

Abstract
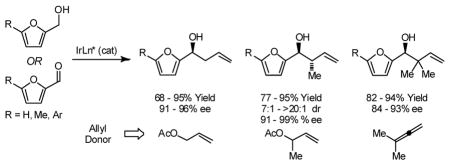
5-Substituted-2-furan methanols 1a–1c are subject to enantioselective carbonyl allylation, crotylation and tert-prenylation upon exposure to allyl acetate, α-methyl allyl acetate or 1,1,-dimethylallene in the presence of an ortho-cyclometallated iridium catalyst modified by (R)-Cl,MeO-BIPHEP, (R)-C3 -TUNEPHOS and (R)-C3-SEGPHOS, respectively. In the presence of isopropanol, but under otherwise identical conditions, the corresponding substituted furfurals 2a–2c are converted to identical products of allylation, crotylation and tert-prenylation. Optically enriched products carbonyl allylation, crotylation and reverse prenylation 3b, 4b and 5b were subjected to Achmatowicz rearrangement to furnish the corresponding γ-hydroxy-β-pyrones 6a–6c, respectively, with negligible erosion of enantiomeric excess.
In the course of studies on C-C bond forming hydrogenations and transfer hydrogenations,1 we found that cyclometallated iridium C,O-benzoates modified by chiral phosphine ligands are effective catalysts for the enantioselective reductive coupling of allyl acetate, α-methyl allyl acetate and 1,1-dimethylallene to carbonyl electrophiles to furnish products of carbonyl allylation,2a,b,e–h crotylation2c,f and tert-prenylation,2d,f respectively. For such “C-C bond forming transfer hydrogenations”, isopropanol serves as the terminal reductant for additions to preformed aldehyde electrophiles. Remarkably, primary alcohols serve dually as hydrogen donors and aldehyde precursors, enabling asymmetric carbonyl allylation, crotylation and tert-prenylation directly from the alcohol oxidation level. Here, hydrogen exchange between an alcohol-unsaturate redox pair enables generation of an electrophile-nucleophile pair. In this way, non-stabilized carbanion equivalents are generated in the absence of stoichiometric metallic reagents. This strategy for carbonyl allylation differs significantly from conventional carbonyl protocols, which exploit stoichiometric quantities of allylmetal reagents or metallic reductants.3,4,5.
In connection with studies toward the syntheses of the mitochondrial electron transport inhibitors ajudazols A and B,6 one of the present authors required a highly enantioselective method for the crotylation of a substituted furfural. Although the enantioselective allylation of furfurals has been achieved using stoichiometric allylmetal reagents,7 Denmark reports the only catalytic method for enantioselective crotylation and reverse prenylation of furfural.7f,n Given the broad utility of furans as building blocks in organic synthesis,8 we sought to further evaluate the scope of our emergent allylation methodology in a systematic study of allylation, crotylation and reverse prenylation substituted furfurals from both the alcohol and aldehyde oxidation levels. Here, we disclose that 5-substituted-2-furan methanols 1a–1c are subject to highly enantioselective carbonyl allylation, crotylation and reverse prenylation upon exposure to allyl acetate, α-methyl allyl acetate or 1,1,-dimethylallene, respectively, in the presence of an ortho-cyclometallated iridium catalyst modified by (R)-Cl,MeO-BIPHEP, (S)-C3-TUNEPHOS and (R)-C3-SEGPHOS, respectively. Under nearly identical conditions, but in the presence of isopropanol, the corresponding substituted furfurals are converted to identical products of allylation, crotylation and reverse prenylation. Achmatowicz rearrangement of the 5-methyl-2-furan methanol adducts 3b, 4b, 5b is described.9
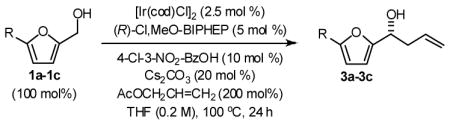
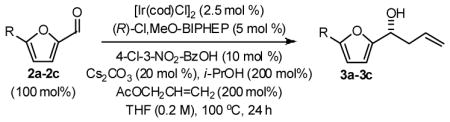
In an initial set of experiments, 5-substituted-2-furan methanols 1a–1c were subjected to conditions for enantioselective iridium catalyzed carbonyl allylation employing an ortho-cyclometallated catalyst generated in situ from [Ir(cod)Cl]2, (R)-Cl,MeO-BIPEHP and 4-chloro-3-nitrobenzoic acid.2a,b,e–h Use of the preformed complex provided comparable yields and selectivities. The products of carbonyl allylation 3a–3c are formed in good isolated yield with uniformly high levels of optical purity using only two equivalents of allyl acetate as the allyl donor (Table 1). Under the same conditions, but in the presence of 2-propanol, the corresponding 5-substituted-2-furfurals 2a–2c are converted to an identical set of carbonyl allylation products 3a–3c in good to excellent isolated yields with uniformly high levels of enantioselectivity. Notably, the parent furans 1a and 2a provide slightly lower yields of the homoallylic alcohol 3a, presumably due to volatility or sensitivity of the furan nucleus with respect to acid promoted degradation during isolation by silica gel chromatography (Table 2).
Table 1.
Enantioselective Carbonyl Allylation of Furan Methanols via Iridium Catalyzed Transfer Hydrogenation.a
 | ||||
|---|---|---|---|---|
| Entry | Furan Methanol | Product | Isolated Yield | ee (%) |
| 1 | R = H, 1a | 3a | 69% | 92 (R) |
| 2 | R = Me, 1b | 3b | 75% | 95 (R) |
| 3 | R=Ar, 1c | 3c | 79% | 94 (R) |
Yields are of material isolated by silica gel chromatography. Enantiomeric excess was determined by chiral stationary phase HPLC analysis. For compound 1c, Ar = 3-chloro-4-methoxyphenyl. See Supporting Information for additional details.
Table 2.
Enantioselective Carbonyl Allylation of Furfurals via Iridium Catalyzed Transfer Hydrogenation.a
 | ||||
|---|---|---|---|---|
| Entry | Furan Methanol | Product | IsolatedYield | ee (%) |
| 1 | R = H, 1a | 3a | 70% | 97 (R) |
| 2 | R = Me, 1b | 3b | 82% | 95 (R) |
| 3 | R = Ar, 1c | 3c | 94% | 97 (R) |
As described for Table 1.
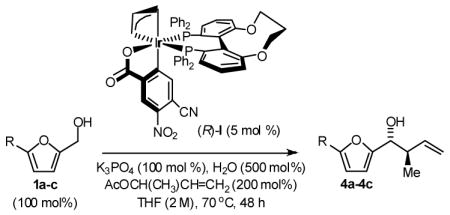
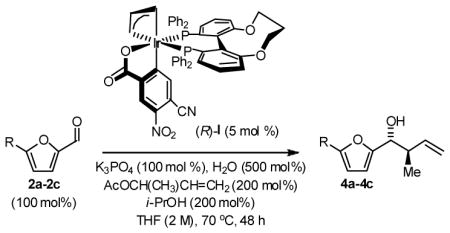
Enantioselective crotylation of 5-substituted-2-furan methanols 1a–1c was explored next.2c,f Here, the preformed ortho-cyclometallated complex generated from [Ir(cod)Cl]2, (S)-C3-TUNEPHOS and 4-cyano-3-nitrobenzoic acid provided enhanced yields and selectivities. A further increase in isolated yield resulted from the use of tribasic potassium phosphate as base in the presence of water. Under these conditions, the 5-substituted-2-furan methanols 1a–1c were converted to the products of crotylation 4a–4c in good to excellent isolated yield with high levels of anti-diastereoselectivity and uniformly high levels of enantioselectivity, as determined by HPLC analysis (Table 3). Under the same conditions, but in the presence of 2-propanol, the corresponding 5-substituted-2-furfurals 2a–2c are converted to an identical set of carbonyl crotylation products 4a–4c with notably enhanced levels of anti-diastereo- and enantioselectivity (Table 4).
Table 3.
Enantioselective Carbonyl Crotylation of Furan Methanols via Iridium Catalyzed Transfer Hydrogenation.a
 | ||||
|---|---|---|---|---|
| Entry | Furan Methanol | Product | Isolated Yield (dr) | ee (%) |
| 1 | R = H, 1a | 4a | 77%, 10:1 | 92 (R,R) |
| 2 | R = Me, 1b | 4b | 76%, 7:1 | 91 (R,R) |
| 3 | R =Ar, 1c | 4c | 95%, 10:1 | 95 (R,R) |
As described for Table 1.
Table 4.
Enantioselective Carbonyl Crotylation of Furfurals via Iridium Catalyzed Transfer Hydrogenation.a
 | ||||
|---|---|---|---|---|
| Entry | Furan Methanol | Product | IsolatedYield(dr) | ee (%) |
| 1 | R = H, 1a | 4a | 72%, >20:1 | 97 (R,R) |
| 2 | R = Me, 1b | 4b | 79%, >20:1 | 94 (R,R) |
| 3 | R = Ar, 1c | 4c | 94%, >20:1 | 99 (R,R) |
As described for Table 1.
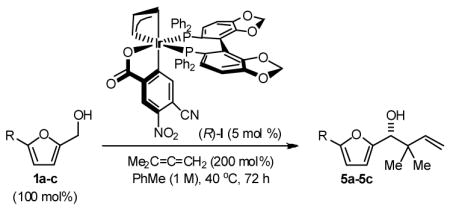
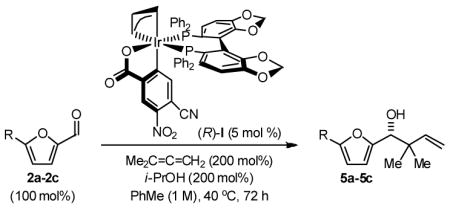
Finally, the enantioselective tert-prenylation of 5-substituted-2-furan methanols 1a–1c was explored using the isolated the ortho-cyclometallated catalyst generated from [Ir(cod)Cl]2, (R)-SEGPHOS and 4-cyano-3-nitrobenzoic acid.2d,f Under remarkably mild conditions, good to excellent isolated yields of 5a–5c were attended by good levels of asymmetric induction (Table 5). Slightly improved isolated yields and enantioselectivities were observed when the tert-prenylated adducts 5a–5c were generated from the corresponding aldehydes 2a–2c (Table 6). Thus, allylation, anti-crotylation and tert-prenylation are achieved from either the alcohol or aldehyde oxidation level with roughly equal facility using 5-substituted-2-furan methanols 1a–1c or 5-substituted-2-furfurals 2a–2c, respectively.
Table 5.
Enantioselective Carbonyl tert-Prenylation Furan Methanols via Iridium Catalyzed Transfer Hydrogenation.a
 | ||||
|---|---|---|---|---|
| Entry | Furan Methanol | Product | Isolated Yield | ee (%) |
| 1 | R = H, 1a | 5a | 82% | 87 (R) |
| 2 | R = Me, 1b | 5b | 84% | 84 (R) |
| 3 | R=Ar, 1c | 5c | 91% | 85 (R) |
As described for Table 1.
Table 6.
Enantioselective Carbonyl tert-Prenylation of Furfurals via Iridium Catalyzed Transfer Hydrogenation.a
 | ||||
|---|---|---|---|---|
| Entry | Furan Methanol | Product | IsolatedYield | ee (%) |
| 1 | R = H, 1a | 5a | 82% | 88 (R) |
| 2 | R = Me 1b | 5b | 89% | 89 (R) |
| 3 | R = Ar, 1c | 5c | 94% | 93 (R) |
As described for Table 1.
The Achmatowicz rearrangement of substituted furan methanols is frequently employing in the total synthesis of natural products.9b Accordingly, the optically enriched products of allylation, crotylation and tert-prenylation 3b, 4b and 5b, respectively, were subjected to conditions for Achmatowicz rearrangement employing N-bromosuccinimide as the oxidant. The corresponding γ-hydroxy-β-pyrones 6a–6c were formed as diastereomeric mixtures at the lactol center. However, negligible erosion of enantiomeric excess was observed at preexisting stereocenters, as determined by chiral stationary phase HPLC analysis (Scheme 1).
Scheme 1.

Achmatowicz rearrangement of optically enriched adducts 3b, 4b and 5b.a
aYields are of material isolated by silica gel chromatography. Enantiomeric excess was determined by chiral stationary phase HPLC analysis.
In summary, we report that 5-substituted-2-furan methanols 1a–1c are subject to enantioselective carbonyl allylation, crotylation and tert-prenylation upon exposure to allyl acetate, α-methyl allyl acetate or 1,1,-dimethylallene in the presence of an ortho-cyclometallated iridium catalyst modified by (R)-Cl,MeO-BIPHEP, (S)-C3-TUNEPHOS and (R)-C3-SEGPHOS, respectively. In the presence of isopropanol, but under otherwise identical conditions, the corresponding substituted furfurals 2a–2c are converted to identical products of allylation, crotylation and tert-prenylation. Optically enriched products carbonyl allylation, crotylation and reverse prenylation 3b, 4b and 5b engage in Achmatowicz rearrangement to furnish the corresponding γ-hydroxy-β-pyrones in good yield and with negligible erosion of enantiomeric excess at the preexisting stereocenter.
Experimental Section
General Procedure for the Preparation of Adducts 3a–3c from Furan Methanols 1a–1c
To a flame dried re-sealable reaction tube purged with argon and containing a magnetic stirrer, was added [Ir(cod)Cl]2 (6.7 mg, 0.010 mmol, 2.5 mol%), (R)-Cl,MeO-BIPHEP (13.0 mg, 0.020 mmol, 5 mol%), 3-nitro-4-chlorobenzoic acid (8.1 mg, 0.040 mmol 10 mol%), Cs2CO3 (26.1 mg, 0.080 mmol, 20 mol%) and the furan methanol (0.40 mmol, 100 mol%). THF (2.0 mL, 0.2 M concentration with respect to the furan methanol) and allyl acetate (86 μL, 0.80 mmol, 200 mol%) were added and the reaction mixture was allowed to stir at 100 °C for 24 hours. The reaction mixture was concentrated in vacuo and purified by flash column chromatography (SiO2) to furnish the corresponding products of allylation 3a–3c.
General Procedure for the Preparation of Adducts 3a–3c from Furfurals 2a–2c
To a flame dried re-sealable reaction tube purged with argon and containing a magnetic stirrer, was added [Ir(cod)Cl]2 (6.7 mg, 0.010 mmol, 2.5 mol%), (R)-Cl,MeO-BIPHEP (13.0 mg, 0.020 mmol, 5 mol%), 3-nitro-4-chlorobenzoic acid (8.1 mg, 0.040 mmol 10 mol%), Cs2CO3 (26.1 mg, 0.080 mmol, 20 mol%) and the furfural (0.40 mmol, 100 mol%). THF (2.0 mL, 0.2 M concentration with respect to the furfural), i-PrOH (61 μL, 0.80 mmol, 200 mol%) and allyl acetate (86 μL, 0.80 mmol, 200 mol%) were added and the reaction mixture was allowed to stir at 100 °C for 24 hours. The reaction mixture was concentrated in vacuo and purified by flash column chromatography (SiO2) to furnish the corresponding products of allylation 3a–3c.
General Procedure for the Preparation of Adducts 4a–4c from Furan Methanols 1a–1c
To a flame dried re-sealable reaction tube purged with argon and containing a magnetic stirrer, was added (R)-Ir-complex I (20.4 mg, 0.020 mmol, 5 mol%), K3PO4 (84.9 mg, 0.40 mmol, 100 mol%), and the corresponding furan methanol (0.40 mmol, 100 mol%). THF (0.2 mL, 2 M concentration with respect to the alcohol), H2O (36 μL, 2.0 mmol, 500 mol%) and α-methyl allyl acetate (86 μL, 0.80 mmol, 200 mol%) were added and the reaction mixture was allowed to stir at 70 °C for 48 hours. The reaction mixture was concentrated in vacuo and purified by flash column chromatography (SiO2) to furnish the corresponding products of crotylation 4a–4c.
General Procedure for the Preparation of Adducts 4a–4c from Furfurals 2a–2c
To a flame dried re-sealable reaction tube purged with argon and containing a magnetic stirrer, was added (R)-Ir-complex I (20.4 mg, 0.020 mmol, 5 mol%), K3PO4 (84.9 mg, 0.400 mmol, 100 mol%), and the corresponding furfural (0.40 mmol, 100 mol%). THF (0.2 mL, 2 M concentration with respect to the aldehyde), H2O (36 μL, 2.0 mmol, 500 mol%), i-PrOH (61 μL, 0.80 mmol, 200 mol%) and α-methyl allyl acetate (86 μL, 0.80 mmol, 200 mol%) were added and the reaction mixture was allowed to stir at 70 °C for 48 hours. The reaction mixture was concentrated in vacuo and purified by flash column chromatography (SiO2), under the conditions noted, to furnish the corresponding products of crotylation 4a–4c.
General Procedure for the Preparation of Adducts 5a–5c from Furan Methanols 1a–1c
To a flame dried re-sealable reaction tube purged with nitrogen and containing a magnetic stirrer, was added (R)-Ir-complex II (5 mol%) and the corresponding furan methanol (100 mol%). Toluene (1 M concentration with respect to the alcohol), 1,1-dimethylallene (200 mol%), and propionaldehyde (5 mol%) were added and the reaction mixture was allowed to stir at 40 °C for 72 hours. The reaction mixture was concentrated in vacuo and purified by flash column chromatography (SiO2) to furnish the corresponding products of tert-prenylation 5a–5c.
General Procedure for the Preparation of Adducts 5a–5c from Furfurals 2a–2c
To a flame dried re-sealable reaction tube purged with nitrogen and containing a magnetic stirrer, was added (R)-Ir-complex II (5 mol%) and the corresponding furfural (100 mol%). Toluene (1 M concentration with respect to the aldehyde), 1,1-dimethylallene (200 mol%), and i-PrOH (200 mol%) were added and the reaction mixture was allowed to stir at 40 °C for 72 hours. The reaction mixture was concentrated in vacuo and purified by flash column chromatography (SiO2) to furnish the corresponding products of tert-prenylation 5a–5c.
General Procedure for the Achmatowicz Rearrangement of Adducts 3b, 4b, and 5b
Alcohols 3b, 4b or 5b (100 mol%) were dissolved in aqueous THF (THF: H2O, 4:1, 0.1 M) and the solution was cooled to 0 °C. N-bromosuccinimide (100 mol%) was added portion-wise while maintaining a temperature of 0 °C. After the reaction had gone to completion as determined by TLC analysis, the reaction mixture was diluted with dichloromethane and washed with KI (10% aqueous solution), Na2S2O4 (15% aqueous solution), and NaHCO3 (10% aqueous solution), and brine. The organic layer was dried (MgSO4), filtered and concentrated in vacuo. The crude residue was purified by flash column chromatography (SiO2) to furnish the corresponding pyrones 6a, 6b or 6c, respectively.
Supplementary Material
Acknowledgments
Acknowledgment is made to the Robert A. Welch Foundation, the ACS-GCI Pharmaceutical Roundtable, and the NIH-NIGMS (RO1-GM069445) for partial support of this research. Dr. Oliver Briel of Umicore is thanked for the generous donation of [Ir(cod)Cl]2. The Landesgraduiertenförderung and the Deutscher Akademischer Austausch Dienst are acknowledged for generous support of Benjamin Bechem.
Footnotes
Supporting Information Available: Spectral data for all new compounds, including scanned images of 1H and 13C NMR spectra. Scanned images of chiral stationary phase HPLC data. Single crystal X-ray diffraction data for the Ir(BIPHEP)(η-C3H5)(C,O-O2C6H3NO2). This material is available free of charge via the internet at http://pubs.acs.org.
References
- 1.For selected reviews on C-C bond forming hydrogenation and transfer hydrogenation, see: Ngai MY, Kong JR, Krische MJ. J Org Chem. 2007;72:1063. doi: 10.1021/jo061895m.Skucas E, Ngai MY, Komanduri V, Krische MJ. Acc Chem Res. 2007;40:1394. doi: 10.1021/ar7001123.Shibahara F, Krische MJ. Chem Lett. 2008;37:1102. doi: 10.1246/cl.2008.1102.Bower JF, Kim IS, Patman RL, Krische MJ. Angew Chem Int Ed. 2009;48:34. doi: 10.1002/anie.200802938.Han SB, Kim IS, Krische MJ. Chem Commun. 2009:7278. doi: 10.1039/b917243m.
- 2.For enantioselective carbonyl allylation, crotylation and reverse prenylation via iridium catalyzed C-C bond forming transfer hydrogenation, see: Kim IS, Ngai MY, Krische MJ. J Am Chem Soc. 2008;130:6340. doi: 10.1021/ja802001b.Kim IS, Ngai MY, Krische MJ. J Am Chem Soc. 2008;130:14891. doi: 10.1021/ja805722e.Kim IS, Han SB, Krische MJ. J Am Chem Soc. 2009;131:2514. doi: 10.1021/ja808857w.Han SB, Kim IS, Han H, Krische MJ. J Am Chem Soc. 2009;131:6916. doi: 10.1021/ja902437k.Lu Y, Kim IS, Hassan A, Del Valle DJ, Krische MJ. Angew Chem Int Ed. 2009;48:5018. doi: 10.1002/anie.200901648.Itoh J, Han SB, Krische MJ. Angew Chem Int Ed. 2009;48:6316. doi: 10.1002/anie.200902328.Lu Y, Krische MJ. Org Lett. 2009;11:3108. doi: 10.1021/ol901096d.Hassan A, Lu Y, Krische MJ. Org Lett. 2009;11:3112. doi: 10.1021/ol901136w.For a recent application in natural product synthesis, see: Harsh P, O’Doherty GA. Tetrahedron. 2009;65:5051. doi: 10.1016/j.tet.2009.03.097.
- 3.For selected reviews on enantioselective carbonyl allylation, see: Yamamoto Y, Asao N. Chem Rev. 1993;93:2207.Ramachandran PV. Aldrichim Acta. 2002;35:23.Kennedy JWJ, Hall DG. Angew Chem Int Ed. 2003;42:4732. doi: 10.1002/anie.200301632.Denmark SE, Fu J. Chem Rev. 2003;103:2763. doi: 10.1021/cr020050h.Yu CM, Youn J, Jung HK. Bull Korean Chem Soc. 2006;27:463.Marek I, Sklute G. Chem Commun. 2007:1683. doi: 10.1039/b615042j.Hall DG. Synlett. 2007:1644.
- 4.For selected reviews of carbonyl allylation based on the reductive coupling of metallo-π-allyls derived from allylic alcohols, ethers or carboxylates, see: Masuyama Y. Palladium-Catalyzed Carbonyl Allylation via π-Allylpalladium Complexes. In: Liebeskind LS, editor. Advances in Metal-Organic Chemistry. Vol. 3. JAI Press; Greenwich: 1994. pp. 255–303.Tamaru Y. Palladium-Catalyzed Reactions of Allyl and Related Derivatives with Organoelectrophiles. In: Negishi E-i, de Meijere A., editors. Handbook of Organopalladium Chemistry for Organic Synthesis. Vol. 2. Wiley; New York: 2002. pp. 1917–1943.Tamaru Y. Novel Catalytic Reactions involving π-Allylpalladium and -Nickel as the Key Intermediates: Umpolung and β-Decarbopalladation of π-Allylpalladium and Nickel-Catalyzed Homoallylation of Carbonyl Compounds with 1,3-Dienes. In: Tsuji J, editor. Perspectives in Organopalladium Chemistry for the XXI Century. Elsevier; Amsterdam: 1999. pp. 215–231.Kondo T, Mitsudo T-a. Curr Org Chem. 2002;6:1163.Tamaru Y. Eur J Org Chem. 2005;13:2647.Zanoni G, Pontiroli A, Marchetti A, Vidari G. Eur J Org Chem. 2007;22:3599.
- 5.For selected examples of carbonyl allylation via catalytic Nozaki-Hiyama-Kishi coupling of allylic halides, see: Fürstner A, Shi N. J Am Chem Soc. 1996;118:2533.Bandini M, Cozzi PG, Umani-Ronchi A. Polyhedron. 2000;19:537.McManus HA, Cozzi PG, Guiry PJ. Adv Synth Catal. 2006;348:551.Hargaden GC, Müller-Bunz H, Guiry PJ. Eur J Org Chem. 2007:4235.Hargaden GC, O’Sullivan TP, Guiry PJ. Org Biomol Chem. 2008;6:562. doi: 10.1039/b715834c.
- 6.Kunze B, Jansen R, Hofle G, Reichenbach H. J Antibiot. 2004;57:151. doi: 10.7164/antibiotics.57.151. [DOI] [PubMed] [Google Scholar]
- 7.For selected examples of enantioselective furfural allylation employing preformed allylmetal reagents, see: Racherla US, Liao Y, Brown HC. J Org Chem. 1992;57:6614.Keck GE, Geraci LS. Tetrahedron Lett. 1993;34:7827.Yanagisawa A, Nakashima H, Ishiba A, Yamamoto H. J Am Chem Soc. 1996;118:4723.Loh TP, Zhou JR. Tetrahedron Lett. 2000;41:5261.Denmark SE, Wynn T. J Am Chem Soc. 2001;123:6199. doi: 10.1021/ja016017e.Denmark SE, Fu J. J Am Chem Soc. 2001;123:6199. doi: 10.1021/ja016017e.Kii S, Maruoka K. Tetrahedron Lett. 2001;42:1935.Yanagisawa A, Nakashima H, Nakatsuka Y, Ishiba A, Yamamoto H. Bull Chem Soc Jpn. 2001;74:1129.Hanawa H, Uraguchi D, Konishi S, Hashimoto T, Maruoka K. Chem Eur J. 2003;9:4405. doi: 10.1002/chem.200305078.Hanawa H, Hashimoto T, Maruoka K. J Am Chem Soc. 2003;125:1708. doi: 10.1021/ja020338o.Xia G, Shibatomi K, Yamamoto H. Synlett. 2004:2437.Kwiatkowski P, Jurczak J. Synlett. 2005:227.Kwiatkowski P, Chaladaj W, Jurczak J. Synlett. 2005:2301.Kwiatkowski P, Chaladaj W, Jurczak J. Tetrahedron. 2006;62:5116.Denmark SE, Fu J, Lawler MJ. J Org Chem. 2006;71:1523. doi: 10.1021/jo052203h.
- 8.For selected review articles on the chemistry of furans, see: Lipshutz BH. Chem Rev. 1986;86:795.Maier Martin E. Nachr Chem. 1993;41:696.Raczko J, Jurczak J. Stud Nat Prod Chem. 1995;16:639.Kappe CO, Murphree SS, Padwa A. Tetrahedron. 1997;53:14179.Rassu G, Zanardi F, Battistini L, Casiraghi G. Chem Soc Rev. 2000;29:109.D’Auria M, Emanuele L, Racioppi R, Romaniello G. Curr Org Chem. 2003;7:1443.Wright DL. Prog Heterocycl Chem. 2005;17:1.Montagnon T, Tofi M, Vassilikogiannakis G. Acc Chem Res. 2008;41:1001. doi: 10.1021/ar800023v.
- 9.Achmatowicz O, Jr, Bukowski P, Szechner B, Zwierzchowska Z, Zamojski A. Tetrahedron. 1971;27:1973.For a review, see: Harris JM, Li M, Scott JG, O’Doherty GA. Strategies Tactics Org Synth. 2004;5:221.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.