

Abstract
Although systems biology is a perfect framework for investigating system-level declines during aging, only a few reports have focused on a comprehensive understanding of system-level changes in the context of aging systems. The present study aimed to understand the most sensitive biological systems affected during aging and to reveal the systems underlying the crosstalk between aging and the ability of calorie restriction (CR) to effectively slow-down aging. We collected and analyzed 478 aging- and 586 CR-related mouse genes. For the given genes, the biological systems that are significantly related to aging and CR were examined according to three aspects. First, a global characterization by Gene Ontology (GO) was performed, where we found that the transcriptome (a set of genes) for both aging and CR were strongly related in the immune response, lipid metabolism, and cell adhesion functions. Second, the transcriptional modularity found in aging and CR was evaluated by identifying possible functional modules, sets of genes that show consistent expression patterns. Our analyses using the given functional modules, revealed systemic interactions among various biological processes, as exemplified by the negative relation shown between lipid metabolism and the immune response at the system level. Third, transcriptional regulatory systems were predicted for both the aging and CR transcriptomes. Here, we suggest a systems biology framework to further understand the most important systems as they age.
Electronic supplementary material
The online version of this article (doi:10.1007/s11357-009-9106-3) contains supplementary material, which is available to authorized users.
Keywords: Aging, Calorie restriction, Systems biology, Transcriptome analysis
Introduction
Biological systems are highly complex and exquisitely regulated networks of cellular components governed by spatial and temporal signals. Most network dynamics are induced by various biological or pathological conditions resulting from the output of enormous combinations of individual cellular components. Recently, new methodological paradigms such as systems biology have facilitated a system-level understanding of biological systems (Hong et al. 2006; Shamir et al. 2005; Das et al. 2006; Slonim et al. 2006; Tanay et al. 2005; Zhou et al. 2005).
Because aging is a most complicated physiological process, caused by changes in multiple biological systems, a systems biology approach can be an efficient analytical probe to better understand altered biological systems that arise during aging.
Here, we apply systems biology approaches to analyze and elucidate correlations among genes involved in aging and CR, the most effective intervention known to slow aging (Chung et al. 2006). We initially screened 478 and 586 differentially expressed genes (DEGs) in mouse for aging and CR, respectively, which we term aging and CR transcriptomes. A statistical method supplemented with pre-annotated biological information on the basis of Gene Ontology (GO) (Gene Ontology Consortium 2006), which is freely accessible from the Mouse Genome Database (MGD) (Blake et al. 2006), presented a broad view of biological systems governed by these aging and CR transcriptomes. This method gave us a comprehensive and better understanding of the immune response, cell adhesion, and lipid metabolism activities most affected by both aging and CR. Specifically, findings show that the immune response and cell adhesion were over-activated by the aging process, while CR attenuated them. In contrast, lipid metabolism was shown to be activated by CR, but suppressed by aging.
In order to investigate the correlations between the aging and CR transcriptomes at system levels, functional modules were constructed using the algorithm of Hong et al. (2006) in conjunction with a number of large-scale expression profiles from the Gene Expression Omnibus (GEO) (Barrett et al. 2005). The data obtained from the functional modules indicated that systems like the immune response and lipid metabolism were closely linked to each other under aging- and CR-related conditions. Additional information obtained from the analysis of functional modules showed possible biological roles for uncharacterized genes by their consistent expression patterns with other genes already characterized by functional modules.
To better understand the correlations between aging and CR transcriptomes at regulatory system levels, we investigated over-represented transcription factor binding sites for both aging and CR transcriptomes. Specifically, we found that 19 (11 up-regulated and eight down-regulated aging-related genes) and 17 (12 up-regulated and five down-regulated CR-related genes) transcription factor binding sites were differentially distributed in the aging and CR transcriptomes. The significance of these transcription factors is strongly supported by the consistent expression patterns between their transcription factors and target genes. The cooperative, regulatory effect of the transcription factor pair was also observed by the consistent expression patterns of genes sharing the transcription factor pair in both transcriptomes. Among the promising transcription factors, members of the CCAAT and forkhead families were found in both the aging and CR transcriptomes.
In present study, we documented systems biology approaches to enhance our understanding of the correlations between aging and CR at system levels. Systems biology supplemented with a mathematical algorithm, and a well-established database from high-throughput data, gives us a more comprehensive understanding and insights into aging and CR processes by shedding light on uncharacterized genes or transcription factors related to aging and CR.
Materials and methods
Identification of the aging and CR transcriptomes
We investigated the aging and CR transcriptomes using two strategies. First, we searched the DEGs for aging and CR as reported in the microarray studies. The four literature references citied are listed in Table 1 for the aging and CR transcriptomes, respectively; and we mapped each gene into the corresponding UniGene cluster in consideration of redundancy. Second, we analyzed the data from aging and CR-related microarray studies that were downloaded from the GEO, a public repository of microarray studies. For the GEO studies, we obtained corresponding annotation files as derived for the DEG study (Table 1). Then, we mapped the probes to the corresponding UniGene clusters and calculated the averaged expression profile for each UniGene cluster to reduce redundancy among probes. We also obtained the DEGs for the aging and CR transcriptomes by unpaired two-class analysis (δ = 2.4) using significance analysis for microarray (SAM) (Tusher et al. 2001).
Table 1.
Differentially expressed genes in aging and CR derived from the literature and GEO (ND not detected)
| Process | Sources | Tissues | Number of genes | PMID/GEO accession |
|---|---|---|---|---|
| Aging | Literature | Retinal pigment epithelium | 29 | 16081535 (Tian et al. 2005) |
| Brain cerebellum | 29 | 10888876 (Lee et al. 2000) | ||
| Brain neocortex | 44 | 10888876 (Lee et al. 2000) | ||
| Heart | 270 | 12419851 (Lee et al. 2002) | ||
| Brain | 36 | 16257272 (Blalock et al. 2005) | ||
| GEO | Brain cerebellum | ND | GDS1008 | |
| Myoblast | 74 | GDS1079 (Beggs et al. 2004) | ||
| Brain | 6 | GDS1311 (Godbout et al. 2005) | ||
| Kidney | 17 | GDS355 | ||
| Kidney | 13 | GDS356 | ||
| Retinal pigment epithelium | ND | GDS396 (Ida et al. 2003) | ||
| Retinal pigment epithelium | ND | GDS397 (Ida et al. 2003) | ||
| Heart | 3 | GDS40 | ||
| CR | Literature | White adipose tissue | 52 | 14688200 (Higami et al. 2004) |
| White adipose tissue | 72 | 16424110 (Higami et al. 2006) | ||
| Brain neocortex | 47 | 10888876 (Lee et al. 2000) | ||
| Heart | 392 | 12419851 (Lee et al. 2002) | ||
| GEO | Lung | 3 | GDS241 (Massaro et al. 2004) | |
| Kidney | 42 | GDS355 | ||
| Kidney | 15 | GDS356 |
Over-represented GO analysis for the aging and CR transcriptomes
We characterized the aging and CR transcriptomes in silico using GO annotation. To this end, we downloaded the GO terminologies from the GO consortium and GO annotations for mouse genes from the MGD. We first reconstructed the GO annotation for mouse UniGene clusters and then we analyzed the annotated frequency of GO terminologies for the whole transcriptome and the transcriptomes of interest, i.e., aging and CR transcriptomes, considering the hierarchical structure of GO terminologies. Using the frequency table for GO terminologies, we investigated GO terminologies heavily annotated in the aging and CR transcriptomes compared against the whole transcriptome. We applied χ2 analysis, an effective method to decipher differential distribution, to determine the biased distribution in this study (P value <10–5).
Microarray studies preprocessing
We downloaded 1,081 microarray studies and 272 microarray annotation files from the GEO repository of expression profiles and pre-processed 381 microarray studies on mice. We mapped the UniGene ID corresponding to each probe on the chips to 1) remove the redundancy caused by the multiple probes corresponding to one gene and 2) standardized the identifier for further processing according to the annotation files. To this end, we calculated the averaged expression profiles from multiple expression profiles of the redundant probes.
For cDNA microarray studies, the expression values were produced as log ratios. However, the expression values in case of an oligochip, such as the Affymetrix chip, were generated as intensity. For this diversity among the types of expression values, we transformed the intensity type expression values into log ratios by dividing the experimental condition profiles by a common reference condition such as ‘wild type’, ‘control’, or ‘0 hour’. If there were no available common references (e.g., microarray for tissue distribution), we standardized the expression profile by computing the z-score.
In a next step, we dropped the less informative microarray studies, which could not sufficiently cover the genes found in 381 microarray studies. Specifically, we measured the coverage rate of microarray studies for genes and then filtered out the microarray study that covered less than 30%. We also dropped the microarray studies containing few experimental conditions, specifically less than three, to avoid generating a simplistic correlation coefficient (e.g., 1 or -1) during further study. Finally, we obtained the most informative 131 microarray studies.
Significant transcription factor analysis
We identified the putative transcription factors found in the upstream regions of aging and CR transcriptomes obtained from UCSC Genome Bioinformatics (http://genome.ucsc.edu/) (Hinrichs et al. 2006). For this, we used the TRANSFAC program, version 10, and its built-in program, MATCH (Matys et al. 2006). For the aging transcriptome, to discriminate the significant transcription factor regulating the up- and down-regulated genes, we applied the statistical method that uses hypergeometric distribution, as illustrated below (P value <0.05).
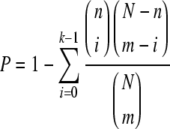 |
Where n transcription factors of interest occur in a transcriptome comprised of N genes, we calculated the P value as the confidence level of having at least k transcription factors of interest in up- or down-regulated groups comprising m.
To ascertain the gene regulation by the putative transcription factors, we analyzed the consistent expression patterns among the transcription factors and the genes that were differentially expressed in the aging and CR transriptomes. For this, we initially collected aging-related microarray studies. Given the microarray studies collected, we filtered out the less informative microarray studies, i.e., those not having many probes, and finally constructed integrated expression profiles by merging the microarray studies, GDS40 (unpublished data), GDS355 (unpublished data), GDS1079 (Beggs et al. 2004) and GDS1311 (Godbout et al. 2005) (ESM File 1). We calculated the Pearson′s correlation coefficient, q, between transcription factors and genes in transcriptome (|q| > 0.8). In order to ensure significance, we constructed random background expression profiles and calculated the significance level of consistent expressional pattern using hypergeometric distribution.
To determine the cooperative activities of transcription factor pairs, we also calculated the correlation coefficient of expression profiles for gene pairs sharing the transcription factor pairs. The significance level was also measured by hypergeometric distribution (P < 10–5) and false discovery rate (FDR) (FDR < 0.01).
Functional modules analysis
Construction of correlation coefficient matrix
We analyzed the functional modules mainly based on the correlation coefficients of the gene expression profiles. For the first step, we constructed correlation coefficient profiles corresponding to the genes found in the individual qualified microarray studies. This procedure gave us the 131 correlation coefficient profiles with a dimension of nC2 × 1 (n = the number of genes in the microarray study) as a result. Given the correlation coefficient profiles, we created a merged correlation coefficient matrix by stacking the 131 correlation coefficient profiles; the resulting matrix had a dimension of m × 131 (m = the number of unions of gene pairs across the 131 correlation coefficient matrix). Given this correlation coefficient matrix, we removed the less informative gene pairs and microarray studies to reduce any unexpected clustering caused by missing values. For this, we defined the less informative gene pairs and microarray studies as: (1) the coverage throughout the overall microarray studies where the gene pairs were less than 70% and 30% of gene pairs and microarray studies, respectively, and (2) the correlation coefficient profiles had abundant low-level correlation coefficients (<0.5); these were discarded.
Construction of functional modules
For the next step, we applied the biclustering approach reported by Shamir et al. (2005) to investigate the functional modules inferred from the correlation coefficient matrices. For this, we used Expander, a freely available program for biclustering, from which we initially obtained 32 and 58 biclusters for the aging and CR transcriptomes, respectively. To be certain of self-homogeneity as functional modules, we computed the secondary correlation coefficients, i.e., correlation coefficients of correlation coefficient profiles in each biclusters, from which the less meaningful quadruplets, specifically those whose secondary correlation coefficient was less than 0.7, were discarded.
We also assessed the self-homogeneity of functional modules. To do that, we parsed the pairs consisting of four different genes, into single genes, termed as ‘module members’ in the functional module. Similarly, the microarray studies in each functional module are termed as ‘module conditions’. Then, we obtained the expression profiles of module members under module conditions. Given the expression profiles of the functional modules, we counted homogeneity, which is defined as the correlation coefficient of expression profiles between genes larger than 0.7. Then, we computed the statistical significance of homogeneity in biclusters by comparison with the random biclusters using χ2 analysis (P < 0.001).
Systems-level associations based on the functional modules
To investigate the systems-level associations, we counted the frequency of the high correlation coefficient (|q| > 0.7) among the biological processes tagged for the genes in the functional modules. The biological meanings of high correlation coefficients were divided into two categories. Positive correlation coefficients identified a positive relation among biological processes, whereas a negative relation among biological processes was accounted by negative correlation coefficients. Then, we applied the statistical significance of relationships between biological processes using χ2 analysis (P < 0.05).
Visualization
Matrices were illustrated using iVici 0.91 (Tarassov and Michnick 2005) and NJ trees were drawn using PHY.FI (Fredslund 2006). Network diagrams were drawn using Cytoscape (Shannon et al. 2003).
Results
Screening for age- and CR-related genes
We identified the DEGs in aging and CR using two different methods. For the age transcriptome, we first collected data on genes previously reported from four valuable microarray studies of various tissues, as listed in Table 1. From these published studies, 383 DEGs in aging were screened. Second, we analyzed microarray data that are freely accessible from GEO to add to the aging-related genes. For this, data from eight microarray studies were collected and analyzed by SAM (δ = 2.4) to identify aging-related genes (Table 1). However, because not every microarray study gave us the DEGs for aging processes, only 108 genes were obtained by the microarray analyses. As a result, we obtained 478 genes, which we collectively termed as the aging transcriptome.
Similar to the definition of the aging transcriptome, CR-related genes—termed the CR transcriptome—were collected using two approaches. Four previous microarray studies were analyzed to identify the CR transcriptome and another three microarray studies were downloaded from GEO, adding to the DEGs in CR (Table 1). We collected 539 and 59 genes from the former and the latter, respectively. As a result, we constructed a CR transcriptome comprising 586 genes in total. Detailed information on aging- and CR-related genes is provided in ESM File 2.
Classification of the age and CR transcriptomes based on GO
To characterize the aging and CR transcriptomes, we analyzed the biological properties in the context of the process and component as suggested by GO. The representative GO terminologies mapped to the aging transcriptome are listed in Fig. 1a, b. We further analyzed the over-represented GO terminologies in the aging transcriptome. Among the 347 genes mapped to GO terminologies, 11 biological processes were over-represented, as shown in Table 2 (P value < 0).
Fig. 1.

a–d Classification of aging and CR transcriptomes in context of Gene Ontology terminologies. a Distribution of the aging transcriptome in respect to biological processes. The major biological processes involved in this study are displayed on the inner bar graph. b Distribution of aging transcriptome in respect to cellular localization. The major compartments are displayed on the inner bar graph. c Distribution of the CR transcriptome in respect to biological processes. The major biological processes involved are displayed on the inner bar graph. d Distribution of the CR transcriptome in respect to cellular localization. The major compartments are displayed on the inner bar graph
Table 2.
Over-represented GO terminologies in the aging and CR transcriptomes (BP biological process, CC cellular component)
| Process | GO context | GO description | Up | Down | Fractions (%) | Fold changec | P valued | |
|---|---|---|---|---|---|---|---|---|
| Aging/CRa | Wholeb | |||||||
| Aging | BP | Muscle development | 9 | 9 | 5.76% | 0.88% | 6.52 | 6.38 × 10-23 |
| Muscle contraction | 4 | 7 | 3.75% | 0.51% | 7.37 | 7.62 × 10-18 | ||
| Cell adhesion | 21 | 8 | 8.36% | 3.88% | 2.15 | 1.21 × 10-5 | ||
| Nervous system development | 9 | 4 | 3.75% | 1.28% | 2.92 | 3.55 × 10-5 | ||
| Cellular protein metabolism | 45 | 30 | 21.61% | 14.30% | 1.51 | 7.76 × 10-5 | ||
| Lipid metabolism | 2 | 10 | 3.46% | 1.25% | 2.77 | 0.0001 | ||
| Immune response | 16 | 4 | 5.76% | 3.13% | 1.84 | 0.0041 | ||
| Amino acid metabolism | 3 | 5 | 2.31% | 1.06% | 2.18 | 0.0212 | ||
| Cell growth | 2 | 3 | 1.44% | 0.57% | 2.54 | 0.0277 | ||
| Cell-cell signaling | 6 | 4 | 3.17% | 1.70% | 1.86 | 0.0316 | ||
| Cytoskeleton organization | 7 | 6 | 4.32% | 2.58% | 1.67 | 0.0383 | ||
| CC | Contractile fiber | 5 | 4 | 2.93% | 0.49% | 5.99 | 5.66 × 10-11 | |
| Mitochondrion | 24 | 22 | 13.49% | 5.80% | 2.32 | 7.52 × 10-10 | ||
| Extracellular space | 54 | 37 | 26.69% | 15.98% | 1.67 | 4.53 × 10-5 | ||
| Lysosome | 8 | 3 | 3.23% | 0.88% | 3.65 | 2.75 × 10-6 | ||
| Peroxisome | 0 | 6 | 1.76% | 0.52% | 3.37 | 0.001296 | ||
| CR | BP | Immune response | 6 | 29 | 7.81% | 3.13% | 2.50 | 6.26 × 10-9 |
| Lipid metabolism | 11 | 6 | 3.79% | 1.25% | 3.03 | 7.84 × 10-7 | ||
| Response to stimulus | 11 | 29 | 9.15% | 4.71% | 1.94 | 6.09 × 10-6 | ||
| Cell proliferation | 8 | 16 | 5.36% | 2.34% | 2.29 | 1.71 × 10-5 | ||
| Glucose catabolism | 4 | 3 | 1.56% | 0.39% | 3.99 | 5.28 × 10-5 | ||
| Cholesterol metabolism | 5 | 2 | 1.56% | 0.40% | 3.90 | 7.13 × 10-5 | ||
| Angiogenesis | 3 | 6 | 2.01% | 0.63% | 3.21 | 0.0001 | ||
| Cell adhesion | 7 | 24 | 6.92% | 3.88% | 1.78 | 0.0007 | ||
| Cell cycle | 10 | 23 | 7.37% | 4.38% | 1.68 | 0.0016 | ||
| Electron transport | 13 | 10 | 5.13% | 2.76% | 1.86 | 0.0017 | ||
| Muscle development | 2 | 7 | 2.01% | 0.88% | 2.27 | 0.0094 | ||
| Cytoskeleton organization | 3 | 17 | 4.46% | 2.58% | 1.73 | 0.0105 | ||
| Chemotaxis | 0 | 7 | 1.56% | 0.68% | 2.31 | 0.0194 | ||
| Amino acid metabolism | 5 | 4 | 2.01% | 1.06% | 1.90 | 0.0451 | ||
| CC | Extracellular space | 38 | 84 | 27.29% | 15.98% | 1.71 | 3.02 × 10-11 | |
| Lysosome | 3 | 11 | 3.13% | 0.88% | 3.55 | 2.27 × 10-7 | ||
| Mitochondrion | 31 | 19 | 11.41% | 5.80% | 1.97 | 2.44 × 10-7 | ||
| Endoplasmic reticulum | 13 | 22 | 8.05% | 3.78% | 2.13 | 1.41 × 10-6 | ||
aThe ratio for the DEGs involved in the terminology among the aging/CR transcriptomes
bThe ratio for the DEGs involved in the terminology among the overall mouse transcriptomes
cThe ratio between the aging/CR fraction and the whole fraction
dP values < 0.05 are considered significant
Interestingly, the genes related to cell adhesion and immune responses were found mostly up-regulated with aging. On the other hand, the genes involved in lipid metabolism showed down-regulation with aging. Regarding cellular compartmentalization, most genes in the aging transcriptome were localized mainly in mitochondria, peroxisomes, and lysosomes. It is worth pointing out that the enrichment of contractile fibers, muscle development/contraction and mitochondria was also shown in cardiac DEGs implicated in the cardiac dysfunction during aging, as reported by Lee et al. (2002).
For the CR transcriptome, a global distribution of biological processes and components are listed in Fig. 1c, d. We further analyzed the representative biological processes and cellular localization for the CR transcriptome. First, we found that similar biological processes were modulated in the CR transcriptome as those found in the aging transcriptome (Table 2). Immune response, lipid metabolism, muscle development, and cell adhesion were all regulated in the CR transcriptome. This finding suggests the overlapping nature between the biological systems of aging and CR. It is important to realize that some of biological processes involved in both transcriptomes are conversely regulated. Specifically, most genes shown to regulate the immune response and cell adhesion were up-regulated with aging, but down-regulated by CR. Whereas, those genes involved in lipid metabolism were mostly up-regulated in the CR transcriptome and down-regulated in the aging transcriptome. It also should be noted that the CR transcriptome was related to more biological processes than the aging transcriptome.
The cellular localization of the CR transcriptome was found to be in mitochondria and lysosomes, as also found for the aging transcriptome. The genes located in lysosomes showed a decreasing tendency for expression under CR compared with a slightly increasing pattern with aging. Interestingly, the genes located in the endoplasmic reticulum (ER), a specialized compartment for protein synthesis, sequestration of Ca2+ and production of steroids and glycogen, were significantly regulated (P = 1.4 × 10-6) by CR. Many of the genes that constitute the extracelluar matrix and cytoskeleton showed a slightly down-regulated pattern under CR.
The correlations between the aging and CR transcriptomes inferred from functional modules
To further investigate the correlations between aging and CR, we constructed functional modules, groups of genes that have similar or closely related biological functions. The functional modules were built by comprehensive and integrative analysis of mouse microarray data compiled from the GEO. For this purpose, initially 381 mouse microarray studies were considered; however, only 82 microarray studies were considered valuable and further processed, while the less informative microarray studies were dropped to reduce noise (ESM File 3).
To construct the functional modules, correlation coefficients between gene pairs were calculated for each microarray study. Biclustering using Expander resulted in 32 biclusters as preliminary functional modules for the aging transcriptome. These preliminary biclusters were further processed to ensure significant coherence in expression patterns (see Materials and methods), resulting in 25 biclusters surviving as functional modules with a confidence P value < 0.001.
The functional modules from the aging transcriptome showed strong correlations in both metabolism (e.g., TCA cycle and lipid metabolism) and the immune response. The module shown in Fig. 2a strongly supports the opposite effects that aging has on metabolism and the immune response, as described in Table 2. In the given module, the genes involved in lipid metabolism and immune response showed opposite expression patterns, seesawing under various biological conditions besides those of aging- or CR-related conditions.
Fig. 2.

a, b Functional modules. a Module 24 for the aging transcriptome. The genes in this module dramatically divided into two different expression profile patterns, and these opposing activity profiles are evident for both the aging CR transcriptomes. b Module 22 for CR transcriptome. The genes in this module are all down-regulated in the CR transcriptome and mainly are involved in immune response
The contrary pattern between transcriptomes was also shown in the CR transcriptome functional module (Fig. 2b). This module, which includes many immune-related genes, shows contrary patterns for three genes in both the aging and CR transcriptomes: Clqb, involved in complement activation, was up-regulated in aging and down-regulated in CR; Ctss was up-regulated in aging (Keppler et al. 2000); and Tmp4 showed a contrary pattern between aging and CR. Many previous reports indicate that both Clqb and Ctss play a role in the immune response, specifically in the interplay with a major histocompatibility complex (MHC) (Denz et al. 2004; Karagiannides et al. 2001). The Tmp4 gene product is an essential component of thin filaments (Denz et al. 2004). Although the expressional patterns of most genes in aging have not been determined, the module shown in Fig. 2b might give clues in predicting expressional changes during aging on the basis of the consistent expression patterns observed across various biological conditions.
The use of functional modules as a whole in the present study allowed us to probe the correlations existing between aging and CR at systems levels consistent with expression patterns among various biological systems. We found that various biological systems are associated with each other (Fig. 3). For instance, lipid metabolism and immune systems were shown to have a significant inverse correlation (P = 6.73 × 10-9). On one hand, lipid metabolism seemed to be inversely related with systems such as the cell cycle (P = 0.00497), cell proliferation (P = 0.00014), and muscle development (P = 0.03642) during aging, while the immune response showed positive correlations with these systems. This comprehensive analysis of functional modules revealed insights into how aging may perturb dynamic and interrelated systems.
Fig. 3.

Functional associations at system levels. Systems in the aging and CR transcriptomes are closely correlated in a positive (red line) or negative (blue line) manner. Lipid metabolism, especially, showed an inverse correlation with many other systems, including immune response, cell cycle, cell proliferation, and muscle development. The systems showing the strongest inverse correlation were lipid metabolism and immune response (P = 6.73 × 10-9). The relationship between lipid metabolism and cytoskeleton organization is somewhat ambiguous (green line), showing positive and negative patterns together
Transcription factor binding sites over-represented in the upstream regions of aging- and CR-related genes
We further analyzed the correlations between the regulatory networks of aging and CR genes by investigating putative transcription factor binding sties that were over-represented in up- or down-regulated genes in both the aging and CR transcriptomes. Classifying genes according to their tendency to up- or down-regulate, we obtained 268 up-regulated genes, 203 down-regulated genes, and seven controversial genes for the aging transcriptome (ESM File 2). For the CR transcriptome, 199 genes were up-regulated and 394 genes were down-regulated. The controversial genes in this transcriptome were discarded. To identify the over-represented transcription factor binding sites in the up- and down-regulated groups, the statistical method of hypergeometric distribution (P value < 0.05) was applied to the transcription factors’ binding sites on 1,000 upstream regions of genes.
In the aging transcriptome, we found 234 upstream for the up-regulated genes and 168 upstream for the down-regulated genes. Of the upstream, 744 and 654 putative transcription factor binding sites were initially found in up- and down-regulated genes, respectively, and a further significance test narrowed these to 11 and eight significant transcription factor binding sites (Table 3). In the CR transcriptome, of the 744 and 654 putative transcription factor binding sites found in up- and down-regulated genes, respectively, the significant test narrowed down to 12 and five overrepresented transcription factor binding sites in up- and down-regulated genes, respectively (Table 3). To ascertain the regulatory effect of the putative transcription factors on target genes, the expressional coherence between transcription factors and their target genes was investigated in four aging-related microarray studies, which we include with this report (ESM File 1). It is important to note that the microarray studies referenced here do not cover all transcription factors due to the incompleteness of microarray studies. Listed in Table 4 are the transcription factors that showed consistent expression profiles with age- or CR-related genes found in aging-related microarray studies.
Table 3.
Transcription factors significantly distributed in up- and down-regulated gene expression in aging and CR
| Process | Overrepresented in | Transcription factor | Up | Down | P value |
|---|---|---|---|---|---|
| Aging | Up-regulated genes | C/EBP δ | 107 | 53 | 0.0027 |
| DEC | 119 | 62 | 0.0036 | ||
| Up | POU6F1 | 15 | 2 | 0.0073 | |
| (234) | GATA-2 | 73 | 35 | 0.0134 | |
| Down | ICSBP (IRF8) | 11 | 1 | 0.0134 | |
| (168) | POU1F1 | 83 | 42 | 0.0162 | |
| Staf | 51 | 22 | 0.0168 | ||
| RFX1 (EF-C) | 187 | 119 | 0.0238 | ||
| TFE | 35 | 14 | 0.0307 | ||
| Freac-7 | 74 | 39 | 0.0405 | ||
| Elk-1 | 161 | 101 | 0.0451 | ||
| Down-regulated genes | Bach2 | 111 | 101 | 0.0078 | |
| Zta | 44 | 49 | 0.01 | ||
| FOXO3 | 4 | 10 | 0.0225 | ||
| CHOP:C/EBP α | 33 | 37 | 0.0271 | ||
| CCAAT box | 127 | 107 | 0.0368 | ||
| FXR/RXR-α | 5 | 10 | 0.0432 | ||
| TGIF | 29 | 32 | 0.0458 | ||
| CR | Up-regulated genes | v-Myb | 120 | 194 | 0.00027 |
| HNF-4α | 54 | 68 | 0.00175 | ||
| Up | TAL1 | 58 | 76 | 0.00219 | |
| (149) | E4BP4 | 10 | 4 | 0.00334 | |
| Down | HLF | 43 | 55 | 0.00799 | |
| (301) | CCAAT box | 107 | 181 | 0.00956 | |
| FOXO1 | 19 | 19 | 0.01833 | ||
| MAZ | 116 | 205 | 0.01963 | ||
| VBP | 13 | 11 | 0.02364 | ||
| Tal-1alpha:E47 | 17 | 17 | 0.02565 | ||
| HNF-3β (FOXA2) | 95 | 163 | 0.03267 | ||
| Max | 7 | 4 | 0.03543 | ||
| Down-regulated genes | IRF-1 | 0 | 9 | 0.02575 | |
| Pax | 106 | 242 | 0.01933 | ||
| PAX6 | 13 | 49 | 0.01821 | ||
| YY1 | 16 | 54 | 0.03019 | ||
| NKX3A | 17 | 54 | 0.0471 |
P values < 0.05 are considered significant
Table 4.
Transcription factors showing the consistent expression patterns with aging- or CR-related genes
| Transcription factors | Genes (downstream) | Correlation coefficient | Changes | Biological process | |
|---|---|---|---|---|---|
| Aging | CR | ||||
| POU6F1 | Mmp15 | 0.81179 | Up | DOWN | Proteolysis |
| GATA-2 | Tpm4 | 0.838773 | Up | DOWN | Muscle development |
| Ctss | 0.7332 | Up | DOWN | Proteolysis | |
| D12Ertd647e | 0.778252 | Up | DOWN | - | |
| Pdha1 | 0.807658 | Down | Up | Glycolysis | |
| 2900073G15Rik | 0.81865 | Up | Down | - | |
| Crat | 0.76079 | Down | Up | Lipid metabolism | |
| ICSBP | Fyn | 0.946193 | Up | Down | Signal transduction |
| (IRF8) | 2410022L05Rik | 0.925995 | Up | Down | - |
| Col4a1 | 0.756036 | Up | Down | Cell adhesion | |
| Usp21 | 0.867904 | Up | Down | Ubiquitin cycle | |
| POU1F1 | Cpt1a | 0.824933 | Down | Up | Lipid metabolism |
| P4ha1 | 0.840611 | Up | Down | Protein metabolism | |
| Ctss | 0.74411 | Up | Down | Proteolysis | |
| Acot1 | 0.79474 | Down | Up | Lipid metabolism | |
| 2900073G15Rik | 0.826971 | Up | Down | - | |
| Cx3cl1 | 0.866131 | Up | Down | Cell adhesion; immune response | |
| Vldlr | 0.800157 | Down | Up | Lipid metabolism | |
| FOXO3 | Tpm4 | 0.879282 | Up | Down | Muscle development |
| P4ha1 | 0.711592 | Up | Down | protein metabolism | |
| D12Ertd647e | 0.749182 | Up | Down | - | |
| Pdha1 | 0.869872 | Down | Up | Glycolysis | |
| 2900073G15Rik | 0.843534 | Up | Down | - | |
| Cx3cl1 | 0.809367 | Up | Down | Cell adhesion; immune response | |
| Vldlr | 0.836079 | Down | Up | Lipid metabolism | |
The importance of these transcription factors during the aging process also has been shown by previous studies. For instance, the regulatory effect of C/EBPδ on aging is supported by Karagiannides et al. (2001), who found that impaired adipogenesis and altered fat tissue function with aging could be caused by the altered expression of the C/EBP families, including C/EBPδ.
Another important transcription factor, Pou1f1, was identified by computation analysis and shows consistent expression patterns with the genes in the aging transcriptome (Table 4). A mutation of the Pou1f1 gene (also named Pit1) results in the Snell dwarf mouse, which shows extended life span. Several lines of evidence using this animal model suggest that an altered management of oxidative stress is strongly related to its increased longevity (Hsieh et al. 2002; Madsen et al. 2004). Supporting evidence for the regulatory effect of Elk-1 is provided in a report by Knebel et al. (2005), who found that basal and inducible phosphorylation of Elk-1 was impaired in cultured fibroblasts of a patient with premature aging syndrome and insulin resistance (Knebel et al. 2005).
The FOXO families are other well-known transcription factors involved in the PI3K-Akt signaling network, which is linked to cancer, metabolism, and aging (Brunet et al. 1999; Biggs et al. 1999). Martin et al. (2006) and Lehtinen et al. (2006) suggested that the FOXO families could modulate the signaling pathway that mediates oxidative stress responses and extends life span. Aging-related microarray studies show that especially FOXO3 exhibits consistent expression patterns with aging- and CR-related genes (Table 4). Notably, the binding site for Irf8 (also known as ICSBP), down-regulated in CR (Fig. 2b), is overrepresented upstream of the up-regulated aging-related genes and shows consistent expression patterns with genes in the aging transcriptome (Table 4). Our finding suggested that Irf8 may be an important transcription factor in modulating the aging and CR processes, although its role and mechanisms are not clear at this time.
We also investigated the cooperative activities of several other transcription factors. Elk-1 and the CCAAT box showed significant cooperative activity in the age-related gene pairs (P value = 7.69 × 10-7, FDR = 0.005) (Table 5), and their effects are also supported by previous studies. Lin et al. (2005) report that Elk-1 and the CCAAT enhancer binding protein (C/EBP) show cooperative activities in the transcriptional regulation of RTP801, a newly discovered stress-response gene. Additionally, Hanlon et al. (2000) also reported that Elk-1 and C/EBP have a synergistic effect on the maximal induction of the c-fos gene. These findings strongly indicate that the cooperative activities of the Elk-1 and C/EBP families might modulate the aging process by the induction of stress-related genes; however, experimental validation remains challenging. Further, Kowenz-Leutz et al. (1997) reported that the combination of v-Myb and the CCAAT box has a synergistic effect on the activation of downstream genes. Mink et al. (1999) suggested an auto-regulatory loop model for the regulation of C/EBP families with v-Myb. HNF-3β also seems to have a synergistic effect on the regulation of downstream genes when combined with the other transcription factors such as Pax and the C/EBP families, although the regulatory mechanisms by aging or CR have not been uncovered.
Table 5.
Transcription factor pairs showing cooperative activities
| Process | Transcription factor pairs | Number of coherencea | Number of gene pairsb | P valuec | FDRd (N = 1,000) |
|---|---|---|---|---|---|
| Aging | Elk-1 : CCAAT box | 27 | 595 | 7.59 × 10-7 | 0.005 |
| CR | v-Myb : MAZ | 125 | 1,485 | 1.6 × 10-28 | 0 |
| MAZ : CCAAT box | 88 | 990 | 1.63 × 10-20 | 0 | |
| v-Myb : CCAAT box | 86 | 1,081 | 4.9 × 10-20 | 0 | |
| Pax : HNF-3beta | 85 | 1,485 | 1.67 × 10-19 | 0 | |
| HNF-3beta : CCAAT box | 83 | 861 | 2.09 × 10-19 | 0 | |
| MAZ : HNF-3beta | 84 | 1,653 | 2.56 × 10-19 | 0 | |
| Pax : MAZ | 77 | 1,225 | 9.57 × 10-18 | 0 | |
| v-Myb : HNF-3beta | 73 | 1,225 | 1.03 × 10-16 | 0 | |
| Pax : CCAAT box | 63 | 780 | 9.94 × 10-15 | 0 | |
| v-Myb : Pax | 54 | 990 | 1.7 × 10-12 | 0 |
aThe number of gene pairs containing the transcription factor pairs and showing the consistent expression patterns
bThe number of gene pairs containing the transcription factor pairs
cThe average P value for 1,000 runs; P values < 0.00001 are considered significant
dAn FDR < 0.01 is considered significant
Discussion
With the massive number of datasets for cellular components, we are often overwhelmed by having more pieces to the puzzle that make up biological phenomena when using so called ‘omic’ studies. Systems biology studies aimed at how the pieces of the puzzle fit together are necessary for all fields of biology in this ‘omics’ era. In the field of aging research, systems biology could be an especially important, new paradigm considering that aging is a biological phenomenon induced by gradual system-level declines.
In this report, we have shown a systems biology approach to further our understanding of the changes that occur during aging and their correlations to the processes of aging and CR—the most powerful intervention known to date to attenuate the aging process. The promising power of this systems approach has been proven valuable in at least three aspects. First, as the present study indicates, this approach could address important questions like what are the driving forces of aging and what are the recovering forces induced by CR at system levels? The answer, as we have suggested, is that the critical systems regulating aging and CR are lipid metabolism, immune response, and cell adhesion, as shown by the results derived from GO analysis (Table 2). Second, changes in systems-level homeostasis could be inferred from the functional modules, a set of genes showing consistent expression patterns. From the functional modules, we have shown remarkable correlations between the homeostasis of lipid metabolism and the immune response under various biological conditions, including aging and CR (Fig. 2a). More systematically, we also constructed a ‘systems-level network’ that reflects perturbed homeostasis among systems based on functional modules (Fig. 3), giving us a possible macroscopic view of the architecture of biological systems. Third, the results of our study give predictions of interesting but uncharacterized genes or transcription factors for their use in future aging research. Although these functional modules contain some incomplete information on the expressional patterns with aging or CR (Fig. 2), it is possible to predict the expressional patterns of genes yet to be characterized with aging or CR according to the overall patterns disclosed by these functional modules. In addition, uncharacterized functional correlations among genes could be suspected by the consistent patterns shown in the functional modules (ESM File 4). In this way, the output derived from systems biology approaches can provide feedback, thereby permitting biology to be better understood in more multilateral contexts.
We documented that various biological systems, as shown in Fig. 4, were influenced by aging or CR. The expression levels of most genes involved in cell adhesion and the immune response were up-regulated with aging, while the expression levels of most genes involved in lipid metabolism were down-regulated with aging. Also, several papers from this laboratory report many aging-induced changes in biological processes. Recently, Zou et al. from our research group (2004) reported that the expression of soluble adhesion molecules, including E-selectin, P-selectin, vascular cell adhesion molecule 1 (VCAM-1), and intercellular adhesion molecule 1 (ICAM-1), increase during aging. Their study suggests that the altered expression of adhesion modules were due to increased oxidative stress with aging. Further evidence comes from our laboratory, Sung et al. (2006), who showed that PPARγ, a key regulator of lipid metabolism, attenuated aging by reducing oxidative stress. In other previous reports, we have also pointed out that certain key players are closely related to inflammatory mediators such as NFκB, IL-1β, IL-6, TNFα, cyclooxygenase-2, adhesion molecules, and inducible NO synthase (Chung et al. 2006).
Fig. 4.

Summary of biological processes and transcription factors involved in aging and CR. The putative transcription factors regulating the mouse aging and CR transcriptomes are described. A total of 11 transcription factors are over-represented in mouse genes that are up-regulated during the aging process. POU6F1 is also over-represented in the common transcriptome, especially those genes that are up-regulated in aging and down-regulated in CR. The putative regulatory elements modulating the genes down-regulated during aging are eight in total. Among these, one regulatory element (i.e., CCAAT box) is a cis-acting regulatory element and the others are trans-acting elements. Interestingly, the CCAAT box is over-represented in the upstream regions of genes that are up-regulated in CR This finding indicates the opposite effect of CCAAT box-related transcription factors on aging and CR. The important regulatory elements involved in the regulation of genes up-regulated in CR are 12 in total. The cis-acting regulatory elements such as CCAAT box and Tal-α:E47 box are also included. It is surprising that two forkhead family transcription factors, FOXO1 and HNF-3β (also known as FOXA2) are involved in the regulation of genes up-regulated in CR. These two transcription factors are also found in some genes in the common transcriptome, especially those down-regulated during the aging process, and contrary to those up-regulated in CR. The transcription factors regulating the genes down-regulated in CR are five. PAX-6 is also an important transcription factor for the regulation of genes up-regulated during aging and down-regulated in CR. There are 14 regulatory elements involved in the regulation of common transcriptome. Among these, six transcription factors are over-represented in genes up-regulated during aging and down-regulated in CR, including POU6F1 and PAX-6, as described above. Eight regulatory elements comprise the various forkhead families involved in the regulation of genes down-regulated during aging and up-regulated in CR, including FOXO1 and HNF-3β described above
The inverse effects of the immune response and lipid metabolism have rarely been addressed in the aging literature. And although a few reports show correlations among biological processes, these studies did not focus on the systems level (Yamada et al. 2003; Wick et al. 1991). In our current study, we found opposite expressional patterns between the immune response and lipid metabolism based on the large-scale expression profiles. The functional modules also supported the inversely related patterns between these two systems under diverse biological conditions. As an additional benefit, these functional modules could shed light on unidentified or uncharacterized genes related to aging or CR, according to their similar or opposite expressional patterns.
We also identified putative transcription factors for aging and CR in this study. Although several lines of evidence support a regulatory role for transcription factors in aging, many transcription factors remain to be defined. Here, we are able to present data on the effects of uncharacterized transcription factors with aging by showing consistent expression patterns between those identified transcription factors and target genes. However, multilateral approaches are necessary to detect other important transcription factors. For instance, we identified PPARα, an important transcription factor in mediating aging-related signaling pathways, using an analysis for detecting conserved transcription factors in human and mouse (P = 0.0216, data not shown); however, the analysis in the present study failed to detect PPARα. On the other hand, although we found other well-known transcription factors such as FOXO (Lehtinen et al. 2006; Martin et al. 2006), GATA (Budovskaya et al. 2008), the C/EBP families (Karagiannides et al. 2001), and Pit-1 (Hsieh et al. 2002; Madsen et al. 2004) by the analysis used in the present study, conservation analysis has failed to detect these transcription factors. Thus, more data collection is required on transcription factors, thereby providing complementary approaches for the prediction of transcription factors in the various strategies. It is also important to point out our present study has some limit because the majority of DEGs data on the cardiac function were obtained from heart. Large-scale data on the common or specific signatures across various aged organs should provide a broader, informative view.
In summary, we present the systems biological approaches to (1) discover the biological systems most likely related to aging or CR, (2) construct a ‘systems-level network’ of the genes involved in the aging process, so as to provide a macroscopic view of the homeostasis of systems during aging, and (3) predict uncharacterized genes or transcription factors involved in aging CR processes. From this study, we found that the immune response, cell adhesion, and lipid metabolism were modulated by aging and that the effects were reversed by CR. We were also able to show predictions of the transcription factors most likely related in the cross-talk between aging and CR. Although further experimental validation is needed to confirm the functional modules and regulatory networks, this approach will be the initial step toward the comprehensive understanding of aging.
Electronic supplementary material
Below is the link to the electronic supplementary material.
List of microarray studies used for coherence analysis. Four microarray studies were analyzed to investigate the consistent expressional patterns between transcription factors and their downstream genes. (DOC 29 kb)
Detailed information on the aging and CR transcriptomes. This file gives detailed information (e.g., gene name, the various IDs, and expressional changes) about the aging and CR transcriptomes and contains two separate sheets for each transcriptome. (XLS 195 kb)
List of microarray studies used for the construction of functional modules. Detailed information about the microarray studies used for the construction of functional modules is contained in this file. The information is referred from the annotations of the GEO platform. (XLS 52 kb)
Consistent expression patterns between Tmp4 and Dck. The expression profiles of Tmp4 and Dck across aging-related conditions listed in ESM File 2 are consistent to each other. The similar expression profiles indicate the close relation in their function; however, few studies on the functional relation between these genes are available. Experimental validation remains challenging. (TIF 891 kb)
Acknowledgements
This research was supported by the Korea Science and Engineering Foundation (KOSEF) [NO.2007-00376 and R01-2007-000-20852-0(2007)]. We thank the Aging Tissue Bank for providing aging research resources.
References
- Barrett T, Suzek TO, Troup DB, Wilhite SE, Ngau WC, Ledoux P, Rudnev D, Lash AE, Fujibuchi W, Edgar R. NCBI GEO: mining millions of expression profiles—database and tools. Nucleic Acids Res. 2005;33:D562–D566. doi: 10.1093/nar/gki022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beggs ML, Nagarajan R, Taylor-Jones JM, Nolen G, Macnicol M, Peterson CA. Alterations in the TGFbeta signaling pathway in myogenic progenitors with age. Aging Cell. 2004;3:353–361. doi: 10.1111/j.1474-9728.2004.00135.x. [DOI] [PubMed] [Google Scholar]
- Biggs WH, 3rd, Meisenhelder J, Hunter T, Cavenee WK, Arden KC. Protein kinase B/Akt-mediated phosphorylation promotes nuclear exclusion of the winged helix transcription factor FKHR1. Proc Natl Acad Sci USA. 1999;96:7421–7426. doi: 10.1073/pnas.96.13.7421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blake JA, Eppig JT, Bult CJ, Kadin JA, Richardson JE. The Mouse Genome Database (MGD): updates and enhancements. Nucleic Acids Res. 2006;34:D562–D567. doi: 10.1093/nar/gkj085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blalock EM, Chen KC, Stromberg AJ, Norris CM, Kadish I, Kraner SD, Porter NM, Landfield PW. Harnessing the power of gene microarrays for the study of brain aging and Alzheimer′s disease: statistical reliability and functional correlation. Ageing Res Rev. 2005;4:481–512. doi: 10.1016/j.arr.2005.06.006. [DOI] [PubMed] [Google Scholar]
- Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS, Anderson MJ, Arden KC, Blenis J, Greenberg ME. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell. 1999;96:857–868. doi: 10.1016/S0092-8674(00)80595-4. [DOI] [PubMed] [Google Scholar]
- Budovskaya YV, Wu K, Southworth LK, Jiang M, Tedesco P, Johnson TE, Kim SK. An elt-3/elt-5/elt-6 GATA transcription circuit guides aging in C. elegans. Cell. 2008;134:291–303. doi: 10.1016/j.cell.2008.05.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung HY, Sung B, Jung KJ, Zou Y, Yu BP. The molecular inflammatory process in aging. Antioxid Redox Signal. 2006;8:572–581. doi: 10.1089/ars.2006.8.572. [DOI] [PubMed] [Google Scholar]
- Das D, Nahle Z, Zhang MQ. Adaptively inferring human transcriptional subnetworks. Mol Syst Biol. 2006;2(2006):0029. doi: 10.1038/msb4100067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denz CR, Narshi A, Zajdel RW, Dube DK. Expression of a novel cardiac-specific tropomyosin isoform in humans. Biochem Biophys Res Commun. 2004;320:1291–1297. doi: 10.1016/j.bbrc.2004.06.084. [DOI] [PubMed] [Google Scholar]
- Fredslund J. PHY.FI: fast and easy online creation and manipulation of phylogeny color figures. BMC Bioinformatics. 2006;7:315. doi: 10.1186/1471-2105-7-315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gene Ontology Consortium The Gene Ontology (GO) project in 2006. Nucleic Acids Res. 2006;34:D322–D326. doi: 10.1093/nar/gkj021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godbout JP, Chen J, Abraham J, Richwine AF, Berg BM, Kelley KW, Johnson RW. Exaggerated neuroinflammation and sickness behavior in aged mice following activation of the peripheral innate immune system. FASEB J. 2005;19:1329–1331. doi: 10.1096/fj.05-3776fje. [DOI] [PubMed] [Google Scholar]
- Hanlon M, Bundy LM, Sealy L. C/EBP beta and Elk-1 synergistically transactivate the c-fos serum response element. BMC Cell Biol. 2000;1:2. doi: 10.1186/1471-2121-1-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higami Y, Pugh TD, Page GP, Allison DB, Prolla TA, Weindruch R. Adipose tissue energy metabolism: altered gene expression profile of mice subjected to long-term caloric restriction. FASEB J. 2004;18:415–417. doi: 10.1096/fj.03-0678fje. [DOI] [PubMed] [Google Scholar]
- Higami Y, Barger JL, Page GP, Allison DB, Smith SR, Prolla TA, Weindruch R. Energy restriction lowers the expression of genes linked to inflammation, the cytoskeleton, the extracellular matrix, and angiogenesis in mouse adipose tissue. J Nutr. 2006;136:343–352. doi: 10.1093/jn/136.2.343. [DOI] [PubMed] [Google Scholar]
- Hinrichs AS, Karolchik D, Baertsch R, Barber GP, Bejerano G, Clawson H, Diekhans M, Furey TS, Harte RA, Hsu F, et al. The UCSC Genome Browser Database: update 2006. Nucleic Acids Res. 2006;34:D590–D598. doi: 10.1093/nar/gkj144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong SE, Rho SH, Yeom YI, Kim do H. HCNet: a database of heart and calcium functional network. Bioinformatics. 2006;22:2053–2054. doi: 10.1093/bioinformatics/btl331. [DOI] [PubMed] [Google Scholar]
- Hsieh CC, DeFord JH, Flurkey K, Harrison DE, Papaconstantinou J. Effects of the Pit1 mutation on the insulin signaling pathway: implications on the longevity of the long-lived Snell dwarf mouse. Mech Ageing Dev. 2002;123:1245–1255. doi: 10.1016/S0047-6374(02)00037-4. [DOI] [PubMed] [Google Scholar]
- Ida H, Boylan SA, Weigel AL, Hjelmeland LM. Age-related changes in the transcriptional profile of mouse RPE/choroid. Physiol Genomics. 2003;15:258–262. doi: 10.1152/physiolgenomics.00126.2003. [DOI] [PubMed] [Google Scholar]
- Karagiannides I, Tchkonia T, Dobson DE, Steppan CM, Cummins P, Chan G, Salvatori K, Hadzopoulou-Cladaras M, Kirkland JL. Altered expression of C/EBP family members results in decreased adipogenesis with aging. Am J Physiol Regul Integr Comp Physiol. 2001;280:R1772–R1780. doi: 10.1152/ajpregu.2001.280.6.R1772. [DOI] [PubMed] [Google Scholar]
- Keppler D, Walter R, Perez C, Sierra F. Increased expression of mature cathepsin B in aging rat liver. Cell Tissue Res. 2000;302:181–188. doi: 10.1007/s004410000269. [DOI] [PubMed] [Google Scholar]
- Knebel B, Avci H, Bullmann C, Kotzka J, Muller-Wieland D. Reduced phosphorylation of transcription factor Elk-1 in cultured fibroblasts of a patient with premature aging syndrome and insulin resistance. Exp Clin Endocrinol Diabetes. 2005;113:94–101. doi: 10.1055/s-2004-830554. [DOI] [PubMed] [Google Scholar]
- Kowenz-Leutz E, Herr P, Niss K, Leutz A. The homeobox gene GBX2, a target of the myb oncogene, mediates autocrine growth and monocyte differentiation. Cell. 1997;91:185–195. doi: 10.1016/S0092-8674(00)80401-8. [DOI] [PubMed] [Google Scholar]
- Lee CK, Weindruch R, Prolla TA. Gene-expression profile of the ageing brain in mice. Nat Genet. 2000;25:294–297. doi: 10.1038/77046. [DOI] [PubMed] [Google Scholar]
- Lee CK, Allison DB, Brand J, Weindruch R, Prolla TA. Transcriptional profiles associated with aging and middle age-onset caloric restriction in mouse hearts. Proc Natl Acad Sci USA. 2002;99:14988–14993. doi: 10.1073/pnas.232308999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehtinen MK, Yuan Z, Boag PR, Yang Y, Villen J, Becker EB, DiBacco S, Iglesia N, Gygi S, Blackwell TK, et al. A conserved MST-FOXO signaling pathway mediates oxidative-stress responses and extends life span. Cell. 2006;125:987–1001. doi: 10.1016/j.cell.2006.03.046. [DOI] [PubMed] [Google Scholar]
- Lin L, Stringfield TM, Shi X, Chen Y. Arsenite induces a cell stress-response gene, RTP801, through reactive oxygen species and transcription factors Elk-1 and CCAAT/enhancer-binding protein. Biochem J. 2005;392:93–102. doi: 10.1042/BJ20050553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madsen MA, Hsieh CC, Boylston WH, Flurkey K, Harrison D, Papaconstantinou J. Altered oxidative stress response of the long-lived Snell dwarf mouse. Biochem Biophys Res Commun. 2004;318:998–1005. doi: 10.1016/j.bbrc.2004.04.126. [DOI] [PubMed] [Google Scholar]
- Martin B, Mattson MP, Maudsley S. Caloric restriction and intermittent fasting: two potential diets for successful brain aging. Ageing Res Rev. 2006;5:332–353. doi: 10.1016/j.arr.2006.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massaro D, Massaro GD, Baras A, Hoffman EP, Clerch LB. Calorie-related rapid onset of alveolar loss, regeneration, and changes in mouse lung gene expression. Am J Physiol Lung Cell Mol Physiol. 2004;286:L896–L906. doi: 10.1152/ajplung.00333.2003. [DOI] [PubMed] [Google Scholar]
- Matys V, Kel-Margoulis OV, Fricke E, Liebich I, Land S, Barre-Dirrie A, Reuter I, Chekmenev D, Krull M, Hornischer K, et al. TRANSFAC and its module TRANSCompel: transcriptional gene regulation in eukaryotes. Nucleic Acids Res. 2006;34:D108–D110. doi: 10.1093/nar/gkj143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mink S, Jaswal S, Burk O, Klempnauer KH. The v-Myb oncoprotein activates C/EBPbeta expression by stimulating an autoregulatory loop at the C/EBPbeta promoter. Biochim Biophys Acta. 1999;1447:175–184. doi: 10.1016/s0167-4781(99)00168-2. [DOI] [PubMed] [Google Scholar]
- Shamir R, Maron-Katz A, Tanay A, Linhart C, Steinfeld I, Sharan R, Shiloh Y, Elkon R. EXPANDER–an integrative program suite for microarray data analysis. BMC Bioinformatics. 2005;6:232. doi: 10.1186/1471-2105-6-232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slonim N, Elemento O, Tavazoie S. Ab initio genotype-phenotype association reveals intrinsic modularity in genetic networks. Mol Syst Biol. 2006;2:2006.0005. doi: 10.1038/msb4100047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung B, Park S, Yu BP, Chung HY. Amelioration of age-related inflammation and oxidative stress by PPARgamma activator: suppression of NF-kappaB by 2, 4-thiazolidinedione. Exp Gerontol. 2006;41:590–599. doi: 10.1016/j.exger.2006.04.005. [DOI] [PubMed] [Google Scholar]
- Tanay A, Steinfeld I, Kupiec M, Shamir R. Integrative analysis of genome-wide experiments in the context of a large high-throughput data compendium. Mol Syst Biol. 2005;1(2005):0002. doi: 10.1038/msb4100005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarassov K, Michnick SW. iVici: Interrelational Visualization and Correlation Interface. Genome Biol. 2005;6:R115. doi: 10.1186/gb-2005-6-13-r115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian J, Ishibashi K, Reiser K, Grebe R, Biswal S, Gehlbach P, Handa JT. Advanced glycation endproduct-induced aging of the retinal pigment epithelium and choroid: a comprehensive transcriptional response. Proc Natl Acad Sci USA. 2005;102:11846–11851. doi: 10.1073/pnas.0504759102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tusher VG, Tibshirani R, Chu G. Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci USA. 2001;98:5116–5121. doi: 10.1073/pnas.091062498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wick G, Huber LA, Xu QB, Jarosch E, Schonitzer D, Jurgens G. The decline of the immune response during aging: the role of an altered lipid metabolism. Ann N Y Acad Sci. 1991;621:277–290. doi: 10.1111/j.1749-6632.1991.tb16986.x. [DOI] [PubMed] [Google Scholar]
- Yamada K, Tokunaga Y, Ikeda A, Ohkura K, Kaku-Ohkura S, Mamiya S, Lim BO, Tachibana H. Effect of dietary fiber on the lipid metabolism and immune function of aged Sprague-Dawley rats. Biosci Biotechnol Biochem. 2003;67:429–433. doi: 10.1271/bbb.67.429. [DOI] [PubMed] [Google Scholar]
- Zhou XJ, Kao MC, Huang H, Wong A, Nunez-Iglesias J, Primig M, Aparicio OM, Finch CE, Morgan TE, Wong WH. Functional annotation and network reconstruction through cross-platform integration of microarray data. Nat Biotechnol. 2005;23:238–243. doi: 10.1038/nbt1058. [DOI] [PubMed] [Google Scholar]
- Zou Y, Jung KJ, Kim JW, Yu BP, Chung HY. Alteration of soluble adhesion molecules during aging and their modulation by calorie restriction. FASEB J. 2004;18:320–322. doi: 10.1096/fj.03-0849fje. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Below is the link to the electronic supplementary material.
List of microarray studies used for coherence analysis. Four microarray studies were analyzed to investigate the consistent expressional patterns between transcription factors and their downstream genes. (DOC 29 kb)
Detailed information on the aging and CR transcriptomes. This file gives detailed information (e.g., gene name, the various IDs, and expressional changes) about the aging and CR transcriptomes and contains two separate sheets for each transcriptome. (XLS 195 kb)
List of microarray studies used for the construction of functional modules. Detailed information about the microarray studies used for the construction of functional modules is contained in this file. The information is referred from the annotations of the GEO platform. (XLS 52 kb)
Consistent expression patterns between Tmp4 and Dck. The expression profiles of Tmp4 and Dck across aging-related conditions listed in ESM File 2 are consistent to each other. The similar expression profiles indicate the close relation in their function; however, few studies on the functional relation between these genes are available. Experimental validation remains challenging. (TIF 891 kb)