

Abstract
Acylation is a frequent means to ensure membrane association of a variety of soluble proteins in living cells. However, many transmembrane proteins are palmitoylated, indicating that this posttranslational modification may also serve as a means to regulate protein trafficking. Based on coarse-grained membrane simulations, we find that protein acylation significantly alters the tilting of transmembrane proteins with respect to the bilayer normal. In addition, the proteins' partitioning behavior and cluster formation ability due to hydrophobic mismatching is strongly altered. Based on our results, we propose that acylation is a potent means to regulate the trafficking of transmembrane proteins along the early secretory pathway.
Introduction
Acylation is a frequent posttranslational modification that transforms soluble proteins into peripheral membrane proteins. Covalently linking, for example, a saturated 16-carbon fatty acid (palmitate) to a cysteine residue (1) promotes the membrane association of a protein (i.e., the palmitate serves as a membrane anchor that penetrates only one leaflet of the lipid bilayer). Other common acylations involve myristyl and farnesyl groups.
Acylation often is a key determinant for the trafficking of peripheral membrane proteins, e.g., in the context of regulating protein transport to nerve terminals and synapses in neurons (2) or when modulating the signaling behavior of oncogenic Ras proteins (3). Indeed, cleavage of a palmitate and subsequent repalmitoylation lead to a cyclic journey of Ras proteins between different cellular organelles, thereby regulating the protein's signaling capacity (4).
Interestingly, many transmembrane proteins are palmitoylated, with their membrane association already ensured by their hydrophobic transmembrane domain (TMD). The role of palmitoylation is here only poorly understood, but has been argued to be a potential tool for protecting transmembrane proteins from the cell's degradation machinery (5,6) or to act as a regulatory mechanism for sorting signals within the cytoplasmic domains (7). A particularly striking example of how palmitoylation can regulate trafficking of transmembrane proteins has been shown very recently for the case of LRP6 (6), a protein that is involved in the secretion of the morphogens Wnt/Wg in mammals and flies, respectively. After translation and clearing the quality control in the endoplasmic reticulum (ER), the transmembrane LRP6 acquires a palmitate that promotes the protein's exit from the ER. Via the Golgi apparatus, LRP6 eventually reaches the cell's plasma membrane. Loss of the palmitoylation lead to retention of LRP6 in the ER. Shortening the TMD of the nonpalmitoylated LRP6 restored the protein's ability to travel to the plasma membrane, whereas palmitoylation in the presence of the shortened TMD led again to a retention in the ER. Based on this data, it was proposed that palmitoylation promotes a tilting of the protein's TMD to reduce the effective hydrophobic mismatch of the TMD of LRP6 with the surrounding lipid bilayer (6).
Hydrophobic mismatch is a central concept for understanding the behavior of transmembrane proteins in different membrane environments (8). If the length of the TMD does not match the thickness of the membrane's hydrophobic core, the local deformation of lipids in the TMD's surrounding can lead to entropy-driven cluster formation (9). A negative mismatch (TMD too short) leads to a local compression of lipids, a positive mismatch (TMD too long) induces a local stretching of lipids. If the mismatch is too large to be compensated by lipid stretching, the protein may tilt relative to the bilayer normal (10). Underlining its impact on biological function, hydrophobic mismatch has been shown experimentally to be an important driving force for protein sorting in vivo (11,12).
Here, we have used coarse-grained membrane simulations to elucidate the role of an acylation, e.g., palmitoylation, of transmembrane proteins. We find that protein acylation significantly alters the orientation of transmembrane proteins (i.e., their tilt with respect to the bilayer normal) as well as their partitioning behavior and cluster formation ability. Based on our numerical results, we propose that (reversible) protein acylation is a key element to regulate the trafficking of transmembrane proteins along the secretory pathway beyond classical sorting strategies like membrane thickness (13) and kin recognition (14).
Method and Model
For the simulations, we have used dissipative particle dynamics (DPD) (15), a coarse-grained simulation technique that lacks the details of molecular dynamics but can cover larger length and timescales. Indeed, in DPD several atoms are combined into effective beads that interact via conservative forces and are driven by a thermostat. In general, two beads i, j at a distance rij = |ri − rj| ≤ r0 interact via a linear repulsive force
(where eij = rij/rij). In this approach, the cutoff length r0 sets the bead size and the basic unit of length for our simulations. The repulsion parameter aij > 0 characterizes the interaction between three types of beads: hydrophobic (T), hydrophilic (H), and water (W) beads. The thermostat is implemented via a dissipative force
(with vij = vi − vj the relative velocity between particles i and j) and a random force
for rij ≤ r0. Here, ξij is a random variable with zero mean and unit variance whereas the parameters γ and σ are related via the fluctuation-dissipation theorem σ2 = 2γ kBT (16).
Larger entities such as lipids and proteins were constructed by connecting individual beads i, j by a harmonic potential
with l0 being the relaxation distance of the spring (17). Linear polymers, e.g., individual lipids, were assigned an additional bending stiffness
All beads were assigned the same mass m, and the mass as well as the thermostat temperature kBT and the interaction cutoff r0 were set to unity; thermostat parameters were γ = 9/2 and σ = 3 in accordance with previous DPD studies (10,15,17–19). In steady state, the chosen parameters yielded a bending energy of the lipid bilayer that is comparable to experimental data. The equations of motion were integrated with a velocity Verlet scheme (time increment Δt = 0.01) using periodic boundary conditions (15). To ensure a tensionless membrane, a barostat was used for the initial 105 timesteps (20). Several observables indicated a proper convergence toward the steady state after this period (Fig. S1, Fig. S2, and Fig. S3 in the Supporting Material). The remaining 106 steps were performed at the fixed equilibrated length of the simulation box. By comparing the membrane thickness and a lipid's diffusion coefficient with experimental data, it is possible to convert DPD units in SI units (r0 ≈ 1 nm, Δt ≈ 80 ps) (19).
Lipids were modeled as semiflexible linear chains of one hydrophilic head (H) and three hydrophobic tail (T) beads, HT3. For homogenous bilayers all lipids were assigned the parameter values kharm = 100 kBT/, l0 = 0.45, and kbend = 10 kBT in accordance with Laradji and Sunil Kumar (18). For inhomogeneous bilayers, 50% of all lipids were assigned a relaxation distance l0 = 0.6 for the Hookean spring, resulting in a second, longer lipid species Ht3 with t denoting the hydrophobic beads of the longer lipid. The repulsion parameters for the various bead types were
| (1) |
The simplest model for a transmembrane protein consisted of a linear HTnH chain of beads with different TMD lengths (n = 4, 5, 6, 7). To one of the head beads (H) a fatty acid chain of m hydrophobic beads was added (Fig. 1 a) as a model for acylation. When considering an H3TnH3 construct, the fatty acid chain was attached to one of the H beads that followed the hydrophobic chain Tn (Fig. 1 a). The acylated forms will be referred to as HTnHA and H3Tn, respectively. In both cases, no bending potential was imposed to achieve a preferential angle θ0 between the acyl chain and the TMD.
Figure 1.

(a) Model of HT5H, H3T5H3, and (HT5H)7 (left to right) proteins with acyl groups of length m = 3 and m = 4, respectively. Hydrophilic head (H) and hydrophobic tail (T) beads are displayed in red and yellow, respectively. The tilt angle φ of the protein with respect to the lipid bilayer and the preferred angle θ0 between the TMD and the acyl chain are indicated on the right. (b) Phase-separated membrane with equal amounts of long (HT3, dark blue and light gray) and short (HT3, gold and light gray) lipids.
To avoid bending and compression of the transmembrane domain especially at the contact zone of the segregated lipid phases, we also considered a more robust construct composed of a hexagonal arrangement of seven HTnH chains, (HTnH)7. With an effective diameter of ∼2r0, i.e., ∼2 nm, this construct may represent well a protein with a stiff α-helical transmembrane domain. As before, the fatty acid modification was a single chain of length m connected to one bead in the upper hydrophilic ring of the protein. Due to the increased TMD stiffness, we also imposed a preferred angle θ0 between the fatty acid and the transmembrane domain with values 20° ≤ θ0 ≤ 90° (Fig. 1 a). For brevity, the acylated construct with θ0 = 90° will be referred to as . Parameters for the connecting Hookean springs in the acyl chain were kharm = 100 kBT/r02, l0 = 0.45, and kbend = 10 kBT.
Results
First, we inspected whether acylation influences the tilting angle φ between the TMD and the bilayer normal. To this end, we considered HTnH, H3TnH3, and (HTnH)7 protein constructs (Fig. 1 a; see also Method and Model). Although HTnH is the simplest possible transmembrane protein, H3TnH3 was used to test to which extent a larger soluble portion can affect the local orientation of the TMD. Due to the DPD simulation setup that is based on soft beads and harmonic springs, HTnH-like proteins may be too flexible to model the behavior of real proteins. We therefore also considered a more rigid, hexagonal protein construct, , with a preferred angle θ0 between the fatty acid and the TMD (Fig. 1 a). In the interest of brevity we report only results for an acylation with θ0 = 90° (construct named in the main text; data for 20° ≤ θ0 < 90° are summarized in Fig. S5).
For all protein constructs, we studied the temporal variation of the tilting angle φ with and without acylation in a homogenous lipid bilayer of HT3 lipids. After equilibration (105 time steps; see also Supporting Material) the tensionless membrane patch (edge length L = 10–15r0) had an average bilayer thickness h = 3.8r0 and φ was monitored for 106 time steps. Representative distributions p(φ) obtained from the time series with and without acylation for n = 7 are shown in Fig. 2 a. As a result, we observed that, in all cases, the width of p(φ) was similar; however, the mean tilting angle 〈φ〉 was shifted toward larger values for acylated proteins. Notably, p(φ) for nonacylated HTnH, H3TnH3, and (HTnH)7 were very similar and had an almost identical mean 〈φ〉.
Figure 2.
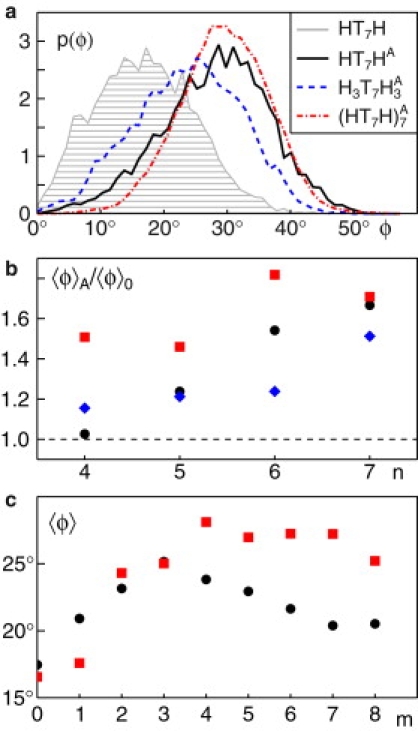
(a) The probability distributions of tilt angles, p(φ), for acylated proteins ((HT7HA, H3T7HA3, and (HT7H)A7; solid black, dashed blue, and dash-dotted red lines) show a strong shift toward larger angles in comparison to a representative nonacylated protein (HT7H; gray shaded). (b) The ratio of average tilt angles with and without acylation, 〈φ〉A and 〈φ〉0, highlights an increased tilting of HTnH (black circles), H3TnH3 (blue diamonds), and (HTnH)7 (red squares) after acylation. Acyl chains had a length m = 3 for HTnH and H3TnH3, whereas m = 4 and θ0 = 90° was chosen for (HTnH)7. In all cases, an increased tilting due to acylation, i.e., a ratio above unity, is clearly visible. (c) The average tilt angles 〈φ〉 of HT7HA (black circles) and (HT7H)A7 (red squares) vary with the length m of the attached acyl chain. For HT7HA a clear maximum is observed at m = 3, but the peak for at m = 4 is much less pronounced.
Consistent with our earlier results (10), we found very small tilting angles 〈φ〉 ≤ 10° for all nonacylated protein constructs in the case of a vanishing or negative hydrophobic mismatch (n ≤ 5). A positive hydrophobic mismatch (n > 5) leads to an increase of 〈φ〉 (Fig. S4). In both regimes, however, tilting was enhanced upon acylation. This is best seen by the ratio of tilt angles for acylated and nonacylated constructs, 〈φ〉A/〈φ〉0, which is consistently above unity and even increases for growing hydrophobic mismatches (Fig. 2 b).
We next studied the influence of the length m of the acyl chain on the tilting angle. We therefore varied m = 1,…,8 and monitored the average tilting angle 〈φ〉 of the TMD for HT7HA and . As can be seen from Fig. 2 c, both constructs showed an increased tilting when m was increased. Although HT7HA showed a maximum effect for m = 3, this peak was less pronounced and shifted to m = 4 for (HT7H)A7. Thus, tilting is maximally enhanced when the acyl chain has a length that roughly matches the thickness of one leaflet of the surrounding bilayer.
Having seen that protein acylation strongly influences how a transmembrane protein is situated in a lipid bilayer, we aimed at probing how hydrophobic mismatch-driven clustering of transmembrane proteins is affected by protein acylation. To this end we determined the radial distribution function g(r) on a tensionless membrane patch of size L ∼30r0 with nine proteins (HTnH or (HTnH)7; see Fig. 1 a). From g(r) we also determined the number of neighboring proteins, Nc, within a distance ≤ 2r0. As a result, we observed that g(r) frequently showed a strong peak for small distances (Fig. 3 a), which indicates clustering. For a vanishing hydrophobic mismatch (n = 5), cluster formation was hardly observed, i.e., Nc < 1 for all considered proteins. For n > 5, cluster formation due to hydrophobic mismatching was well observed for (HTnH)7. In addition, HTnH showed a significant, yet less pronounced clustering (Fig. 3 b). In general, addition of an acyl chain caused abandoning of cluster formation. Only (HT7H)7 showed a slight increase of Nc. A reduced ability for cluster formation was also seen for (HTnH)7 on inhomogeneous membranes with two domains of different thickness (see also below). Thus, cluster formation due to hydrophobic mismatching is, in general, reduced via acylation.
Figure 3.

(a) Representative radial distribution function g(r) for (HTnH)7 with n = 6 (dashed) and n = 7 (solid). Acylated proteins (red lines) show a lower tendency to form clusters than their nonacylated counterparts (black lines). (b) Average number of neighboring proteins Nc for nonacylated (black symbols) and acylated (red symbols) proteins (with HTnH and (HTnH)7 shown as circles, slightly shifted laterally for better visibility, and squares, respectively). Increasing the hydrophobic mismatch by extending the TMD length n yields an increase in Nc for both proteins. Upon acylation, cluster formation is, in general, abandoned. Open squares (shifted laterally for better visibility) indicate values found for (HTnH)7 (black) and (HTnH)7A (red) in an inhomogeneous bilayer (see Fig. 1b). Again, a clear suppression of cluster formation is observed. (c) Fraction of short lipids, fs, surrounding (HTnH)7 (black) and (HTnH)7A (red) within a distance ≤ 3r0 in an inhomogeneous bilayer. Although the decrease of fs for (HTnH)7 highlights a preferred partitioning into the domain with the least hydrophobic mismatch, acylation yields a nearly uniform sampling of both domains, as evidenced by fs → 50%.
To explore the possible role of acylation on the partitioning behavior of transmembrane proteins in inhomogeneous membranes, we implemented a model membrane that consisted of two lipid species that differed in length (see Method and Model). These membranes show a spontaneous segregation of lipids with a coexistence of two lipid phases of different thickness (Fig. 1 b). The membrane thickness in the two phases was measured to be 4r0 and 5.5r0, respectively. As the simple HTnH model protein proved to be unnaturally compressible and prone to bending especially when being subject to the forces at the domain border, we only used the protein with an acyl-chain length m = 4 and its nonacylated counterpart.
Several studies have highlighted that a hydrophobic mismatch drives partitioning of a protein to the lipid phase with the least mismatch (9–11). Indeed, we observed that proteins with a short TMD did spend more time in the thinner domain, whereas long proteins preferred regions with longer lipids. To quantify this effect, we monitored the average fraction fs of short lipids (HT3) in a distance ≤3r0 around the protein (Fig. 3 c). In agreement with the notion that longer TMDs preferentially localize to the thicker domain of the bilayer, we observed a decrease of fs for the nonacylated constructs. This strong partitioning was considerably softened upon acylation, i.e., fs tended toward 50%, hence allowing the protein to explore both domains equally.
We also measured the radial distribution function of proteins in the inhomogeneous bilayer. In similarity to the case of bilayers with a homogenous thickness, acylation here also decreased the clustering ability of the proteins (i.e., Nc decreased (Fig. 3 b)).
Discussion
Using DPD as a simulation tool, we have found that acylation of transmembrane proteins significantly alters the tilting of the TMD with a clear dependence on the TMD length. Moreover, the presence of a fatty acid modification was found to reduce hydrophobic mismatch-induced clustering on homogenous and inhomogeneous lipid bilayers. In addition, acylated transmembrane proteins showed a significantly different partitioning behavior on phase-separated bilayers with regions of different thickness: whereas nonacylated proteins always partitioned into the phase that matched best the length of their TMD, the addition of a fatty acid chain significantly increased the probability of a protein to also visit regions in which the TMD experiences a stronger hydrophobic mismatch.
In our simulations, we have used two models for transmembrane proteins, HTnH and (HTnH)7, which varied in the rigidity and diameter of the TMD. Both models showed similar phenomena, with some changes in the actual numbers, hence underlining that the observed effects of acylation are fairly robust and not very specific to the chosen model. The higher flexibility of the single-chain construct HTnH, however, may be inconsistent with the expected rigidity of natural α-helical TMDs, which rely very much on extensive hydrogen bonding. We therefore consider the (HTnH)7 construct to be a more realistic model.
The results and predictions of our simulations may be tested directly using artificial membranes (e.g., supported bilayers or giant unilamellar vesicles) and purified transmembrane peptides with a designed TMD length and acylation state. The predicted partitioning behavior of (acylated) proteins may be studied, for example, by simple time-lapse fluorescence microscopy on phase-separating ternary lipid mixtures. The inhibition of cluster formation due to hydrophobic mismatching may be studied on homogenous membranes with fluorescence resonance energy transfer, which can test directly the oligomeric state of fluorescently labeled proteins. An alternative would be fluorescence (cross) correlation spectroscopy, which relies on the change in the (co-)diffusional mobility due to oligomerization.
In addition to these biophysically testable predictions, our numerical results also support the notion that acylation can be used as a potent means to regulate the transport behavior of transmembrane cargo proteins. Using acylation, cells may prevent, for example, hydrophobic mismatch-induced clustering of transmembrane proteins, hence regulating their localization along the early secretory pathway. Given that clustering of transmembrane cargo molecules has indeed been shown to modulate the turnover kinetics of the COPI and COPII vesicle machineries (21–23), we therefore predict that acylation counteracts the sorting of transmembrane proteins into transport vesicles. This prediction may be tested by adding palmitoylation sites to simple transmembrane peptides with a well-characterized length of the TMD. The transport behavior of such tailored cargo proteins has been studied in detail (11), and therefore provides a suitable approach for testing our prediction. This prediction, however, assumes that ER membranes are fairly homogenous in thickness, i.e., proteins do not partition diffusively into a domain with the least hydrophobic mismatch. Owing to the complexity of ER membranes, the existence of thinner and thicker domains is conceivable. In this case, acylation actually may promote ER export above a basal level by allowing proteins to explore the other domains more easily. Which of the two scenarios is applicable can only be decided by inspecting the particular protein construct. In addition, the lipid composition of the ER may be altered by affecting the sterol contents (24). This caveat is also supported by recent data on LRP6 (6) which, at first glance, relate the TMD length and acylation state to its transport behavior. The reported experimental data highlight an additional ER retention mechanism via ubiquitination of some, but not all of the studied LRP6 mutants. It is hence well anticipated that additional cellular factors will influence the transport of (acylated) transmembrane proteins, yet the effects described here are very likely a crucial ingredient that can tip the balance toward the desired trafficking behavior.
Supporting Material
Five figures are available at http://www.biophysj.org/biophysj/supplemental/S0006-3495(09)01742-1.
Supporting Material
Acknowledgments
Support by Deutsche Forschungsgemeinschaft grant No. WE 4335/2-1 and the Institute for Modeling and Simulation in the Biosciences in Heidelberg is gratefully acknowledged.
References
- 1.Bijlmakers M.J., Marsh M. The on-off story of protein palmitoylation. Trends Cell Biol. 2003;13:32–42. doi: 10.1016/s0962-8924(02)00008-9. [DOI] [PubMed] [Google Scholar]
- 2.el-Husseini Ael.-D., Bredt D.S. Protein palmitoylation: a regulator of neuronal development and function. Nat. Rev. Neurosci. 2002;3:791–802. doi: 10.1038/nrn940. [DOI] [PubMed] [Google Scholar]
- 3.Rocks O., Peyker A., Bastiaens P.I. An acylation cycle regulates localization and activity of palmitoylated Ras isoforms. Science. 2005;307:1746–1752. doi: 10.1126/science.1105654. [DOI] [PubMed] [Google Scholar]
- 4.Goodwin J.S., Drake K.R., Kenworthy A.K. Depalmitoylated Ras traffics to and from the Golgi complex via a nonvesicular pathway. J. Cell Biol. 2005;170:261–272. doi: 10.1083/jcb.200502063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Linder M.E., Deschenes R.J. Palmitoylation: policing protein stability and traffic. Nat. Rev. Mol. Cell Biol. 2007;8:74–84. doi: 10.1038/nrm2084. [DOI] [PubMed] [Google Scholar]
- 6.Abrami L., Kunz B., van der Goot F.G. Palmitoylation and ubiquitination regulate exit of the Wnt signaling protein LRP6 from the endoplasmic reticulum. Proc. Natl. Acad. Sci. USA. 2008;105:5384–5389. doi: 10.1073/pnas.0710389105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stöckli J., Rohrer J. The palmitoyltransferase of the cation-dependent mannose 6-phosphate receptor cycles between the plasma membrane and endosomes. Mol. Biol. Cell. 2004;15:2617–2626. doi: 10.1091/mbc.E03-11-0808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jensen M.O., Mouritsen O.G. Lipids do influence protein function-the hydrophobic matching hypothesis revisited. Biochim. Biophys. Acta. 2004;1666:205–226. doi: 10.1016/j.bbamem.2004.06.009. [DOI] [PubMed] [Google Scholar]
- 9.Mouritsen O.G., Bloom M. Mattress model of lipid-protein interactions in membranes. Biophys. J. 1984;46:141–153. doi: 10.1016/S0006-3495(84)84007-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schmidt U., Guigas G., Weiss M. Cluster formation of transmembrane proteins due to hydrophobic mismatching. Phys. Rev. Lett. 2008;101:128104. doi: 10.1103/PhysRevLett.101.128104. [DOI] [PubMed] [Google Scholar]
- 11.Ronchi P., Colombo S., Borgese N. Transmembrane domain-dependent partitioning of membrane proteins within the endoplasmic reticulum. J. Cell Biol. 2008;181:105–118. doi: 10.1083/jcb.200710093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Munro S. An investigation of the role of transmembrane domains in Golgi protein retention. EMBO J. 1995;14:4695–4704. doi: 10.1002/j.1460-2075.1995.tb00151.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bretscher M.S., Munro S. Cholesterol and the Golgi apparatus. Science. 1993;261:1280–1281. doi: 10.1126/science.8362242. [DOI] [PubMed] [Google Scholar]
- 14.Nilsson T., Slusarewicz P., Warren G. Kin recognition. A model for the retention of Golgi enzymes. FEBS Lett. 1993;330:1–4. doi: 10.1016/0014-5793(93)80906-b. [DOI] [PubMed] [Google Scholar]
- 15.Nikunen P., Karttunen M., Vattulainen I. How would you integrate the equations of motion in dissipative particle dynamics simulations? Comput. Phys. Commun. 2003;153:407–423. [Google Scholar]
- 16.Español P., Warren P. Statistical mechanics of dissipative particle dynamics. Europhys. Lett. 1995;30:191. [Google Scholar]
- 17.Shillcock J.C., Lipowsky R. Equilibrium structure and lateral stress distribution of amphiphilic bilayers from dissipative particle dynamics simulations. J. Chem. Phys. 2002;117:5048–5061. [Google Scholar]
- 18.Laradji M., Sunil Kumar P.B. Dynamics of domain growth in self-assembled fluid vesicles. Phys. Rev. Lett. 2004;93:198105. doi: 10.1103/PhysRevLett.93.198105. [DOI] [PubMed] [Google Scholar]
- 19.Guigas G., Weiss M. Size-dependent diffusion of membrane inclusions. Biophys. J. 2006;91:2393–2398. doi: 10.1529/biophysj.106.087031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jakobsen A.F. Constant-pressure and constant-surface tension simulations in dissipative particle dynamics. J. Chem. Phys. 2005;122:124901. doi: 10.1063/1.1867374. [DOI] [PubMed] [Google Scholar]
- 21.Lanoix J., Ouwendijk J., Nilsson T. Sorting of Golgi resident proteins into different subpopulations of COPI vesicles: a role for ArfGAP1. J. Cell Biol. 2001;155:1199–1212. doi: 10.1083/jcb.200108017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Weiss M., Nilsson T. A kinetic proof-reading mechanism for protein sorting. Traffic. 2003;4:65–73. doi: 10.1034/j.1600-0854.2003.40202.x. [DOI] [PubMed] [Google Scholar]
- 23.Forster R., Weiss M., Pepperkok R. Secretory cargo regulates the turnover of COPII subunits at single ER exit sites. Curr. Biol. 2006;16:173–179. doi: 10.1016/j.cub.2005.11.076. [DOI] [PubMed] [Google Scholar]
- 24.Runz H., Miura K., Pepperkok R. Sterols regulate ER-export dynamics of secretory cargo protein ts-O45-G. EMBO J. 2006;25:2953–2965. doi: 10.1038/sj.emboj.7601205. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.