

Abstract
Blockade of the CD40/CD154 signaling pathway using anti-CD154 antibodies has shown promise in attenuating the alloimmune response and promoting long-term graft survival in murine model systems. Unfortunately, thromboembolic side effects observed in humans have hampered its progression through clinical trials. Appropriately designed anti-CD40 antibodies may provide a suitable alternative. We investigated two isoforms of a novel monoclonal rat anti-mouse CD40 antibody (7E1) for characteristics and effects mirroring those of anti-CD154: 7E1-G1 (an IgG1 isotype) and 7E1-G2b (an IgG2b isotype). In vitro proliferation assays to measure the agonist properties of the two anti-CD40 antibodies revealed similar responses when plate-bound. However, when present as a soluble stimulus, 7E1-G1 but not 7E1-G2b led to proliferation. Importantly, 7E1-G2b was as effective as anti-CD154 when administered in vivo in concert with CTLA4-Ig in promoting both allogeneic bone marrow chimerism and skin graft survival, while 7E1-G1 was not. The protection observed with 7E1-G2b was not due to depletion of CD40 bearing antigen presenting cells. These data suggest that an appropriately designed anti-CD40 antibody can promote graft survival as well as anti-CD154, making 7E1-G2b an attractive substitute in mouse models of costimulation blockade-based tolerance regimens.
Keywords: CD40, CD154, costimulation, transplantation tolerance
Introduction
The generation of an optimal T cell response requires signals delivered via T cell receptor engagement (signal 1) as well as accessory signals in the form of costimulatory molecules (signal 2) (1). When signal 1 is delivered in the absence of signal 2, T cell activation can be severely altered, resulting in anergy, apoptosis, abortive proliferation, or immunoregulation (2-4). The potential of inducing these states using agents that interrupt these signaling events has captivated the interest of the transplant community. Several signaling pathways have been targeted for blockade with some measure of success including ICOS, OX40, 4-1BB and CD70 (5-8). In particular, inhibition of the earliest identified and most well studied costimulatory pathways, CD28 and CD40, have shown impressive synergy in mouse models for the prevention of graft rejection (9). Blockade of the CD28 pathway with CTLA4-Ig (abatacept) and LEA29Y (belatacept), a second generation CD28 blocker with an increased affinity for CD80/86, proved efficacious in non-human primate models (10-12), paving the way for the use of belatacept in human clinical trials of kidney transplantation (13).
Blockade of the CD40 signaling pathway also has shown great promise in non-human primates (14, 15). However, clinical trials using anti-CD154 monoclonal antibodies (mAb) were halted following evidence of unanticipated thromboembolic side effects (16-18). It now is appreciated that CD154, expressed by platelets as well as present in soluble form, is intimately involved in the formation and stabilization of thrombi (19, 20). Clinical development of traditional anti-CD154 mAbs has been largely suspended. To continue exploring the inhibition of the CD40 pathway as a therapeutic modality, alternative means must be sought to interrupt this pivotal costimulation pathway without the potential for precipitating dysregulated thrombostasis.
The CD40 cascade provides essential signals to many aspects of immunity. Studies in vitro and utilizing CD40 deficient mice have described its role in, among others, T cell-dependent immunoglobulin class switching and germinal center formation, CD8 T cell priming, dendritic cell longevity and cytokine production, and endothelial activation (21-25). In fully allogeneic models of cardiac and islet, but not skin, transplantation, genetic deficiency of CD40 in both the donor and the recipient results in long-term graft survival (26-28). Investigations outside of the field of transplantation using anti-CD40 mAb have generally focused on their agonistic properties in order to augment immune responses to thymus-independent antigens or to boost anti-tumor and anti-virus activity (22, 29-32). There are, however, reports of successful immune attenuation achieved in mouse and non-human primate kidney allograft models employing anti-CD40 mAb (33-36). These anti-CD40 antibodies have shown synergy with CD28 pathway blockers (both a CTLA-4 Ig fusion protein and anti-CD86 mAbs) in promoting renal and islet allograft survival in non-human primates while also preserving established antiviral immunity (33, 35). The mechanisms behind anti-CD40 mediated immune suppression and how it compares to anti-CD154 mediated suppression remain unclear.
Herein we report two novel murine anti-CD40 binding antibodies: 7E1-G1, a rat IgG1 isotype, and its natural isotype switch variant 7E1-G2b, a rat IgG2b isotype. Following standard hybridoma selection and purification techniques, the clone 7E1-G1 was selected as the lead candidate based on its specificity for murine CD40, ability to inhibit ligand binding, and paucity of agonism (37). It was hypothesized that an antibody which strongly induced both complement fixation and fragment crystallizable (Fc) receptor interactions would be the most favorable for promoting graft survival. Such an antibody would potentially deplete CD40 expressing antigen-presenting cells before the priming of an effective response could occur. However, rat IgG1 antibody isotypes have been shown in various models to poorly fix complement and interact weakly with Fc receptors (38-40). Thus, an antibody having the same specificity as 7E1-G1 but possessing an IgG2b isotype was sought. To this end, the isotype switch variant 7E1-G2b was generated by the sib-selection technique (37, 41).
In this study, we found that when used in concert with CTLA4-Ig, the anti-CD40 antibody 7E1-G2b, but not 7E1-G1, was as effective as anti-CD154 in promoting allogeneic bone marrow chimerism and skin graft survival. The mechanism underlying this prolongation did not involve depletion of CD40-expressing cell subsets, as numbers of antigen presenting cells (APCs) remained unchanged among the treatment groups. Furthermore, 7E1-G1 but not 7E1-G2b functioned as a potent CD40 agonist in vivo, suggesting the involvement of Fc receptors in mediating these effects. However, in our system, proinflammatory signals delivered by activating Fc receptors were not responsible for the immunostimulatory properties of 7E1-G1, nor were inhibitory signals through FcγRIIb found to be involved in the immunosuppressive effects observed with 7E1-G2b.
Methods
Ab generation
A recombinant mouse CD40 immunoglobulin fusion protein consisting of the extracellular region of mouse CD40 fused to the hinge CH2 and CH3 domains of a mouse IgG2a antibody was used to immunize Lewis rats. Three days following the last immunization, leukocytes were fused with X63-Ag8.653 mouse myeloma cells to create rat/mouse heterohybridomas. Supernatants were tested for reactivity with mouse CD40 and for the ability to inhibit the binding of soluble mouse CD40 to its ligand CD154. Stimulatory properties were measured by proliferation of splenic B cells in the presence of anti-IgM. Suppression of antibody production was measured by immunization of mice with sheep red blood cells. The clone 7E1-G1 was selected as the lead anti-CD40 mAb candidate for murine studies (37).
A natural isotype switch variant of 7E1-G1, from an IgG1 to an IgG2b, was generated by the sib-selection technique (41). Briefly, an anti-CD40 mAb of the IgG2b isotype was identified by ELISA among supernatants of 96 well plates that had been seeded at 1000 cells/well with the original 7E1-G1 hybridoma. Subsequent rounds of plating and identification of IgG2b positive wells at seeding densities of 200 and then 20 cells/well followed by two rounds of cloning by limiting dilution led to the isolation of the clonal IgG2b switch variant, 7E1-G2b.
Mice
Adult male 6- to 8-wk-old C57BL/6, BALB/c, C3H/HeJ, B6.129S2-Igh-6tm1Cgn/J, and B6.129P2-Fcgr3tm1Sjv/J, were obtained from The Jackson Laboratory (Bar Harbor, ME). Act-mOVA mice were generously provided by M. Jenkins (University of Minnesota, Minneapolis, MN). B6.129P2-Fcer1gtm1Rav N12 and B6.129S4-Fcgr2btm1TtK N12 mouse strains were obtained from Taconic Farms (Germantown, NY). Animals received humane care in specific pathogen-free housing conditions in accordance with Emory University Institutional Animal Care and Use Committee guidelines.
Competitive binding and affinity assays
The M12 B cell lymphoma cell line was generously provided by N. Iwakoshi (Emory University, Atlanta, GA). To compare the binding affinities of the antibodies, M12 B cells were incubated at 4°C for 30 minutes with 8 pg/mL to 1 mg/mL of 7E1-G1, 7E1-G2b, isotype matched control or FGK4.5. Isotype control antibodies and FGK4.5, a rat IgG2a isotype (42), were purchased from BioXCell (West Lebanon, NH). Cells were washed with FACS buffer (phosphate buffered saline (PBS) supplemented with 0.5% bovine serum albumin and 1 mM EDTA, pH 7.2) and bound antibody was then detected with PE-conjugated F(ab')2 goat anti-rat IgG (Jackson ImmunoResearch, West Grove, PA) at 4°C for 30 minutes. For the competitive binding assay, cells were incubated with the same titrations of antibodies as noted above, along with 10 μg/mL of FLAG-tagged soluble recombinant mouse CD154 (Axxora, San Diego, CA) at 4°C for 30 minutes. Cells were washed with FACS buffer and bound CD154 was detected with FITC-conjugated anti-FLAG M2 antibody (Sigma-Aldrich, St. Louis, MO) at 4°C for 30 minutes. Nonviable cells were excluded by the addition of 7AAD 10 minutes prior to sample acquisition (BD Biosciences, San Jose, CA).
In vitro proliferation assay
To test the 7E1 variants as an immobilized stimulus, Immulon 4 HBX plates (Daigger, Vernon Hills, IL) were incubated overnight with 1 ng/mL to 1 mg/mL of either 7E1-G1, 7E1-G2b, isotype-matched control, or FGK4.5. For provision of a soluble stimulus, Costar cell culture plates (Sigma-Aldrich, St. Louis, MO) were blocked with 10% bovine serum albumin in PBS prior to the addition of the same titrated concentrations of antibodies as noted above. B cell responders from C57BL/6 mice were enriched from spleen and lymph node with Lymphocyte Separation Media (Mediatech, Herndon, VA) then purified using the Mouse B cell Isolation Kit and VarioMACS Separator (Miltenyi Biotec, Auburn, CA). Purity was confirmed to be >98% B220+ B cells. Purified B cells were added to both immobilized and soluble stimulus plates in parallel and, following 72 hours in culture, were pulsed for 12 hours with 0.5 μCi/well of [methyl-3H]thymidine. Cells were harvested and [methyl-3H]thymidine incorporation was measured on a standard β counter microplate reader (Biotek, Winooski, VT).
Bone marrow chimerism
BALB/c donor bone marrow was flushed from tibiae and femora and C57BL/6 recipient mice were transfused on days 0 and 6 with 2×107 donor marrow cells i.v, along with a single 500 mg dose of busulfan on day 5. On days 0, 2, 4, and 6, mice received 500 μg i.p. of human CTLA4-Ig (Bristol-Myers Squibb, New York, NY) and either 7E1-G1, 7E1-G2b, isotype matched control or hamster anti-mouse CD154 mAb (MR1: BioXCell, West Lebanon, NH). Chimerism was monitored by flow cytometric analysis of H-2Kd-expressing donor cells.
Skin grafting
Full thickness ear and tail skin grafts (∼1cm2) were transplanted onto the dorsal thorax of recipient mice and secured with a plastic adhesive bandage for 6 days. As indicated, recipients received 500 μg i.p. of CTLA4-Ig and either 7E1-G1, 7E1-G2b, isotype matched control, or anti-CD154 on days 0, 2, 4, and 6. Graft survival was monitored by daily visual inspection and rejection was defined as the complete loss of viable epidermal tissue.
BrdU labeling for in vivo proliferation
Mice were treated on days 0, 2, 4 and 6 with 500 μg i.p. of either 7E1-G1, 7E1-G2b, isotype matched control or FGK4.5. On day 7, mice were pulsed with 2 mg i.p. of 5-bromo-2-deoxyuridine (BrdU) then sacrificed 4 hrs later. BrdU incorporation was detected using the APC-BrdU Flow Kit according to the manufactures protocol (BD Biosciences).
Flow cytometry for phenotype and absolute number
At the indicated time points, animals were sacrificed, spleens harvested and single cell suspension prepared in FACS buffer. Unless previously stated, antibodies obtained from BD Biosciences included the following: anti-CD8-, CD11b-, CD19-, CD43-, CD44-, CD80-, CD86-, H-2Kd-, I-Ab-FITC, CD4-, CD19-, CD43-, CD45R-, CD70-, CD86-, 41BBL-, Vβ5-, Vβ8-, Vβ11-PE, CD3-, CD4-, CD11c-, CD19-, CD25-, CD44-, CD80-APC, CD3-, CD4-Pacific Blue, CD8-PerCP, CD11b-, CD45R-, CD69-PerCP-Cy5.5, and I-Ab-biotin.
Antibodies obtained from Invitrogen (Carlsbad, CA) included: anti-CD8-, CD45R-, and streptavidin-Pacific Orange. Antibodies obtained from eBioscience (San Diego, CA) included anti-CD19-Pacific Blue. Where noted, absolute numbers of splenocyte subsets were determined by TruCount bead analysis according to the manufacturer's instructions (BD Biosciences). Flow cytometric data was analyzed using FlowJo Software (Treestar, Ashland, OR).
Statistical analysis
Time to skin graft rejection was represented by Kaplan-Meier survival curves and comparison of graft survival was calculated using log-rank assessment. Titration curves were compared by nonlinear regression best curve fit using a sigmoidal dose-response (variable slope) equation over the exponential portion of each data set. Additional statistical analysis was performed using two-tailed unpaired Student's t-test. A p-value of less than 0.05 was considered significant for all tests. (GraphPad Software, La Jolla, CA).
Results
7E1-G1 and 7E1-G2b possess equivalent affinities for murine CD40 and both effectively inhibit the interaction of CD154 with CD40
Preliminary characterization during the development of the 7E1 variants suggested that 7E1-G2b was a legitimate isotype switch variant of 7E1-G1 and thus should possess the same specificity and affinity for CD40 (37). In order to confirm this, three sets of tests were performed. First, N-terminal peptide sequencing of the heavy chain showed that both antibodies were identical for the first 35 amino acids. Second, PCR primers specific for the variable heavy chain CDRs of 7E1-G1 yielded a band of the appropriate size from cDNA obtained from either 7E1 antibody, but not from two unrelated antibodies. Finally, ELISA-based measurement of the binding activity of purified 7E1-G1 and 7E1-G2b to immobilized murine CD40 gave essentially identical titration curves (37). In order to corroborate this finding, we compared their binding affinities to CD40 in vitro, utilizing the B cell line M12 that constitutively expresses a high level of murine CD40 on its surface (data not shown). Cultured M12 cells were incubated with increasing concentrations of 7E1-G1, 7E1-G2b, or FGK4.5, a strong mouse CD40 agonist (42), and bound antibody was subsequently detected with a fluorescently labeled polyclonal anti-rat IgG secondary antibody. The resulting titration curves for the 7E1 variants were essentially overlapping (EC50 = 81.63 ng/mL), while the titration curve for the unrelated antibody FGK4.5 was distinct from either of the 7E1 variants (EC50 = 95.00 ng/mL) (Figure 1a). These results suggested that any potential alterations in the idiotypic region of the 7E1 variants did not significantly affect its affinity for mouse CD40.
Figure 1. 7E1-G1 and 7E1-G2b have an equivalent affinity for murine CD40 and effectively inhibit the binding of CD154.

(A) M12 B cells were incubated with the indicated concentrations of 7E1-G1, 7E1-G2b or FGK4.5. Bound antibody was detected with a PE-labeled polyclonal anti-rat IgG secondary antibody by flow cytometry. The values are presented as the percentage of maximum fluorescence observed for each antibody. (B) M12 B cells were incubated with the indicated concentrations of 7E1-G1, 7E1-G2b or FGK4.5 along with 10 μg/mL of Flag-tagged soluble CD154 fusion protein. A FITC-labeled anti-Flag secondary antibody was used to detect bound CD154. The values represent a percentage of the maximum fluorescence observed with sCD154 alone. Data points represent four independent experiments with triplicate samples.
We next sought to assess the ability of the 7E1 variants to compete with soluble CD154 for binding to cell-bound CD40. We hypothesized that both 7E1-G1 and 7E1-G2b would inhibit this interaction to a similar degree. To test this, M12 B cells were incubated with increasing concentrations of 7E1-G1, 7E1-G2b, or FGK4.5, along with a constant amount of Flag-tagged soluble CD154 fusion protein (sCD154). Bound sCD154 then was detected with a fluorescently labeled anti-FLAG secondary antibody. The mean fluorescence intensity (MFI) observed at each concentration of competitor antibody was divided by the MFI of M12 cells cultured with sCD154 alone. The competitive inhibition curves generated for the 7E1 variants were again essentially overlapping (EC50 = 0.238 μg/mL) and distinct from FGK4.5 (EC50 = 1.362 μg/mL) (Figure 1b). These results further confirmed that the idiotypic region of 7E1-G2b had not undergone significant changes that impacted CD154 binding, as well as showed that both 7E1-G1 and 7E1-G2b had the same ability to block the interaction between CD40 and CD154.
Anti-CD40 treatment combined with CTLA4-Ig promotes allogeneic chimerism and skin graft survival
Previous murine studies from our laboratory and others have demonstrated the efficacy of costimulatory blockade-based protocols for promoting hematopoietic chimerism and skin graft survival (9, 43-45). These have involved the use of an anti-CD154 binding mAb paired with the CD28 pathway-blocking reagent CTLA4-Ig. Here we sought to determine whether an anti-CD40 mAb could effectively substitute for anti-CD154 in costimulation blockade-based tolerance induction regimens. First, groups of C57BL/6 mice were grafted with allogeneic BALB/c skin and treated with CTLA4-Ig and either 7E1-G1, 7E1-G2b, anti-CD154, or isotype-matched control on days 0, 2, 4, and 6. Untreated recipients promptly rejected their grafts with a median survival time (MST) of 11 days. Recipients treated with CTLA4-Ig/7E1-G2b or anti-CD154 showed a modest prolongation of skin graft survival (MST = 27 and 23 days, respectively) compared to CTLA4-Ig/7E1-G1 (MST = 18 days) in the stringent BALB/c to C57BL/6 model, indicative of the ‘costimulation blockade resistant’ phenotype (46) seen with this particular strain combination of donor and recipient (Figure 2a). In the more permissive BALB/c to C3H to model (47), graft survival also was prolonged with CTLA4-Ig combined with 7E1-G2b or anti-CD154 treatment (MST > 75 days), as compared to CTLA4-Ig/7E1-G1 treatment (MST = 29 days) or untreated recipients (MST=19 days) (Figure 2b).
Figure 2. 7E1-G2b promotes allogeneic bone marrow chimerism and skin graft survival equivalent to anti-CD154 when used in concert with CTLA4-Ig.

C57BL/6 mice were treated with 500 μg of CTLA4-Ig and 7E1-G1, 7E1-G2b, anti-CD154, or rat isotype IgG control, i.p. on days 0, 2, 4, and 6. Recipients treated with CTLA4-Ig and 7E1-G2b or anti-CD154 show a modest prolongation of skin graft survival in (A) the stringent BALB/c to C57BL/6 model, and long-term survival in (B) the more permissive BALB/c to C3H model. (C, D, E) C57BL/6 mice were treated with 500 μg of CTLA4-Ig and 7E1-G1, 7E1-G2b, anti-CD154, or rat isotype IgG control, i.p. on days 0, 2, 4, and 6. Busulfan was given i.v. on day 5, 600 mg per mouse, along with allogeneic BALB/c bone marrow, 2×107 cells i.v. on days 0 and 6, and skin graft on day 0. The percentage of H-2Kd+ donor B220+ B cells (C) and CD4+ T cells (D) reached similar levels in 7E1-G2b and anti-CD154 treated groups, while 7E1-G1 and isotype treated animals failed to sustain chimerism. (E) Chimeric mice are tolerant to donor skin grafts, as both 7E1-G2b and anti-CD154 treated recipients had prolonged graft survival not seen in the 7E1-G1 and isotype treated groups. (F) The percentage of Vβ11+ and Vβ5.1/5.2+ CD4+ T cells in the isotype and 7E1-G1 treated groups were consistent with wild-type B6 levels. In contrast, CD4+ T cells from 7E1-G2b and anti-CD154 treated groups show significant deletion of Vβ11+ and Vβ5.1/5.2+. Vβ deletion was specific as the use of Vβ8.1/8.2+ by CD4+ T cells remained comparable in all groups. Data is shown from day 58-post transplant.
Next, we sought to test the 7E1 variants in a model of allogeneic chimerism induction. C57BL/6 mice received BALB/c bone marrow (2×107 cells i.v. on days 0 and 6) and BALB/c skin grafts on day 0, as well as busulfan, an alkylating agent toxic to hematopoietic stem cells, on day 5. Mice received CTLA4-Ig and 7E1-G1, 7E1-G2b, anti-CD154, or isotype matched control on days 0, 2, 4, and 6. Peripheral blood was monitored for the presence of donor-derived B and T cells. The percentage of donor B220+ B cells (Figure 2c) and CD4+ T cells (Figure 2d) reached similar levels in 7E1-G2b and anti-CD154 treated groups, while 7E1-G1 and isotype treated animals completely failed to sustain significant donor chimerism. The successful induction of hematopoietic chimerism directly correlated to skin graft survival, as both 7E1-G2b and anti-CD154 treated recipients enjoyed prolonged graft survival (MST > 75 days). In contrast, 7E1-G1 treated recipients rapidly rejected their skin grafts (MST = 16 days) (Figure 2e).
We also tracked the frequency of donor reactive Vβ11+ and Vβ5.1/5.2+ CD4+ T cells in these chimeric recipients. T cells possessing these Vβ segments are normally deleted by negative selection in the thymus of BALB/c mice due to their high affinity for endogenous retroviral superantigens bound to I-E MHC molecules. Wild-type C57BL/6 mice do not express I-E; Vβ11+ and Vβ5.1/5.2+ CD4+ T cells constitute roughly 4-5% and 2-3% of the peripheral CD4+ T cell compartment, respectively. The deletion of these specific Vβ expressing T cells in our chimeric mice thus serves as a surrogate measure of successful donor tolerance (48). As shown (Figure 2f) on day 58, the percentage of Vβ11+ and Vβ5.1/5.2+ CD4+ T cells in the CTLA4-Ig/7E1-G1-treated recipients (striped bar) were similar to levels observed in the CTLA4-Ig/isotype-treated controls (black bar). In contrast, CD4+ T cells from CTLA4-Ig/7E1-G2b (grey bar) and CTLA4-Ig/anti-CD154 (white bar) treated groups show a significant reduction in the frequency of Vβ11+ and Vβ5.1/5.2+ T cells in the periphery. This Vβ deletion was specific for donor-reactive T cells, as the use of Vβ8.1/8.2+ by CD4+ T cells remained comparable in all groups.
The in vitro agonist potential of the 7E1 anti-CD40 antibodies is dependent on both the isotype and the mode of stimulation
The data presented above demonstrated that although the 7E1 variants possessed equal abilities to block CD154 binding to CD40, they showed dramatically different abilities to synergize with CTLA4-Ig in promoting allogeneic graft survival. We hypothesized that this is due to differences in their agonist properties. To test this, we used the induction of B cell proliferation in vitro as an indirect measure of CD40 agonism.
Splenic B cells were isolated from C57BL/6 mice, purified by density gradient and magnetic bead separation, and cultured with either soluble or plate-bound antibody. For provision of an immobilized stimulus, high binding affinity culture plates were pre-coated with increasing concentrations of 7E1-G1, 7E1-G2b, isotype matched control antibody or FGK4.5 at concentrations ranging from 1ng/mL to 100 μg/mL. Alternatively, the ability of soluble antibody to induce B cell proliferation was measured by incubation in conventional culture plates that were pre-blocked prior to the addition of the same concentrations of antibody as described above. We found that when 7E1-G1 or 7E1-G2b was presented as a plate bound stimulus, similar levels of proliferation as compared to FGK4.5 were observed (Figure 3a). However, when the antibodies were free in solution, the ability of 7E1-G2b to induce proliferation was significantly reduced. In contrast, soluble 7E1-G1 continued to promote B cell expansion, although not as robustly as FGK4.5 (Figure 3b). These results demonstrated that although both 7E1-G1 and 7E1-G2b had equivalent affinities for CD40 and could induce B cell proliferation when present as immobilized stimuli, only 7E1-G1 retained its agonistic properties when soluble. This therefore raised the possibility that the Fc-portion of the molecule was playing a prominent role in whether the antibody functioned as an agonist or antagonist.
Figure 3. 7E1-G2b induces B cell proliferation in vitro only when present as an immobilized stimulus.

(A) Purified B cells were added to high binding affinity 96 well plates that had been coated with the indicated concentrations of either 7E1-G1, isotype IgG1 control, 7E1-G2b, isotype IgG2b control, or FGK4.5. In parallel, (B) purified B cells were added to conventional 96 well plates, pre-blocked with bovine serum albumen, to which the same antibody concentrations were added. Following 72-hour incubation at 37°C, all plates were pulsed for an additional 18 hours with 0.5 μCi/well of [methyl-3H]thymidine. Actively dividing cells which had taken up the radioactive probe were quantified by scintillation counter. Representative data with triplicate samples, and experiments were repeated three times.
7E1 treatment does not cause depletion of CD40 expressing cells
We next explored the in vivo effects of treatment with the 7E1 variants. We hypothesized that the CD40-binding mAbs act by depleting CD40-expressing cells, resulting in a dearth of cells remaining to stimulate graft rejection. To test this, mice were injected on days 0, 2, 4, and 6 with either 7E1-G1, 7E1-G2b, isotype matched control antibody, FGK4.5 or were left untreated. On day 7, mice were sacrificed and the absolute numbers of various cell subsets were enumerated from the spleen by TruCount analysis. We found that treatment with 7E1-G2b did not lead to B cell, dendritic cell, or T cell depletion, as the absolute numbers of each did not significantly differ from isotype control or untreated animals (Figure 4a). Assessment of cell numbers in the lymph nodes, bone marrow and blood showed trends similar to those observed in the spleen (data not shown). However, mice treated with 7E1-G1 showed a statistically significant three-fold increase in CD8+ T cells and B cells and an approximately two-fold increase in dendritic cells, with numbers close to those seen with FGK4.5.
Figure 4. In vivo proliferation induced through CD40 ligation is dependent upon the specific isotype of the anti-CD40 antibody.
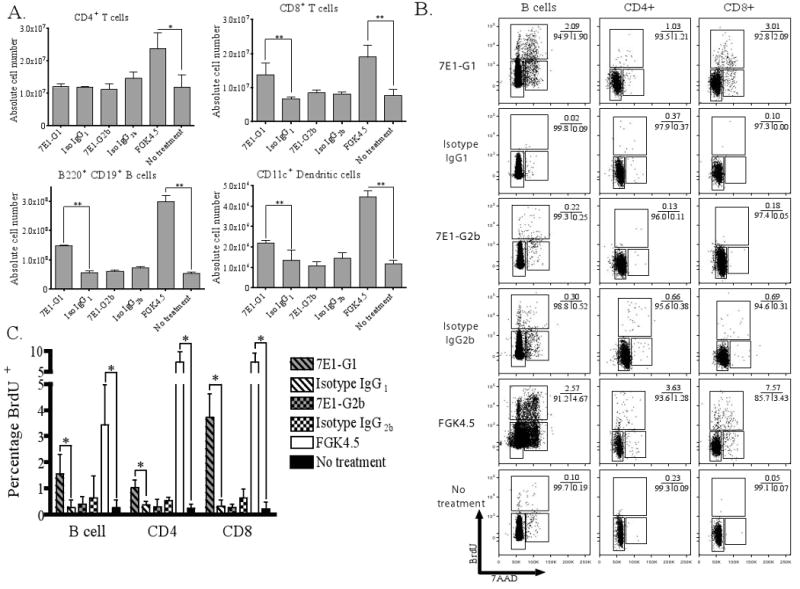
C57BL/6 mice were injected on days 0, 2, 4, and 6 with 500 μg of either 7E1-G1, isotype IgG1 control, 7E1-G2b, isotype IgG2b control, FGK4.5, or left untreated. (A) The absolute number of splenic cell subsets was enumerated by TruCount analysis. No significant differences were noted for any cell subsets between 7E1-G2b and isotype control. 7E1-G1 treated animals showed a significant increase in the total number of CD8+ T cells, B220+ B cells and CD11c+ dendritic cells in the spleen, although to a lesser extent than that observed with FGK4.5. (B) Similarly treated animals were injected with 2 mg of BrdU i.p. and spleens were harvested 4 hours later. The cell cycle phases and total DNA synthetic activities of splenic B cells, CD4+ and CD8+ T cells can be determined by the expression of total DNA using 7-AAD and detection of newly formed DNA by BrdU incorporation. These data are representative examples of three independent experiments with three mice per group. (C) A summary of the percentage of BrdU+ cells from the three independent experiments described above. The percentage of CD8+ T cells was statistically significantly increased between 7E1-G1 and isotype IgG1 control treated groups. No significant difference was noted between 7E1-G2b and isotype IgG2b control treatment.
To determine if these increases in cell numbers were due to increased trafficking to the spleen or de novo cell division, groups of mice were given the same treatment as above and pulsed on day 7 with 2 mg of BrdU for 4 hours. BrdU, when paired with the DNA stain 7AAD, can reveal both the presence of dividing cells and their specific state in the cell cycle. B cells, CD8+ T cells and, to a lesser extent, CD4+ T cells show a clear population of cells progressing through the cell cycle with 7E1-G1 and FGK4.5 treatment, but not in 7E1-G2b or isotype control treated animals (Figure 4b). This brief snapshot of division represents a 5-fold increase in proliferating B cells, a 3-fold increase in proliferating CD4+ T cells and an 11-fold increase in proliferating CD8+ T cells with 7E1-G1 as compared to isotype control treatment (Figure 4c). Thus, the ability of 7E1-G2b to promote graft survival is not due to the depletion of CD40-expressing cells. In contrast, 7E1-G1 treatment leads to a significant increase in the total number of T cells, B cells and dendritic cells in the spleen.
7E1-G1, but not 7E1-G2b, induces the expression of activation markers on antigen presenting cells
Due to the findings that 7E1-G1 induced proliferation in vitro and in vivo, we hypothesized that administration of 7E1-G1, but not 7E1-G2b, results in the maturation of antigen presenting cells. To examine this, C57BL/6 mice were given a single injection of either 7E1-G1, 7E1-G2b, isotype matched control antibody or FGK4.5. Mice were sacrificed at various time points and splenocytes were stained for the presence of activation markers on CD11c+ dendritic cells. 7E1-G1 treatment led to a three-fold increase in MHC class II expression, a four-fold increase in CD80 expression and a seven-fold increase in the expression of CD86 on dendritic cells, closely mirroring the level of expression observed for FGK4.5 treated animals (Figure 5a). The percentage of dendritic cells expressing CD70 increased by over 100-fold, 41BBL increased by 12-fold, and OX40L increased by 15-fold in 7E1-G1 and FGK4.5 but not 7E1-G2b-treated animals.
Figure 5. 7E1-G1, but not 7E1-G2b, increases expression of co-stimulatory molecules and matures antigen presenting cells.

C57BL/6 mice were given a single injection with 500 μg of either 7E1-G1, isotype IgG1 control, 7E1-G2b, isotype IgG2b control, FGK4.5 or left untreated. For analysis of (A) CD11c+ dendritic cells, animals were sacrificed after 6, 24, 36, 60 hours, and 7 days. For analysis of (B) B220+CD19+ B cells, animals were sacrificed after 3, 5, 7 and 14 days. The expression is displayed as a fold change compared to untreated animals of either the mean fluorescence intensity or the percentage of positive cells, as noted on axes. These data are representative examples of three independent experiments with three mice per group.
B220+ CD19+ B cells also were measured for activation markers in these same treated animals. As noted for dendritic cells, B cells in 7E1-G1 treated animals showed a three-fold increase in the expression of MHC class II, and an almost two-fold increase in both CD80 and CD86 (Fig. 5b). The percentage of CD69-expressing B cells increased roughly two-fold immediately following 7E1-G1 treatments. This was in contrast to what was observed in 7E1-G2b treated animals, in which expression patterns did not differ from isotype treated animals.
Increased expression of activation markers on T cells following treatment with 7E1-G1, but not 7E1-G2b
We hypothesized that the effects of the anti-CD40 antibodies on antigen presenting cells would subsequently cause T cell activation as well. To test this, C57BL/6 mice were given a single injection of either 7E1-G1, 7E1-G2b, isotype matched control antibody or FGK4.5. Mice were sacrificed at the indicated time points and stained for the expression of activation markers on CD3+ T cell subsets. CD4+ T cells from 7E1-G1 treated mice showed an almost two-fold increase in the percentage of cells expressing CD25 and CD43, which peak by day 7 (Figure 6a). The percentage of cells expressing CD44 and CD69 increased by two-fold, which can be seen as early as day 3. In contrast, the percentage of CD4+ T cells expressing each of these activation markers for 7E1-G2b treated animals was at or below what was observed for isotype control treated animals. The percentage of CD8+ T cells from 7E1-G1 treated animals expressing CD25 increased by four-fold, CD43 by almost 20-fold, CD44 by three-fold and CD69 by five-fold, all of which peak by day 7 (Figure 6b). 7E1-G2b did not induce any notable activation marker expression on CD8+ T cells beyond that seen for isotype treated animals.
Figure 6. Isotype-specific CD40 agonism translates to T cell activation.

C57BL/6 mice were given a single injection with 500 μg of either 7E1-G1, isotype IgG1 control, 7E1-G2b, isotype IgG2b control, FGK4.5 or left untreated. For analysis of (A) CD4+ T cells and (B) CD8+ T cells, animals were sacrificed after 3, 5, 7 and 14 days and stained for the indicated markers. The expression is displayed as a fold change of the percentage of positive cells compared to untreated animals. These data are representative examples of three independent experiments with three mice per group.
The presence of activated B cells alone cannot explain the differences observed between the two isotypes of anti-CD40 antibodies
Resting B cells make up a large percentage of the total cells present in the spleen and express a high basal level of CD40 (49). As we have shown, B cells aggressively responded to 7E1-G1 both in vivo and in vitro, up-regulating activation markers and robustly proliferating. We therefore hypothesized that the prominent activation of B cells by 7E1-G1 created an overall stimulated environment that supercedes the effects of CTLA4-Ig, thus leading to prompt graft rejection. To test this, we made use of a different model of skin graft rejection than previously presented herein. Untreated C57BL/6 mice reject BALB/c skin grafts with a MST of 11 days, while costimulation blockade modestly extends survival to 23 days (Figure 2a). To investigate potentially subtle differences between 7E1-G1 and 7E1-G2b treatment in the following experiments, we used a single minor mismatch skin graft rejection model. The mOVA-C57BL/6 mouse constitutively expresses full length, membrane-bound chicken ovalbumin driven by the β-actin promoter (50). When used as skin graft donors for untreated C57BL/6 recipients, these single mismatched grafts were rejected with a MST of 18 days (Fig. 7a). Costimulation blockade consisting of CTLA4-Ig and 7E1-G2b or anti-CD154 extended graft survival indefinitely (MST > 200 days) (Figure 7a and (51). Isotype matched control antibody and CTLA4-Ig prolonged graft survival for > 100 days in about half of the recipients tested (MST=55 days), highlighting the significant contribution of blocking the CD28 pathway in this model. Interestingly, when 7E1-G1 was substituted for anti-CD154, the beneficial effects of CTLA4-Ig were ablated, the MST was reduced to 22 days and all grafts were rejected.
Figure 7. Fc-receptor and B cell involvement in mediating the differences seen between 7E1-G1 and 7E1-G2b.

(A) Wild-type C57BL/6 mice were transplanted with minor antigen mismatched mOVA (C57BL/6 background) skin grafts and treated with 500 μg of CTLA4-Ig and either 7E1-G1, isotype IgG1 control, 7E1-G2b, isotype IgG2b control, or anti-CD154 i.p on days 0, 2, 4, and 6. (B) B cell deficient mice were used as recipients of mOVA skin grafts with the same treatment regime as noted above, and showed graft survival similar to that of wild-type recipients. (C) FcγRIII deficient recipients did not show significant prolongation of mOVA skin graft survival when treated with CTLA4-Ig and 7E1-G1. (D) FcεRγI deficient recipients also did not show significant differences in the prolongation of mOVA skin graft survival as compared to wild-type recipients. (E) FcγRIIb deficient recipient mice, lacking the inhibitory Fc receptor, exhibited no diminution in the long-term graft survival observed in 7E1-G2b treated mice, while all other treatment groups promptly rejected their grafts.
We obtained B cell deficient mice for use as recipients of mOVA skin grafts to test the hypothesis that the failure of 7E1-G1 to extend graft survival was due to the activation of B cells. B cell deficient recipients were treated with 500 μg of CTLA4-Ig and 7E1-G1 or isotype matched control i.p. on days 0, 2, 4, and 6, and received an mOVA skin graft on day 0. In the absence of B cells, the MST of the CTLA-4 Ig/7E1-G1 treated animals was 21 days, not significantly different from that observed for wild type recipients. Untreated recipients promptly reject grafts with an MST of 13 days, while CTLA-4 Ig/isotype control recipients demonstrated long-term survival (MST=65 days) (Figure 7b).
The involvement of individual Fc receptors cannot fully account for the differences observed between the two isotypes of anti-CD40 antibodies
We next sought to explore the possibility that Fc receptors could account for the difference in efficacy observed between the 7E1 variants. To test the hypothesis that activating signals delivered by Fc receptors were responsible for the graft rejection observed with 7E1-G1, FcγRIII deficient mice were used as recipients of mOVA skin grafts. FcγRIII is the activating Fc receptor for which the murine IgG1 antibody isotype has the greatest affinity (52). Recipients were treated with 500 μg of CTLA4-Ig and 7E1-G1, 7E1-G2b or isotype matched control i.p. on days 0, 2, 4, and 6, along with a mOVA skin graft on day 0. The MST of CTLA-4 Ig/7E1-G1-treated FcγRIII deficient animals was 20 days, which was not significantly different when compared to CTLA-4 Ig/isotype matched control or similarly treated wild type recipients (MST = 17 days) (Figure 7c). CTLA-4 Ig/7E1-G2b-treated FcγRIII deficient animals show no diminution in their long-term graft survival (MST > 75 days).
Several other activating Fc receptors have been identified, for which IgG1 isotype antibodies have varying affinities. In order to determine if interactions with other activating Fc receptors, besides FcγRIII, contributed to the graft rejection seen for 7E1-G1 treated animals, we obtained FcRγ deficient mice. FcRγ, also referred to as FcεRIγ, is the common signaling chain that associates with and is responsible for the surface expression of all of the currently reported murine activating Fc-receptors (53). In the absence of all activating Fc receptors, 7E1-G1 treated animals again did not exhibit prolongation of graft survival and exhibited an MST of 20 days, compared to the long-term survival seen for 7E1-G2b treated animals (MST > 75 days). Approximately 50% of the animals treated with CTLA-4 Ig and isotype control Ab experienced long term graft survival (MST 44 and 56 days), while untreated animals rejected their grafts with an MST of 11 days (Figure 7d).
Another potential hypothesis for the involvement of Fc receptors was that the ligation of inhibitory Fc receptors was necessary for the prolongation of skin graft survival with 7E1-G2b treatment. To test this hypothesis, we obtained FcγRIIb deficient mice, which lack expression of the sole murine inhibitory Fc receptor (52). Following the same protocol outlined above, we observed no difference in long-term graft survival between FcγRIIb deficient and wild type recipients when treated with CTLA4-Ig and 7E1-G2b (MST > 75 days). Interestingly, the 50% long-term graft survival that was observed in wild-type recipients of mOVA skin grafts treated with CTLA4-Ig alone (MST 55 days) was abrogated in the FcγRIIb deficient hosts, as 100% of the grafts were rejected with a MST of 20 days (Figure 7e).
Discussion
Interruption of the CD40/CD154 signaling pathway remains a potent means of attenuating the rejection response and an attractive therapeutic target for the prolongation of graft survival. Anti-CD154 mAbs have been widely used in many murine models, and are among the most effective agents tested for the inhibition of rejection. However, the use of anti-CD154 antibodies in human clinical trials led to thromboembolic events, likely due to the unappreciated expression of CD154 on platelets and its involvement in the stabilization of thrombi (16-20). Despite this set-back, anti-CD40 mAbs have shown efficacy in non-human primate models of both renal and islet transplantation, tested in clinical models for tumor immunotherapy and thus far have been free of the side effects observed with the use of anti-CD154 mAbs (33-35, 54, 55). For these reasons, blockade of the CD40/CD154 pathway remains an area of intense interest and potential opportunity in the transplant community. However, the field is in need of experimental evidence to guide the selection of candidate therapeutic antibodies for potential clinical development.
Understanding the mechanisms by which anti-CD40 mAbs function to mediate either immunosuppression or immune activation is critical for the rational design of agents for use in transplantation. Our study assessed two isotypes of a novel murine anti-CD40 antibody, 7E1-G1 and 7E1-G2b, for characteristics mirroring anti-CD154 blockade. We found that, when used in concert with CTLA4-Ig, 7E1-G2b, but not 7E1-G1, effectively promoted allogeneic bone marrow chimerism and skin graft survival, similarly to the effects observed with anti-CD154 (Figure 2). Because similar outcomes were observed by blocking either the ligand or the receptor, our results suggest that blockade of the CD40/CD154 pathway may be the critical effect of both anti-CD40 and anti-CD154 mAbs. Other mechanisms that have been postulated in the literature include the possibility that antibodies bound to CD154 serve to deplete activated T cells via complement-mediated mechanisms or antibody-dependent cellular cytotoxicity (56). Still another alternative explanation may be that anti-CD154 induces reverse negative signaling upon the T cell itself (57). While our results do not formally exclude these possibilities, they suggest that the inhibition of positive signaling events propagated through CD40 is an important mechanism of action of both anti-CD40 and anti-CD154 mAbs.
This concept is well-supported in the literature, in that interrupting CD40-mediated signals delivered to APCs during T cell priming has been shown to profoundly impact the dynamics of the T cell:APC interaction. Among these effects are limitation of the up-regulation of activation markers such as CD80 and CD86 (58), and a decrease in the production of proinflammatory cytokines such as IL-8, IL-12, MIP-1α and TNFα (59, 60). CD40 signaling is also important for APC survival, mediating the up-regulation of the anti-apoptotic molecules Bcl-2 and Bcl-xl (61-63). Although we did not observe active depletion of CD40-expressing APCs following treatment with 7E1-G2b, the blockade of survival signals normally provided by the CD40 pathway may be detrimental to APC longevity and the duration of antigen presentation (23, 64, 65). The contributions of these effects may culminate in the deviation of responding T cells from an effector phenotype and direct them towards an alternative program of anergy (66), abortive proliferation (67), apoptosis (68) or a regulatory cell phenotype (69). The extent to which these mechanisms play a role in transplant tolerance mediated by either anti-CD154 or 7E1-G2b remains an important area of future investigation.
We found that the anti-CD40 mAb 7E1-G2b effectively synergized with CTLA-4 Ig in order to prolong graft survival (Figure 2). In contrast, treatment with 7E1-G1 failed to prolong graft survival (Figure 2), despite the fact that these antibodies possess identical idiotypic regions. These results highlight the ability of an appropriately designed anti-CD40 mAb to be used therapeutically as either immunosuppression/tolerance induction agents or immunostimulatory adjuvants. Interestingly, both antibodies have equivalent affinity for their antigen and are able to inhibit the interaction between CD40 and CD154 to the same extent (Figure 1a,b). Thus, the dichotomy in their functionality may be explained by the fact that while both antibodies behaved as agonists when plate-bound (Figure 3a), 7E1-G1 but not 7E1-G2b acted as a CD40 agonist to increase B cell proliferation and activation when present in soluble form both in vitro and in vivo (Figure 3b, 4, 5, 6). Prior studies have focused for the most part on differences in idiotypic regions as being the primary determinant which conferred either activating or antagonistic properties upon a particular monoclonal antibody; however, our results suggest that monoclonals with identical epitope specificity can possess agonist or antagonist properties in vivo depending on the isotype alone. Our results also suggest that assessment of the behavior of an anti-CD40 mAb as an agonist with regard to B cell proliferation in soluble form may be a useful in vitro screening assay to help guide the selection of candidate anti-CD40 mAbs likely to function as blocking agents in vivo. We would propose that any candidate anti-CD40 mAbs should be evaluated based on their 1) ability to prevent ligand binding in vitro, and 2) failure to induce B cell proliferation when used in soluble form in vitro. These results therefore demonstrate the therapeutic potential of an appropriately designed anti-CD40 blocking agent in synergizing with CTLA-4 Ig to promote long-term graft survival. It should be noted that while these results help to establish the efficacy of anti-CD40 mAbs in transplantation and provide a springboard for mechanistic studies of these antibodies, mouse models are sometimes limited in their ability to predict side effects related to antibody administration in human subjects (17, 70-72). Detailed safety studies of each individual antibody clonotype are warranted prior to clinical translation.
Despite the findings that 7E1-G1 but not 7E1-G2b induced B cell proliferation and activation both in vitro and in vivo, we found that B cells were not required for the pro-rejection phenotype associated with the 7E1-G1 mAb, as 7E1-G1 still exhibited this pro-rejection phenotype in B cell deficient recipients (Figure 7). Likewise, we found no evidence of significant depletion of B cells (nor other CD40-expressing cells) with either 7E1 variant; therefore, our data further suggest that depletion of CD40-expressing APC is not the major mechanism by which 7E1-G2b promotes long-term graft survival (Figure 4). While studies investigating the effects of Chi220, a chimeric anti-human CD40 mAb, did demonstrate significant depletion of B cells in non-human primates, those studies also indicated that B cell depletion was not the primary mechanism of immunosuppression in their model (73).
Due to the fact that the idiotypic regions are identical between these two antibodies, their differences in functionality likely lie within the Fc portion of the antibody. One potential mechanism that could underlie these differences in isotype is their differential ability to fix complement and mediate the depletion of CD40-bearing cells. However, we reject this hypothesis on the basis of two observations: 1) no depletion of CD40-bearing cells was observed in vivo, and 2) the differential effects of the antibodies on B cell proliferation were observed in an in vitro, complement-free system. An alternate hypothesis for how isotype might impact functionality is through their binding to Fc receptors.
With regard to Fc receptor binding, one might envision that there are two possibilities to explain the immunostimulatory properties observed with 7E1-G1: either the mAb binds activating FcγRIII receptors and induces positive Fc receptor-mediated signaling into the APC, or the mAb binding to any FcR simply acts as a scaffold to induce crosslinking of CD40 molecules on the surface of the APC, resulting in the delivery of CD40-mediated signals. While the role of Fc receptors in conferring either immunosuppressive or immunostimulatory effects on a given anti-CD40 mAb remains to be fully explored, in our system, proinflammatory signals delivered by activating Fc receptors were not responsible for immunostimulatory properties of 7E1-G1, as FcεRγI deficient mice exhibited similar graft survival as compared to wild-type mice (Figure 7d). Likewise, one also could imagine two analogous scenarios to explain the immunosuppressive effects of 7E1-G2b: the mAb is functioning primarily to block CD40 engagement of CD154, or the mAb is binding to inhibitory FcγRIIb receptors to induce negative signaling to B cells or other APC. We favor the former, as inhibitory signals through FcγRIIb were not found to be involved in the immunosuppressive effects observed with 7E1-G2b, as graft survival was unchanged between FcγRIIb deficient and wild-type animals (Figure 7e). Our results are consistent with the hypothesis that Fc-receptors cross-link surface-bound 7E1-G1 mAb to a greater extent than the 7E1-G2b mAb and thus contributes to the agonistic signal delivered uniquely by 7E1-G1. Future experiments using in vivo treatment with Fc-blocking antibodies or delivery of F(ab')2 fragments could further delineate the role of Fc receptors in the in vivo effects of these antibodies.
In conclusion, we have demonstrated that CD28 blockade can effectively synergize with CD40 blockade using either anti-CD154 or an appropriately designed anti-CD40 monoclonal antibody. Although clinical translation of anti-CD154 has encountered difficulties, this should not belie the clear utility that can be gained by further studying the inhibition of the CD40 pathway in transplantation. Understanding the cellular and subcellular mechanisms that underlie the ability of blockade of the CD40/CD154 interaction to attenuate donor-reactive T cell responses and promote long-term graft survival is crucial to the development of novel therapeutics to target this pathway.
References
- 1.Jenkins MK. The ups and downs of T cell costimulation. Immunity. 1994;1:443–446. doi: 10.1016/1074-7613(94)90086-8. [DOI] [PubMed] [Google Scholar]
- 2.Wood KJ, Sakaguchi S. Regulatory T cells in transplantation tolerance. Nat Rev Immunol. 2003;3:199–210. doi: 10.1038/nri1027. [DOI] [PubMed] [Google Scholar]
- 3.Jenkins MK, Taylor PS, Norton SD, Urdahl KB. CD28 delivers a costimulatory signal involved in antigen-specific IL-2 production by human T cells. J Immunol. 1991;147:2461–2466. [PubMed] [Google Scholar]
- 4.Schwartz RH. A cell culture model for T lymphocyte clonal anergy. Science. 1990;248:1349–1356. doi: 10.1126/science.2113314. [DOI] [PubMed] [Google Scholar]
- 5.Yamada A, Salama AD, Sho M, Najafian N, Ito T, Forman JP, Kewalramani R, Sandner S, Harada H, Clarkson MR, Mandelbrot DA, Sharpe AH, Oshima H, Yagita H, Chalasani G, Lakkis FG, Auchincloss H, Jr, Sayegh MH. CD70 signaling is critical for CD28-independent CD8+ T cell-mediated alloimmune responses in vivo. J Immunol. 2005;174:1357–1364. doi: 10.4049/jimmunol.174.3.1357. [DOI] [PubMed] [Google Scholar]
- 6.Ozkaynak E, Gao W, Shemmeri N, Wang C, Gutierrez-Ramos JC, Amaral J, Qin S, Rottman JB, Coyle AJ, Hancock WW. Importance of ICOS-B7RP-1 costimulation in acute and chronic allograft rejection. Nat Immunol. 2001;2:591–596. doi: 10.1038/89731. [DOI] [PubMed] [Google Scholar]
- 7.Demirci G, Amanullah F, Kewalaramani R, Yagita H, Strom TB, Sayegh MH, Li XC. Critical role of OX40 in CD28 and CD154-independent rejection. J Immunol. 2004;172:1691–1698. doi: 10.4049/jimmunol.172.3.1691. [DOI] [PubMed] [Google Scholar]
- 8.Wang J, Guo Z, Dong Y, Kim O, Hart J, Adams A, Larsen CP, Mittler RS, Newell KA. Role of 4-1BB in allograft rejection mediated by CD8+ T cells. Am J Transplant. 2003;3:543–551. doi: 10.1034/j.1600-6143.2003.00088.x. [DOI] [PubMed] [Google Scholar]
- 9.Larsen CP, Elwood ET, Alexander DZ, Ritchie SC, Hendrix R, Tucker-Burden C, Cho HR, Aruffo A, Hollenbaugh D, Linsley PS, Winn KJ, Pearson TC. Long-term acceptance of skin and cardiac allografts after blocking CD40 and CD28 pathways. Nature. 1996;381:434–438. doi: 10.1038/381434a0. [DOI] [PubMed] [Google Scholar]
- 10.Kirk AD, Harlan DM, Armstrong NN, Davis TA, Dong Y, Gray GS, Hong X, Thomas D, Fechner JH, Knechtle SJ. CTLA4-Ig and anti-CD40L prevent renal allograft rejection in primates. Proc Natl Acad Sci USA. 1997;94:8789–8794. doi: 10.1073/pnas.94.16.8789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Levisetti MG, Padrid PA, Szot GL, Mittal N, Meehan SM, Wardrip CL, Gray GS, Bruce DS, Thistlethwaite JR, Jr, Bluestone JA. Immunosuppressive effects of human CTLA4Ig in a non-human primate model of allogeneic pancreatic islet transplantation. Journal of Immunology. 1997;159:5187–5191. [PubMed] [Google Scholar]
- 12.Larsen CP, Pearson TC, Adams AB, Tso P, Shirasugi N, Strobertm E, Anderson D, Cowan S, Price K, Naemura J, Emswiler J, Greene J, Turk LA, Bajorath J, Townsend R, Hagerty D, Linsley PS, Peach RJ. Rational development of LEA29Y (belatacept), a high-affinity variant of CTLA4-Ig with potent immunosuppressive properties. Am J Transplant. 2005;5:443–453. doi: 10.1111/j.1600-6143.2005.00749.x. [DOI] [PubMed] [Google Scholar]
- 13.Vincenti F, Larsen C, Durrbach A, Wekerle T, Nashan B, Blancho G, Lang P, Grinyo J, Halloran PF, Solez K, Hagerty D, Levy E, Zhou W, Natarajan K, Charpentier B. Costimulation blockade with belatacept in renal transplantation. N Engl J Med. 2005;353:770–781. doi: 10.1056/NEJMoa050085. [DOI] [PubMed] [Google Scholar]
- 14.Kirk AD, Burkly LC, Batty DS, Baumgartner RE, Berning JD, Buchanan K, Fechner JH, Jr, Germond RL, Kampen RL, Patterson NB, Swanson SJ, Tadaki DK, TenHoor CN, White L, Knechtle SJ, Harlan DM. Treatment with humanized monoclonal antibody against CD154 prevents acute renal allograft rejection in nonhuman primates. Nat Med. 1999;5:686–693. doi: 10.1038/9536. [DOI] [PubMed] [Google Scholar]
- 15.Kenyon NS, Fernandez LA, Lehmann R, Masetti M, Ranuncoli A, Chatzipetrou M, Iaria G, Han D, Wagner JL, Ruiz P, Berho M, Inverardi L, Alejandro R, Mintz DH, Kirk AD, Harlan DM, Burkly LC, Ricordi C. Long-term survival and function of intrahepatic islet allografts in baboons treated with humanized anti-CD154. Diabetes. 1999;48:1473–1481. doi: 10.2337/diabetes.48.7.1473. [DOI] [PubMed] [Google Scholar]
- 16.Kanmaz T, Fechner JJ, Jr, Torrealba J, Kim HT, Dong Y, Oberley TD, Schultz JM, Bloom DD, Katayama M, Dar W, Markovits J, Schuler W, Hu H, Hamawy MM, Knechtle SJ. Monotherapy with the novel human anti-CD154 monoclonal antibody ABI793 in rhesus monkey renal transplantation model. Transplantation. 2004;77:914–920. doi: 10.1097/01.tp.0000116392.72152.75. [DOI] [PubMed] [Google Scholar]
- 17.Kawai T, Andrews D, Colvin RB, Sachs DH, Cosimi AB. Thromboembolic complications after treatment with monoclonal antibody against CD40 ligand. Nat Med. 2000;6:114. doi: 10.1038/72162. [DOI] [PubMed] [Google Scholar]
- 18.Weaver TA, Charafeddine AH, Kirk AD. Costimulation blockade: towards clinical application. Front Biosci. 2008;13:2120–2139. doi: 10.2741/2829. [DOI] [PubMed] [Google Scholar]
- 19.Inwald DP, McDowall A, Peters MJ, Callard RE, Klein NJ. CD40 is constitutively expressed on platelets and provides a novel mechanism for platelet activation. Circ Res. 2003;92:1041–1048. doi: 10.1161/01.RES.0000070111.98158.6C. [DOI] [PubMed] [Google Scholar]
- 20.Andre P, Prasad KS, Denis CV, He M, Papalia JM, Hynes RO, Phillips DR, Wagner DD. CD40L stabilizes arterial thrombi by a beta3 integrin--dependent mechanism. Nat Med. 2002;8:247–252. doi: 10.1038/nm0302-247. [DOI] [PubMed] [Google Scholar]
- 21.Kawabe T, Naka T, Yoshida K, Tanaka T, Fujiwara H, Suematsu S, Yoshida N, Kishimoto T, Kikutani H. The immune responses in CD40-deficient mice: impaired immunoglobulin class switching and germinal center formation. Immunity. 1994;1:167–178. doi: 10.1016/1074-7613(94)90095-7. [DOI] [PubMed] [Google Scholar]
- 22.Bennett SRM, Carbone FR, Karamalis F, Flavell RA, Miller JFAP, Heath WR. Help for cytotoxic-T-cell responses is mediated by CD40 signalling. Nature. 1998;393:478–480. doi: 10.1038/30996. [DOI] [PubMed] [Google Scholar]
- 23.Miga AJ, Masters SR, Durell BG, Gonzalez M, Jenkins MK, Maliszewski C, Kikutani H, Wade WF, Noelle RJ. Dendritic cell longevity and T cell persistence is controlled by CD154-CD40 interactions. European journal of immunology. 2001;31:959–965. doi: 10.1002/1521-4141(200103)31:3<959::aid-immu959>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- 24.Lazarevic V, Myers AJ, Scanga CA, Flynn JL. CD40, but not CD40L, is required for the optimal priming of T cells and control of aerosol M. tuberculosis infection. Immunity. 2003;19:823–835. doi: 10.1016/s1074-7613(03)00324-8. [DOI] [PubMed] [Google Scholar]
- 25.Hollenbaugh D, Mischel-Petty N, Edwards CP, Simon JC, Denfeld RW, Kiener PA, Aruffo A. Expression of functional CD40 by vascular endothelial cells. J Exp Med. 1995;182:33–40. doi: 10.1084/jem.182.1.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Phillips NE, Markees TG, Mordes JP, Greiner DL, Rossini AA. Blockade of CD40-mediated signaling is sufficient for inducing islet but not skin transplantation tolerance. J Immunol. 2003;170:3015–3023. doi: 10.4049/jimmunol.170.6.3015. [DOI] [PubMed] [Google Scholar]
- 27.Raisky O, Spriewald BM, Morrison KJ, Ensminger S, Mohieddine T, Obadia JF, Yacoub MH, Rose ML. CD8(+) T cells induce graft vascular occlusion in a CD40 knockout donor/recipient combination. J Heart Lung Transplant. 2003;22:177–183. doi: 10.1016/s1053-2498(02)00465-5. [DOI] [PubMed] [Google Scholar]
- 28.Nathan MJ, Mold JE, Wood SC, Csencsits K, Lu G, Eichwald EJ, Bishop DK. Requirement for donor and recipient CD40 expression in cardiac allograft rejection: induction of Th1 responses and influence of donor-derived dendritic cells. J Immunol. 2004;172:6626–6633. doi: 10.4049/jimmunol.172.11.6626. [DOI] [PubMed] [Google Scholar]
- 29.Schoenberger SP, Toes REM, van der Voort EIH, Offringa R, Melief CJM. T-cell help for cytotoxic T lymphocytes is mediated by CD40-CD40L interactions. Nature. 1998;393:480–483. doi: 10.1038/31002. [DOI] [PubMed] [Google Scholar]
- 30.Staveley-O'Carroll K, Schell TD, Jimenez M, Mylin LM, Tevethia MJ, Schoenberger SP, Tevethia SS. In vivo ligation of CD40 enhances priming against the endogenous tumor antigen and promotes CD8+ T cell effector function in SV40 T antigen transgenic mice. J Immunol. 2003;171:697–707. doi: 10.4049/jimmunol.171.2.697. [DOI] [PubMed] [Google Scholar]
- 31.Maxwell JR, Campbell JD, Kim CH, Vella AT. CD40 activation boosts T cell immunity in vivo by enhancing T cell clonal expansion and delaying peripheral T cell deletion. Journal of Immunology. 1999;162:2024–2034. [PubMed] [Google Scholar]
- 32.Dullforce P, Sutton DC, Heath AW. Enhancement of T cell-independent immune responses in vivo by CD40 antibodies. Nat Med. 1998;4:88–91. doi: 10.1038/nm0198-088. [DOI] [PubMed] [Google Scholar]
- 33.Haanstra KG, Ringers J, Sick EA, Ramdien-Murli S, Kuhn EM, Boon L, Jonker M. Prevention of kidney allograft rejection using anti-CD40 and anti-CD86 in primates. Transplantation. 2003;75:637–643. doi: 10.1097/01.TP.0000054835.58014.C2. [DOI] [PubMed] [Google Scholar]
- 34.Haanstra KG, Sick EA, Ringers J, Wubben JA, Kuhn EM, Boon L, Jonker M. Costimulation blockade followed by a 12-week period of cyclosporine A facilitates prolonged drug-free survival of rhesus monkey kidney allografts. Transplantation. 2005;79:1623–1626. doi: 10.1097/01.tp.0000158426.64631.ed. [DOI] [PubMed] [Google Scholar]
- 35.Adams AB, Shirasugi N, Jones TR, Durham MM, Strobert EA, Cowan S, Rees P, Hendrix R, Price K, Kenyon NS, Hagerty D, Townsend R, Hollenbaugh D, Pearson TC, Larsen CP. Development of a chimeric anti-CD40 monoclonal antibody that synergizes with LEA29Y to prolong islet allograft survival. J Immunol. 2005;174:542–550. doi: 10.4049/jimmunol.174.1.542. [DOI] [PubMed] [Google Scholar]
- 36.Masunaga T, Yamashita K, Sakihama H, Hashimoto T, Hua N, Imai A, Inobe M, Miyazaki T, Todo S, Uede T. Dimeric but not monomeric soluble CD40 prolongs allograft survival and generates regulatory T cells that inhibit CTL function. Transplantation. 2005;80:1614–1622. doi: 10.1097/01.tp.0000181093.50141.6c. [DOI] [PubMed] [Google Scholar]
- 37.Aruffo AA, Hollenbaugh D, Siadak AW, Berry KK, Harris L, Thorne BA, Bajorath J. Methods of using antibodies against human CD40. U. S. P. Office, ed. Bristol-Myers Squibb Company; United States: 1998. [Google Scholar]
- 38.Hale G, Clark M, Waldmann H. Therapeutic potential of rat monoclonal antibodies: isotype specificity of antibody-dependent cell-mediated cytotoxicity with human lymphocytes. J Immunol. 1985;134:3056–3061. [PubMed] [Google Scholar]
- 39.Bruggemann M, Teale C, Clark M, Bindon C, Waldmann H. A matched set of rat/mouse chimeric antibodies. J Immunol. 1989;142:3145–3150. [PubMed] [Google Scholar]
- 40.Nimmerjahn F, Ravetch JV. Divergent immunoglobulin g subclass activity through selective Fc receptor binding. Science. 2005;310:1510–1512. doi: 10.1126/science.1118948. [DOI] [PubMed] [Google Scholar]
- 41.Hale G, Cobbold SP, Waldmann H, Easter G, Matejtschuk P, Coombs RR. Isolation of low-frequency class-switch variants from rat hybrid myelomas. J Immunol Methods. 1987;103:59–67. doi: 10.1016/0022-1759(87)90242-0. [DOI] [PubMed] [Google Scholar]
- 42.Rolink A, Melchers F, Andersson J. The SCID but not the RAG-2 gene product is required for S mu-S epsilon heavy chain class switching. Immunity. 1996;5:319–330. doi: 10.1016/s1074-7613(00)80258-7. [DOI] [PubMed] [Google Scholar]
- 43.Adams AB, Durham MM, Kean L, Shirasugi N, Ha J, Williams MA, Rees PA, Cheung MC, Mittelstaedt S, Bingaman AW, Archer DR, Pearson TC, Waller EK, Larsen CP. Costimulation blockade, busulfan, and bone marrow promote titratable macrochimerism, induce transplantation tolerance, and correct genetic hemoglobinopathies with minimal myelosuppression. J Immunol. 2001;167:1103–1111. doi: 10.4049/jimmunol.167.2.1103. [DOI] [PubMed] [Google Scholar]
- 44.Salomon B, Bluestone JA. Complexities of CD28/B7: CTLA-4 costimulatory pathways in autoimmunity and transplantation. Annu Rev Immunol. 2001;19:225–252. doi: 10.1146/annurev.immunol.19.1.225. [DOI] [PubMed] [Google Scholar]
- 45.Wekerle T, Kurtz J, Bigenzahn S, Takeuchi Y, Sykes M. Mechanisms of transplant tolerance induction using costimulatory blockade. Curr Opin Immunol. 2002;14:592–600. doi: 10.1016/s0952-7915(02)00378-3. [DOI] [PubMed] [Google Scholar]
- 46.Trambley J, Bingaman AW, Lin A, Elwood ET, Waitze SY, Ha J, Durham MM, Corbascio M, Cowan SR, Pearson TC, Larsen CP. Asialo GM1(+) CD8(+) T cells play a critical role in costimulation blockade-resistant allograft rejection. J Clin Invest. 1999;104:1715–1722. doi: 10.1172/JCI8082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Williams MA, Trambley J, Ha J, Adams AB, Durham MM, Rees P, Cowan SR, Pearson TC, Larsen CP. Genetic characterization of strain differences in the ability to mediate CD40/CD28-independent rejection of skin allografts. J Immunol. 2000;165:6849–6857. doi: 10.4049/jimmunol.165.12.6849. [DOI] [PubMed] [Google Scholar]
- 48.Tomita Y, Khan A, Sykes M. Role of intrathymic clonal deletion and peripheral anergy in transplantation tolerance induced by bone marrow transplantation in mice conditioned with a nonmyeloablative regimen. J Immunol. 1994;153:1087–1098. [PubMed] [Google Scholar]
- 49.Grewal IS, Flavell RA. CD40 and CD154 in cell-mediated immunity. Annual Review of Immunology. 1998;16:111–135. doi: 10.1146/annurev.immunol.16.1.111. [DOI] [PubMed] [Google Scholar]
- 50.Ehst BD, Ingulli E, Jenkins MK. Development of a novel transgenic mouse for the study of interactions between CD4 and CD8 T cells during graft rejection. Am J Transplant. 2003;3:1355–1362. doi: 10.1046/j.1600-6135.2003.00246.x. [DOI] [PubMed] [Google Scholar]
- 51.Ford ML, Wagener ME, Hanna SS, Pearson TC, Kirk AD, Larsen CP. A critical precursor frequency of donor-reactive CD4+ T cell help is required for CD8+ T cell-mediated CD28/CD154-independent rejection. J Immunol. 2008;180:7203–7211. doi: 10.4049/jimmunol.180.11.7203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ravetch JV, Bolland S. IgG Fc receptors. Annu Rev Immunol. 2001;19:275–290. doi: 10.1146/annurev.immunol.19.1.275. [DOI] [PubMed] [Google Scholar]
- 53.Nimmerjahn F, Ravetch JV. Fcgamma receptors: old friends and new family members. Immunity. 2006;24:19–28. doi: 10.1016/j.immuni.2005.11.010. [DOI] [PubMed] [Google Scholar]
- 54.Robak T. Novel monoclonal antibodies for the treatment of chronic lymphocytic leukemia. Current cancer drug targets. 2008;8:156–171. doi: 10.2174/156800908783769319. [DOI] [PubMed] [Google Scholar]
- 55.Fanale MA, Younes A. Monoclonal antibodies in the treatment of non-Hodgkin's lymphoma. Drugs. 2007;67:333–350. doi: 10.2165/00003495-200767030-00002. [DOI] [PubMed] [Google Scholar]
- 56.Monk NJ, Hargreaves RE, Marsh JE, Farrar CA, Sacks SH, Millrain M, Simpson E, Dyson J, Jurcevic S. Fc-dependent depletion of activated T cells occurs through CD40L-specific antibody rather than costimulation blockade. Nat Med. 2003;9:1275–1280. doi: 10.1038/nm931. [DOI] [PubMed] [Google Scholar]
- 57.Blair PJ, Riley JL, Harlan DM, Abe R, Tadaki DK, Hoffmann SC, White L, Francomano T, Perfetto SJ, Kirk AD, June CH. CD40 ligand (CD154) triggers a short-term CD4(+) T cell activation response that results in secretion of immunomodulatory cytokines and apoptosis. The Journal of experimental medicine. 2000;191:651–660. doi: 10.1084/jem.191.4.651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hancock WW, Sayegh MH, Zheng XG, Peach R, Linsley PS, Turka LA. Costimulatory function and expression of CD40 ligand, CD80, and CD86 in vascularized murine cardiac allograft rejection. Proc Natl Acad Sci USA. 1996;93:13967–13972. doi: 10.1073/pnas.93.24.13967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cella M, Scheidegger D, Palmer LK, Lane P, Lanzavecchia A, Alber G. Ligation of CD40 on dendritic cells triggers production of high levels of interleukin-12 and enhances T cell stimulatory capacity: T-T help via APC activation. J Exp Med. 1996;184:747–752. doi: 10.1084/jem.184.2.747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Koch F, Stanzl U, Jennewein P, Janke K, Heufler C, Kampgen E, Romani N, Schuler G. High level IL-12 production by murine dendritic cells: upregulation via MHC class II and CD40 molecules and downregulation by IL-4 and IL-10. J Exp Med. 1996;184:741–746. doi: 10.1084/jem.184.2.741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chen M, Huang L, Shabier Z, Wang J. Regulation of the lifespan in dendritic cell subsets. Mol Immunol. 2007;44:2558–2565. doi: 10.1016/j.molimm.2006.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dallman C, Johnson PW, Packham G. Differential regulation of cell survival by CD40. Apoptosis. 2003;8:45–53. doi: 10.1023/a:1021696902187. [DOI] [PubMed] [Google Scholar]
- 63.Nopora A, Brocker T. Bcl-2 controls dendritic cell longevity in vivo. J Immunol. 2002;169:3006–3014. doi: 10.4049/jimmunol.169.6.3006. [DOI] [PubMed] [Google Scholar]
- 64.Obst R, van Santen HM, Melamed R, Kamphorst AO, Benoist C, Mathis D. Sustained antigen presentation can promote an immunogenic T cell response, like dendritic cell activation. Proc Natl Acad Sci U S A. 2007;104:15460–15465. doi: 10.1073/pnas.0707331104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Obst R, van Santen HM, Mathis D, Benoist C. Antigen persistence is required throughout the expansion phase of a CD4(+) T cell response. The Journal of experimental medicine. 2005;201:1555–1565. doi: 10.1084/jem.20042521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kurtz J, Ito H, Wekerle T, Shaffer J, Sykes M. Mechanisms involved in the establishment of tolerance through costimulatory blockade and BMT: lack of requirement for CD40L-mediated signaling for tolerance or deletion of donor-reactive CD4+ cells. Am J Transplant. 2001;1:339–349. doi: 10.1034/j.1600-6143.2001.10409.x. [DOI] [PubMed] [Google Scholar]
- 67.Quezada SA, Bennett K, Blazar BR, Rudensky AY, Sakaguchi S, Noelle RJ. Analysis of the underlying cellular mechanisms of anti-CD154-induced graft tolerance: the interplay of clonal anergy and immune regulation. J Immunol. 2005;175:771–779. doi: 10.4049/jimmunol.175.2.771. [DOI] [PubMed] [Google Scholar]
- 68.Wells AD, Li XC, Li Y, Walsh MC, Zheng XX, Wu Z, Nunez G, Tang A, Sayegh M, Hancock WW, Strom TB, Turka LA. Requirement for T-cell apoptosis in the induction of peripheral transplantation tolerance. Nature Medicine. 1999;5:1303–1307. doi: 10.1038/15260. [DOI] [PubMed] [Google Scholar]
- 69.Graca L, Honey K, Adams E, Cobbold SP, Waldmann H. Cutting edge: anti-CD154 therapeutic antibodies induce infectious transplantation tolerance. J Immunol. 2000;165:4783–4786. doi: 10.4049/jimmunol.165.9.4783. [DOI] [PubMed] [Google Scholar]
- 70.Ford ML, Wagener ME, Gangappa S, Pearson TC, Larsen CP. Antigenic disparity impacts outcome of agonism but not blockade of costimulatory pathways in experimental transplant models. Am J Transplant. 2007;7:1471–1481. doi: 10.1111/j.1600-6143.2007.01826.x. [DOI] [PubMed] [Google Scholar]
- 71.Yu XZ, Albert MH, Martin PJ, Anasetti C. CD28 ligation induces transplantation tolerance by IFN-gamma-dependent depletion of T cells that recognize alloantigens. J Clin Invest. 2004;113:1624–1630. doi: 10.1172/JCI20940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Suntharalingam G, Perry MR, Ward S, Brett SJ, Castello-Cortes A, Brunner MD, Panoskaltsis N. Cytokine storm in a phase 1 trial of the anti-CD28 monoclonal antibody TGN1412. N Engl J Med. 2006;355:1018–1028. doi: 10.1056/NEJMoa063842. [DOI] [PubMed] [Google Scholar]
- 73.Pearson TC, Trambley J, Odom K, Anderson DC, Cowan S, Bray R, Lin A, Hollenbaugh D, Aruffo A, Siadak AW, Strobert E, Hennigar R, Larsen CP. Anti-CD40 therapy extends renal allograft survival in rhesus macaques. Transplantation. 2002;74:933–940. doi: 10.1097/00007890-200210150-00006. [DOI] [PubMed] [Google Scholar]