylglyoxylate (1) is crucial for diastereocontrol; however, diastereomerically pure diols of 4 can be isolated following a single recrystallization. Additionally, it has been shown that enantioselective variants are possible using simple enantiopure amino alcohols to mediate chirality transfer. It is noteworthy that the identical functional group arrays present in 416 and 817 have been utilized in natural product synthesis, illustrating the potential value of these processes.
Supplementary Material
Acknowledgments
We are grateful to the National Institutes of Health (National Institute of General Medical Sciences, GM068443) for support of this research. D.A.N. acknowledges a fellowship from Eli Lilly and an ACS Division of Organic Chemistry Fellowship sponsored by Novartis. J.S.J. is an Eli Lilly Grantee and a 3M Nontenured Faculty Awardee. Xin Linghu is thanked for experimental assistance.
Footnotes
Supporting Information Available: Experimental details and analytical data for all new compounds, CSP-SFC traces of racemic 7 and enantioenriched 7, and all stereochemical proofs. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Moser WH. Tetrahedron. 2001;57:2065. [Google Scholar]
- 2.Brook AG. Acc Chem Res. 1974;7:77. [Google Scholar]
- 3.(a) Reich HJ, Holtan RC, Bolm C. J Am Chem Soc. 1990;112:5609. [Google Scholar]; (b) Takeda K, Ohnishi Y. Tetrahedron Lett. 2000;41:4169. [Google Scholar]; (c) Degl’Innocenti A, Ricci A, Mordini A, Reginato G, Colotta V. Gazz Chim Ital. 1987;117:645. [Google Scholar]
- 4.(a) Nicewicz DA, Yates CM, Johnson JS. Angew Chem, Int Ed. 2004;43:2652. doi: 10.1002/anie.200353354. [DOI] [PubMed] [Google Scholar]; (b) Linghu X, Nicewicz DA, Johnson JS. Org Lett. 2002;4:2957. doi: 10.1021/ol0263649. [DOI] [PubMed] [Google Scholar]; (c) Linghu X, Johnson JS. Angew Chem, Int Ed. 2003;42:2534. doi: 10.1002/anie.200250554. [DOI] [PubMed] [Google Scholar]
- 5.Takeda K, Tanaka T. Synlett. 1999:705. [Google Scholar]
- 6.Linghu X, Potnick JR, Johnson JS. J Am Chem Soc. 2004;126:3070. doi: 10.1021/ja0496468. [DOI] [PubMed] [Google Scholar]
- 7.Bolm C, Kasyan A, Heider P, Saladin S, Drauz K, Guenther K, Wagner C. Org Lett. 2002;4:2265. doi: 10.1021/ol025911n. [DOI] [PubMed] [Google Scholar]
- 8.Kuwajima I, Kato M. Tetrahedron Lett. 1980;21:623. [Google Scholar]
- 9.Reich HJ, Eisenhart EK, Olson RE, Kelly MJ. J Am Chem Soc. 1986;108:7791. doi: 10.1021/ja00284a051. [DOI] [PubMed] [Google Scholar]
- 10.Yamaguchi M, Hayashi A, Minami T. J Org Chem. 1991;56:4091. [Google Scholar]
- 11.(a) Frantz DE, Fässler R, Carreira EM. J Am Chem Soc. 2000;122:1806. [Google Scholar]; (b) Anand NK, Carreira EM. J Am Chem Soc. 2001;123:9687. doi: 10.1021/ja016378u. [DOI] [PubMed] [Google Scholar]
- 12.ZnBr2 and ZnCl2 also effected the transformation; however, higher temperatures and reaction times were required.
- 13.See Supporting Information for details.
-
14.The yield of propargyl alcohol 6 was diminished by small amounts (<10%) of Meerwein–Ponndorf–Verley reduction of 1a, presumably by intermediate A:
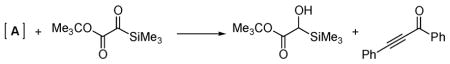 However, neither side product was observed when the title reaction reaction was run using the conditions described in eq 1, further supporting our mechanistic interpretations.
However, neither side product was observed when the title reaction reaction was run using the conditions described in eq 1, further supporting our mechanistic interpretations.
- 15.Only two examples of enantioenriched glycolate aldolates of this type have been prepared, to our knowledge. One was by a chiral auxiliary method: Murata Y, Kamino T, Hosokawa S, Kobayashi S. Tetrahedron Lett. 2002;43:8121.The other was prepared by a direct catalytic method involving chiral zinc complexes: Kumagai N, Matsunaga S, Kinoshita T, Harada S, Okada S, Sakamoto S, Yamaguchi K, Shibasaki M. J Am Chem Soc. 2003;125:2169. doi: 10.1021/ja028926p.
- 16.Carreira EM, Du Bois J. J Am Chem Soc. 1995;117:8106. [Google Scholar]
- 17.Trost BM, Probst GD, Schoop A. J Am Chem Soc. 1998;120:9228. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.