

Abstract
The most commonly used procedures to induce arterial injury in mice are carotid artery ligation with cessation of blood flow and mechanically-induced denudation of endothelium in the carotid or the femoral arteries. Both procedures result in neointimal hyperplasia after two to three weeks. A survey of various inbred strain of mice shows that strain-specific differences in susceptibility to injury-induced neointimal hyperplasia are different than those for susceptibility to diet-induced atherosclerosis, with strains identified as susceptible to both neointimal hyperplasia and atherosclerosis, resistant to both, susceptible to atherosclerosis but resistant to neointimal hyperplasia, or resistant to atherosclerosis but susceptible to neointimal hyperplasia. Inflammatory cells such as T and B lymphocytes, which are contributory to atherosclerosis, are protective against injury-induced neointimal hyperplasia. In contrast, the infiltration of monocytes into the site of injury and their differentiation to macrophages favor neointimal hyperplasia similar to their pathogenic role in atherosclerosis. The regulatory role of lymphocytes and macrophages in neointimal hyperplasia is related to the production of cytokines such as interferon-γ and tumor necrosis factor-α, respectively. Interestingly, inducible nitric oxide synthase (iNOS) activity appears to inhibit neointimal hyperplasia in the endothelial denudation model but contributes to neointimal hyperplasia when arterial injury is induced by periadventitial cuff placement. The difference appears to be due to the time required for endothelial recovery and the participation of inflammatory cells. Thus, although arterial injury-induced neointimal hyperplasia results in similar vascular occlusion as progressive atherosclerosis, the pathology and mechanism of the two disease processes are quite different.
Keywords: Neointimal Hyperplasia, Mouse Genetics, Nitric Oxide, Lymphocytes, Smooth Muscle Cells, Macrophages, Bone Marrow Progenitor Cells
Background and Introduction
Despite recent advances in treatment strategies to significantly reduce the risk of heart disease, myocardial infarction and other forms of ischemic heart failure due to atherosclerosis remain the major causes of death in industrialized countries [1]. In a recent report examining the association of risk factors with coronary heart disease in 350,000 patients, almost 90% of the patients had prior history of diabetes, smoking, hypercholesterolemia, and/or hypertension [2]. This study convincingly documented that exposure to one or more of these risk factors is causative for atherosclerosis. However, it is important to note that not everyone with exposure to these risk factors has atherosclerotic disease. The current hypothesis is that atherosclerosis is a chronic inflammatory disease initiated by vascular endothelial injury as a consequence of hyperlipidemia, hypertension, and constant exposure to cytopathic agents including cigarette smoke and other risk factors such as homocysteine and infectious microorganisms [3]. The injury-induced endothelial dysfunction increases leukocyte and platelet adherence, thus inducing procoagulant activities and the release of vasoactive cytokines and growth factors [3]. Circulating monocytes recruited to the injured endothelium also differentiate into macrophages thereupon their endocytosis of modified LDL leading to foam cell formation. The inflammatory response also leads to activation of the normally quiescent medial smooth muscle cells, resulting in their migration to the intima and their proliferation to form an intermediate lesion. If this process is unabated, or if endothelial injury persists without repair, the cycle continues and the lesion expands with formation of fibrous tissues that overlay the core of lipid and necrotic materials forming an occlusive plaque [3]. It is the balance between endothelial repair and chronic vascular inflammation with endothelial dysfunction that determines progression and severity of atherosclerosis.
Once the atherosclerotic plaque develops sufficiently to impede blood flow, the only current treatments available to alleviate vascular occlusion are limited to surgical procedures such as transluminal coronary angioplasty, percutaneous delivery of balloon-expanded stents, directional coronary atherectomy, and coronary bypass surgery. Unfortunately, re-occlusion of the target vessel occurs frequently after these procedures, thus limiting the potential success for achieving long-term vascular patency [4]. The pathogenic mechanism of surgery-induced arteriosclerosis shares common features as well as distinct characteristics as that of progressive atherosclerosis. Like atherosclerosis, the etiology of surgery-induced arteriosclerosis is also endothelial dysfunction and denudation. However, the accelerated forms of surgery-induced arteriosclerosis are different from progressive atherosclerosis in that lipid deposition and macrophage foam cell appearance are late events [5-9]. Interestingly, the pathology of various forms of surgery-induced arteriosclerosis is also different depending on the intervention strategy used [10-12]. For example, neointimal hyperplasia with smooth muscle cell migration from the tunica media to the intima and intimal cell proliferation are the major contributing factors in vascular occlusion after bypass grafts and cardiac transplant patients. In contrast, restenosis from balloon angioplasty is primarily due to vessel elastic recoil, negative constrictive remodeling, thrombus formation at the site of injury, smooth muscle cell migration and proliferation, and excessive extracellular matrix deposition. Although vascular stent placement has eliminated the possibility of vessel elastic recoil and constrictive remodeling, neointimal hyperplasia with smooth muscle cell migration and proliferation and excessive extracellular matrix deposition remains the major factor of in-stent restenosis [10, 13]. More recently, drug-eluting stents have shown tremendous promise for alleviating acute in-stent restenosis [14, 15]. Unfortunately, post drug-eluting stent restenosis still occurs in a number of cases and the latest studies have questioned the safety of drug-eluting stents, highlighting the increased frequency of thrombotic events occurring far beyond that observed with bare-metal stents [16, 17]. Moreover, coated stents cannot be used to treat all forms of neointimal hyperplasia. For example, neointimal hyperplasia with smooth muscle cell migration and proliferation and excessive extracellular matrix deposition is also a common feature of accelerated arteriosclerosis after bypass surgery or allograft cardiac transplant. Venous neointimal hyperplasia resulting in vascular occlusion is also common in hemodialysis grafts [10, 12, 13, 18, 19]. Neointimal hyperplasia as a result of these surgical procedures or as a result of progressive atherosclerosis cannot be alleviated with coated stents. Thus, an alternative strategy needs to be designed to alleviate these vascular complications due to neointimal hyperplasia. The accomplishment of this goal requires a thorough understanding of the mechanism of neointimal hyperplasia after endothelial injury. Mouse models have become the experimental animal model of choice due to their well-defined genetics as well as availability of numerous inbred strains and the ability to manipulate the mouse genome. The goal of this article is to summarize some of the key findings, highlighting similarities and differences between diet-induced progressive atherosclerosis and injury-induced neointimal hyperplasia in mice.
Arterial Injury in Mice
The two most commonly used procedures to induce endothelial denudation in mice are carotid artery ligation and mechanical denudation of the endothelial layer in the vessel wall. In the arterial ligation model, blood flow in the common carotid artery is disrupted by ligation near the distal bifurcation [20]. This procedure results in dramatic reduction of vessel diameter and the decreased blood flow that results in normalizing wall shear stress, leading to the formation of an extensive smooth muscle cell-rich neointima after 2-4 weeks. One advantage of this model is its reproducibility due to the ease of operation. However, the disadvantage of this model is that it does not resemble the physiological setting of vascular intervention, thus the mechanism responsible for neointimal formation due to arterial ligation may be different from neointimal formation due to endothelial denudation as most often seen in clinical settings such as balloon angioplasty and intravascular stent implementation.
A mechanically-induced endothelial denudation model was initially developed by Lindner and colleagues by passing a flexible guide wire through the carotid artery three times [21]. The removal of the endothelial cell layer results in the covering of the denuded surface with a platelet monolayer and the activation of the underlying medial smooth muscle cells. Neointimal formation is typically observed within 2 weeks after injury. The advantage of this experimental procedure is its resemblance to angioplasty and its maintenance of normal blood flow. The disadvantage of this procedure is the relatively challenging operational procedure, which leads to difficulty in achieving reproducible results. In many instances, insertion and passage of the guide wire through the vessel wall results in tearing and disruption of the elastic lamina, thus causing exuberant smooth muscle cell response.
Our laboratory has modified the endothelial denudation procedure substituting the guide wire with an epon resin probe made by forming epon beads slightly larger than the diameter of the carotid artery on a 3-0 nylon suture (Fig. 1). The epon resin probe can be custom made according to the diameter of the carotid artery of each animal. In our procedure, the mice are anesthetized and immobilized. The fur covering the neck from sternum to chin is removed with lotion hair remover before exposing the entire length of one carotid artery. The exposed artery is ligated immediately proximal from the point of bifurcation with a 7-0 suture. Another 7-0 suture is placed around the common carotid artery immediately distal from the branch point of the external carotid. A transverse arteriotomy is then made between the 7-0 sutures for the insertion of the resin probe, which is then advanced toward the aortic arch and withdrawn five times, as diagrammed in Figure 1. At the end of the procedure, the probe is removed and the proximal 7-0 suture is ligated. Blood flow is restored through the carotid branch points and the incision closed with a 5-0 suture. The entire procedure usually is performed within 20 min. Complete endothelial cell denudation can be confirmed 1 hr after injury by en face Evans Blue staining of the whole vessel and Von Willebrand Factor (VWF) immunohistochemical staining of cross sections obtained from the injured vessel and the contralateral uninjured carotid artery (Fig. 1). In a typical experiment, we usually allow the experimental animals to recover and then housed them under normal conditions for 14 days prior to their sacrifice for analysis. Characterization of neointimal hyperplasia is typically performed with paraffin embedded tissues. In this procedure, mice are anesthetized and perfusion fixed with 10% buffered formalin for 20 min at a constant pressure of 100 mm Hg. The entire neck is usually dissected from each mouse, fixed in 10% buffered formalin for an additional 48 hr, and then decalcified for 48 hr prior to embedding in paraffin. We usually make identical whole-neck cross sections of 5 μm each, starting from the distal side of the neck beginning at the point of the distal 7-0 ligature. The whole neck sections allow the evaluation of the injured artery in comparison with the uninjured artery serving as the internal control. Representative photomicrographs of whole neck sections showing both the uninjured and injured carotid arteries of a mouse are shown in Figure 2. For each animal, four levels of serial sections are obtained at 500-μm intervals. Parallel sections are subjected to Hematoxylin and eosin staining and for Verhoeff VanGieson (VVG) staining of the elastic lamina. Unstained serial sections from each level can be used for immunohistochemistry.
Figure 1.
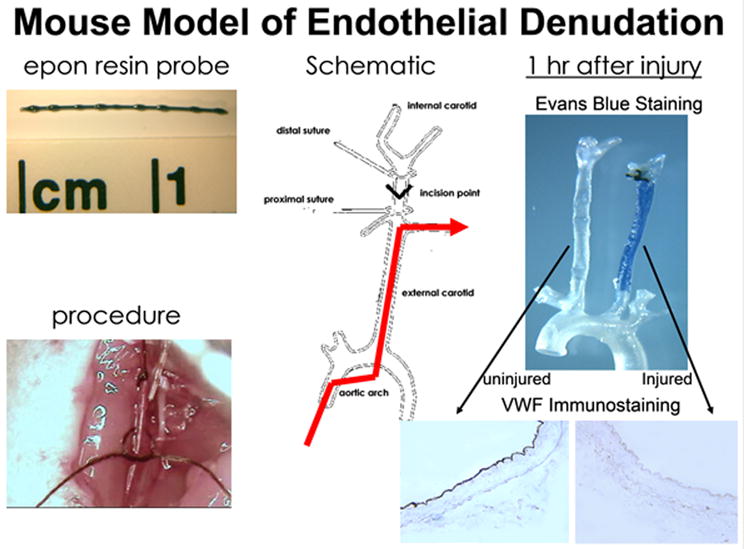
Mouse model of endothelial denudation. The top left panel shows the epon resin probe used to induce endothelial denudation of the carotid arteries in mice. The bottom left panel is a photomicrograph of the actual procedure by which the epon resin probe is inserted into the carotid artery. A schematic of the procedure is shown in the middle panel. The red line shows the restoration of blood flow after the vascular injury procedure. The right hand panel shows results of endothelial denudation 1 hr after the procedure. The top right panel is the Evans Blue staining of the left uninjured artery and the right injured artery, with the bottom right panel showing VWF immunostaining of the sections dissected from the respective carotid arteries.
Figure 2.

Representative photomicrographs of a whole-neck section with Hematoxylin and Eosin staining. Panel A shows the whole neck section with both the injured left carotid artery and the uninjured right carotid artery. Scale bar, 100 μm. Panels b and c show the magnified versions of the ininjured and injured arteries, respectively. Scale bar, 50 μm. The arrows indicate the external elastic laminae and the arrowheads indicate the internal elastic laminae in each section. This figure is reprinted from ref. 29 with permission from the American Society for Investigative Pathology.
In our laboratory, morphometric analysis is performed on elastin-stained sections. For each animal, four whole neck cross sections with both injured and uninjured carotid arteries are measured. Images are digitized and captured with video camera connected to a personal computer. Measurements are performed at a magnification of ×200 using a Scion Image analysis computer program. For each artery, luminal area, area inside the internal elastic lamina, and the area encircled by external elastic lamina are measured. Medial area is calculated as area encircled by external elastic lamina-area inside the internal elastic lamina and intimal area is calculated as area inside the internal elastic lamina--luminal area. To calculate the medial thickness for each vessel cross-section, the linear distance between internal elastic lamina and external elastic lamina is measured independently in four places, each separated at 90° apart and then averaged. The ratio of intimal area and medial area and the percent of luminal stenosis are calculated based on these measurements.
Genetic Influence on Vascular Response to Injury
Several studies have shown that genetic background significantly influences vascular response to injury in mice. In an early report, Carmeliet et al [22] showed that C57BL/6 mice are less responsive than C57BL/6 × 129 mixed background mice in vascular occlusion after injury. Our laboratory extended this study by examining 5 different inbred strains of mice, namely C57BL/6J, C57L/J, FVB/N, 129sv, and C3H/He, for their response to resin-probe induced injury of the carotid arteries. Our results showed that, whereas the thickness of the media layer increased 14 days after arterial injury in all 5 inbred strains of mice, only 2 strains, the C57L/J and FVB/N, were identified as susceptible strains with massive neointimal area and 30-50% stenosis of the injured arteries after endothelial denudation [23]. The other 3 strains of mice were resistant with minimal neointimal area and <10% arterial stenosis 14 days after injury. Interestingly, the C57BL/6 mice, which are well characterized as a susceptible strain to diet-induced atherosclerosis, were found to be resistant to neointimal hyperplasia after endothelial denudation. In contrast, the FVB/N mice, which are resistant to diet-induced atherosclerosis, were found to be susceptible to neointimal hyperplasia after mechanical denudation of their vascular endothelium [23]. The C57L/J mice are susceptible to both diet-induced atherosclerosis and injury-induced neointimal hyperplasia, whereas the C3H/He mice are resistant to both. These results clearly demonstrated genetic variation as a determinant for vascular stenosis in response to mechanical denudation of the endothelium. Importantly, the data documented that genetic determinants for injury-induced neointimal hyperplasia are distinct from those that determine susceptibility to diet-induced atherosclerosis.
In addition to the survey of the inbred strains of mice, we have evaluated vascular stenosis after endothelial denudation in mice derived from F1 cross between the susceptible C57L/J and the resistant C57BL/6J mice. Examination of over 40 F1 mice showed minimal neointimal hyperplasia and vascular stenosis 14 days after endothelial denudation [23]. These results suggested that a recessive gene that is homozygous in C57L/J mice confers susceptibility to neointimal hyperplasia. Additional experiments performed in N2 hybrid mice generated by backcrossing the F1 hybrid mice to the susceptible C57L/J mice revealed a wide range of neointimal sizes, spanning from the resistant phenotype to ones with severe neointimal hyperplasia similar to that observed in the susceptible C57/LJ parental strain. Of 77 mice examined in the study, 56 mice (72.7%) of the N2 mice were resistant to injury-induced neointimal hyperplasia and 8 (10.4%) were found to be highly susceptible. Thirteen N2 mice (16.9%) displayed neointimal size at the intermediate level. Therefore, the distribution of neointima-susceptible and -resistant mice in the N2 hybrid mice suggests that there are at least 2 genes within the C57BL/6 genome that dictate resistance to injury-induced neointimal hyperplasia. The identity of these genes remains unknown and requires additional detailed genetic mapping studies.
The carotid ligation model has also been used to determine strain-specific differences in vascular response to injury. In a study comparing 11 different strains of inbred mice, Lindner and colleagues showed a wide variation in response with some strains showing vessel enlargement (positive remodeling) while other strains displaying reduction in vessel diameter (negative remodeling) [24]. Interestingly, carotid ligation model revealed luminal narrowing in all 11 inbred strains of mice. Nevertheless, strain-specific differences remained noticeable. The FVB/N mice, which are most susceptible to neointimal hyperplasia in the mechanical denudation model, along with the SJL/J mice were found to be the most susceptible to luminal area reduction after carotid artery ligation [24]. However, the determinants of luminal area appear to be different between FVB/N and SJL/J mice. Whereas the FVB/N mice displayed massive neointima after carotid ligation, similar to that observed with the injury-induced endothelial denudation model, very little neointima was observed in SJL/J mice and their luminal narrowing phenotype was found to be due to negative remodeling of the vessel wall [24]. The examination of other strains revealed that some strains achieved luminal size reduction by inward negative remodeling without increased intimal mass, others adjust by neointimal hyperplasia alone yet some strains respond with both neointimal hyperplasia and inward remodeling. The authors proposed that the inward remodeling component in the ligation model is likely a physiological response and the neointimal formation is probably a pathological response. Taken together, these observations indicate a genetic component in determining both qualitative and quantitative luminal response to blood flow cessation. The development of massive neointima, involving enhanced smooth muscle cell migration and proliferation, in FVB/N mice following both endothelial denudation and blood flow cessation models of arterial injury suggest that the FVB/N strain harbors gene(s) that promote smooth muscle cell activation and response.
Role of ApoE and Hypercholesterolemia on Vascular Response to Injury
A commonly used mouse model to study vascular occlusive disease is the apoE knockout mouse. The popularity of this animal model owes in large part to their hypercholesterolemia [25, 26] and altered immune responses [27] due to the lack of apoE-mediated lipid transport and suppression of inflammatory response [28]. The importance of apoE in limiting neointimal hyperplasia after endothelial denudation was illustrated by two independent studies showing that the C57BL/6J mice, which are normally resistant to injury-induced neointimal hyperplasia, displayed massive neointima after vascular injury in the absence of a functional apoE gene [29, 30]. In another neointima-resistant strain, the C3H mice, apoE gene deletion has no effect on its resistance to injury-induced neointimal formation under chow fed conditions but a small neointima was detected when the animals were fed a Western type high fat/high cholesterol diet [30]. The latter study, along with studies showing neointimal formation in the hyperlipidemic LDLR-/- mice [31], suggested that hyperlipidemia is a contributing factor toward neointimal formation. The observation that hypercholesterolemia further exacerbates neointimal formation in apoE-/- mice is consistent with this hypothesis [30]. However, it should be noted that pathology by which hyperlipidemia and apoE gene deletion contributes to the neointimal formation process appear to be quite different. Whereas hyperlipidemia exacerbates neointimal growth by promoting macrophage foam cell formation and inflammation [31], very few if any macrophage foam cells are detected in the intima of apoE-/- mice after arterial injury [32]. Therefore, the protective effects of apoE against neointimal hyperplasia are independent of its cholesterol transport properties and its ability to limit macrophage foam cell formation. The latter conclusion is supported by studies showing that increasing circulating apoE levels via hepatic apoE transgenic expression inhibits neointimal hyperplasia without altering plasma lipid levels in the neointimal hyperplasia-susceptible FVB/N strain [29]. The ability of apoE to limit neointimal hyperplasia may be directly related to its ability to inhibit smooth muscle cell migration and proliferation [33]. Previous studies have shown that apoE inhibition of smooth muscle cell migration and proliferation through different mechanisms [34, 35]. In vitro studies with primary smooth muscle cells isolated from human, mouse, and rat aortas have indicated that apoE inhibition of smooth muscle cell proliferation in response to growth factors and oxidized LDL does not required its binding to cell surface receptor proteins [35], but its interaction with cell surface heparin sulfate proteoglycans results in activation of inducible nitric oxide synthase (iNOS) expression and activity, thereby inhibiting cell growth [34]. The activation of iNOS is not required for apoE inhibition of smooth muscle cell migration toward PDGF [34]. In contrast, apoE binding to LDL receptor-related protein-1 (LRP-1) on the surface of smooth muscle cells is necessary for its anti-migratory properties [36]. The binding of apoE to LRP-1 triggers cell signaling pathway(s) that lead(s) to intracellular cAMP accumulation and activation of protein kinase A [36]. These latter events are directly responsible for apoE suppression of PDGF-directed smooth muscle cell migration.
Nitric Oxide
The capacity of apoE to iinhibit neointimal hyperplasia in mice requires its recruitment to the site where endothelial injury occurs [37]. Presence of apoE at the site of endothelial denudation results in activation of inducible nitric oxide synthase (iNOS) activity in the neointima-resistant C57BL/6 mice but not in the neointima-susceptible FVB/N mice [38]. However, increased level of circulating apoE and its recruitment to the denuded artery promotes iNOS induction, which corresponds to the reduction of neointimal hyperplasia in this susceptible strain [38].
The studies with apoE transgenic and knockout mice clearly indicated that nitric oxide synthase protects against neointimal hyperplasia and vascular occlusion after endothelial injury. These in vivo observations are consistent with in vitro studies demonstrating nitric oxide inhibition of vascular smooth muscle cell proliferation [39]. Interestingly, neointimal hyperplasia was not observed after mechanically-induced endothelial denudation of the carotid arteries in iNOS gene knockout C57BL/6J mice [38]. Nevertheless, the iNOS-knockout mice displayed significant increases in both medial area and medial thickness after arterial injury [38]. We interpret these data to indicate that injury-induced hyperplasia of the medial smooth muscle cells in iNOS-knockout mice but cell migration to the intima remains suppressed in this neointima-resistant strain even in the absence of iNOS. This interpretation is consistent with data obtained from the carotid ligation model, in which iNOS-knockout mice responded to the cessation of blood flow with significantly elevated constrictive remodeling and limited neointimal formation [40]. In other models where neointimal hyperplasia is prominent, including transplant arteriosclerosis [41] and chronic adventitial stimulation with interleukin-1β [42], iNOS activity was shown to be protective with iNOS gene deletion enhancing and iNOS activation inhibiting neointimal hyperplasia.
In addition to iNOS, which is expressed and synthesizes nitric oxide in a transient manner during stimulation, nitric oxide is also synthesized in the vessel wall by endothelial nitric oxide synthase (eNOS) expressed in endothelial cells. Using an endothelial denudation model by balloon injury in rats, Janssens et al [43] showed that increasing eNOS expression and activity inhibits neointimal hyperplasia. In the mouse model, neointimal hyperplasia induced by cuff placement around the femoral artery is greater in eNOS-knockout mice than wild type mice [44]. The neointima formed after carotid artery ligation is also significantly larger in eNOS-knockout mice than their wild type counterparts [40]. The accelerated neointimal hyperplasia in eNOS-knockout mice is not due to thrombus formation or changes in blood pressure as antiplatelet and antihypertensive therapies cannot reduce the size and progression of the neointima [44]. In contrast to the iNOS-knockout models, eNOS knockout mice do not show constrictive remodeling after carotid artery ligation [40], illustrating the distinct roles of iNOS and eNOS in protection against vascular lesion formation. Importantly, these studies showed that both iNOS and eNOS activities are important in vascular protection after injury, suggesting that agents that promote their activities may be beneficial. In this regard, recent studies showed that increasing vascular nitric oxide level via localized delivery of S-nitrosocysteine crosslinked hydrogel [45], or suppression of the endogenous NOS inhibitor asymmetric dimethylarginine via transgenic elevation of dimethylarginine dimethylaminohydrolase expression (46) promotes endothelial repair and inhibits neointimal hyperplasia after vascular injury.
Although the vast majority of the literature supports the importance of eNOS and iNOS in limiting occlusion after vascular injury, there are data that show different results. Chyu et al [47] induced vascular injury by placement of a nonocclusive plastic cuff around the carotid artery and reported reduced neointimal thickening in the iNOS-knockout mice compared to control. The difference between this and other studies is not immediately apparent, but may be related to differences in the time points for the analysis of vascular pathology and/or differences in the procedure used to induce vascular injury. The periadventitial placement of plastic cuff invokes a significant inflammatory cell participation but inflammatory cell infiltration appears to be an early protective event in the mechanically-induced endothelial denudation model [32]. Whether this or other differences between the two experimental models may account for the different results with the iNOS-knockout mice remain to be determined.
Role of T and B Lymphocytes in Vascular Response to Injury
In contrast to the extensive literature on the role of cellular and humoral immunity in progressive atherosclerosis, the effects of inflammatory cells in injury-induced neointimal hyperplasia have received considerably less attention. Studies in the rat model, both with athymic nude rats and T-lymphocytes-depleted rats yielded opposite results regarding the role of T lymphocytes in neointimal hyperplasia [48, 49]. Additionally, although interferon-γ (IFN-γ) secreted by activated T lymphocytes inhibits smooth muscle cell proliferation in vitro, and IFN-γ also limits arterial proliferative lesions in response to arterial injury in vivo [50], cyclosporine suppression of lymphocyte activation had no effect on injury-induced neointimal proliferation in cholesterol-clamped rabbits [51]. While cyclosporine may have other effects in addition to its suppression of lymphocyte activation, nevertheless, these studies illustrated the complexity of the issue. It is possible that subsets of T lymphocytes may also affect injury-induced neointimal hyperplasia in different manners.
Our laboratory has compared wild type and RAG2-/- mice with both T and B lymphocyte deficiencies for their vascular response to injury to determine the importance of these inflammatory cells in neointimal hyperplasia. The data showed that lymphocyte infiltration is one of the earliest events in wild type mice after endothelial denudation, with both CD4+ and CD8+ cells adhering to the denuded vessel wall within 1 hr of injury [32]. This process is transient as no immune cells were detectable after 14 days. In contrast to the contributory role of lymphocytes in progressive atherosclerosis, the T and B lymphocytes present at the site of endothelial denudation serve a protective role in limiting massive smooth muscle cell response to vascular injury. The absence of T and B lymphocytes in the RAG2-/- mice results in massive neointimal formation, with corresponding increased number of proliferating smooth muscle cells present in the injured arteries [32]. Thus, our data are consistent with studies in the rat model reported by Hansson and colleagues, which showed a protective role of T lymphocytes against neointimal hyperplasia in vivo [49]. These results are also consistent with data obtained with the periadventitial cuff placement model of arterial injury, in which another immune cell deficient model, the RAG1-/- mice, were shown to have increased intimal thickening compared to wild type mice [52]. The latter study extended these observations with results showing adoptive transfer of B cells to RAG1-/- mice inhibited carotid ligation-induced intimal thickening [52]. Taken together, these studies suggest that both T and B lymphocytes, as well as IFN-γ secreted by T cells, have the capability of protection against neointimal hyperplasia.
A very interesting study was presented in late 2005 using the cuff injury model to explore the details of T lymphocytes and IFN-γ in modulation of vascular response to injury. In that study, Dimayuga et al compared wild type mice and RAG1-/- mice with or without reconstitution with T cell-enriched splenocytes in vascular response at various time points after periadventitial cuff placement [53]. Their results revealed a bimodal role of T lymphocytes and IFN-γ as both inhibitors and contributors toward neointimal hyperplasia. During the early phase of arterial repair, the T lymphocytes present at the site of injury inhibits intimal thickening. The infusion of exogenous IFN-γ to RAG1-/- mice starting at the day of injury also inhibited intimal thickening caused by periadventitial cuff placement [53]. Thus, T lymphocytes and IFN-γ secreted by these cells are protective against neointimal hyperplasia. Surprisingly, the infusion of IFN-γ neutralizing antibodies to the mice starting at 7 days after carotid artery injury resulted in decreased intimal thickening, suggesting that cytokine promotes intimal hyperplasia in the later phase [53]. The infusion of IFN-γ antibodies to RAG1-/- mice at 7 days after injury also promoted intimal thickening, indicating that T and B lymphocytes are not responsible for producing the IFN-γ that promotes intimal thickening at the late phase of vascular response to injury. Thus, it is possible that macrophages and natural killer (NK) cells may be responsible for this late-stage effects of IFN-γ in neointimal hyperplasia. Additional studies showed that the late IFN-γ effects on neointimal hyperplasia are mediated via enhanced IFN regulatory factor-1 induced iNOS expression [53]. Thus, this finding offers a potential explanation for differences in protection versus contributory role of iNOS in neointimal formation observed using the mechanical endothelial denudation and the periadventitial cuff placement models of injury. As discussed earlier, immune cells are detected only during the early phase of arterial injury in the endothelial denudation model and prolonged presence of immune cells is reported in the cuff placement model. The contribution of NK T cells in periadventitial collar-induced neointimal formation is highlighted in a recent study showing that defective lipid antigen presentation to natural killer T cells due to Cd1d gene ablation significantly reduced neointimal area by >60% [54].
Role of Macrophages in Vascular Response to Injury
In addition to the recruitment of T and B lymphocytes, endothelial denudation also recruits monocytes to the site of injury as a consequence of increased monocyte chemoattractant protein 1 (MCP-1) expression when the underlying smooth muscle cells are exposed to growth factors. MCP-1 also binds to the CC chemokine receptor-2 (CCR2) to induce intracellular signals associated with cell growth and migration [55, 56]. The deletion of CCR2 gene in mice decreases intimal hyperplasia after vascular injury induced by guidewire denudation of the femoral artery [9]. At an early time point of 5 days after vascular injury, smooth muscle cell proliferation was found to be reduced by almost 60% in CCR2-/- mice compared to littermate controls. The difference in lesion development remains prominent after 4 weeks [9]. However, CCR2 is not expressed by either mouse or human smooth muscle cells [9, 57]. Therefore, it is unlikely that MCP-1 and CCR2 contribute to neointimal hyperplasia by direct smooth muscle cell signaling and activation. It is more likely that MCP-1 and CCR2 affect neointimal hyperplasia via an indirect effect. Previous studies have shown that CCR2-/- mice are defective in leukocyte adhesion [58], monocyte recruitment [58, 59], and reduced IFN-γ production [60]. Since lymphocytes and IFN-γ are protective against endothelial denudation-induced neointimal hyperplasia, the reduced neointimal hyperplasia observed in the injured arteries of CCR2-/- mice is unlikely related to their defective leukocyte adhesion and reduced IFN-γ phenotypes. These studies suggest that even though monocytes are present in the injured arteries only in limited number [9, 32, 61], monocytes and monocyte-derived macrophages play a contributory role in modulating neointimal hyperplasia after endothelial denudation. Thus, in contrast to the T and B lymphocytes, which play opposite roles in promoting or inhibiting atherosclerosis and injury-induced neointimal hyperplasia, monocyte recruitment and their differentiation into macrophages appear to be key factors in both of these vascular occlusive diseases.
The importance of monocyte recruitment toward injury-induced neointimal hyperplasia is supported by independent observations showing that targeted disruption of P-selectin abolished macrophage infiltration and neointimal formation after endothelial denudation of either the carotid or the femoral arteries in mice [62, 63]. Additional studies revealed that monocyte recruitment is triggered by activated platelet secretion of the chemokine RANTES and the blocking of RANTES receptors attenuates endothelial denudation-induced macrophage infiltration and neointimal hyperplasia [64]. The RANTES receptor CCR5 is involved with this process as CCR5-deficient mice are protected against injury-induced neointimal hyperplasia [65]. The deficiency of CCR5 results in up-regulation of the anti-inflammatory cytokine interleukin 10 (IL-10) in neointimal smooth muscle cells, which in turn is responsible for reduction of macrophage recruitment and the suppression of neointimal hyperplasia. A direct role for the protective effect of IL-10 against neointimal hyperplasia is evident from studies showing IL-10 deficient mice, in the hypercholesterolemic apoE*3-Leiden mouse background, displayed a significant increase of neointimal formation whereas over-expression of IL-10 results in a reduction of neointima after periadventitial cuff placement [66].
Tumor Necrosis Factor
The mechanism by which activated macrophages recruited to the site of injury contributes to neointimal hyperplasia has not been definitively defined. However, proinflammatory cytokines produced by both activated macrophages and smooth muscle cells are direct contributors to vascular occlusive diseases. In particular, tumor necrosis factor-α (TNF-α) has been shown to stimulate smooth muscle cell proliferation [67] and induces apoptosis of both smooth muscle and endothelial cells in vitro [68, 69]. In rats, the blockade of TNF-α inhibits neointimal thickening by accelerating endothelial repair [70]. The TNF-α effects on rats have been recapitulated in mice, with additional studies showing that TNF-α inhibits neointimal thickening by modulating expression of the transcription factor E2F1 in vascular smooth muscle and endothelial cells [71]. In smooth muscle cells, TNF-α has no effect on cell proliferation or apoptosis but induces apoptosis in E2F1-overexpressing cells [71]. In contrast, in endothelial cells, TNF-α inhibits E2F1 expression and activity to induce cell cycle arrest and enhances apoptosis of proliferating cells [71]. The divergent effects of TNF-α on E2F1 activity and function in endothelial and smooth muscle cells are responsible for its promotion of neointimal hyperplasia after vascular injury.
Possible Involvement of Bone Marrow-derived Progenitor Stem Cells in Neointimal Hyperplasia
There is existing evidence as well as controversy regarding the possible role of bone marrow-derived progenitor stem cells in vascular pathology in response to endothelial damage. The ability of hematopoietic stem cells to differentiate into vascular smooth muscle cells was initially suggested by in vitro experiments showing the ability of hematopoietic stem cells seeded on vascular smooth muscle cells to express smooth muscle specific markers such as smooth muscle α-actin and calponin [72]. In a detailed study examining the origin of intima cells in models of post-angioplasty restenosis, graft vasculopathy, and hyperlipidemia-induced atherosclerosis, Sata et al showed that a significant number of intimal cells are derived from bone marrow cells [72]. Interestingly, the amount of bone marrow-derived stem cells that differentiate into intimal smooth muscle cells differs depending on the procedure used to induce endothelial damage and/or dysfunction. In mechanically-induced endothelial denudation model, approximately 60% of the intimal smooth muscle cells were shown to be derived from bone marrow stem cells, whereas close to 80% of the intimal smooth muscle cells in graft vasculopathy were shown to be bone marrow derived [72]. In contrast, only 40% of the smooth muscle cells in hyperlipidemia-induced atherosclerotic plaques were identified as bone marrow-derived cells in the Sata study [72]. The detection of circulating smooth muscle cells in vascular lesions led these and other investigators to conclude that these progenitor stem cells contribute to atherosclerosis and vascular stenotic lesions [72-74].
The theory that intimal smooth muscle cells include at least in part bone marrow-derived progenitor cells has been challenged by numerous recent studies that failed to confirm the earlier observations. In one study to examine the origin of the intimal smooth muscle cells associated with vein graft transplants, vena cava segments were isografted to carotid arteries between 4 types of transgenic mice, including wild type, smooth muscle-specific LacZ transgenic mice, smooth muscle-specific LacZ mice in apoE-null background, and ROSA26 expressing β-galactosidase in all tissues. Double staining for β-gal and cell nuclei showed that ∼40% of the smooth muscle cells in the intima are originated from the host and 60% from the donor vessel. Interestingly, no bone marrow-derived cells are observed in the intima area of any recipient mice [75]. In another study examining the origin of neointimal smooth muscle cells in the intima of hyperlipidemia-induced atherosclerotic plaque in irradiated apoE-/- mice after reconstitution of the bone marrow with cells from eGFP+apoE-/- mice, detailed examination of ∼10,000 smooth muscle cells in the intima failed to identify any bone marrow-derived cells [76]. The latter study also failed to detect any bone marrow-derived cells in the intima after arterial graft transplant-induced atherosclerosis, thus concluding that smooth muscle cells in the intima are exclusively derived from the local vessel wall [76]. These results are consistent with other studies showing minimal bone marrow-derived cell contribution to the intimal smooth muscle cells in the systemic hypoxia-induced arteriogenesis model [77]. The difference between these studies showing minimal, if any, contribution of bone marrow-derived cells to the intima with earlier studies showing bone marrow cell involvement has been attributed to the methods used to characterize the cell type in the lesion area. As stated in an editorial review by Hoofnagle et al [78], it is critically important to use highly sophisticated confocal or high-resolution z-axis sectioning microscopy technique to examine a large number of cells in the intima, and that previous studies using low resolution immunomicroscopy technique to examine small numbers of intimal smooth muscle cells is not adequate. In particular, the use of smooth muscle α-actin immunoreactivity for smooth muscle cells is also unreliable as this protein is now known to be expressed by other lineages in addition to smooth muscle cells [79]. Accordingly, the more recent in-depth characterization revealed that bone marrow-derived cells contribute minimally to intimal smooth muscle cells in diet-induced atherosclerosis and graft transplanted arteriosclerosis. Since endothelial denudation-induced neointimal hyperplasia elicit very different vascular responses than atherosclerosis and graft transplant-induced arteriosclerosis, as discussed earlier, whether bone marrow progenitor cells participate in the denudation-induced neointimal hyperplasia remains unclear and requires additional in depth characterization with highly sensitive techniques.
Concluding Remarks
In summary, mechanically-induced arterial injury in mice has provided valuable information regarding the mechanism of neointimal hyperplasia. The current data illustrated significant differences in the pathology and mechanism of injury-induced neointimal hyperplasia compared to lesion formation observed in hypercholesterolemia-induced progressive atherosclerosis. Although there are discrepancies in the literature regarding the involvement of nitric oxide and immune cells in neointimal hyperplasia, most if not all of the differences can be explained by the methodologies used to induce arterial injury, the genetic background of the mice, and the time of lesion analysis after arterial injury. Investigators interested in exploring the importance of a particular gene product in injury-induced neointimal hyperplasia should consider these variables in selecting the appropriate model for their studies.
Acknowledgments
Work in the author's laboratory described in this article is supported by NIH Grant HL61332.
Abbreviations Used
- apoE
apolipoprotein E
- IFN-γ
interferon-γ
- iNOS
inducible nitric oxide synthase
- eNOS
endothelial nitric oxide synthase
- TNF-α
tumor necrosis factor-α
References
- 1.Lusis AJ. Nature. 2000;407:233–241. doi: 10.1038/35025203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Greenland P, Knoll MD, Stamler J, Neaton JD, Dyer AR, Garside DB, Wilson PW. JAMA. 2003;290:891–897. doi: 10.1001/jama.290.7.891. [DOI] [PubMed] [Google Scholar]
- 3.Ross R. New Engl J Med. 1999;340:115–126. doi: 10.1056/NEJM199901143400207. [DOI] [PubMed] [Google Scholar]
- 4.Ferns GA, Avades TY. Int J Exp Pathol. 2000;81:63–88. doi: 10.1046/j.1365-2613.2000.00143.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Clowes AW, Clowes MM, Fingerle J, Reidy MA. J Cardiovasc Pharmacol. 1989;14:s12–s15. [PubMed] [Google Scholar]
- 6.Reidy MA, Fingerle J, Lindner V. Circulation. 1992;86(Suppl III):43–46. [PubMed] [Google Scholar]
- 7.Rogers C, Edelman ER, Simon DI. Proc Natl Acad Sci USA. 1998;95:10134–10139. doi: 10.1073/pnas.95.17.10134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Roque M, Fallon JT, Badimon JJ, Zhang WX, Taubman MB, Reis ED. Arterioscler Thromb Vasc Biol. 2000;20:335–342. doi: 10.1161/01.atv.20.2.335. [DOI] [PubMed] [Google Scholar]
- 9.Roque M, Kim WJH, Gazdoin M, Malik A, Reis ED, Fallon JT, Badimon JJ, Charo IF, Taubman MB. Arterioscler Thromb Vasc Biol. 2002;22:554–559. doi: 10.1161/hq0402.105720. [DOI] [PubMed] [Google Scholar]
- 10.Virmani R, Farb A. Curr Opin Lipidol. 1999;10:499–506. doi: 10.1097/00041433-199912000-00004. [DOI] [PubMed] [Google Scholar]
- 11.Moreno PR, Palacios IF, Leon MN, Rhodes J, Fuster V, Fallon JT. Am J Cardiol. 1999;84:462–466. doi: 10.1016/s0002-9149(99)00334-3. [DOI] [PubMed] [Google Scholar]
- 12.Virmani R, Kolodgie FD, Burke AP, Farb A, Schwartz SM. Arterioscler Thromb Vasc Biol. 2000;20:1262–1275. doi: 10.1161/01.atv.20.5.1262. [DOI] [PubMed] [Google Scholar]
- 13.Lowe HC, Oesterle SN, Khachigian LM. J Am Coll Cardiol. 2002;39:183–193. doi: 10.1016/s0735-1097(01)01742-9. [DOI] [PubMed] [Google Scholar]
- 14.Moses JW, Leon MB, Popma JJ, Fitzgerald PJ, Holmes DR, O'Shaughnessy C, Caputo RP, Kereiakes DJ, Williams DO, Teirstein PS, Jaeger JL, Kuntz RE, the SIRIUS Investigators N Engl J Med. 2003;349:1315–1323. doi: 10.1056/NEJMoa035071. [DOI] [PubMed] [Google Scholar]
- 15.Stone GW, Ellis SG, Cox DA, Hermiller J, O'Shaughnessy C, Mann JT, Turco M, Caputo R, Bergin P, Greenberg J, Popma JJ, Russell ME, the TAXUS-IV Investigators N Engl J Med. 2004;350:221–231. doi: 10.1056/NEJMoa032441. [DOI] [PubMed] [Google Scholar]
- 16.Stone GW, Moses JW, Ellis SG, Schofer J, Dawkins KD, Morice MC, Colombo A, Schampaert E, Grube E, Kirtane AJ, Cutlip DE, Fahy M, Pocock SJ, Mehran R, Leon MB. N Engl J Med. 2007;356:998–1008. doi: 10.1056/NEJMoa067193. [DOI] [PubMed] [Google Scholar]
- 17.Joner M, Finn AV, Farb A, Mont EK, Kolodgie FD, Ladich E, Kutys R, Skirija K, Gold HK, Virmani R. J Am Coll Cardiol. 2006;48:193–202. doi: 10.1016/j.jacc.2006.03.042. [DOI] [PubMed] [Google Scholar]
- 18.Mehra MR, Ventura HO, Stapleton DD, Smart FW. J Heart Lung Transplant. 1995;14:S207–S211. [PubMed] [Google Scholar]
- 19.Roy-Chaudhury P, Kelly BS, Miller MA, Reaves A, Armstrong J, Nanayakkara N, Heffelfinger SC. Kidney Int. 2001;59:2325–2334. doi: 10.1046/j.1523-1755.2001.00750.x. [DOI] [PubMed] [Google Scholar]
- 20.Kumar A, Lindner V. Arterioscler Thromb Vasc Biol. 1997;17:2238–2244. doi: 10.1161/01.atv.17.10.2238. [DOI] [PubMed] [Google Scholar]
- 21.Lindner V, Fingerle J, Reidy MA. Circ Res. 1993;73:792–796. doi: 10.1161/01.res.73.5.792. [DOI] [PubMed] [Google Scholar]
- 22.Carmeliet P, Moons L, Collen D. Cardiovasc Res. 1998;39:8–33. doi: 10.1016/s0008-6363(98)00108-4. [DOI] [PubMed] [Google Scholar]
- 23.Kuhel DG, Zhu B, Witte DP, Hui DY. Arterioscler Thromb Vasc Biol. 2002;22:955–960. doi: 10.1161/01.atv.0000017994.77066.75. [DOI] [PubMed] [Google Scholar]
- 24.Harmon KJ, Couper LL, Lindner V. Am J Pathol. 2000;156:1741–1748. doi: 10.1016/S0002-9440(10)65045-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang SH, Reddick RL, Piedrahita JA, Maeda N. Science. 1992;258:468–471. doi: 10.1126/science.1411543. [DOI] [PubMed] [Google Scholar]
- 26.Plump AS, Smith JD, Hayek T, Aalto-Setala K, Walsh A, Verstuyft JG, Rubin EM, Breslow JL. Cell. 1992;71:343–353. doi: 10.1016/0092-8674(92)90362-g. [DOI] [PubMed] [Google Scholar]
- 27.Laskowitz DT, Lee DM, Schmechel D, Staats HF. J Lipid Res. 2000;41:613–620. [PubMed] [Google Scholar]
- 28.Ali K, Middleton M, Pure E, Rader DJ. Circ Res. 2005;97:922–927. doi: 10.1161/01.RES.0000187467.67684.43. [DOI] [PubMed] [Google Scholar]
- 29.Zhu B, Kuhel DG, Witte DP, Hui DY. Am J Pathol. 2000;157:1839–1848. doi: 10.1016/S0002-9440(10)64823-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shi W, Pei H, Fischer JJ, James JC, Angle JF, Matsumoto AH, Helm GA, Sarembock IJ. J Lipid Res. 2004;45:2008–2014. doi: 10.1194/jlr.M400254-JLR200. [DOI] [PubMed] [Google Scholar]
- 31.Tian J, Pei H, Sanders JM, Angle JF, Sarembock IJ, Matsumoto AH, Helm GA, Shi W. Biochem Biophys Res Commun. 2006;345:1004–1009. doi: 10.1016/j.bbrc.2006.04.180. [DOI] [PubMed] [Google Scholar]
- 32.Zhu B, Reardon CA, Getz GS, Hui DY. Arterioscler Thromb Vasc Biol. 2002;22:450–455. doi: 10.1161/hq0302.105377. [DOI] [PubMed] [Google Scholar]
- 33.Ishigami M, Swertfeger DK, Granholm NA, Hui DY. J Biol Chem. 1998;273:20156–20161. doi: 10.1074/jbc.273.32.20156. [DOI] [PubMed] [Google Scholar]
- 34.Ishigami M, Swertfeger DK, Hui MS, Granholm NA, Hui DY. Arterioscler Thromb Vasc Biol. 2000;20:1020–1026. doi: 10.1161/01.atv.20.4.1020. [DOI] [PubMed] [Google Scholar]
- 35.Swertfeger DK, Hui DY. J Biol Chem. 2001;276:25043–25048. doi: 10.1074/jbc.M102357200. [DOI] [PubMed] [Google Scholar]
- 36.Zhu Y, Hui DY. J Biol Chem. 2003;278:36257–36263. doi: 10.1074/jbc.M303171200. [DOI] [PubMed] [Google Scholar]
- 37.Moore ZWQ, Zhu B, Kuhel DG, Hui DY. Am J Pathol. 2004;164:2109–2116. doi: 10.1016/S0002-9440(10)63769-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Moore ZWQ, Hui DY. J Lipid Res. 2005;46:2083–2090. doi: 10.1194/jlr.M500177-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Garg UC, Hassid A. J Clin Invest. 1989;83:1774–1777. doi: 10.1172/JCI114081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yogo K, Shimokawa H, Funakoshi H, Kandabashi T, Miyata K, Okamoto S, Egashira K, Huang P, Akaike T, Takeshita A. Arterioscler Thromb Vasc Biol. 2000;20:e96–e100. doi: 10.1161/01.atv.20.11.e96. [DOI] [PubMed] [Google Scholar]
- 41.Koglin J, Glysing-Jensen T, Mudgett JS, Russell ME. Circulation. 1998;97:2059–2065. doi: 10.1161/01.cir.97.20.2059. [DOI] [PubMed] [Google Scholar]
- 42.Fukumoto Y, Shimokawa H, Kozai T, Kadokami T, Kuwata K, Yonemitsu Y, Kuga T, Egashira K, Sueishi K, Takeshita A. Circulation. 1997;96:3104–3111. doi: 10.1161/01.cir.96.9.3104. [DOI] [PubMed] [Google Scholar]
- 43.Janssens S, Flaherty D, Nong Z, Varenne O, VP N, Haustermans C, Zoldhelyi P, Gerard R, Collen D. Circulation. 1998;97:1274–1281. doi: 10.1161/01.cir.97.13.1274. [DOI] [PubMed] [Google Scholar]
- 44.Moroi M, Zhang L, Yasuda T, Virmani R, Farb A, Gold HK, Fishman MC, Huang PL. J Clin Invest. 1998;101:1225–1232. doi: 10.1172/JCI1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Masters KS, Lipke EA, Rice EE, Liel MS, Myler hA, Zygourakis C, Tulis DA, West JL. J Biomater Sci Ed. 2005;16:659–672. doi: 10.1163/1568562053783722. [DOI] [PubMed] [Google Scholar]
- 46.Konishi H, Sydow K, Cooke JP. J Am Coll Cardiol. 2007;49:1099–1105. doi: 10.1016/j.jacc.2006.10.068. [DOI] [PubMed] [Google Scholar]
- 47.Chyu KY, Dimayuga P, Zhu J, Nilsson J, Kaul S, Shah PK, Cercek B. Circ Res. 1999;85:1192–1198. doi: 10.1161/01.res.85.12.1192. [DOI] [PubMed] [Google Scholar]
- 48.Ferns GAA, Reidy MA, Ross R. Am J Pathol. 1991;138:1045–1057. [PMC free article] [PubMed] [Google Scholar]
- 49.Hansson GK, Holm J, Holm S, Fotev Z, Hedrich HJ, Fingerle J. Proc Natl Acad Sci USA. 1991;88:10530–10534. doi: 10.1073/pnas.88.23.10530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hansson GK, Holm J. Circulation. 1991;84:1266–1272. doi: 10.1161/01.cir.84.3.1266. [DOI] [PubMed] [Google Scholar]
- 51.Andersen HO, Hansen BF, Holm P, Stender S, Nordestgaard BG. Arterioscler Thromb Vasc Biol. 1999;19:1687–1694. doi: 10.1161/01.atv.19.7.1687. [DOI] [PubMed] [Google Scholar]
- 52.Dimayuga P, Cercek B, Oguchi S, Fredrikson GN, Yano J, Shah PK, Jovinge S, Nilsson J. Arterioscler Thromb Vasc Biol. 2002;22:644–649. doi: 10.1161/01.atv.0000012455.62765.bf. [DOI] [PubMed] [Google Scholar]
- 53.Dimayuga PC, Li H, Chyu KY, Fredrikson GN, Nilsson J, Fishbein MC, Shah PK, Cercek B. Arterioscler Thromb Vasc Biol. 2005;25:2528–2534. doi: 10.1161/01.ATV.0000190606.41121.00. [DOI] [PubMed] [Google Scholar]
- 54.Strom A, Wigren M, Hultgardh-Nilsson A, Saxena A, Gomez MF, Cardell S, Fredrikson GN, Nilsson J. Arterioscler Thromb Vasc Biol. 2007;101:e83–e89. doi: 10.1161/CIRCRESAHA.107.160705. [DOI] [PubMed] [Google Scholar]
- 55.Sozzani S, Locati M, Zhou D, Rieppi M, Luini W, Lamorte G, Gianchi G, Polentarutti N, Allavena P, Mantovani A. J Leukoc Biol. 1995;57:788–194. doi: 10.1002/jlb.57.5.788. [DOI] [PubMed] [Google Scholar]
- 56.Yen H, Zhang Y, Penfold S, Rollins BJ. J Leukoc Biol. 1997;61:529–532. doi: 10.1002/jlb.61.4.529. [DOI] [PubMed] [Google Scholar]
- 57.Schecter AD, Rollins BJ, Zhang YJ, Charo IF, Fallon JT, Rossikhina M, Giesen PL, Nemerson Y, Taubman MB. J Biol Chem. 1997;272:28568–28573. doi: 10.1074/jbc.272.45.28568. [DOI] [PubMed] [Google Scholar]
- 58.Kuziel WA, Morgan SJ, Dawson TC, Griffin S, Smithies O, Ley K, Maeda N. Proc Natl Acad Sci USA. 1997;94:12053–12058. doi: 10.1073/pnas.94.22.12053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kurihara T, Warr G, Loy J, Bravo R. J Exp Med. 1997;186:1757–1762. doi: 10.1084/jem.186.10.1757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Boring L, Gosling J, Chensue SW, Kunkel SL, Farese RVJ, Broxmeyer HE, Charo IF. J Clin Invest. 1997;100:2552–2561. doi: 10.1172/JCI119798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Roque M, Fallon JT, Badimon JJ, Zhang WX, Taubman MB, Reis ED. Arterioscler Thromb Vasc Biol. 2000;20:335–342. doi: 10.1161/01.atv.20.2.335. [DOI] [PubMed] [Google Scholar]
- 62.Manka D, Collins RG, Ley K, Beaudet AL, Sarembock IJ. Circulation. 2001;103:1000–1005. doi: 10.1161/01.cir.103.7.1000. [DOI] [PubMed] [Google Scholar]
- 63.Smyth SS, Reis ED, Zhang W, Fallon JT, Gordon RE, Coller BS. Circulation. 2001;103:2501–2507. doi: 10.1161/01.cir.103.20.2501. [DOI] [PubMed] [Google Scholar]
- 64.Schober A, Manka D, von Hundelshausen P, Huo Y, Hanrath P, Sarembock IJ, Ley K, Weber C. Circulation. 2002;106:1523–1529. doi: 10.1161/01.cir.0000028590.02477.6f. [DOI] [PubMed] [Google Scholar]
- 65.Zernecke A, Liehn EA, Gao JL, Kuziel WA, Murphy PM, Weber C. Blood. 2006;107:4240–4243. doi: 10.1182/blood-2005-09-3922. [DOI] [PubMed] [Google Scholar]
- 66.Eefting D, Schepers A, de Vries MR, Pires NMM, Grimbergen JM, Lagerweij T, Nagelkerken LM, Monraats PS, Jukema JW, van Bockel JH, Quax PHA. Atherosclerosis. 2006;193:335–342. doi: 10.1016/j.atherosclerosis.2006.09.032. [DOI] [PubMed] [Google Scholar]
- 67.Rectenwald JE, Moldawer LL, Huber TS, Seeger JM, Ozaki CK. Circulation. 2000;102:1697–1702. doi: 10.1161/01.cir.102.14.1697. [DOI] [PubMed] [Google Scholar]
- 68.Geng YJ, Wu Q, Muszynski M, Hansson GK, Libby P. Arterioscler Thromb Vasc Biol. 1996;16:19–27. doi: 10.1161/01.atv.16.1.19. [DOI] [PubMed] [Google Scholar]
- 69.Spyridopoulos I, Brogi E, Kearney M, Sullivan AB, Cetrulo C, Isner JM, Losordo DW. J Mol Cell Cardiol. 1997;29:1321–1330. doi: 10.1006/jmcc.1996.0365. [DOI] [PubMed] [Google Scholar]
- 70.Krasinski K, Spyridopoulos I, Kearney M, Losordo DW. Circulation. 2001;104:1754–1756. doi: 10.1161/hc4001.098046. [DOI] [PubMed] [Google Scholar]
- 71.Goukassian DA, Kishore R, Krasinski K, Dolan C, Luedemann C, Yoon Ys, Kearney M, Hanley A, Ma H, Asahara T, Isner JM, Losordo DW. Circ Res. 2003;93:162–169. doi: 10.1161/01.RES.0000082980.94211.3A. [DOI] [PubMed] [Google Scholar]
- 72.Sata M, Siura A, Kunisato A, Tojo A, Okada S, Tokuhisa T, Hirai H, Makuuchi M, Hirata Y, Nagai R. Nature Medicine. 2002;8:403–409. doi: 10.1038/nm0402-403. [DOI] [PubMed] [Google Scholar]
- 73.Saiura A, Sata M, Hirata Y, Nagai R, Makuuchi M. Nature Medicine. 2001;7:382–383. doi: 10.1038/86394. [DOI] [PubMed] [Google Scholar]
- 74.Han Cl, Campbell GR, Campbell JH. J Vasc Res. 2001;38:113–119. doi: 10.1159/000051038. [DOI] [PubMed] [Google Scholar]
- 75.Hu Y, Mayr M, Metzler B, Erdel M, Davison F, Xu Q. Circ Res. 2002;91:13e–20. doi: 10.1161/01.res.0000037090.34760.ee. [DOI] [PubMed] [Google Scholar]
- 76.Bentzon JF, Weile C, Sondergaard CS, Hindkjaer J, Kassem M, Falk E. Arterioscler Thromb Vasc Biol. 2006;26:2696–2702. doi: 10.1161/01.ATV.0000247243.48542.9d. [DOI] [PubMed] [Google Scholar]
- 77.O'Neill TJ, Wamhoff BR, Owens GK, Skalak TC. Circ Res. 2005;97:1027–1035. doi: 10.1161/01.RES.0000189259.69645.25. [DOI] [PubMed] [Google Scholar]
- 78.Hoofnagle MH, Thomas JA, Wamhoff BR, Owens GK. Arterioscler Thromb Vasc Biol. 2006;26:2579–2581. doi: 10.1161/01.ATV.0000249623.79871.bc. [DOI] [PubMed] [Google Scholar]
- 79.Owens GK, Kumar MS, Wamhoff BR. Physiol Rev. 2004;84:767–801. doi: 10.1152/physrev.00041.2003. [DOI] [PubMed] [Google Scholar]