

Abstract
Oxidative stress is implicated in the pathogenesis of a variety of human diseases, including neurodegenerative disease, atherosclerosis and cancer, as well as progressive and even normal aging processes. Increased generation of free radicals derived primarily from molecular oxygen has also been associated with neuronal damage induced by a variety of environmental agents. However, measuring oxidative stress in biological systems is complex and requires accurate quantification of either free radicals or damaged biomolecules. One method to quantify oxidative injury is to measure lipid peroxidation. Lipids are readily attacked by free radicals, resulting in the formation of a number of peroxidation products. F2-isoprostanes (F2-IsoPs) are one group of these compounds, which are derived by the free radical peroxidation of arachidonic acid (AA). The F2-IsoPs, prostaglandine F2-like compounds, have been shown as the most accurate measure of oxidative damage in vivo. This review summarizes current methodology used to quantify F2-IsoPs and discusses the utility of these and other prostaglandine (PG)-like compounds as in vivo biomarkers of oxidative stress in neuronal tissues.
Keywords: F2-isoprostanes, oxidative damage, lipid peroxidation, neuroprostanes
Introduction
Oxidative stress is characterized by an imbalance between free radical production, principally derived from oxygen, and antioxidant defenses, consist of antioxidant enzymes and small molecular weight antioxidants, such as glutathione. Reactive radicals readily attack a variety of critical biological molecules, including DNA and essential cellular proteins. However, the high content of unsaturated lipids in the brain leads to pronounced lipid peroxidation, the central feature of oxidant injury in the brain. In addition, the brain is considered to be exceedingly sensitive to oxidative stress, because of its great consumption of oxygen, glucose and energy, as well as relatively low levels of antioxidants (Floyd, 1997; Simonian and Coyle, 1996).
Lipid peroxidation is the mechanism by which lipids are attacked by chemical species that have sufficient reactivity to abstract a hydrogen atom from a methylene carbon in their chain. Lipid peroxidation in vivo, through a free radical pathway, requires a polyunsaturated fatty acid (PUFA) and a reactant oxidant inducer that together form a free-radical intermediate. The free radical intermediate subsequently reacts with oxygen to generate a peroxyl radical, which with unpaired electrons may additionally abstract a hydrogen atom from another PUFA, thus initiating a propagation reaction that spreads like a brushfire. Hence, greater the number of double bonds in the molecule and higher instability of hydrogen atom adjacent to the double bond explains why unsaturated lipids are particularly susceptible to peroxidation (Pratico et al., 2004; Gao et al., 2006). Such reactions have been long recognized, but the biological importance of lipid peroxidation has been explored only in the last three decades (Gutteridge and Halliwell, 1990).
There have been a number of analytical approaches, which permit quantification of lipid peroxidation, or free radical-catalyzed damage to DNA or proteins (Lee and Blair, 2001). However, many of these techniques suffer from lack of sensitivity and specificity, especially when used to assess oxidant stress status in vivo. In a recent multi-investigator study, termed the Biomarkers of Oxidative Stress Study (BOSS), sponsored by the National Institutes of Health, it was found that the quantification of F2-IsoPs represents the most accurate method to assess oxidative stress status in vivo (Kadiiska et al., 2005). Accordingly, we highlight this method in the present review and address it advantages and shortcomings. We will commence the review with a discussion on the nature of the F2-IsoPs. This will be followed by a description on the methodology for quantification of F2-IsoPs and concrete examples on measurements of F2-IsoPs in studies conducted by our laboratory.
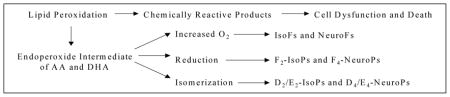
F2-IsoPs are prostaglandin-like compounds which are produced by a noncyclooxygenase free radical-catalyzed mechanism involving the peroxidation of the PUFA, arachidonic acid (AA, C20:4, ω-6). Formation of these compounds initially involves the generation of four positional peroxyl radical isomers of arachidonate, which undergo endocyclization to PGG2-like compounds. These intermediates are reduced to form four F2-IsoP regioisomers, each of which can consist of eight racemic diastereomers (Morrow et al., 1990). In contrast to cyclooxygenase (COX)-derived prostaglandins (PGs), non-enzymatic generation of F2-IsoPs favors the formation of compounds in which the stereochemistry of the side chains is oriented cis in relation to the prostane ring. A second important difference between F2-IsoPs and PGs is that F2-IsoPs are formed primarily in situ, esterified to phosphplipids and subsequently released by a phospholipases (Morrow et al., 2002; Gao et al., 2006; Famm and Morrow, 2003), whereas PGs are generated only from free arachidonic acid (Morrow et al., 1990).
F2-IsoPs analogues may be formed by peroxidation of other polyunsaturated fatty acid substrates, such as eicosapentaenoic acid (EPA), that lead to the production of F3-IsoPs, and docosahexaenoic acid (DHA), which generates F4-IsoPs. The latter compounds are also termed neuroprostanes (F4-NeuroPs), due to the high levels of their precursor in brain (Roberts et al., 1998).
Several methods have been developed to quantify the F2-IsoPs from biological materials (Basu, 2008). Our laboratory uses a gas chromatography/mass spectrometry (GC/MS) to quantify the F2-IsoPs, the methodology that was established at our University (by the pioneering work of Dr. Roberts and Dr. Morrow, Vanderbilt University Medical School) (Morrow et al., 1990). More specifically, after isolation and derivatization of the F2-IsoPs, we take advantage of stable isotope dilution, negative ion chemical ionization (NICI) GC/MS with select ion monitoring (SIM) for quantification. This methodology allows the lower limit of detection of the F2-IsoPs to be in the low picogram range. These properties, along with the assay’s high sensitivity and specificity, allow the F2-IsoPs to be an excellent biomarkers of and the most robust and sensitive measure of oxidative stress in vivo.
Quantification of F2-IsoPs
Measurement of F2-IsoPs has revolutionized our ability to quantify oxidative injury in vivo. F2-IsoPs are stable, robust molecules and are detectable not only in neuronal tissues but also in other tissues and biological fluids, such as plasma, urine, cerebrospinal fluid and bronchoalveolar lavage fluid. As F2-IsoPs can be readily generated ex vivo in biological materials containing arachidonoyl-containing lipids, it is important to process the samples immediately after isolation or assure their immediate storage at −70 °C for later quantification. Formation of F2-IsoPs does not occur if a free radical scavenging agent like butylated hydroxytoluene (BHT) is added to the organic solvent during extraction of phospholipids (Morrow and Roberts, 1999) or if the samples are rapidly frozen in liquid nitrogen prior to placement at −70 °C.
I. Lipid extraction and hydrolysis of F2-IsoPs-containing phospholipids in tissue samples
Formation of F2-IsoPs occurs in situ in the phospholipid bilayer and then subsequently released in free form. This creates two forms of F2-IsoPs, one that remains esterified in the membrane and a second that is hydrolyzed and released in free form. To quantify total F2-IsoPs formation, both free and esterified F2-isoPs are analyzed. It is necessary to extract the phospholiopids from the tissue and release the F2-isoPs from the phospholipids via base hydrolysis.
-
Fresh or frozen samples (0.05–0.25 g) are added in ice-cold 5 ml of Folch solution (chloroform:methanol, 2:1, vol/vol) containing 0.005% BHT in a polypropylene culture tube with cap. The tissue is then homogenized with a blade homogenizer (Kinematica PolytronR PT 10–35; Brinkmann Instruments, Westbury, NY) for approximately 30 sec. The second aliquot of ice-cold Folch solution, added to a separate culture tube, is used to wash the blade homogenizer and to ensure that all sample tissue is recovered as tissue can adhere to or become lodged inside the blade of the homogenizer. The two aliquots are then combined, covered with a nitrogen blanket, and mixed every 10 min over 30 min at 25 °C to allow maximal extraction of lipids from homogenized tissue.
Presence of butylated hydroxytuluene (BHT) (Sigma-Aldrich, cat. no. B1378) during extraction and hydrolysis is important in order to inhibit ex vivo formation of F2-IsoPs; Since polystyrene is not resistant to chloroform and its subsequent interference with the analytical procedures, it is recommended that lipid extraction be carried out in polypropylene tubes; Keep on ice. -
The lipid extracts are mixed vigorously with 2.0 ml NaCl (0.9%, wt/vol), and the phases separated by centrifugation at 300 × g for 10 min at 25 °C. After centrifugation, the upper aqueous layer is discarded and the lower organic layer carefully separated from the intermediate semisolid proteinaceous layer. Organic layer is then evaporated to dryness under a stream of nitrogen.
The organic layer and the proteinaceous layer can be readily separated by carefully pouring off the organic layer into a new culture tube. If the proteinaceous layer is small, because of the type and size of the tissue sample, it is often easier to remove the aqueous and proteinaceous layers simultaneously via suction. However, care must be taken not to compromise the organic phase if this approach is used. -
Total lipids are dissolved in 0.5 ml methanol containing BHT (0.005%), stored at 80 °C or if further processed, 0.5 ml of aqueous KOH (15%) added to the residue, and thus lipid extracts are saponified to release esterified isoprostanes. The mixture was sonicated and mixed vigorously until thoroughly suspended, and heated at 37 °C for 30 min to affect hydrolysis and release of the F2-IsoPs. The mixture is then acidified to pH 3 with 1 M HCl (cca 1.2 ml) and diluted to a final volume of 10 ml with pH 3 water in preparation for purification of F2-IsoPs with solid-phase extraction (SPE).
It is important to dilute the methanol in this solution to 5% or less to ensure proper column extraction of free F2-IsoPs in the subsequent purification procedure.
II. Sample purification for mass spectrometric analysis
-
Following acidification of the sample to pH 3 with 1 M HCl, 200 to 1000 pg of deuterated standard is added. The mixture is vortexed and F2-IsoPs isolated using reversed-phase and normal-phase solid-phase extractions (SPE)
The internal standard is a deuterium-labeled isoprostane, [2H4]15-F2t-IsoP (8-iso-PGF2a) (Cayman Chemical, Ann Arbor, MI, cat. no. 316351). The amount of internal standard added depends on the levels of F2-IsoPs in the sample as well as the sensitivity of the mass spectrometer. For low-level samples such as cerebrospinal fluid (CSF), less internal standard needs to be added. Samples that consist of a particularly large amount of tissue will require more internal standard. This is because complex tissues such as brain, despite our best purification efforts, will still contain some unwanted compounds that may potentially have the same m/z (mass-to-charge) ratio as the internal standard when analyzed by GC/MS. Increasing the amount of internal standard to 1000 pg in these samples minimizes the variability in the internal standard ion channel due to contamination in the tissue sample. A 10-ml plastic syringe (Laboratory Supply, cat. no. SMJ512878) is used to elute the sample and subsequent solvents through the Sep-Pak cartridge. For reverse phase, Sep Pak Plus C18 columns (Waters Associates, Milford, MA, cat. no. WAT03657; each cartridge contains 500 mg of C18) are preconditioned with 5 ml methanol (flow rate, ~1.0 ml/min) and 7.0 ml H2O (adjusted to pH 3.0 with 1.0N HCl). Once the sample has been added, the column is washed sequentially with 10 ml of water (pH 3) and 10 ml of heptane, which removes nonpolar contaminates including un-oxidized AA. The F2-IsoPs are eluted with 10 ml of ethyl acetate/heptane (50:50, vol/vol) into a 20-ml scintillation vial.
-
The ethyl acetate/heptane eluate from the C18 Sep-Pak is then dried over anhydrous Na2SO4 and applied to a silica Sep-Pak cartridge (Waters Associates, Milford, MA, cat. no. WAT036580; each cartridge contains 500 mg of silica), which has been preconditioned with 5 ml of ethyl acetate. Once the sample has been added, the column is washed with 5 ml of ethyl acetate and the F2-IsoPs are eluted with 5 ml of ethyl acetate/methanol (50:50, vol/vol) into a 5-ml glass react-a-vial (with Teflon-lined cap, Supelco, cat. No.33299). For normal-phase SPE, Sep Pak Plus Silica (Waters) columns are used with a flow rate of ~0.5 ml/min throughout.
Drying of ethyl acetate/heptane eluate should be completed promptly, as Na2SO4 has been shown to adsorb lipids to some degree. Care must be taken not to transfer any Na2SO4 to the silica Sep-Pak cartridge.
III. Conversion of F2-IsoPs to corresponding pentafluorbenzyl (PFB) esters
Isoprostanes isolated in ethyl acetate/methanol eluate by SPE are dried at 37 °C under a nitrogen stream, and derivatized to pentafluorbenzyl esters. Samples are vigorously mixed with 40 μl pentafluorobenzyl bromide: anhydrous acetonitrile (10:90, vol/vol) plus 20 μl diisopropylethylamine: anhydrous acetonitrile (10:90, vol/vol). Following reaction at 37 °C for 20 min, the esters are dried under a nitrogen stream, and dissolved in 50 μl chloroform:methanol (2:3, vol/vol).
Do not work outside of a well-ventilated hood because pentafluorobenzyl bromide (PFBB, Sigma-Aldrich, cat. no. 10105-2) is a potent lachrymator. N,N′-diisopropylethylamine (DIPE, Sigma-Aldrich, cat. no. D3887).
IV. Tin layer chromatography
Thin-layer chromatography (TLC) is accomplished with 5 cm × 20 cm glass plates covered with a 250μm layer of silica gel particles 60 A in diameter (Partisil LK6D; Whatman, Maidstone, England, cat. no. WC486562IV). Just before use, the plates are washed with ethyl acetate:ethanol (90:10, vol/vol), activated at 95 °C for 20 min, and cooled in a dessicator. A TLC chamber is lined with filter paper and conditioned 30 min with 100 ml chloroform: ethanol (90:10, vol/vol).
Dissolved samples in chloroform: methanol (50 μl) are applied to the upper half of pre-adsorbent in four pre-scored lanes, and dried 5–10 s with a hair dryer. Sample plates are added to both ends of the chamber. In contrast, TLC standard (5 μg of the methyl ester of PGF2α/5 μl CH3OH; Cayman Chemical, Ann Arbor, MI, cat. no. 16011) is applied to a separate plate that is positioned towards the center of the TLC chamber. After the chamber is rapidly closed, solvent is allowed to migrate 13 cm, and the plates removed.
Samples are scraped from silica plates in the region of the TLC standard visualized by spraying with a 10% solution of phosphomolybdic acid in ethanol (Sigma Chemical Co., St. Louis, MO, cat. no. P4869) followed by heating. The areas 1 cm below and 1 cm above PGF2α (Rf ~0.15) are scraped and extracted from the silica with 1 ml of ethyl acetate.
-
Following centrifugation at 13,000 × g for 3 min at 4 °C, isoprostane pentafluorobenzyl esters in the ethyl acetate are transferred into a virgin microcentrifuge tube and stored at 80 °C or samples further processed for the GC-MS analysis.
Ethyl acetate should be carefully removed without disrupting the silica pellet in the bottom of the tube (silica may affect instrument sensitivity). Avoid applying samples to the first 1 cm of the plate and do not spray the sample plate.
V. Formation of trimethylsilyl eter derivatives and quantification of F2-IsoPs
Once dried under a nitrogen stream, samples are dissolved in 8 μl dimethylformamide (DMF, Sigma-Aldrich, cat. no. 6407) and mixed with 20 μl bis(trimethylsilyl)trifluoroacetamide (BSTFA, Supelco, cat. no. 33084) to covert the residue to the trimethylsilyl ether derivatives.
-
After heating for 5.0 min at 37 °C, silylated samples are dried at 37 °C under a nitrogen stream, redissolved in 20 μl of undecane, which has been dried over calcium hydride, and transferred into autosampler vial for GC-MS analysis.
DMF should be stored over calcium hydride to prevent water accumulation. Similarly to the amount of internal standard that is added to a sample, consideration should be given to the amount of undecane that is used to dissolve the derivatized sample. The amount added will depend on the levels of F2-IsoPs. Samples that are rich in F2-IsoPs will require greater amounts of undecane to keep them from overloading the column during GC. Likewise, low-level samples will require less undecane in order for the GC/MS signal to be of sufficient intensity for optimal quantification. For quantification of F2-IsoPs, we routinely use a Hewlett Packard 5982A GC/MS system interfaced with an IBM Pentium computer. GC is performed using a 15-m, 0.25-mm diameter, 0.25-μm film thickness, DB1701 fused silica capillary column (Fisons, Folsum, CA). The column temperature is programmed from 190 to 290° at 20°/min. Methane is used as the carrier gas for NICI at a flow rate of 1 ml/min. Ion source temperature is 250 °C, electron energy is 70 eV, and the filament current is 0.25 mA.
The major ions generated in the NICI mass spectra of the pentafluorobenzyl ester, tris-trimethylsilyl ether derivatives of F2-IsoP are m/z 569 and corresponding ion for the [2H4]15-F2t-IsoP internal standard, m/z 573 (Figure 2). For quantification purposes we compare the height of the pick containing derivatized F2-IsoPs (m/z 569) with the height of the deuterated internal standard peak (m/z 573). Quantification of the F2-IsoPs levels may be also achieved by comparing the areas of the appropriate peaks in the m/z 569 SIM chromatogram of the F2-IsoPs to that of the peak of the internal standard in the m/z 573 SIM chromatogram (Figure 2).
Figure 2.

Chromatograms for F2-IsoPs and F4-NeuroPs from the mouse cerebrum. All chromatograms plot abundance vs. time (min). (A) m/z 569 chromatogram showing F2-IsoPs, (B) m/z 573 chromatogram showing internal standard, (C) m/z 593 chromatogram showing F4-NeuroPs.
The coefficient of variance for the assay is routinely less than 8%.
Timing and troubleshooting
In general, 12–15 samples can be assayed for F2-IsoPs in approximately 10 hrs by an experienced investigator. Homogenization, lipid extraction and hydrolysis of this number of samples requires ~3 hrs; Sep-Pac purifications takes ~ 2 hrs; drying, derivatization and TLC purification requires ~ 3 hrs; and drying and silylation requires ~ 2 hrs. Though compared to other assays of oxidative stress the time requirement for this assay is relatively large, it is noteworthy that the present assay has the greatest sensitivity and specificity for the detection of lipid peroxidation. Mass spectrometric analysis is automated and each sample requires ~ 15 min of instrument time. If the peak signal is low or if no peaks are detected by the mass spectrometer, the samples should be removed from autosampler vial, washed by ethyl acetate, dried under nitrogen and procedure for conversion to silylether derivative should be repeated. If the internal standard is detected at m/z 573 but there is low or non existent peak at 569, than levels of F2-IsoPs are below the limit of detection. The limit of detection for the assay is ~ 5 pg.
Alternative methods for measuring F2-IsoPs
GC-MS was the first technique used in early discovery and quantification of isoprostanes by investigators at our University (Morrow et al., 1990). Over the past 20 years, other methods, such as enzyme immunoassays, radioimmuniassays, liquid chromatography (LC)-mass spectrometry, GC-MS-MS, and LC-MS-MS (Basu, 1998; Wang et al., 1995: Liang, et al., 2003) have been developed and used to exploit the impact of this biomarker on human health and disease (Morrow et al., 1990; Wang et al., 1995; Pratico et al., 1998; Basu, 2007). The mass spectrometry-based methods are very sensitive and specific, but necessitate a skilled technical stuff and are laborious to execute, because of extensive purification and appropriate derivatization procedures. Immunoassays, although less specific and quantitative than GC-MS methods, have been found to be helpful tools for new discoveries in medical and pharmaceutical science. Immunoassays have a huge sample-analyzing capacity at fairly low expense. Thus, a well-validated technique could be a significant tool for evaluating free radical-mediated reactions in clinical research, where a large number of samples must be analyzed at an affordable cost. Specific antibodies against isoprostanes can also be used for in situ localization by immunostaining in oxidative stress-injured tissues. Immunostaining with specific antibodies opens possibilities for therapeutic application of various radical scavengers in disease-related damage (Basu, 2007).
Formation of isoprostanes from other polyunsaturated fatty acids
Similar studies of lipid peroxidation products have been performed for other substrate lipids. Of particular interest are oxidation products of docosahexaenoic acid (DHA, C22:6, ω-3), which have been termed F4-neuroprostanes (F4-NeuroPs). In contrast to AA, which is evenly distributed in all cell types in all tissues, DHA is highly concentrated in neuronal membranes (Salem et al., 1986). Thus, while the measurement of F2-IsoPs provides an index of global oxidative damage in the brain, integrating data from both glial and neuronal cells, determination of F4-NeuroPs permits the specific quantification of oxidative damage to neuronal membranes in vivo. In fact, to our knowledge, F4-NeuroPs are the only quantitative in vivo marker of oxidative damage that is selective for neurons. This is particularly important because of the implication of oxidative damage and lipid peroxidation being causative factors in numerous neurodegenerative diseases (Montine et al., 2004; Milatovic et al., 2005).
The assay for quantification of F4-NeuroPs is similar to the F2-IsoPs assay. A single sample can be used to measure both families of compounds by splitting the sample before the TLC step. After conversion to PFB esters, the sample is dissolve in twice the amount of methanol: chloroform (3:2, vol/vol) (100 μl). Then 50 μl are spotted on two separate lanes and the TLC plates are scraped accordingly, 1 cm above and below the methyl ester PGF2α standard for the F2-IsoPs and 1 cm below and 4 cm above the standard for the F4-NeuroPs. Quantification of F4-NeuroPs levels is achieved through SIM and comparing the area of the appropriate peaks in the m/z 593 SIM chromatogram of the F4-NeuroPs to that of the single peak of the internal standard in the m/z 573 SIM chromatogram (Figure 2).
In addition, three major structural isomers of IsoPs are formed from a common intermediate: F-ring IsoPs formed by reduction and D/E-ring IsoPs formed by isomerization (Morrow et al., 1998). The ratio of F-ring to D/E-ring compounds reflects the reducing environment in which F2-IsoPs form, with greater reducing equivalents favoring a higher ratio of F- to D/E-ring compounds (Morrow et al., 1998). More recently, it has been shown that in the presence of increased oxygen tension in the microenvironment in which lipid peroxidation occurs, an additional oxygen insertion step may take place (Fessel et al., 2002). This step diverts the IsoP pathway to form tetrahydrofuran ring-containing compounds termed isofurans (IsoFs), which are functional markers of lipid peroxidation under conditions of increased oxygen tension. Thus, measurements of IsoFs represent a much more robust indicator of hyperoxia-induced lung injury than measurements of F2-IsoPs. Like IsoPs, the IsoFs are chemically and metabolically stable so are well suited to act as in vivo biomarkers of oxidative damage.
Another F-ring IsoPs have been shown to generate from the peroxidation of eicosapentaenoic acid (EPA, C20:5, ω-3) which have been termed F3-IsoPs. Levels of IsoPs generated from EPA can significantly exceed those of F2-IsoPs generated from AA, perhaps because EPA contains more double bonds and is therefore more easily oxidizable (Gao et al., 2006).
F2-isoprostanes levels in neuronal tissues
Next we will provide some concrete examples on the utility of F2-IsoPs measurements from oxidative stress studies conducted in our laboratory. We have explored cerebral oxidative damage in a several models of neurodegeneration including excitotoxicity generated by kainic acid (KA) (Milatovic et al., 2005; Zaja-Milatovic et al., 2008), neurotoxicity associated with anticholinesterase agents (Milatovic et al., 2006; Gupta et al., 2007) and innate immune activation by lipopolysaccharide (LPS) (Milatovic et al., 2003, 2004). We routinely measured two markers of free radical damage to lipids, F2-IsoPs and F4-NeuroPs, using methodologies analogous to those described above; the former reflecting oxidative damage to all cerebral tissue elements and the latter providing a relatively selective window into peroxidation of neuronal membranes. For glial innate immune response we have used a single intracerebroventricular (ICV) injection of LPS, a major component of Gram negative bacterial cell walls. We have previously shown that LPS-activated glial innate immune response leads to indirect neuronal oxidative damage and synaptodendritic degeneration exclusively through a CD14-dependent mechanism free of behavioral or febrile response, and that LPS itself has no direct toxic effect on neurons (Montine et al., 2002; Milatovic et al., 2004). Control mice have received ICV PBS, which led to no change in cerebral F2-IsoPs or F4-NeuroPs over 24 h following ICV PBS vehicle; average (+S.D.) basal F2-IsoPs = 3.2 + 0.3 ng/g and basal F4-NeuroPs = 12.6 + 2.7 ng/g. ICV LPS generated delayed cerebral oxidative damage with no significant increase in cerebral F2-IsoPs even 10 h after injection. However, following this delay, cerebral oxidative damage peaked at about 24 h following ICV LPS and then decreased to near baseline levels by 36 h. An identical time course has been observed for F4-NeuroPs following LPS administration (Figure 3) (Milatovic et al., 2005).
Figure 3.

Time course for cerebral oxidative damage following ICV injection of LPS. Data are F2-IsoPs and F4-NeuroPs expressed as ng per g tissue following injection at time = 0 (n≥5 per time point). * p<0.01 compared to untreated control.
In contrast to indirect neuronal damage induced with ICV LPS, there was a rapid onset of detectable oxidative damage upon ICV KA that was greatest at the earliest time point (30 min) following injection, returned to baseline by 60 min, and remained at baseline levels 24 h after injection (Zaja-Milatovic et al., 2008). Kainate is a rigid analog of glutamate, and is a very potent stimulant of a subset of ligand-gated ion channels called KA receptors (Ben-Ari and Cossart, 2000; Milatovic et al., 2005). While mice entered status epilepticus within a minute of ICV KA injection, KA-induced seizures did not lead to activated resident inflammatory cells in brain until several days (>4) after exposure, outside of the time frame used in our experiments. Thus, cerebral oxidative damage from these two toxins followed very different temporal profiles. KA led to the rapid onset of cerebral oxidative damage while LPS led to delayed oxidative damage. In both instances, the magnitude of cerebral oxidative damage achieved in these two models was equivalent and comparable to that observed in diseased regions of brain from patients with Alzheimer’s disease (Reich et al., 2001).
We have also evaluated metal-induced alterations in biomarker of oxidative damage, F2-IsoPs, in primary astrocytes and neuronal cultures as well as peripheral nervous system. Our preliminary data show that primary rat cortical neurons exposed to 0.5 mM of manganese for 6 hours showed structural damage to neurons and significant increase in F2-IsoPs levels (78.6 pg/mg protein) compared to controls (52.2 pg/mg protein). At the same experimental condition, primary astrocytes cultures showed 51% increase in F2-IsoPs levels compared to control (137.3 pg/mg protein) (Milatovic et al., 2007). Our previous study also showed that N,N-diethyldithiocarbamate (DEDC) mediates lipid oxidation and elevation of total copper in peripheral nerve. F2-isoprostane levels in rat sciatic nerve were significantly elevated in the 2-week DEDC-exposed group (1.9 ng/g of sciatic nerve) over controls (1.2 ng/g of sciatic nerve) (Viquez et al., 2008). This relationship is consistent with copper-mediated oxidative stress contributing to the myelinopathy. Combined, the F2-IsoPs method has been shown reliable and reproducible in plethora of condition associated with oxidative stress, providing an early and exquisitely sensitive biomarker for lipid peroxidation.
Conclusion
The discovery of the F2-IsoPs as products of non-enzymatic lipid peroxidation has been a major breakthrough in the filed of lipid peroxidation and free radical chemistry. The quantification of F2-IsoPs has extended our understanding the role of free radicals in physiological processes and has established the occurrence of oxidative stress in a wide variety of research models and disease states. In addition to F2-IsoPs, knowledge of the lipid peorxidation products of DHA, F4-NeuroPs have provided a unique insight into oxidative damage occurring within neurons. Thus, availability of sensitive assays for quantification of F2-IsoPs and F4-NeuroPs levels in neuronal tissues provides powerful approach to advance our understanding of the role of oxidative damage in diseases of the CNS and the role of different cell types in this process. The method is reproducible and reliable and serves as an excellent platform for future studies on the role of oxidative stress in mediating human disease. Furthermore, the sensitivity of the method offers unique potential for surveying F2-IsoPs concentrations as potential biomarkers of early disease, as they can be reliably and accurately measured in a plethora of biological media.
Figure 1.

Schematic presentation of procedure used for the extraction, purification, derivatization and mass spectrometric analysis of F2-IsoPs from neuronal tissues
Acknowledgments
This study was supported by grants from NIH NS057223 (DM), NIEHS ES 007331, NIEHS 10563 and DoD W81XWH-05-1-0239 (MA).
Literature Cited
- Basu S. Radioimmunoassay of 8-iso-prostaglandin F2alpha: an index of oxidative injury via free radical catalysed lipid peroxidation. Prostaglandins Leukot Essent Fatty Acids. 1998;58:319–325. doi: 10.1016/s0952-3278(98)90042-4. [DOI] [PubMed] [Google Scholar]
- Basu S. F2-isoprostanes in human health and diseases: from molecular mechanisms to clinical implications. Antioxidants and Redox Signaling. 2008;10:1405–1434. doi: 10.1089/ars.2007.1956. [DOI] [PubMed] [Google Scholar]
- Ben-Ari Y, Cossart R. Kainate, a double agent that generates seizures: two decades of progress. Trends Neurosci. 2000;23:580–587. doi: 10.1016/s0166-2236(00)01659-3. [DOI] [PubMed] [Google Scholar]
- Crow JA, Borazjani A, Potter PM, Ross MK. Hydrolysis of pyrethroids by human and rat tissue: Examination of intestinal, liver, and serum carboxylesterases. Toxicol Appl Pharmacol. 2007;221:1–12. doi: 10.1016/j.taap.2007.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Famm SS, Morrow JD. The isoprostanes: Unique products of arachidonic acid oxidation- a review. Curr Med Chem. 2003;10:1723–1740. doi: 10.2174/0929867033457115. [DOI] [PubMed] [Google Scholar]
- Fessel JP, Porter NA, Moore KP, Sheller JR, Roberts LJ., 2nd Discovery of lipid peroxidation products formed in vivo with a substituted tetrahydrofuran ring (isofurans) that are favored by increased oxygen tension. Proc Natl Acad Sci U S A. 2002;99:16713–16718. doi: 10.1073/pnas.252649099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Floyd RA. Protective action of nitrone-based free radical traps against oxidative damage to the central nervous system. Adv Pharmacol. 1997;38:361–378. doi: 10.1016/s1054-3589(08)60991-6. [DOI] [PubMed] [Google Scholar]
- Gao L, Yin H, Milne GL, Porter NA, Morrow JD. Formation of F-ring Isoprostane-like Compounds (F3-Isoprostanes) in Vivo from Eicosapentaenoic Acid. J Biol Chem. 2006;281:14092–14099. doi: 10.1074/jbc.M601035200. [DOI] [PubMed] [Google Scholar]
- Gupta RC, Milatovic S, Dettbarn WD, Aschner M, Milatovic D. Neuronal oxidative injury and dendritic damage induced by carbofuran: Protection by memantine. Toxicol Appl Pharmacol. 2007;219:97–105. doi: 10.1016/j.taap.2006.10.028. [DOI] [PubMed] [Google Scholar]
- Gutteridge JMC, Halliwell B. The measurement and mechanism of lipid peroxidation in biological systems. Trends Biochem Sci. 1990;15:129–1365. doi: 10.1016/0968-0004(90)90206-q. [DOI] [PubMed] [Google Scholar]
- Kadiiska MB, Gladen BC, Baird DD, Germolec D, Graham LB, Parker CE, Nyska A, Wachsman JT, Ames BN, Basu S, Brot N, Fitzgerald GA, Floyd RA, George M, Heinecke JW, Hatch GE, Hensley K, Lawson JA, Marnett LJ, Morrow JD, Murray DM, Plastaras J, Roberts LJ, II, Rokach J, Shigenaga MK, Sohal RS, Sun J, Tice RR, Van Thiel DH, Wellner D, Walter PB, Tomer KB, Mason RP, Barrett JC. Biomarkers of Oxidative Stress Study II: Are oxidation products of lipids, proteins, and DNA markers of CCl4 poisoning? Free Radic Biol Med. 2005;38:698–710. doi: 10.1016/j.freeradbiomed.2004.09.017. [DOI] [PubMed] [Google Scholar]
- Lee SH, Blair IA. Oxidative DNA damage and cardiovascular disease. Trends Cardiovasc Med. 2001;11:148–155. doi: 10.1016/s1050-1738(01)00094-9. [DOI] [PubMed] [Google Scholar]
- Liang Y, Wei P, Duke RW, Reaven PD, Harman SM, Cutler RG, Heward CB. Quantification of 8-iso-prostaglandin-F(2alpha) and 2,3-dinor-8-iso-prostaglandin-F(2alpha) in human urine using liquid chromatography-tandem mass spectrometry. Free Radic Biol Med. 2003;34:409–418. doi: 10.1016/s0891-5849(02)01018-3. [DOI] [PubMed] [Google Scholar]
- Milatovic D, Gupta RC, Aschner M. Anticholinesterase Toxicity and Oxidative Stress. The Scientific World Journal. 2006;6:295–310. doi: 10.1100/tsw.2006.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milatovic D, Milatovic S, Montine K, Shie FS, Montine TJ. Neuronal oxidative damage and dendritic degeneration following activation of CD14-dependent innate immunity response in vivo. J Neuroinflamm. 2004;1:20. doi: 10.1186/1742-2094-1-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milatovic D, Zaja-Milatovic S, Montine KS, Horner PJ, Montine TJ. Pharmacologic suppression of neuronal oxidative damage and dendritic degeneration following direct activation of glial innate immunity in mouse cerebrum. J Neurochem. 2003;87:1518–1526. doi: 10.1046/j.1471-4159.2003.02120.x. [DOI] [PubMed] [Google Scholar]
- Milatovic D, VanRollins M, Li K, Montine KS, Montine TJ. Suppression of cerebral oxidative damage from excitotoxicity and innate immune response in vivo by α- or γ-tocopherol. J Chromatography B. 2005;827:88–93. doi: 10.1016/j.jchromb.2005.03.037. [DOI] [PubMed] [Google Scholar]
- Milatovic D, Yin Z, Gupta RC, Sydoryk M, Albrecht J, Aschner JL, Aschner M. Manganese induces oxidative impairment in cultured rat astrocytes. Toxicol Science. 2007;98:198–205. doi: 10.1093/toxsci/kfm095. [DOI] [PubMed] [Google Scholar]
- Montine TJ, Milatovic D, Gupta RC, Morrow JD, Breyer R. Neuronal oxidative damage from activated innate immunity in EP2 receptor-dependent. J Neurochem. 2002;83:463–470. doi: 10.1046/j.1471-4159.2002.01157.x. [DOI] [PubMed] [Google Scholar]
- Montine KS, Quinn JF, Zhang J, Fessel Jp, Roberts LJ, 2nd, Morrow JD, Montine TJ. Isoprostanes and related products of lipid peroxidation in neurodegenerative diseases. Chem Phys Lipids. 2004;128:117–718. doi: 10.1016/j.chemphyslip.2003.10.010. [DOI] [PubMed] [Google Scholar]
- Morrow JD, Hill KE, Burk RF, Nammour TM, Badr KF, Roberts LJ., 2nd A series of prostaglandin F2-like compounds are produced in vivo in humans by a non-cyclooxygenase, free radical-catalyzed mechanism. Proc Natl Acad Sci U S A. 1990;87:9383–9387. doi: 10.1073/pnas.87.23.9383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrow JD, Awad JA, Boss HJ, Blair IA, Roberts LJ., II Non-cyclooxygenase derived prostanoids (F2-isoprostanes) are formed in situ on phospholipids. Proc Natl Acad Sci USA. 1992;89:10721–10725. doi: 10.1073/pnas.89.22.10721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrow JD, Roberts LJ. Mass spectrometric quantification of F2-isoprostanes in biological fluids and tissues as a measure of oxidant stress. Methods Enzymol. 1999;300:3–12. doi: 10.1016/s0076-6879(99)00106-8. [DOI] [PubMed] [Google Scholar]
- Morrow JD, Harris TM, Roberts LJ., 2nd Noncyclooxygenase oxidative formation of a series of novel prostaglandins: analytical ramifications for measurement of eicisanoids. Analyt Biochem. 1990;184:1–10. doi: 10.1016/0003-2697(90)90002-q. [DOI] [PubMed] [Google Scholar]
- Morrow JD, Roberts LJ, Daniel VC, Awad JA, Mirochnitchenko O, Swift LL, Burk RF. Comparison of formation of D2/E2-isoprostanes and F2-isoprostanes in vitro and in vivo--effects of oxygen tension and glutathione. Arch Biochem Biophys. 1998;353:160–171. doi: 10.1006/abbi.1998.0645. [DOI] [PubMed] [Google Scholar]
- Pratico D, Barry OP, Lawson JA, Adiyaman M, Hwang SW, Khanapure SP, Iuliano L, Rokach J, Fitzgerald GA. IPF2alpha-I: an index of lipid peroxidation in humans. Proc Natl Acad Sci USA. 1998;95:3449–3454. doi: 10.1073/pnas.95.7.3449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pratico D, Rokach J, Lawson J, FitzGerald GA. F2-isoprostanes as indices of lipid peroxidation in inflammatory diseases. Chem Phys Lipids. 2004;128:165–171. doi: 10.1016/j.chemphyslip.2003.09.012. [DOI] [PubMed] [Google Scholar]
- Reich EE, Markesbery WR, Roberts LJ, II, Swift JD, Morrow JD, Montine JT. Brain Regional Quantification of F-Ring and D-/E-Ring Isoprostanes and Neuroprostanes in Alzheimer’s Disease. Am J Pathol. 2001;158:293–297. doi: 10.1016/S0002-9440(10)63968-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts LJ, II, Montine TJ, Markesbery WR, Tapper AR, Hardy P, Chemtob S, Dettbarn WD, Morrow JD. Formation of Isoprostane-like Compounds (Neuroprostanes) in Vivo from Docosahexaenoic Acid. J Biol Chem. 1998;273:13605–13612. doi: 10.1074/jbc.273.22.13605. [DOI] [PubMed] [Google Scholar]
- Salem N, Kim HY, Lyergey JA. Docosahexaenoic acid: membrane function and metabolism. In: Martin RE, editor. Health effects of polyunsaturated acids in seafoods. Academic Press; New York: 1986. pp. 263–317. [Google Scholar]
- Simonian NA, Coyle JT. Oxidative stress in neurodegenerative diseases. Annu Rev Pharmacol Toxicol. 1996;36:83–106. doi: 10.1146/annurev.pa.36.040196.000503. [DOI] [PubMed] [Google Scholar]
- Viquez OM, Valentine HL, Amarnath K, Milatovic D, Valentine WM. Copper accumulation and lipid oxidation precede inflammation and myelin lesions in N,N-diethyldithiocarbamate peripheral myelinopathy. Toxicol Appl Pharmacol. 2008;229:77–85. doi: 10.1016/j.taap.2008.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Ciabattoni G, Creminon C, Lawson J, Fitzgerald GA, Patrono C, Maclouf J. Immunological characterization of urinary 8-epi-prostaglandin F2 alpha excretion in man. J Pharmacol Exp Ther. 1995;275:94–100. [PubMed] [Google Scholar]
- Zaja-Milatovic S, Gupta RC, Aschner M, Montine TJ, Milatovic D. Pharmacologic suppression of oxidative damage and dendritic degeneration following kainic acid-induced excitotoxicity in mouse cerebrum. Neurotoxicol. 2008;29:621–627. doi: 10.1016/j.neuro.2008.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]